
Abstract
Introduction
p62 Is a Substrate for Selective Autophagy
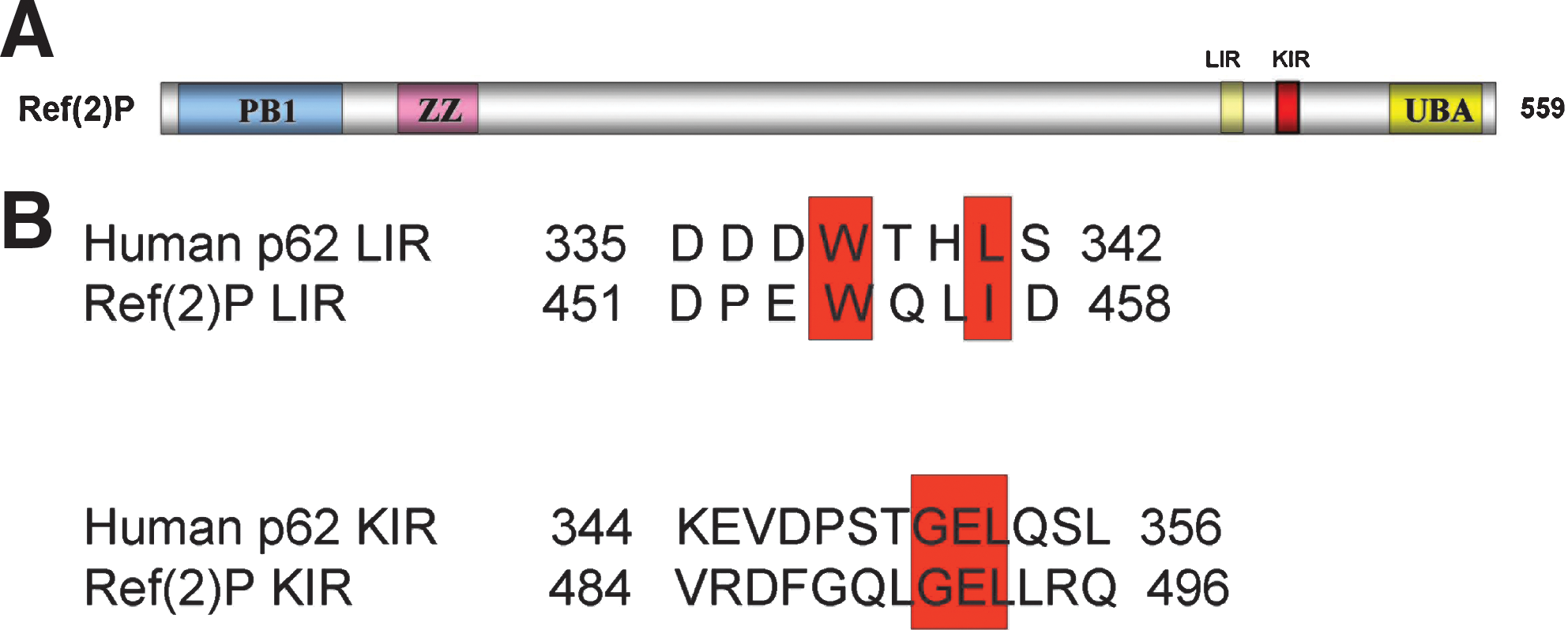
The human p62 protein has 440 amino acid residues and contains several functional motifs (12) (Fig. 1). At the N-terminus, it contains a Phox and Bem1p (PB1) domain, which is responsible for di- and multi-merization of the protein as well as interaction with the structurally related autophagy receptor neighbor of Brca1 gene (NBR1) and the protein kinases extracellular signal-regulated kinase (ERK), mitogen-activated protein kinase kinase kinase 3 (MEKK3), mitogen-activated protein extracellular-signal-regulated kinase kinase 5 (MEK5), ζ (zeta) isotype of protein kinase C (PKCζ), and λ/ι (lamda/iota) isotype of protein kinase C (PKCλ/ι) (12, 28, 29). The PB1 domain is followed by a ZZ-type zinc finger domain, which contains the binding site for receptor-interacting serine-threonine kinase 1 (RIP1) (28, 29) and a TNF receptor-associated factor 6 (TRAF6) binding domain (TB) domain, which contains the binding site for the E3 ubiquitin-protein ligase TRAF6 (28, 29). Nuclear localization signals and nuclear export signal are also present (35). Importantly, p62 harbors a LC3 interacting region (LIR) and a KEAP1 interacting region (KIR) motif that are responsible for the interaction with human microtubule-associated protein 1 light chain 3 (LC3) and Kelch-like ECH-associated protein 1 (KEAP1), respectively (10, 12, 19). The C-terminus of p62 contains a ubiquitin-associated (UBA) domain required for its binding to mono- and poly-ubiquitin (2, 12) (Fig. 1). The Drosophila single p62 homolog, Ref(2)P (refractory to Sigma P, ref(2)P/CG10360), was characterized in a screen for modifiers of sigma virus multiplication (3). Ref(2)P has been shown to interact with Drosophila atypical protein kinase C and participate in the Toll-signaling pathway (1, 13). Ref(2)P is 599 amino acids long and contains an N-terminal PB1 domain followed by a ZZ-type zinc finger domain and a C-terminal UBA domain. Bioinformatics analysis of the sequence of Ref(2)P reveals the presence of putative LIR and KIR motifs, but their functional role has to be experimentally tested in vitro and in vivo (Fig. 2). We have shown that Ref(2)P is a major component of protein aggregates formed during normal aging in Drosophila adult brain (31). Ref(2)P was also shown to be a major component of protein aggregates in flies that are defective in autophagy, in flies that have impaired proteasomal function, and in Drosophila models of human neurodegenerative diseases (31) (Fig. 3). Both the abilities of Ref(2)P to oligo- and multimerize (through its PB1 domain) and to bind ubiquitinated proteins (through its UBA domain) were shown to be required during the in vivo formation of protein aggregates in the adult brain of Drosophila (31). Given the fact that Drosophila is an easily genetically modifiable model organism and that it is an excellent model for higher animals, further studies should be implemented to investigate the in vivo roles of Ref(2)P.


p62 is a component of inclusion bodies found in protein aggregation diseases in the brain and liver, and p62 knock-out mice exhibit a phenotype of neurodegeneration (36). p62 sequesters into cytoplasmic inclusion bodies, called sequestosomes, which are not surrounded by a membrane (2). Importantly, p62 localizes into membrane-confined autophagosomal and lysosomal structures (2). Additionally, p62 localizes in distinct nuclear bodies (2). The formation of p62 bodies was shown to be dependent on both the ubiquitin-binding ability of the UBA domain and the polymerizing ability of the PB1 domain. The autophagic marker protein LC3 was shown to localize to p62 bodies, and LC3 was shown to co-immunoprecipitate with p62 (2). In a following study, it was found that p62 directly binds to LC3, via its LIR domain, which spans between amino acids 321 and 342 of the human p62 protein. The LIR domain of p62 was shown to be responsible for autophagic degradation of p62 bodies and the constitutive autophagic degradation of ubiquitinated proteins (12). Similar results were reported for murine p62 protein where the same LC3 interacting motif was identified and was named LRS (LC3 recognition sequence). Its domain extending between amino acids 334 and 344 was found to be sufficient for binding to LC3 (14).
In a recent study, it was reported that self-oligomerization of p62 is important for its localization to the autophagosome formation site that is associated to the endoplasmic reticulum (ER) and that this process occurs independently of LC3 (16). The authors tested whether LC3 is required for the association of p62 with autophagic membranes using mouse embryonic fibroblasts. They found that p62 is recruited to early autophagic structures independentently of LC3. p62 localized in ER-associated autophagosomal formation sites and colocalized with serine/threonine protein kinase unc51-like kinase 1, mammalian homolog of Atg1 (ULK1) and autophagy related protein 14 (ATG14) at these sites. This localization was shown to occur independently of the 200 kDa family kinase interacting protein FIP200. Importantly, it was shown that the PB1 domain of p62 and its self-oligomerization is important for its localization to the autophagosome formation site. A model was proposed in which oligomerized p62 molecules are targeted to the autophagosome formation sites and are consequently incorporated into the formed autophagosomes via p62's interaction with LC3 (16).
The presence of p62 as a component in most of the protein aggregates/inclusion bodies found in human diseases (12), its striking accumulation when autophagy is inhibited, and its ability to bind mono- and poly-ubiquitin suggests a model where p62 first serves as a collector of presumably not functional ubiquitinated proteins and consequently as an adaptor for targeting them for autophagic degradation. These molecules can form aggregates via p62's ability to multimerize through its PB1 domain. These aggregates can be targeted for autophagic degradation through both LIR and PB1 domains. Interestingly, p62 was recently reported to be required for the autophagic clearance of a nonubiquitinated substrate as well (45). Therefore, p62 appears to be important for the clearance of proteins through autophagy and consists of the link between autophagy and protein degradation.
p62 Is an Emerging Regulator of NRF2 KEAP1 Oxidative Stress Signaling
The KEAP1–NRF2 pathway plays a central role in the protection of the cell under oxidative and electrophilic stresses. The transcription factor NRF2 induces the expression of many cytoprotective genes (43). Target genes of NRF2 are involved in (i) glutathione synthesis (glutamate-cysteine ligase), (ii) elimination of reactive oxygen species (ROS) (thioredoxin reductase 1, peroxiredoxin 1), (iii) detoxification of xenobiotics (NAD(P)H dehydrogenase quinone 1, glutathione S-transferase gene family), and (iv) drug transport (multidrug resistance-associated protein gene family) (43). KEAP1 is an essential regulator of NRF2 activity. Under normal conditions, KEAP1 directly interacts with the NRF2-ECH homology domain 2 (Neh2) domain located in the N-terminal region of NRF2 (43). KEAP1 also associates with Cullin 3 to form an ubiquitin E3 ligase complex, in which KEAP1 serves as the substrate adaptor. KEAP1 mediates polyubiquitination of NRF2 by the Cullin3-KEAP1 ubiquitin E3 ligase complex. This results in constitutive degradation of NRF2 by the proteasome (43). KEAP1 contains multiple reactive cysteines whose reactive thiols have been shown to be modified by several electrophilic reagents (18, 43). Modified cysteines of KEAP1 impair the structural integrity of the KEAP1–Cullin3 E3 ligase complex, resulting in reduced ubiquitination and proteasomal degradation of NRF2. Thus, in the presence of electrophiles or ROS, NRF2 accumulates in nuclei, where it heterodimerizes with small avian musculoaponeurotic fibrosarcoma protooncogene (MAF) proteins and activates target genes for cytoprotection through antioxidant response element (ARE) / electrophile response element (17, 43). Importantly, it has been shown that NRF2 and KEAP1 have been found to be frequently mutated in various cancer cases, such as lung, head, neck, and gallbladder cancers (32, 33, 40 –42) (Table 1). These mutations abolish the NRF2–KEAP1 interaction. This results in constitutive activation of NRF2 followed by the induction of its target genes that help the tumor cells to resist oxidative stress and anticancer agents. Interestingly, it was also shown that expression of the oncoproteins K-RasG12D, B-RafV619E, and myelocytomatosis viral oncogene (Myc)ERT2 activates NRF2, lowers intracellular ROS, and thereby mediates tumorigenesis (5).
KEAP1 and NRF2 have been found mutated in several human cancer samples. p62 has been found to accumulate in human cancer samples. See the main text for further details.
KEAP1, Kelch-like ECH-associated protein 1; NRF2, NF-E2-related factor 2.
Intriguingly, several groups have recently reported an unexpected role for p62 in the NRF2/KEAP1 pathway. p62 was shown to interact with KEAP1 at the NRF2 binding site, thereby competing with KEAP1 binding and thus promoting NRF2 release from KEAP1 and inducing antioxidant gene expression (4, 8, 10, 19, 20). Komatsu and colleagues in a comprehensive study demonstrated that the DGR and CTR domains of KEAP1 (DC domain of KEAP1), which contain the double glycine repeat or kelch repeat (DGR, residues 315–598) and the carboxy terminal region (CTR, residues 599–624), are responsible for interaction with p62 (19). The KEAP1 DC domain is shown to be essential for the interaction between KEAP1 and the Neh2 domain of NRF2 (27). They also showed that p62's amino acids residues 345–359 are essential and sufficient for the interaction between p62 and KEAP1-DC and named this domain as the KEAP1 interacting region (KIR) (19). Zhang's group further reported that amino acids 349–354 are sufficient for the interaction between p62 and KEAP1 (20). KIR is located close to the C terminus of p62 and adjacent to LIR/LRS (12, 14). p62 binding to LC3/GABAA receptor–associated protein (GABARAP) family proteins can be completed by KEAP1 in vitro, suggesting that in a single p62 molecule, binding to either the LIR or the KIR is mutually exclusive (10). However, since p62 can homodimerize and polymerize itself, both KEAP1 and LC3 may bind to p62 homodimer or polymer (12). KEAP1 was found to be sequestered in ubiquitin-positive inclusion bodies and autophagosomes in a p62-dependent manner (10). Interestingly, Johansen's group further reported that p62 is a target gene for NRF2. The p62 promoter contains an ARE where NRF2 binds and activates the transcription of the p62 gene. This creates a positive feedback loop where p62 expression is regulated by NRF2, and p62, in turns, regulates NRF2 stabilization and degradation (10). Therefore, p62 regulates its own transcription by controlling NRF2 activation. These results open new avenues for elucidating the role of p62 in oxidative stress signaling. On the other hand, starvation has been reported to induce ROS formation, which, in turns, triggers autophagy (39). This creates a negative feedback whereby, under conditions of starvation, ROS formation stimulates autophagy, which will result in p62 degradation and attenuation of NRF2 activation.
p62 Is an Emerging Regulator of Tumorigenesis
p62 has important physiological roles in mammals. The first indication of its physiological function is related to Paget's disease of the bone, a disease characterized by increased bone remodeling at discrete sites throughout the cytoskeleton, and is based on the finding that p62 contains several mutations associated with this disease (21). Analyses of mice lacking p62 show that it regulates osteoclastogenesis and bone homeostasis, through its binding partner TRAF6 by activating the transcription factor nuclear factor-kappa-B (NF-κB) (6). Additionally, p62 knock-out mice develop late-onset obesity that leads to impaired glucose tolerance and insulin resistance (37). p62 is also reported to regulate ERK1 in metabolism (23). In addition to these physiological roles, there is accumulating evidence that p62 also has a role in tumorigenesis (28). p62 mRNA levels are significantly increased in several tumor types, especially human lung cancers, as indicated by expression microarray data from tumor samples (7). p62 protein levels are also elevated in cells transformed by an activated mutant of the Harvey Rat Sarcoma gene (H-Ras) oncoprotein, which is a strong driver of tumorigenesis (7). Depletion of p62 inhibits Ras-induced cell transformation in cell cultures and in a mouse model of Ras-induced lung carcinogenesis due to the impaired activation of NF-κB (7). Additionally, it was reported that metabolic stress combined with autophagy deficiency and p62 deregulation results in oxidative stress and increased tumorigenicity (26). High levels of p62 due to autophagy defects is sufficient to alter NF-κB regulation and gene expression and to promote tumorigenesis. Therefore, the fact that p62 seems to be an activator of NF-κB, which is known to play a critical role in proinflammatory signaling and cancer, may explain the role of p62 in tumorigenesis.
Conversely, p62 may also play a role in tumor suppression through its control of cell death and cell cycle progression. p62 was shown to form cytosolic signaling speckles that regulate caspase-8 activation (11). When the proapoptotic ligand TNF-related apoptosis-inducing ligand (TRAIL) binds to its death receptors 4/5 (DR4/5), it recruits the adaptor protein FAS-associated protein with death domain (FADD). FADD, in turns, recruits caspase-8. Caspase-8 is then poly-ubiquitinated by the neddylated form of the E3 ubiquitin ligase Cullin3. Once poly-ubiquitinated, caspase-8 recruits p62, which regulates caspase-8 activation (11). Enhanced caspase-8 oligomerization and activation were also reported to be promoted through its interaction with p62 and LC3 upon or after proteasomal inhibition (34). It is worth mentioning that it has been shown that formation of aggregates containing p62, TRAF6 and the interleukin-1 receptor associated kinase IRAK controls the activation of NF-κB (38). p62's ability to be sequestered or organize different functional signaling aggregates will, therefore, determine whether the cell survives or dies. Since p62 is degraded by autophagy, the availability of p62 protein pools would be crucial for controlling the formation of signaling aggregates/hubs under normal or abnormal conditions as, for example, cancer.
Recent data by Moscat's group highlight a previously unknown role of p62 in cell cycle regulation. The authors showed that p62 is phosphorylated at early mitosis by the crucial cell cycle regulator cyclin dependent kinase 1 (CDK1) at threonine-269 and serine-272 (24). Additionally, p62 interacts with CDK1. When these phosphorylation sites were mutated and the phosphorylation-defective mutant p62 protein expressed in cells, an altered G2/M transition was observed. Phosphorylation of p62 was shown to be required for the maintenance of appropriate cyclin B1 levels and the levels of CDK1 necessary to allow cells to properly enter and exit mitosis. Importantly, lack of p62 phosphorylation by CDK1 leads to enhanced cell proliferation and tumorigenesis in Ras-transformed cells. Phosphorylation of p62 at threonine-269 and serine-272 has also been implicated in nuclear-cytoplasmic shuttling of p62 and its localization to promyelocytic leukemia nuclear body (PML) bodies, which are nuclear sites of proteolysis (35). Interestingly, it is known that CDK1 and cyclin B1 form an active complex within the cytoplasm, which is translocated into the nucleus during late prophase (9). It is tempting to speculate that p62 could regulate CDK1/cyclin B1 degradation in PML bodies. Taken together, the data just cited suggest that p62 is an emerging regulator of cell survival and cell cycle.
The literature analyzed so far clearly demonstrates that p62 has an important role in regulation of autophagy, oxidative stress signaling, and tumorigenesis. However, the interplay between these processes has been elusive. Recent work by Mizushima's and Komatsu's groups has added an important aspect about the role of p62 in tumorigenesis in the context of autophagy and oxidative stress. They showed that hepatocellular carcinoma cells exhibit accumulation of p62, which results in persisting activation of NRF2 (15, 44). The authors initially found that liver-specific autophagy related protein 5 (ATG5) or autophagy related protein 7 (ATG7) deficiency causes the development of multiple tumors in the livers of mutant mice. This phenomenon was associated with age, as only old mice developed tumors. Autophagy mutant mice started to develop small tumors only after six to nine months of age. The number and size of tumors increased with age, and the liver was occupied by multiple tumors after 19 months of age. These tumors exhibited no sign of malignancy and were diagnosed as benign tumors (adenomas) (15, 44). Histological analyses of these tumors showed the presence of swollen and deformed mitochondria and genomic instability markers. ATG7-deficient livers showed accumulation of p62 into inclusion bodies that also contained KEAP1. This p62 accumulation resulted in the activation of NRF2 and the increased expression of NRF2 target genes, such as NAD(P)H dehydrogenase quinone 1 and glutathione S-transferase mu 1 (15). p62 and KEAP1 double-positive aggregates were also found in human liver disorders, including hepatitis, liver cirrhosis, and hepatocellular carcinomas. Quantitative analysis showed that 25% of hepatocellular human carcinomas contained p62 and KEAP1 positive aggregates and that there was a significant induction of the NRF2 target gene NAD(P)H dehydrogenase quinone 1 in 8 out of 15 hepatocellular carcinomas examined, showing that p62 accumulation is associated with NRF2 activation (15) (Table 1). Furthermore, deletion of p62 suppressed anchorage-independent growth in a hepatocellular carcinoma cell line and aggregate formation. Taken together, these results indicate a role of p62 in tumor development by NRF2 activation.
NRF2 activation has recently been found to be driven by the expression of the oncogenes K-RasG12D, B-RafV619E, and MycERT2 (5). Additionally, NRF2 and KEAP1 mutations found in various cancer cases (32, 33, 40 –42) (Table 1) abolish the NRF2–KEAP1 interaction, resulting in constitutive activation of NRF2 followed by the induction of its target genes, which help the tumor cells resist oxidative stress and anticancer agents. In a similar manner, p62-dependent sequestration of KEAP1 under conditions of dysfunctional autophagy leads to NRF2 activation, and this is an additional activation mechanism of NRF2 and a novel pathway of tumor promotion. Thus, p62 is a stress response protein and is directly linked to an oxidative stress response signaling pathway, which leads to tumorigenesis. Taken together, these results suggest that the NRF2-mediated antioxidant program represents an emerging mediator of tumorigenesis.
Given the evidence implicating p62 as an oncoprotein, it is puzzling that this protein also has potential tumor suppressor properties. An interesting aspect that may solve the role of p62 in tumor suppression in the context of KEAP1 regulation is the finding that KEAP1 also mediates downregulation of NF-κB by targeting IKKβ for degradation (22). IKKα and IKKβ are the main upstream activators of the canonical NF-κB pathway, which phosphorylate the NF-κB inhibitory protein IκB. This results in the activation of NF-κB and its translocation to the nucleus where it promotes transcriptional activation of its target genes. p62 was shown to regulate the IKKβ activators, atypical protein kinase C (PKC) and TRAF6 (29). Therefore, p62 controls the interplay between the NRF2/KEAP1 pathway and the NF-κB pathway (Fig. 4).
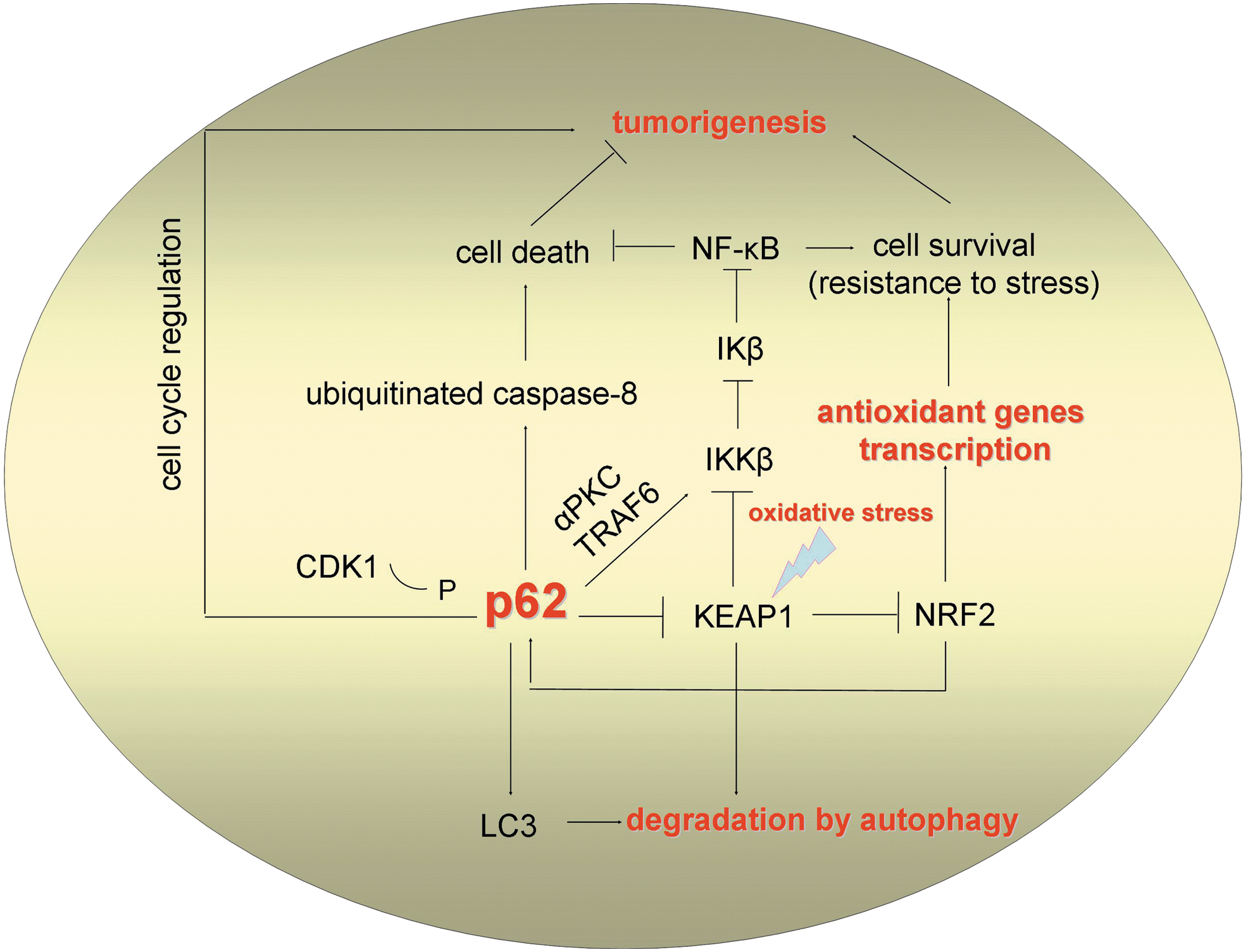
It is worth mentioning that KEAP1 may regulate ROS production and apoptosis through its binding partner phosphoglycerate mutase family member 5 (PGAM5). PGAM5 is an outer mitochondrial membrane protein that binds to the substrate binding pocket in the Kelch domain of KEAP1, and its C-terminal domain binds to the anti-apoptotic protein B-cell lymphoma-extra large protein (BCL-XL) (25). In addition, p62 has been found to be associated to dysfunctional mitochondria (30). These findings suggest for a complex interplay between p62, NRF2/KEAP1, and mitochondrial homeostasis.
Concluding Remarks and Future Directions
As just mentioned, p62 appears to implement remarkably important and, at the same time, diverse functions in the cell, resembling a cellular “swiss army knife” (Fig. 4). This ability arises from its unique functional motifs and protein-protein interaction properties (12, 28, 29). The literature reviewed here demonstrates that p62 plays a significant role in the regulation of autophagy, oxidative stress signaling, and tumorigenesis. Application of a systems biology approach would elucidate the interplay between these processes. It is surprising that p62 is not crucial for organism survival in mammals and flies (3, 6, 31). This either means that p62 has important but not absolutely vital roles or suggests the presence of other proteins with redundant functions, such as NBR1, with regard to autophagy in mammals (12).
Selective degradation by autophagy constitutes a key mechanism in regulating the function of p62 and may have applications in fighting aggregation-related diseases, such as neurodegenerative diseases as well as cancer. p62's ability to collect ubiquitinated proteins and target them for degradation may have a clinical impact. It would be interesting to test whether induced expression of p62 ameliorates phenotypes related to neurodegeneration in vivo. It will also be important to elucidate whether small or large aggregates are removed per se, or whether soluble, ubiquitinated or nonubiquitinated proteins can also be selectively degraded by autophagy. The important findings that p62 directly interacts with KEAP1 and that p62 is a target gene for NRF2 show the presence of a positive feedback loop for p62 regulation (10). It would be challenging to examine whether the p62 gene is transcriptionally regulated by other transcriptional factors as well, in a nonself-regulative manner. The emerging data showing a role for p62 in tumorigenesis through persistant activation of NRF2 (15) highlight the crucial roles of autophagy and p62 in tumor promotion. However, autophagy appears to have a dual role in cancer. First, it acts as a tumor suppressor during the early stages of tumorigenesis through mitigation of stress and genome damage and possibly through programmed cell death of the tumor cells and second, it induces tumor progression during late stages of tumorigenesis through survival of tumor cells under conditions of low nutrient accessibility (26). Therefore, it will be important to carefully test the role of p62 in each context.
In conclusion, recent research on autophagy and p62 offers a fertile ground for studying the molecular mechanisms of human diseases as neurodegeneration and cancer. Future studies will hopefully contribute to the identification of drug targets for these diseases.
Footnotes
Acknowledgments
I.P.N. and H.S. acknowledge support from the European Research Council. The authors apologize for not citing important articles due to space restrictions.