
Abstract
Introduction
In part due to the eukaryotic nature of the parasites, antibacterial and antiviral drugs are ineffective against them. To minimize the toxicity of the anti-parasitic drugs to the higher eukaryotic hosts, the logical approach is to capitalize on parasite-specific proteins and pathways that are absent in or significantly different from the host. In this article, we review and examine parasitic-specific Cys modifications that are essential for protein folding and, hence, may serve as potential drug targets.
Directed by their amino-acid sequences, nascent polypeptide chains form short secondary structures, such as α helices and β strands, often flanked by flexible loops, which are subsequently folded into stable three-dimensional structures. The inability of a polypeptide chain to fold into its native higher order conformation impairs its functions and increases its degradation (38, 55). Although the primary structure of a polypeptide largely governs its folding potential into a native tertiary structure, other factors, including van der Waals interactions, chaperons, hydrogen bonding, and disulfide bonding, also play critical roles in the folding process (19, 22, 39).
Two Cys residues residing in different parts of a polypeptide chain or in two different polypeptide chains but are adjacent in the tertiary or quarternary structure can be oxidized to form a disulfide (S-S) bond (22, 55) (Fig. 1). This electron transfer reaction requires an oxidative micro-environment, which, in eukaryotes, is provided by the lumen of the endoplasmic reticulum (ER) (45, 80, 81). Small-molecule oxidants, such as glutathione, cysteamine disulfides, dehydroascorbate and vitamin K epoxide as well as redox enzymes, such as protein disulfide isomerase (PDI) and sulfhydryloxidase, serve as electron acceptors in disulfide bond formation. In this Forum Review article, we focus on a few specialized parasitic enzymes that affect or catalyze disulfide reactions, while other articles in this series cover the rest.
Protein Disulfide Isomerases
A resident of the ER, PDI is a multi-domain multifunctional enzyme that belongs to the thioredoxin (Trx) superfamily and catalyzes the reversible formation of disulfide bonds between Cys residues within proteins (74, 76), which are often rate-limiting steps in protein folding, thus accelerating the correct folding of their client proteins (21 –23). The prototype PDI contains the domain structure a-b-b′-a′; whereas the a and a' domains show sequence similarity to Trx and contain two Cys-X-X-Cys (CXXC) active sites; the b, b' domains are redox inactive and show structural similarity to Trx without significant sequence homology (27, 34).
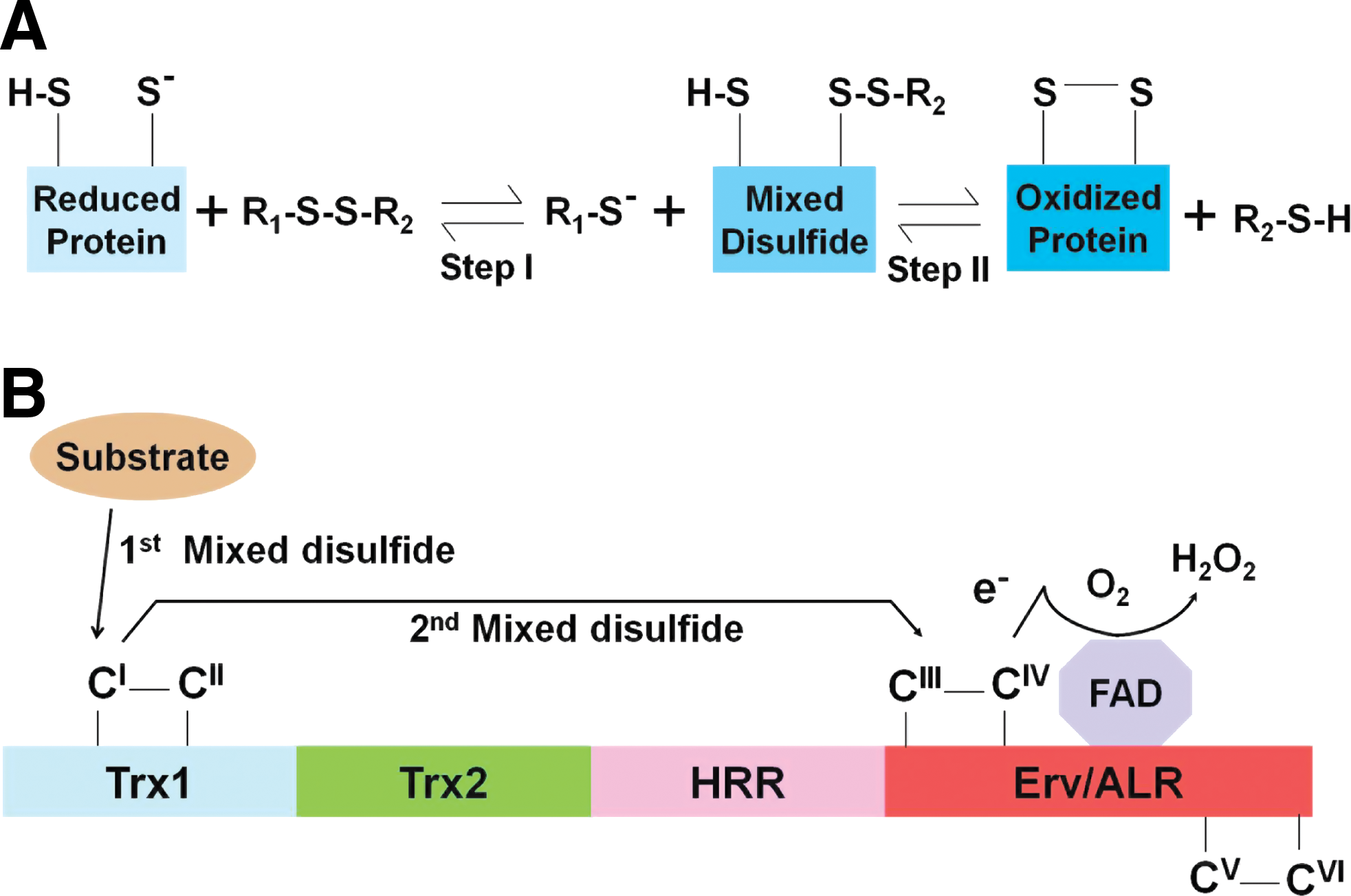
In general, unfolded proteins are not only functionally suboptimal or fully useless but also activate ER stress via the unfolded protein response, which can be detrimental to cell survival (36). Disulfide bond formation during protein folding is mediated by two thiol-disulfide exchange reactions (21, 23, 38). In the first step, a reactive thiolate anion (S−) in the reduced form of a protein displaces one sulfur atom from an oxidized (disulfide-bonded) redox reagent, resulting in the formation of a mixed disulfide bond between the protein and the redox reagent. In the second step, the remaining thiol anion attacks the mixed disulfide bond, leading to the formation of the oxidized protein (Fig. 1A) (21, 23, 38). Disulfide bond formation is also mediated by a process called disulfide reshuffling in which cysteine thiolate anion of a protein attacks an existing disulfide bond on the same protein to generate a new disulfide bond.
PDI itself undergoes reversible Cys disulfide formation, such that its reduced (dithiol) form catalyzes reduction of mispaired thiol residues of the substrate and acts as an isomerase. PDI, therefore, essentially promotes the posttranslational modification by disulfide exchange. It is to be noted that in prokaryotes, the Dsb family enzymes catalyze the same reaction and reside in the periplasmic space, through which proteins traffic to their extracytoplasmic destinations (49, 65, 76). Moreover, in eukaryotes, the flavin adenine dinucleotide (FAD)-linked sulfhydrol oxidase, Erv1, which resides in the intermembrane mitochondrial space and forms a redox cycle with Mia40 through a disulfide relay, plays an essential role in the maturation of cytoplasmic proteins through the formation of disulfide bonds (18, 23, 29, 32, 77). The major additional task of PDI is to function as a chaperone, that is, assist misfolded proteins to attain a correctly folded state without enzymatic disulfide shuffling. Studies in yeast have revealed the essentiality of PDI for cell survival (31, 66).
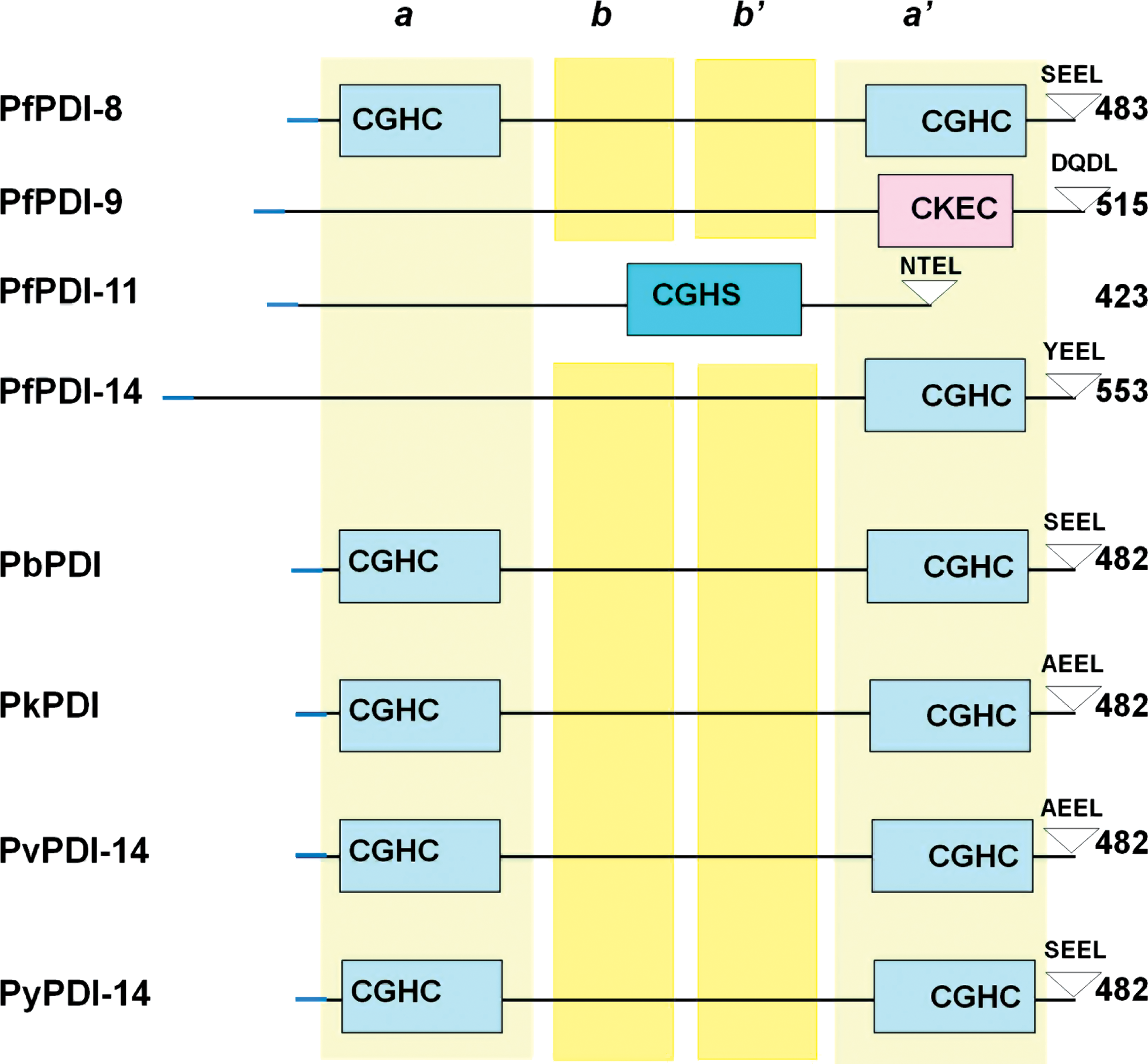
In a leading study (54), nine PDI-like sequences were noted in five species of malaria parasites, including Plasmodium falciparum and Plasmodium vivax (both human), Plasmodium knowlesi (monkey), and Plasmodium berghei and Plasmodium yoelii (both mouse). PlasmoDB identifies four PDI-like sequences in the P. falciparum genome that are located on four chromosomes 8, 9, 11, and 14, and named accordingly: PfPDI-8, PfPDI-9, PfPDI-11, and PfPDI-14 (Fig. 2). Significant sequence similarity of plasmodial PDI with the PDI of other species together with the experimental demonstration of PDI activity of a number of recombinant PfPDI (see below) suggest that all PDI sequences in the parasite may be functional. All four PfPDIs have cleavable N-terminal signal peptides, one or two thioredoxin reductase (TR) domains, and a C-terminal ER-retention signal (such as SEEL in PfPDI-8), suggesting that all locate to ER (Fig. 2). Only PfPDI-8 seems to conform to the classical PDI structure a-b-b'-a', consisting of two Trx domains, each with a pair of active Cys sites as a part of the Cys-Gly-His-Cys (CGHC) active site motif. PfPDI-9, 11, and 14, in contrast, have only one Trx domain with the active site motif Cys-Lys-Glu-Cys (CKEC) and CGHC, respectively, closely resembling the a' (C-terminal) domain of other organisms (Fig. 2). In another variation, the single Trx domain of PfPDI-11 aligns with the a (N-terminal) domain (54).
PfPDI-8 was expressed and biochemically characterized in two laboratories (54, 63). The protein exhibited typical biochemical functions of a PDI: oxidase/isomerase and reductase activities, as well as a chaperone-like behavior on a denaturated protein substrate. The protein was found in the parasitic ER and is absent in the Golgi. The antiplasmodial compound DS61 inhibited the recombinant PfPDI oxidase/isomerase activity but not that of the human recombinant PDI, pointing to structural differences between the two and suggesting that PfPDI is a potential target for novel antimalarials that may be structurally related to DS61.
Multiplicity of PDI homologs is not unique to P. falciparum but is rather common in all organisms; yeast, for example, has at least 6 (33), and humans have 14 (28). This is most likely because each PDI enzyme has unique, nonredundant functions related to their substrate specificity, cellular location, regulation, and association with specific accessory proteins, thus allowing their involvement in different pathways. Lastly, multiple PDI sequences of various domain structures, including the PfPDI-8 prototype (Fig. 2), are also found in other protozoan parasites (54), notably Toxoplasma, Crytosporidium, Giardia (58), Trypanosoma (73), and Leishmania (68). Based on what we know about PDI in Plasmodium, the PDI of these parasites are also likely to be essential for infection and, hence, useful as potential drug targets.
A large number of exported parasite proteins, essential for virulence and pathogenesis, transit through the ER for proper folding (54, 63). They also contain multiple Cys residues at conserved positions within well-defined protein domains, disulfide linkages of which are essential for functionality. Such domains include the F2-domain of EBA-175 (82, 86), epidermal growth factor (EGF)-like domains of merozoite surface protein-1 (MSP-1) (41), the ectodomain of apical merozoite antigen-1 (AMA-1) (7, 26, 44), and host-cell-receptor binding (cytoadherence) domains of the erythrocyte membrane protein 1 (PfEMP1) (11, 59). All these proteins are likely substrates for one or more PfPDI.
Next, we describe three of these–AMA-1, EMP1, and MSP-1–in some detail.
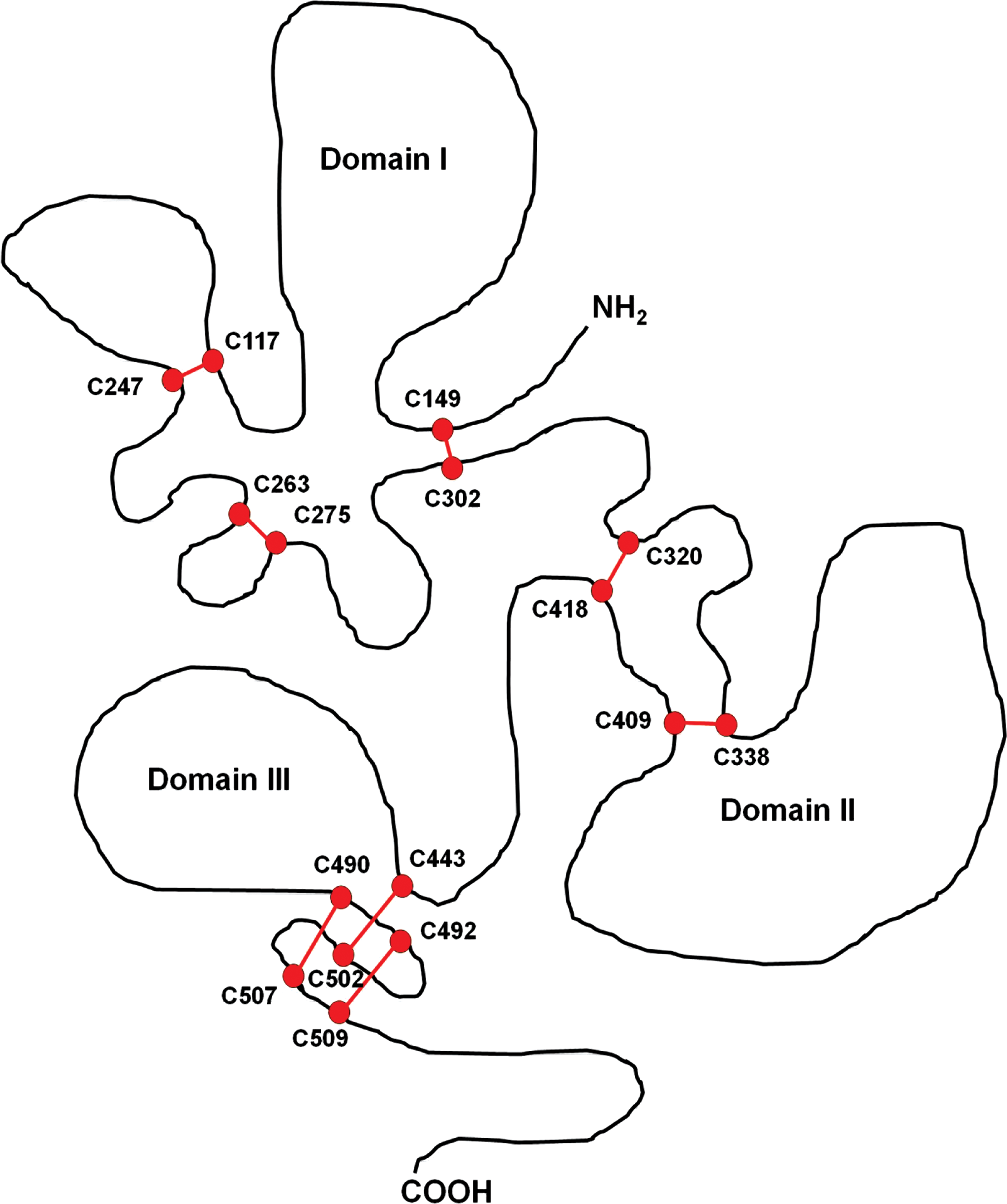
The best studied of these is the AMA-1 protein, which requires eight intramolecular disulfide bonds to attain its final conformation and is essential for the erythrocyte invasion (44). The crystal structures of three AMA-1 proteins from different Plasmodium species, namely P. falciparum, P. chabaudi, and P. vivax have been determined so far (9, 44, 70). The AMA1 proteins in all Plasmodia contain 16 invariant Cys residues in the ectoplasmic region, and analysis of the disulfide-bond pattern suggests a division into three distinct domains (9, 44, 57, 64, 70), containing three, two, and three disulfide bridges, respectively (Fig. 3). Correct disulfide pairing in the recombinant AMA-1 protein is indeed essential for protective immune response (7), which underscores the need for redox-assisted folding for AMA1-based vaccine development.
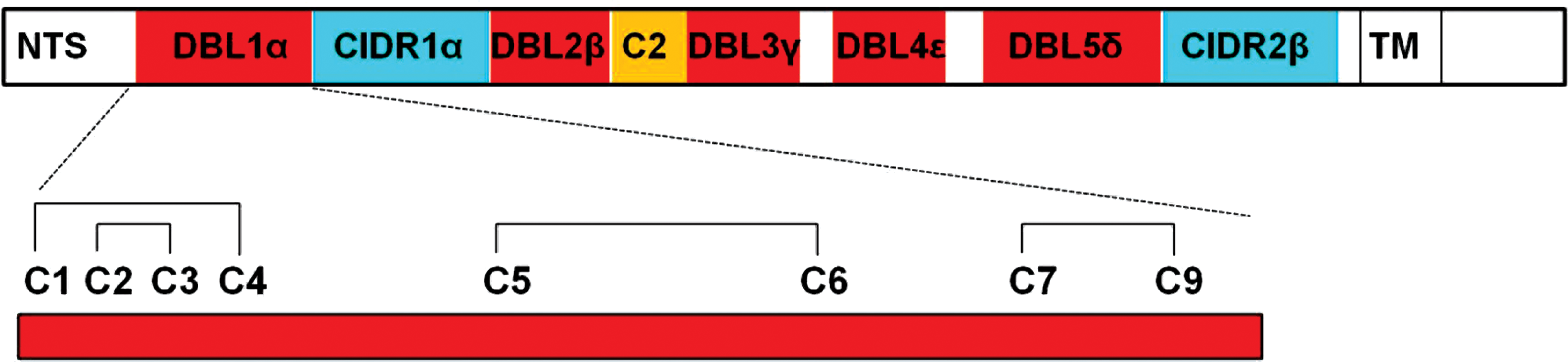
PfEMP1 is responsible for a phenomenon named “rosetting”, in which P. falciparum causes infected red blood cells (RBC) to form rosettes with uninfected RBC, which is characteristic of severe malaria (15, 72). The protein is exported to the erythrocyte surface via ER (2, 85) and requires correct folding of its Cys-rich interdomain region (CIDR) for cyto-adhesiveness. PfEMP1 contains the following major domains (Fig. 4): N-terminal segment (NTS), Duffy binding-like (DBL), and CIDR (48). The α-class DBL1, which follows NTS, has been directly implicated in rosette formation, and forms multiple Cys-Cys bridges (pair of oxidized Cys joined by disulfide bridge [S-S]) (Fig. 4), which change the structure of the NTS-DBLα module (48).
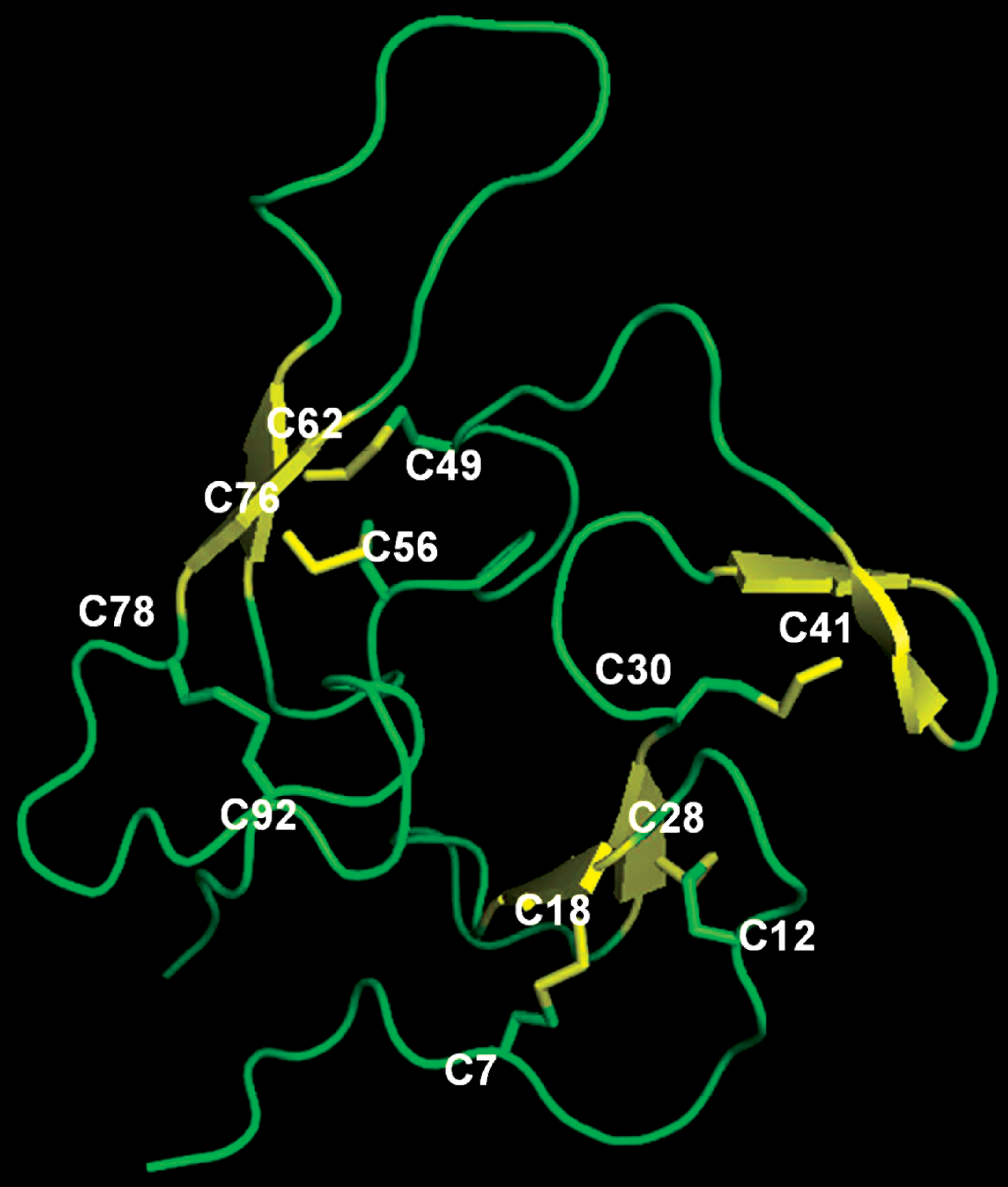
Finally, the P. falciparum MSP-1 contains two closely packed EGF-like domains, each stabilized by three disulfide bonds. The structure of this domain, as determined for P. falciparum and P. knowlesi, (37, 47, 60), revealed all six disulfide bonds that are required for correct folding of the dual domain protein (Fig. 5). These studies are highly relevant, partly because MSP-1 is a leading vaccine candidate and its native conformation is important for invasion (41).
Quiescin-Sulfhydryl Oxidase
Sulfydryl oxidases (also see Ero1 below) are believed to be required for the generation of disulfide bonds in a variety of organisms (38). The Quiescin-sulfhydryl oxidase (QSOX) family of flavoenzymes (42, 50) catalyzes the direct and facile insertion of disulfide bonds into unfolded or denatured proteins with concomitant reduction of oxygen to hydrogen peroxide (H2O2). The reaction can be summarized as follows: 2R-SH+O2→R-S-S-R+H2O2 (Fig. 1A). In contrast to other flavoprotein sulfhydryl oxidases, QSOX directly oxidizes a wide range of reduced proteins and peptides without mediation of PDI or small molecules.
The QSOX members are FAD-linked enzymes expressed in a wide variety of eukaryotic organisms except in fungi and yeast (50). A prototype QSOX is comprised of four domains: two Trx domains (Trx1 and Trx2), followed by a helix-rich region of unknown functions and the C-terminal Erv/ALR domain that serves as the oxidative catalytic engine that forms an FAD-mediated connection between and thiol/disulfide and molecular oxygen (18, 21, 22, 39). Interestingly, QSOX enzymes of protozoan parasites (see below), green algae, and higher plants lack the Trx2 domain partially or completely (19, 22, 39). Four potential redox-active groups—one FAD and three CXXC disulfide motifs—are found in QSOX proteins (55). The solvent exposed C
Ero1 Class of Sulfhydryl Oxidase
Very similar to QSOX, the Ero1 family members are also flavin-dependent sulfhydryl oxidases, found in a number of parasites. Unlike QSOX, the Ero1 family is found in yeast and higher eukaryotes (49). Both families lack an obvious ER retention signal, but it is possible that they may be retained in the ER lumen by association with a PDI homolog, where Ero1 can promote re-oxidation of the reduced PDI (67, 75, 84, 46). Both T. brucei and P. falciparum contain a single homolog of Ero1 (54), and sequence alignment revealed that similar to human Ero1α, this molecule has 15 cysteine residues with two conserved Cys triads that are critical for Ero1α function, suggesting that malaria thiol-oxidation pathway utilizes a mechanism that is similar to other eukaryotes. However, neither protein has been biochemically characterized. Giardia lamblia genome, while encoding five PDI homologs, lacks Ero1 (58, 61).
Cyclophilin A of Schistosoma: Folding Regulated by Intramolecular Disulfide
Helminths are multicellular eukaryotic parasites that infect over one-fourth of the world population and are believed to induce type 2 T cell immunity, leading to atopic diseases, such as allergy and asthma (56). As mentioned earlier, Schistosoma is a major helminth that causes schistosomiasis and human morbidity in tropical countries (5). Since the extracellular parasite is exposed to reactive oxygen species derived from the host immune cells as well as from its own oxygen metabolism, Schistosoma has developed an efficient antioxidant defense mechanism comprising thioredoxin peroxidase-1 (Tpx-1), Trx, and thioredoxin reductase (TrxR) (5, 52). Since this organism lacks catalase and expresses low levels of glutathione peroxidase, it utilizes Tpx-1 for the detoxification of H2O2 and other hydroperoxides (5). Importantly, the Trx/TrxR system also provides reducing equivalents to disulfide-mediated protein folding in this organism (8). The immune suppressant, cyclosporin A (CsA), which targets cyclophilins (Cyps) (see below), is widely used for the treatment of schistosomiasis (5).
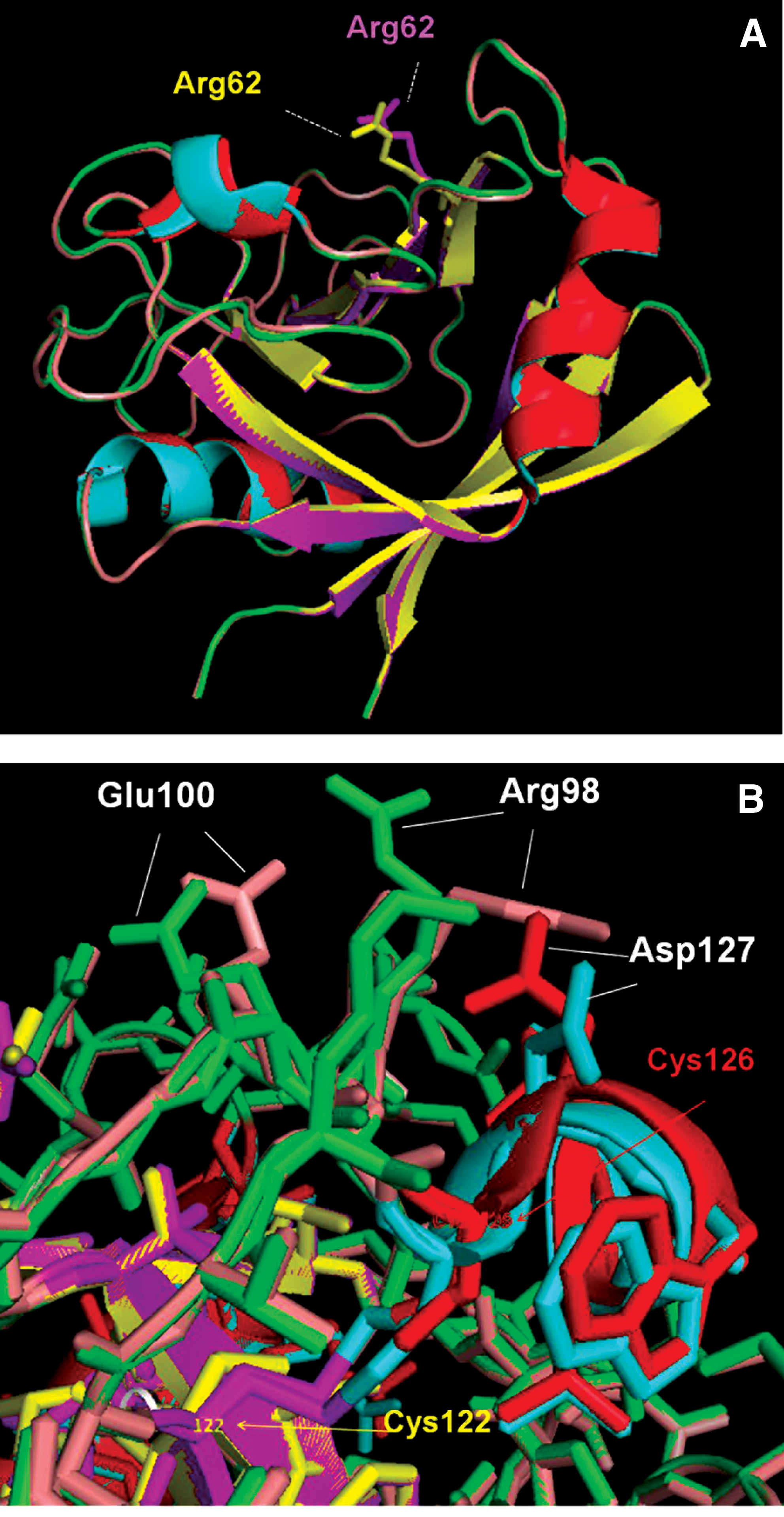
The CyP family represents ubiquitous chaperones that help a large cohort of client proteins to fold properly (1, 10). Their inherent PPIase activity and biological function are pharmacologically inhibited by CsA, a cyclic undecapeptide, first isolated from the fungus Tolypocladium inflatum by Sandoz researchers in Norway nearly half a century ago (13). Classic crystallographic studies revealed that CsA functions by tightly binding the PPIase catalytic pocket (83). However, it remains largely unknown whether the structure or function of CyP itself is regulated. A recent study of Cyclophilin A of Schistosoma mansoni (SmCyPA) revealed an interesting mechanism of regulation. SmCyPA uniquely contains two closely spaced Cys residues, namely Cys122 and Cys126; although the first Cys is conserved in other CyPs, the second is not, and is often replaced by a Thr. The juxtaposition of Cys122 and Cys126 allows reversible disulfide bond formation between the two and the resultant formation of an oxidized or a reduced state of SmCyPA, a phenomenon not reported for any other CyP. Parallel functional studies suggested a mechanism for regulation of SmCypA activity via oxidation of its thiol groups, such that oxidized SmCypA was inactive, whereas reduced SmCypA was found to be an efficient isomerase, inhibited by low nanomolar CsA. The mechanism of this dramatic difference in catalytic activity was revealed when high-resolution X-ray crystal structures were obtained for both the oxidized and the reduced forms (40). The Cys122 and Cys126 were found to be located in the vicinity of the active site and at a distance of 3.8 Å from each other. Formation of disulfide bridge between the two resulted in significant alteration of three major structural pockets lined by functionally important residues. To illustrate, we present representative superimposed structures of the two forms (Fig. 6). Comparison of the ribbon presentations (Fig. 6A) shows that there was little or no change in the overall protein backbone due to the disulfide bridge. However, when the amino-acid side chains were compared, those of key amino acids were found to be shifted. Such residues included not only the catalytic residue Arg62 (Fig. 6A), but also many others such as Glu100, Arg98, and Asp127 (Fig. 6B). Thus, SmCyPA is a unique chaperone whose own folding and enzymatic activity is reversibly regulated by an intramolecular disulfide bridge, which is, in turn, regulated by the redox environment of the parasitic cytoplasm. Lastly, the confidence gained from these structural studies may allow the development of alternative and more effective schistosomiasis inhibitors targeting the dual Cys-thiol region of SmCyPA, as it is absent in all human CyPs.
Cys Disulfide-Assisted Folding of Trypanosome Variable Surface Glycoproteins
A major role of redox-assisted folding in unicellular parasites is exemplified by the variable surface glycoproteins (VSGs) of trypanosomes. Many parasites, such as Trypanosoma and Plasmodia, have evolved a common mechanism to evade host immune response and maintain a low level of chronic infection in their natural hosts (20). This is achieved through antigenic variation of the VSGs that constitute the coat proteins of the parasite. As implied, the coat is highly immunogenic and elicits a robust antibody response. However, the parasite genome contains roughly a thousand VSG genes that are constantly switched on and off, such that the VSG makeup of the coat keeps changing, allowing evasion of host immunity and perpetuation of infection.
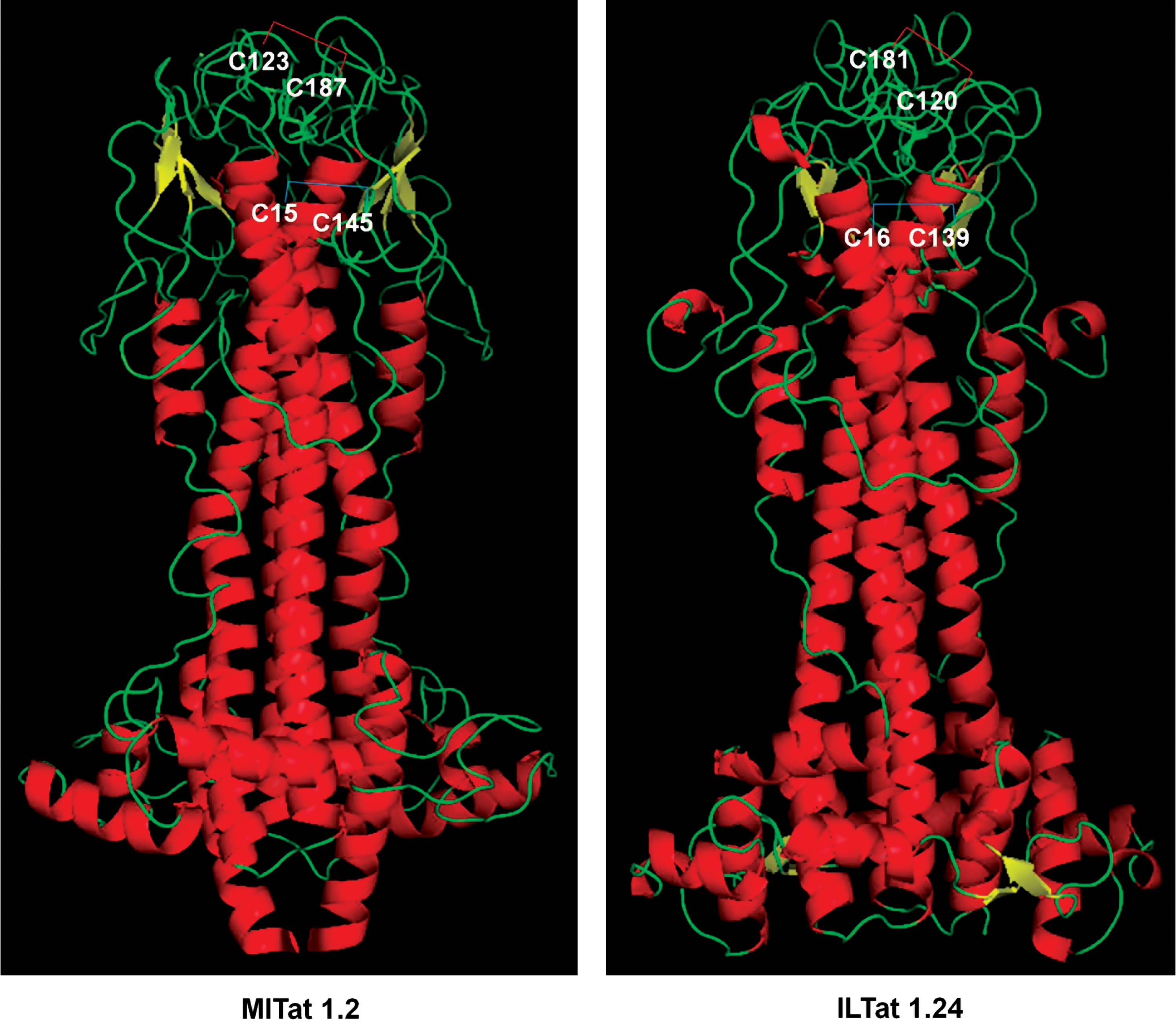
An average VSG polypeptide is a little more than 400 amino acids long and consists of a single large N-terminal domain of 350–400 residues and one or two smaller C-terminal domains (6, 14, 16). Interestingly, in addition to being heavily glycosylated, each domain contains a string of conserved Cys residues, many of which are disulfide bonded in a characteristic pattern, even though the overall sequence, especially that of the N-terminal domain, is highly variable. A representative example (Fig. 7) shows two Cys disulfide bridges in the Type A VSGs. The experimentally determined three-dimensional structure of the same proteins (12, 17, 35) revealed the close proximity of the bridging Cys residues (Fig. 8). The overall importance of Cys in the structural integrity of VSG was tested by site-directed mutagenesis of selected C-terminal Cys residues to Ala. The 1H-NMR spectra of the purified recombinant mutants showed considerably less dispersion of resonances, indicating that no mutant retained the structure of wild-type VSG.

The exact redox system responsible for VSG disulfide formation is yet to be identified. Nonetheless, Trypanosoma are known to have an unusual redox biology, in which a bis-glutathione derivative, trypanothione, plays a major role (30). As mentioned here and in other articles in this Forum, three flavin-linked sulfhydryl oxidases can be identified in the T. brucei genome, namely TbALR, Ero1, and the QSOX described earlier. Of these, the last two are found in the ER, and, thus, likely to participate in disulfide-promoted folding of the VSGs that traffic through ER for glycosylation as well.
Secreted Redox Chaperones and Their Extracellular Immune Roles
Accumulating literature has provided evidence for an intriguingly novel role of chaperones as a potential extracellular entity. Many molecular chaperones, including those involved in redox-assisted protein folding, are secreted from cells and function as signals for a variety of cells, such as the macrophages (43). The major extracellular chaperones involved in parasite infection are Trx and peroxiredoxin, as briefly described next.
The Trx superfamily consists of redox enzymes that contain the CXXC motif and catalyze oxidative protein folding through disulfide formation. The prototype full-length Trx is a roughly 100 amino acid-long polypeptide of ∼12 kDa, whereas a naturally truncated form of Trx is known that contains only 80–84 N-terminal residues and, hence, is named Trx80. The latter is produced and released by monocytes, and was originally discovered as an eosinophil cytotoxicity-enhancing factor in the blood of patients with schistosomiasis (78). The exact mechanism of the generation and function of Trx80 and the role of the parasite in these processes, if any, remain to be elucidated. However, Trx and Trx reductases are now known in a number of platyhelminth parasites, such as S. mansoni, Echinococcus granulosus, and Taenia crassiceps (3, 5, 71), all of which cause chronic immune pathologies. It would be interesting to find out whether similar Trx-related products also serve as extracellular immune regulators in these parasite infections.
Peroxiredoxin was, in fact, initially known as macrophage 23-kDa stress protein and has been found to be an essential intracellular inhibitor of macrophage activation by lipopolysaccharide (LPS) (87). In chronic parasitic infections, macrophages are activated and are associated with a polarized Th2 response. This could be replicated in a laboratory mouse model of latent as well as chronic infection by the liver fluke, Fascila hepatica, a trematode. F. hepatica is the causative agent of fasciolosis, a zoonosis that affects cattle and causes major losses in agriculture and dairy. The alternative activation state of macrophages was induced by both live flukes and parasite excretory–secretory products, the active component of which was found to be peroxiredoxin. In a direct experiment, purified recombinant F. hepatica peroxiredoxin alternatively activated mouse macrophage cells as evidenced by the production of interleukin (IL)-10, prostaglandin E2, and correspondingly low IL-12 levels (25).
Conclusion
Reversible Cys disulfide bridges clearly regulate the structure and function of a large variety of proteins in protozoan and metazoan parasites. There is a now a growing body of evidence that these proteins play essential roles in not only parasite growth but also host-parasite signaling and immune response. The parasitic enzymes that catalyze the thiol redox reaction possess conserved as well as unique domains, making them lucrative targets for new drug development. Nonetheless, various aspects of regulation and function of these enzymes are still unknown and will certainly constitute major research areas of the future.
Footnotes
Acknowledgments
This work was supported in part by a grant from the Michael J. Fox Foundation for Parkinson's Research (SB), a State of Ohio Third Frontier grant (SB), and U.S. National Institutes of Health (NIH) grants 1R43 GM090383 (SB) and R56 AI083638 (SJH). The authors wish to thank DeLano Scientific for making the PyMol program freely available.