
Abstract
Introduction: Primary Lessons from Yeast
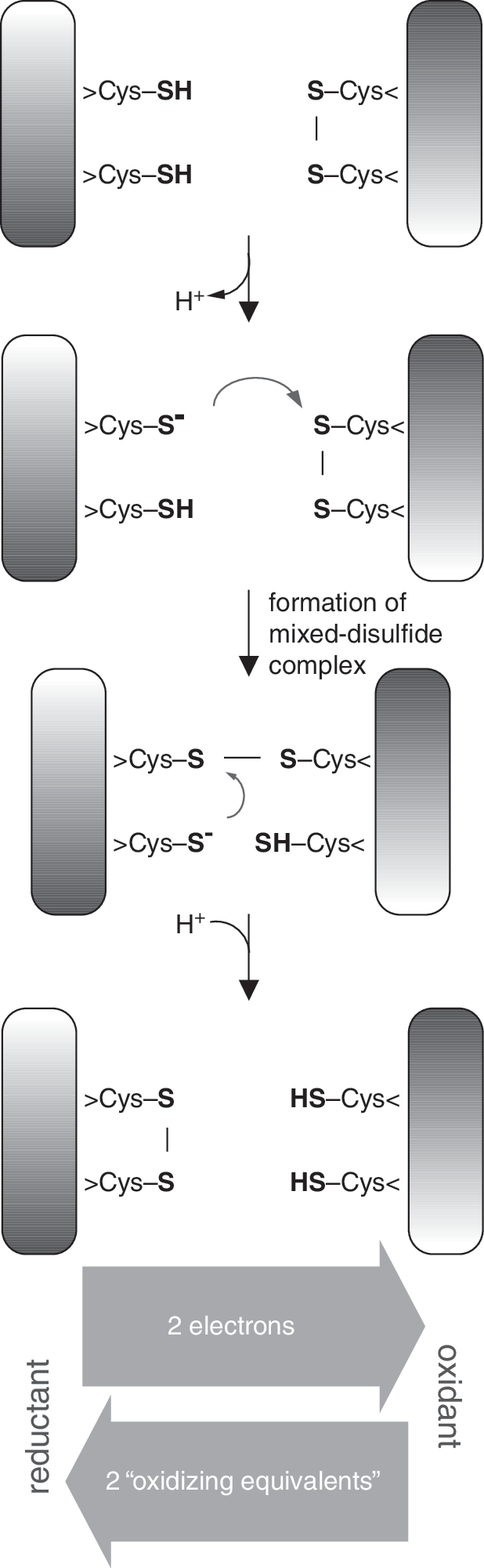
ERO1 has first been described in baker's yeast as an essential gene (20, 44). Its product Ero1p is an ER-resident glycoprotein and a critical determinant for the oxidizing capacity of the yeast cell (20, 44). It possesses two redox-active di-cysteine active sites. The “inner active site” is oxidized by a proximally bound flavin adenine dinucleotide (FAD) cofactor (25, 50), which itself receives oxidizing equivalents by reducing O2 to hydrogen peroxide (H2O2) (26). The resulting disulfide bond is then transferred from the core of the protein to the “outer active site” (47), which is located in a flexible peptide loop (25). Via its outer active site—also termed the “shuttle disulfide”—Ero1p can directly and specifically oxidize one of the two active-site cysteine pairs in PDI, the archetypal member of the PDI family (21, 22, 50). This disulfide relay from Ero1p to PDI enables oxidized PDI to subsequently introduce disulfide bonds into folding substrates (Fig. 2) (21, 50).

Since the resolution of mismatched substrate disulfides by reduced PDIs is a fundamental component of oxidative protein folding, an unregulated (hyper-) oxidation of PDI by Ero1p would be undesirable. Thus, a redox-sensitive shutdown mechanism represented by noncatalytic, intramolecular disulfide bonds effectively impairs Ero1p activity (27, 48). As the oxidation state of these cysteine pairs is controlled by the ER redox poise, a regulatory feedback loop ensues in which Ero1p is solely active when new disulfides are required (48). Taken together, many of the conserved features of Ero1 sulfhydryl oxidases including their fold, mechanism of action, physiological relevance, substrate specificity, and the principle of their tunable activation status have been unraveled in experiments with the yeast enzyme.
What Yeast Has Failed to Teach Us
Orthologs of Ero1p exist in all eukaryotes (8). Mammalian genomes harbor two Ero1-like genes, ERO1L and ERO1LB, which encode Ero1α and Ero1β, respectively (14, 42), and will be the subject of this review. Interestingly, the study of Ero1α and Ero1β has not only unveiled many parallels to the yeast enzyme, but also a number of important differences. Most prominent among these, the mammalian Ero1 genes appear to be nonessential, as evidenced by the viability of mice carrying mutated copies of both ERO1L and ERO1LB (55). The mutated genes feature an intronic “gene trapping” insertion containing a strong viral splice acceptor. As a consequence, the vast majority of mRNAs derived from the ERO1L/ERO1LB loci will give rise to nonfunctional, truncated proteins.
Lipopolysaccharide-activated spleen cells (LPS blasts) isolated from the mutant mice are nearly indistinguishable from their wild-type counterparts with regard to the efficiency of oxidative folding of immunoglobulin M (IgM) (55, 56). With one known exception (the oxidative folding of proinsulin in the β-cells of the pancreas, discussed further below), it is therefore reasonable to assume that the overall pace of disulfide-bond formation is not severely compromised in ERO1L/ERO1LB mutant mice and does not phenocopy the fatal situation in Ero1p-deficient yeast cells.
These observations can be explained by alternative mechanisms for de novo disulfide-bond generation in the mammalian ER (13) and/or by incomplete gene trapping, which would allow the low-level synthesis of operational Ero1 molecules. Indeed, residual amounts of Ero1β were detected on mRNA level in cardiomyocytes (16) and on protein level in pancreatic tissue and LPS blasts from Ero1 double mutant mice (55). In all cell types studied so far, only a minor portion of Ero1α and, presumably, Ero1β is maintained in an active form (6, 11, 16) (see also below). Consequently, when shifted to increased activity through redox regulation, the residual levels of Ero1β in ERO1L/ERO1LB compound mutant mice might actually suffice to support disulfide-bond formation. Along the same lines, yeast cells can proliferate in the presence of very low amounts of Ero1p. Sufficient levels of Ero1p to allow for growth of an ero1-null strain can be provided by a plasmid encoding ERO1 under the control of a galactose-inducible promoter even when cells are grown in glucose (where the promoter is largely repressed) (Carolyn S. Sevier, pers. comm.). It is therefore important to consider that the designation of ERO1L/ERO1LB mutant mice as “knockout animals” can provoke an underestimation of the functional significance of Ero1 enzymes in mammals. Interestingly, the high proliferation rate of immortalized mouse embryonic fibroblasts is profoundly affected by the ERO1L/ERO1LB compound mutation (Ester Zito, David Ron, and Christian Appenzeller-Herzog, unpublished observations), suggesting that under in vitro conditions, normal levels of Ero1 enzymes are required for optimal cell growth. On the other hand, flies homozygous for a nonsense allele of their single ERO1L gene develop almost normally (49), which strongly suggests the existence of ERO1L-independent pathways for disulfide-bond formation.
In addition, while both Ero1p and Ero1α (and, most likely, Ero1β) are soluble ER proteins, which are peripherally membrane-associated (14, 20, 42), only Ero1p possesses a C-terminal membrane-targeting domain (43). Further differences between Ero1p and the mammalian Ero1 enzymes include a distinct set of regulatory disulfide bonds for the shutdown of oxidase activity and different mechanisms of substrate recognition and oxidation. These issues will be covered in subsequent sections.
Ero1 Enzymes Are Feedback-Regulated Sulfhydryl Oxidases
Of note, not all of the cysteines within the primary sequence of Ero1 are positionally conserved from yeast to mammals. While the inner and outer active-site cysteines (Cys394-Cys397 and Cys94-Cys99 in human Ero1α) and the long-range disulfide connecting the two active-site peptides (Cys85-Cys391 in Ero1α) are preserved, intramolecular disulfide bonds homologous to the regulatory Cys143-Cys166 and Cys150-Cys295 in Ero1p are absent from both Ero1α and Ero1β (Fig. 3). Amino acid substitution and mass spectrometry analysis revealed that Ero1α in its most oxidized redox state forms a disulfide bond between Cys94 and Cys131 (6, 10). As Cys94 is a constituent of the outer active site, Cys94-Cys131 has to be resolved to allow disulfide shuttling to PDI. Interestingly, also Cys99 forms a “nonactive-site” disulfide with Cys104 in the shut-off state of Ero1α (10). Like Cys131, Cys104 has no equivalent in Ero1p.

The identification of these new types of regulatory disulfide, which are likely to be present in Ero1β, too (53), has implications for the redox-driven mechanisms of enzyme (in)activation. Thus, in an oxidizing ER environment when reduced substrates for Ero1α are scarce, newly produced disulfide bonds arising from the inner active site will be “stored” as Cys94-Cys131 and Cys99-Cys104. Molecularly, a likely scenario would be the nucleophilic attack of the shuttle disulfide by Cys131 (giving rise to Cys94-Cys131) followed by the transfer of a second disulfide from the inner active site via the transient formation of Cys99-Cys394 [in analogy to Ero1p; (47)], which is then resolved through nucleophilic attack by Cys104. As to the reactivation under reducing conditions, the situation is less straightforward. Two electrons are required to break either of the two regulatory disulfide bonds, before the shuttle disulfide can be reformed through nucleophilic attack. The finding that the concentration of reduced PDI in the ER influences the extent of Cys94-Cys131 formation suggests PDI as the reductant (6). However, PDI has proven to be an ineffective activator of Ero1α in a reconstituted reaction (9, 10, 15, 52). In keeping with the predominantly inactive state of Ero1α in the ER (6, 11, 16), this relative resistance of Ero1α toward PDI-mediated reduction could be a critical determinant of ER redox control rather than a manifestation of its poor catalytic proficiency. Indeed, when the thiol load of the ER is maximized by treatment with a strong reducing agent, the Ero1α-dependent re-formation of disulfides is exceptionally fast upon washout of the reductant (7). These findings are consistent with the concept that Ero1α is an environment-dependent sulfhydryl oxidase, the activity of which is governed by the redox state of its own substrate(s).
In contrast to the aforementioned disulfide bonds, the long-range disulfide, which is conserved in all Ero1 orthologs, does not involve any active-site cysteines (Fig. 3). Two alternative views exist on the role of this disulfide during catalysis. Based on the finding that purified Ero1α C85A-C104A-C131A-C391A—although well-folded—is less active in an oxidase assay than Ero1α C104A-C131A (9), one opinion holds that Cys85-Cys391 must be intact for efficient substrate oxidation. The second view, which we tend to favor, suggests a rearrangement of this disulfide during enzyme activation. Thus, an as yet unidentified cysteinyl thiolate anion might attack, for example, Cys85 and thereby free Cys391 to create a new short-range disulfide in Ero1α that facilitates the communication between inner and outer active site. It can also be speculated that the presumed isomerization reaction is not readily triggered through intramolecular attack, but instead catalyzed by a thiol-disulfide isomerase such as ERp44 or another PDI that could initially resolve the long-range disulfide (for discussion of mixed-disulfide interactions of Ero1, see below). In potential support of this second view, activated Ero1α (10) and Ero1β (53) virtually co-migrate with the fully reduced forms on nonreducing gels. In addition, presumably catalytic mixed-disulfide complexes between Ero1α or Ero1β and PDI trapped in living cells after treatment with a reductant display markedly decreased gel mobility—indicative of long-range disulfide resolution—as compared with the complexes trapped at steady state (7). Intriguingly, the crystal structure of a hyperactive Ero1α C104A-C131A mutant, which still harbors Cys85-Cys391, does not reveal any obvious pathway for O2 to reach the protein-embedded FAD moiety (29). We speculate that structural flexibility upon disruption of Cys85-Cys391 will be instrumental for the emergence of such an aqueous O2 channel. A detailed study on the reductive activation of Ero1p has also indicated that a subfraction of the long-range disulfide is resolved prior to the catalysis of substrate oxidation (27).
Mechanisms of selection of specific sulfhydryl substrates
Among the total of ∼20 PDI-like proteins in humans (5), PDI itself has been shown to be the major substrate for Ero1-mediated oxidation. Numerous cues in favor of this view exist including data from both cell culture and in vitro assays (6, 7, 29, 53). Ero1α recognizes PDI by a lock-and-key principle irrespective of whether or not the redox-active cysteines are present in PDI (29, 41). Thus, initial binding prior to the formation of a catalytic mixed disulfide occurs through noncovalent interactions. It has been conclusively demonstrated that these interactions are of hydrophobic nature (29). They involve a protruding β-hairpin in Ero1α, which contains a critical tryptophan residue at its very tip (Trp272), and a hydrophobic cleft in the substrate-binding domain of PDI (36). Even though experimental data were exclusively generated with Ero1α, both the hairpin structure and the crucial tryptophan are present in Ero1β as well (Fig. 4). Accordingly, the principle of substrate recognition is probably conserved among the mammalian isoforms. In addition, as hinted by in silico complex modeling (36), this mode of interaction presumably facilitates the specific thiol-disulfide exchange between the C-terminal active-site domain of PDI and the shuttle disulfide in Ero1α (7, 9, 10, 15, 52). In contrast, Ero1p harbors no tryptophan-containing β-hairpin (25) and preferentially oxidizes the N-terminal active-site domain of yeast PDI (51).

As opposed to the bona fide substrate PDI, its homolog ERp44 can efficiently bind to Ero1α even in the absence of the β-hairpin (36). Further, equal amounts of ERp44—Ero1α mixed disulfides are detected with all single-cysteine mutants of Ero1α (12), indicating that ERp44 can attack at least one disulfide other than the shuttle disulfide with its active-site cysteine. Besides these, also other PDI-family members—although inferior substrates for oxidation (29, 53)—form mixed disulfides with Ero1α and Ero1β within cells under steady-state conditions (7, 30, 46). As pointed out for PDI (7), these complexes most likely involve an oxidized (noncatalytic) form of Ero1α. In addition, they are not strictly dependent on the presence of an intact shuttle disulfide (7, 12) so that they might be formed in analogy to the ERp44–Ero1α interaction. The physiological roles of these covalent complexes are currently unclear.
Ero1α and Ero1β: Functional substitutes or brothers in arms?
While the above sections have highlighted many shared catalytic characteristics between Ero1α and Ero1β, the two homologs have also diverged in a number of features. For instance, the transcriptional regulation of ERO1L and ERO1LB is different, which undoubtedly contributes to the distinct expression profiles in human tissues (42) (Fig. 5). The high Ero1β levels in insulin-producing β-cells of the pancreas are maintained by the key pancreatic transcription factor PDX1 (31). The importance of Ero1β in these cells is highlighted by the finding that the oxidative maturation of proinsulin is delayed in pancreatic islets from ERO1LB mutant mice, which manifests in a diabetic phenotype (55). Intriguingly, this phenotype is neither complemented by a compensatory increase in Ero1α levels nor exacerbated by additional mutation of ERO1L, which argues against redundancy of Ero1 isoforms in the endocrine pancreas of mice (55). ERO1LB is also a target of the ER-stress-responsive transcription factor ATF6α (1) that is preferentially activated under reducing ER conditions (39).

ERO1L, on the other hand, is a transcriptional target of HIF1α that is upregulated in response to hypoxia or hypoglycemia (23, 37). Indeed, Ero1α is instrumental in counteracting ER hypo-oxidation brought about by hypoxic treatment of cells (16). In addition, the expression of ERO1L is enhanced during adipogenesis, which depends on the nuclear hormone receptor peroxisome proliferator-activated receptor gamma (PPARγ) (45). Finally, Ero1α is induced during a late stage of ER stress signaling through the binding of C/EBP homologous protein (CHOP; also known as Gadd153) to its promotor (34) (see below).
Ectopic expression of Ero1β—in contrast to Ero1α that has no discernible effect—moderately increases the oxidation of ER oxidoreductases and glutathione (6). This is likely owing to the relative lability of the regulatory disulfide bonds in Ero1β, since mutation of Cys100 and Cys130 in Ero1β does not activate the purified enzyme to the same extent as the equivalent mutations in Ero1α (53). Further, wild-type Ero1β shows higher rates of oxygen consumption during in vitro substrate oxidation compared with Ero1α (53). At the same time, however, the initial slope of glutathione re-oxidation after complete chemical reduction is less pronounced in Ero1β- than in Ero1α-overexpressing cells (7). Thus, the catalytic turnover rates of Ero1α and Ero1β differ in a context-dependent manner.
Can these disparities be explained at the molecular level? One of the two intercalating amino acids between the inner active-site cysteines in Ero1α and Ero1β is different (Fig. 4). When mutated to the Ero1β active-site sequence, the catalytic turnover number of Ero1α increases (53). Moreover, Ero1β harbors an additional cysteine at position 262 (Fig. 4), which has been proposed to form a unique type of regulatory disulfide together with Cys100 (53). However, as predictable by homology to Ero1α, Cys262 is located at the end of one of the four core α-helices and immediately upstream of the PDI-interacting β-hairpin loop (Fig. 4). We therefore consider it unlikely that this region adopts a fundamentally different conformation in Ero1β. In a structural homology model, the side chain of Cys262 is located ∼30 Å away from Cys100 and buried in the structure (data not shown). Finally, Ero1β harbors three unique N-glycosylation sites in the peptide surrounding Cys130 (Fig. 4). In the model, the glycosylated asparagines are solvent exposed and positioned between the PDI-binding site and the shuttle disulfide (data not shown), raising the possibility that substrate recruitment might be modulated by the presence of bulky oligosaccharides. Overall, Ero1β—basally expressed at low levels (Fig. 5) and an early target of the ER stress response (1)—represents an effective stress oxidase that can counteract ER hypo-oxidation. Ero1α, on the other hand, likely fulfills a tightly regulated housekeeping function with regard to disulfide generation. Whether and to what extent the two isoforms functionally and/or physically interact in tissues where they are co-expressed remains to be examined.
Ero1α Is Critical for ER-Stress-Induced Apoptosis
Besides the regulated production of disulfides, Ero1α has additional roles. A major fraction of Ero1α molecules localizes to so-called mitochondria-associated membranes (MAMs), a subdomain of the ER that is tethered to mitochondria (3, 24). When activated by exogenous reductants or under hypoxic conditions, Ero1α relocates to the bulk of the ER (24), suggesting that its function at the MAM might be independent of its activity as an oxidase. The excitable calcium channels of the inositol 1,4,5-trisphosphate receptor (IP3R) family are also enriched in MAMs, and IP3R-facilitated calcium shuttling to cytosol and mitochondria is a critical branch of apoptotic cell death signaling during severe ER stress (17). One of the switches that positively regulate such calcium flow is Ero1α (32), which is induced by the ER stress-dependent, apoptogenic transcription factor CHOP (34). Since the channeling activity of IP3R subtype 1 is impeded by reversible binding of ERp44 (28), elevated levels of Ero1α—a preferred ligand of ERp44 (2, 36)—likely activate IP3R1 during ER stress by sequestering ERp44 (in analogy to the mechanism described in the next section). Conversely and similar to the knockdown of IP3R1, silencing of Ero1α in macrophages inhibits ER-stress-induced apoptosis by lowering ER calcium release (32). Recently, knockdown of Ero1α in HeLa cells has been demonstrated to predominantly hamper the accumulation of calcium in mitochondria upon stimulation of IP3Rs (and only marginally in the cytosol), which is in agreement with the enrichment of Ero1α in MAMs (3).
An alternative, although less plausible possibility is that Ero1α does not enhance IP3R1 activity by lowering the availability of ERp44 but by hyperoxidizing the ER. The inhibitory association with ERp44 depends on two reduced cysteinyl thiols in IP3R1 (28), the oxidation of which upon ER stress could consequently lead to channel activation. In potential support of this, antioxidant treatment mimicked the inhibitory effect of Ero1α knockdown on ER calcium release (32). However, as the CHOP–Ero1α–IP3R1 signaling pathway induces NADPH oxidase 2-derived reactive oxygen species (ROS), which in turn further amplify CHOP (33), the application of antioxidants did not necessarily exert its effect by counteracting Ero1α activity. Moreover, given the tight regulatory mechanisms that prevent excessive disulfide and H2O2 synthesis by Ero1α (see above), its increased expression does not per se hyperoxidize the ER (6, 7). The probably minor contribution of Ero1-derived ROS to ER-stress-induced cell death has recently been evaluated in detail elsewhere (4).
Role of Ero1α in ERp44-Mediated ER Retention/Retrieval
Apart from their involvement in calcium signaling, Ero1α and ERp44 orchestrate the secretion of diverse disulfide-linked dimers and higher order oligomers by the reversible formation of mixed-disulfide ERp44–protein adducts (Fig. 6). ERp44 binds to substrates via its nonclassical CXXS active-site motif (2), which lacks a resolving cysteine and therefore renders these intermolecular disulfide adducts longer-lived than typical catalytic complexes. Moreover, despite its C-terminal ER retrieval signal ERp44 is atypically enriched in the ER-Golgi intermediate compartment (ERGIC)/cis-Golgi (35, 54), which favors a model in which ERp44 retrieves substrates from ERGIC/cis-Golgi to ER.

What is the role of Ero1α in the intracellular retention of secretory proteins? Increased expression of Ero1α displaces ERp44 from most of its endogenous substrates (2), indicating it to be a preferred mixed-disulfide ligand of ERp44. Accordingly, the secretion of ERp44 retention substrates such as IgM (2), the adipose-derived hormone adiponectin (54), or sulfatase modifying factor 1 (SUMF1; also known as formylglycine-generating enzyme) (19, 35) is enhanced upon overexpression of Ero1α (Fig. 6). In the case of adipocytes, endogenous induction of Ero1α by PPARγ agonists is physiologically relevant during differentiation and stimulation (45, 54). Although speculative, vascular endothelial growth factor (VEGF) might also add to the list of ERp44 substrates, as its secretion, which is prominent in hypoxic tumors, is positively regulated by Ero1α (37) (Fig. 6).
All secretory ERp44 substrates identified so far (and VEGF) undergo cysteine-dependent dimerization/oligomerization to be secreted. The cysteines engaged in this process have, in case of retention, previously been linked to ERp44 (Fig. 6), which is consistent with the concept that ERp44 traps incompletely assembled subunits. Although this concept is commonly referred to as “thiol-mediated retention,” it is still unclear how these trapping interactions are formed. As ERp44 comprises a single-cysteine active site, the oxidizing equivalents to join two thiol groups would either have to be contributed by the binding partner (e.g., through an intramolecular disulfide or glutathionylation) or by ERp44 itself. The latter possibility could involve a disulfide-linked homodimer or a heterodimer composed of Ero1α and ERp44. However, as detailed above, abundant Ero1α–ERp44 complexes rather inhibit than promote “thiol-mediated retention.”
Conclusions and Perspectives
Ero1 enzymes are an integral part in our understanding of redox maintenance in the ER. The two mammalian isoforms, which differ from the yeast enzyme in a number of aspects, fulfill similar roles in regulated disulfide production, but also display distinctive features. While recent work has begun to link these features to isoform-specific in vivo functions, there is certainly more room for discovery regarding the physiological roles of Ero1α and Ero1β. An eminent question for the future is how redundant these roles are in mammals. Given the viability of ERO1L/ERO1LB gene trap mice that still harbor detectable levels of the stress oxidase Ero1β (55), it is possible that Ero1β can largely substitute for Ero1α deficiency. Motivated by the finding that cardiomyocytes show decreased excitability due to lowered calcium transients in ERO1L mutant mice (16), however, it will be important to more closely look at, for example, ER-stress-induced apoptosis or the secretion of ERp44 substrates in these mice. Moreover, the question whether ERO1L/ERO1LB knockout mice are also viable remains to be answered.
Since the reduction of O2 by Ero1 oxidases produces cytotoxic H2O2 in the ER, the function of ER-resident H2O2-degrading peroxidases and their crosstalk with the PDI family are likely to be fundamental (4, 13). For instance, the activity of peroxiredoxin IV can produce disulfides and channel them into oxidative folding (56). As for Ero1-derived H2O2, it will be most interesting to carry out loss-of-function analyses with two other PDI peroxidases, GPx7 and GPx8, which physically interact with Ero1α (40).
Many additional features of mammalian Ero1 enzymes still require further investigation. The functional significance of covalent Ero1 dimerization (18) that apparently involves Cys166 in Ero1α (29) is completely obscure. Likewise, the molecular basis of membrane association of Ero1α is not known, and it remains to be clarified whether its ER retention/MAM localization is mostly mediated by interaction with the ER membrane (14, 42) or with ERp44 and other PDI family members (41). In addition, the conservation among metazoans of the N-terminal segment including two disulfides in Ero1α (but not Ero1β, Fig. 3) suggests some as yet enigmatic function for this stretch of amino acids. It will also be instrumental to molecularly describe the mode(s) of interaction between Ero1α and ERp44 including the cysteine connectivities and subcellular localization of the complex (ER, ERGIC, or MAM) and compare it to the complex involving PDI (36). Since the catalytic Ero1α–PDI interaction plays a critical role during the PDI-assisted translocation of the cholera toxin A1 subunit from the ER to the cytosol (38), it also remains to be explored, if endogenous ER-associated degradation substrates require this interplay. Further, the mechanism of redox-driven activation of Ero1α/β remains to be elucidated, which—in conjunction with the aforementioned—will set the stage for more exciting Ero1 news in the future.
Footnotes
Acknowledgments
We thank Lars Ellgaard for comments on the article. Work in the authors' laboratory is funded by the Swiss National Science Foundation, the University of Basel, and the August Collin-Fonds. T.R. is a recipient of a Ph.D. fellowship by the Boehringer Ingelheim Fonds.