
Abstract
Introduction
Innovation
Nitric oxide (NO) enhanced oxidative/nitrosative stress, Rho kinase, p53 and cell death receptor expression, and cell death in cultured hepatoma cells. Hydrogen peroxide (H2O2) supplementation increased NO synthase-3 (NOS-3) serine/threonine phosphorylation ratio, NOS-3 activity, NO production, and cell death in 4TO-NOS cells. NOS-3 overexpression or intratumoral administration of NO donor increased p53 and cell death receptor expression and apoptosis, and reduced tumor volume in tumors obtained after subcutaneous implantation of hepatoma cells in a xenograft mouse model. The clinical application of the study is related to the potential sensitization of hepatoma cells by NO to chemotherapy through increased p53 and cell death receptor expression.
CD95 (48 kDa) is a type I transmembrane receptor expressed in activated lymphocytes, in a variety of tissues of lymphoid or nonlymphoid origin, as well as in tumor cells. CD95 ligand (40 kDa) (CD95L/FasL) and anti-CD95 agonist induce trimerization of the receptor with generation of the death-inducing signaling complex (DISC) constituted by a death domain-containing adaptor molecule named Fas-associated death domain (FADD), 2 isoforms of procaspase-8 (procaspase-8/a and procaspase-8/b), procaspase-10, and the cellular FADD-like IL-1 beta-converting enzyme (FLICE) inhibitory protein long and short isoforms (cFLIPL/S). An autoproteolytic multistep activation results in the formation of a caspase-8 heterotetramer containing two large subunits (p18) and two small subunits (p10), which is released into the cytosol to propagate the apoptotic signal. The CD95 promoter has p53-responsive elements (15, 43) that are involved in the upregulation of CD95 receptor expression in lung and colon cancer cell lines (14). The overexpression of p53 increases CD95 receptor expression and apoptosis in human glioma cells (5). Data described above suggest that the increase of p53 expression by NO may induce CD95-dependent cell death. However, it has also been observed that NO reduces CD95-induced apoptosis by cGMP-dependent and independent mechanism in T lymphocytes (52), as well as prevent detachment of cFLIP to the DISC in bronchial epithelial cell line (7) or trophoblast (8). The reduced expression of the CD95 receptor and CD95L and failure of transformed cells to undergo apoptosis have been frequently observed in hepatocarcinoma, especially in advanced stages of cell dedifferentiation (22, 36). Several genetic metabolic defects leading to increased oxidative stress have been related to fatty liver progressing to steatohepatitis and cancer (45). In particular, the lack of transaldolase, critical enzyme in the nonoxidative branch of pentose phosphate pathway, leads to the reduction of hepatic GSH, which appears to be essential in mitochondrial dysfunction, reduction of NO, resistance to CD95-dependent cell death, and development of experimental hepatocarcinogenesis (20). The increase of CD95-mediated apoptosis may be a promising anticancer therapy (55).
We examined the effect of increased intracellular NO concentration using NO donor or NOS-3 overexpression on CD95-dependent cell death in different hepatoma cells lines. The antitumoral properties of NO have also been evaluated in the xenograft mouse model. The study showed that the NO donor increased CD95 expression and caspase-8 and 3 activities in HepG2, Huh7, and Hep3B cells. NOS-3 overexpression increased oxidative/nitrosative stress, p53 and CD95 expression, cFLIPL and cFLIPS shift, and cell death in HepG2 (4TO-NOS) cells. The lack of effect of catalase in 4TO-NOS cells suggests that hydrogen peroxide (H2O2) is not mediating cell death induced by NOS-3 overexpression. However, the additional supplementation with H2O2 increased NOS activity and exacerbated cell death in 4TO-NOS cells. The subcutaneous implantation of 4TO-NOS cell or the intratumoral administration of NO donor reduced tumor cell growth and increased the expression of p53 and cell death receptors in tumors developed in a xenograft mouse model.
Results
NO induced CD95 expression and cell death in HepG2 cells
NO donor such as 2,2′-(hydroxynitrosohydrazino)bis-ethanamide (NONOate) increased CD95 protein (Fig. 1A) expression and caspase-8 (Fig. 1B) and caspase-3 (Fig. 1C) activities in HepG2, Huh7, and Hep3B cells (p≤0.05). The mRNA and protein expression of NOS-3, but not NOS-1 and NOS-2, were routinely detected in the control and cytokine-stimulated HepG2 cell line (data not shown). NOS-3 overexpression increased NOS-3 mRNA (Fig. 2A) and protein (Fig. 2B) expression, as well as p53 protein expression (Fig. 2C) in HepG2 cells. The reduction of NOS-3 protein expression in empty-vector-transfected cells (4TO) compared to wild-type cells may be due to a transfection-related stress cell response in HepG2 (Fig. 2B) cells. In concordance with the effect of NONOate (Fig. 1A), NOS-3 overexpression also increased CD95 protein (Fig. 3A) expression and caspase-8 (Fig. 3B) and caspase-3 (Fig. 3C) activities in the 4TO-NOS cell line (p≤0.05).


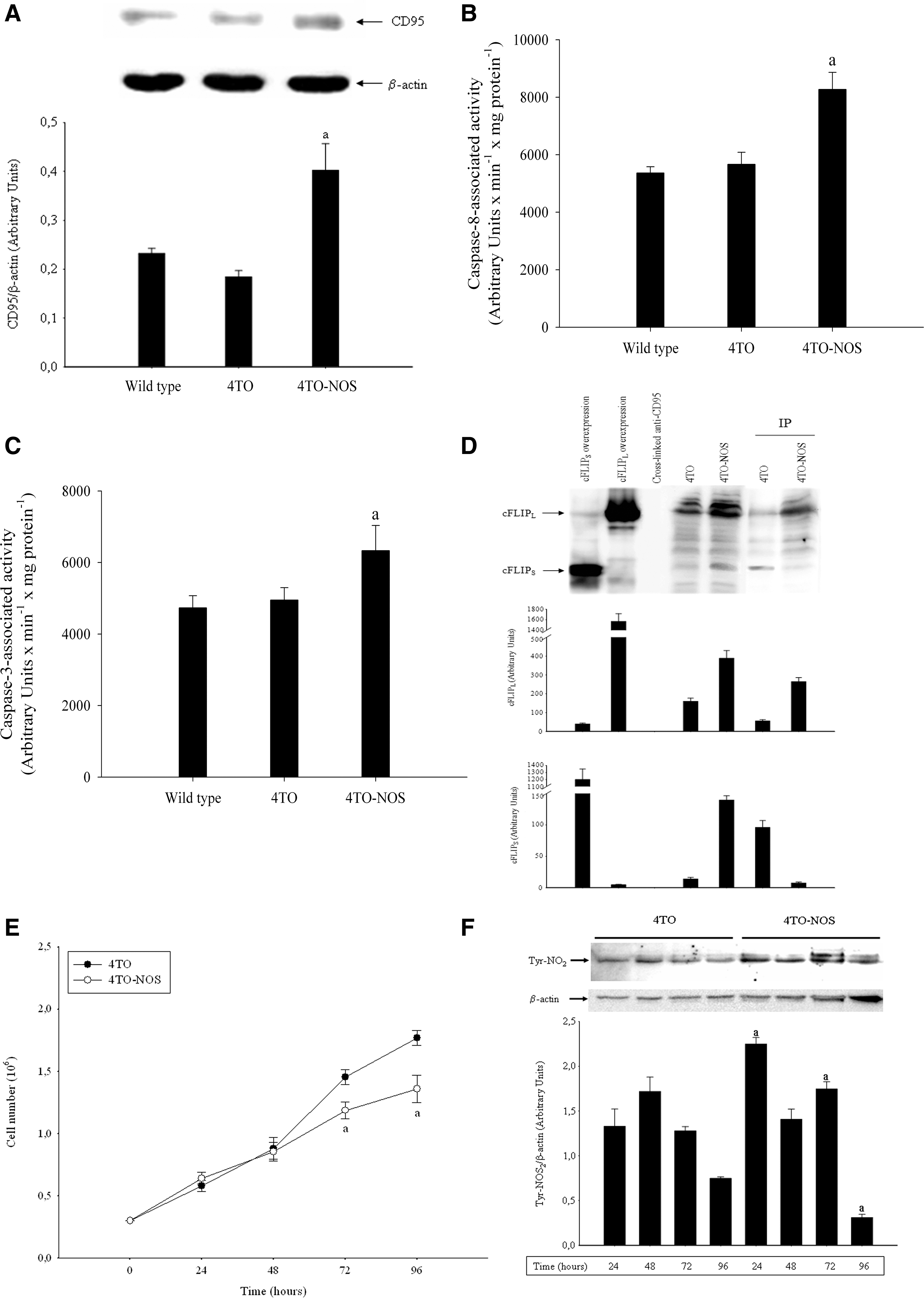
The protein expression of cFLIPL and cFLIPS was assessed in cell lysates and CD95-immunoprecipitated fractions from 4TO and 4TO-NOS cells. The cross-linking procedure effectively prevented the disruption of anti-human CD95 antibodies and protein G Sepharose (Fig. 3D, third lane). NOS-3 overexpression increased cFLIPL and cFLIPS protein expression in cell lysate (Fig. 3D, fifth lane). NOS-3 overexpression increased cFLIPL and reduced cFLIPS protein expression in the CD95-immunoprecipitated fraction (Fig. 3D, seventh lane). The induction of cell death by NOS-3 overexpression was related to an alteration of the total cell number and protein Tyr-nitration in cultured 4TO-NOS cells. In this sense, the total cell number was significantly reduced in 4TO-NOS compared to control (4TO) cells at 72 and 96 h (Fig. 3E) (p≤0.05). The level of protein Tyr-nitration was dramatically increased in 4TO-NOS at all time studied (Fig. 3F).
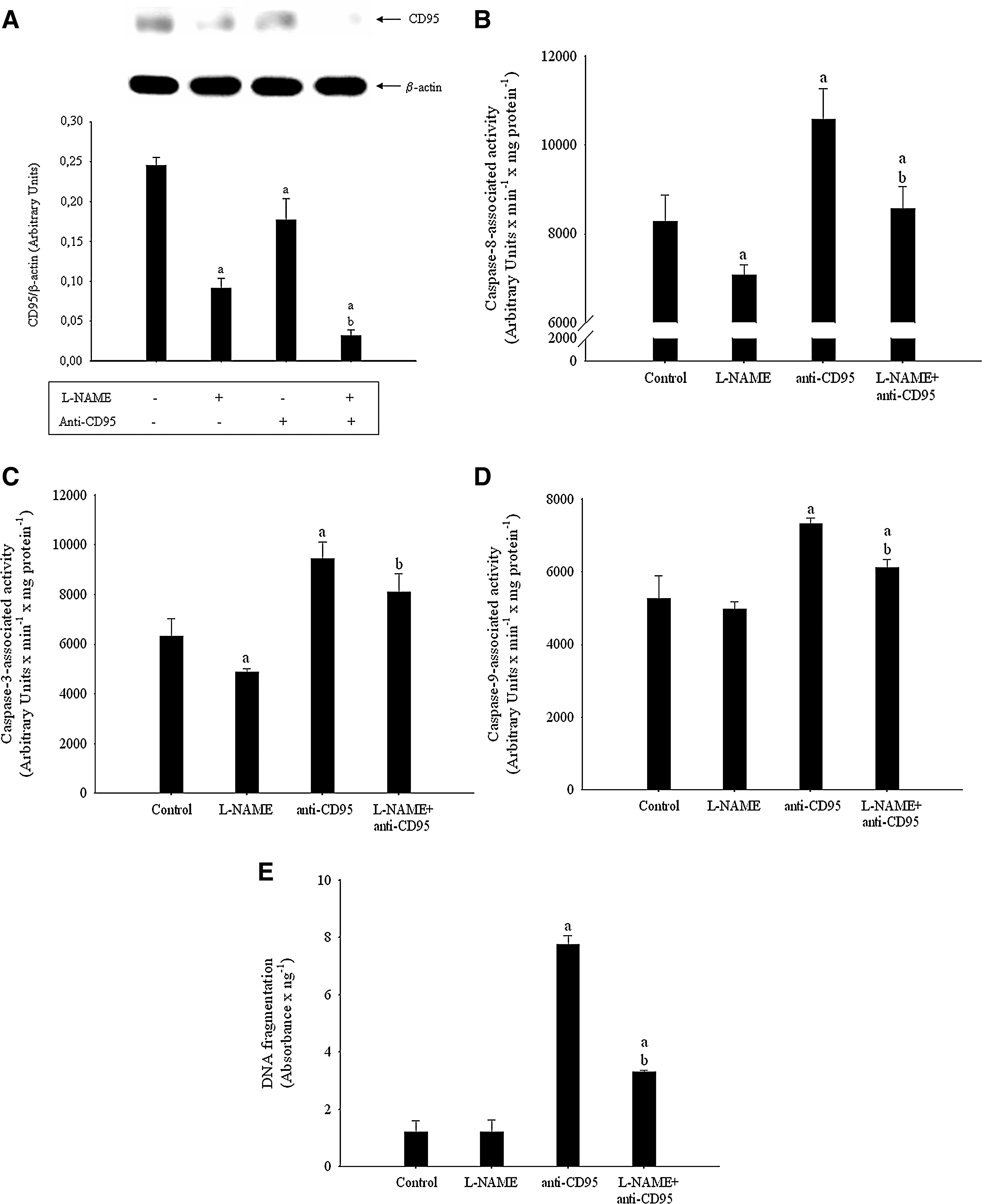
The inhibition of NO production by Nω-Nitro-
Regulation of RhoA kinase and p53 expression in NOS-overexpressed cells
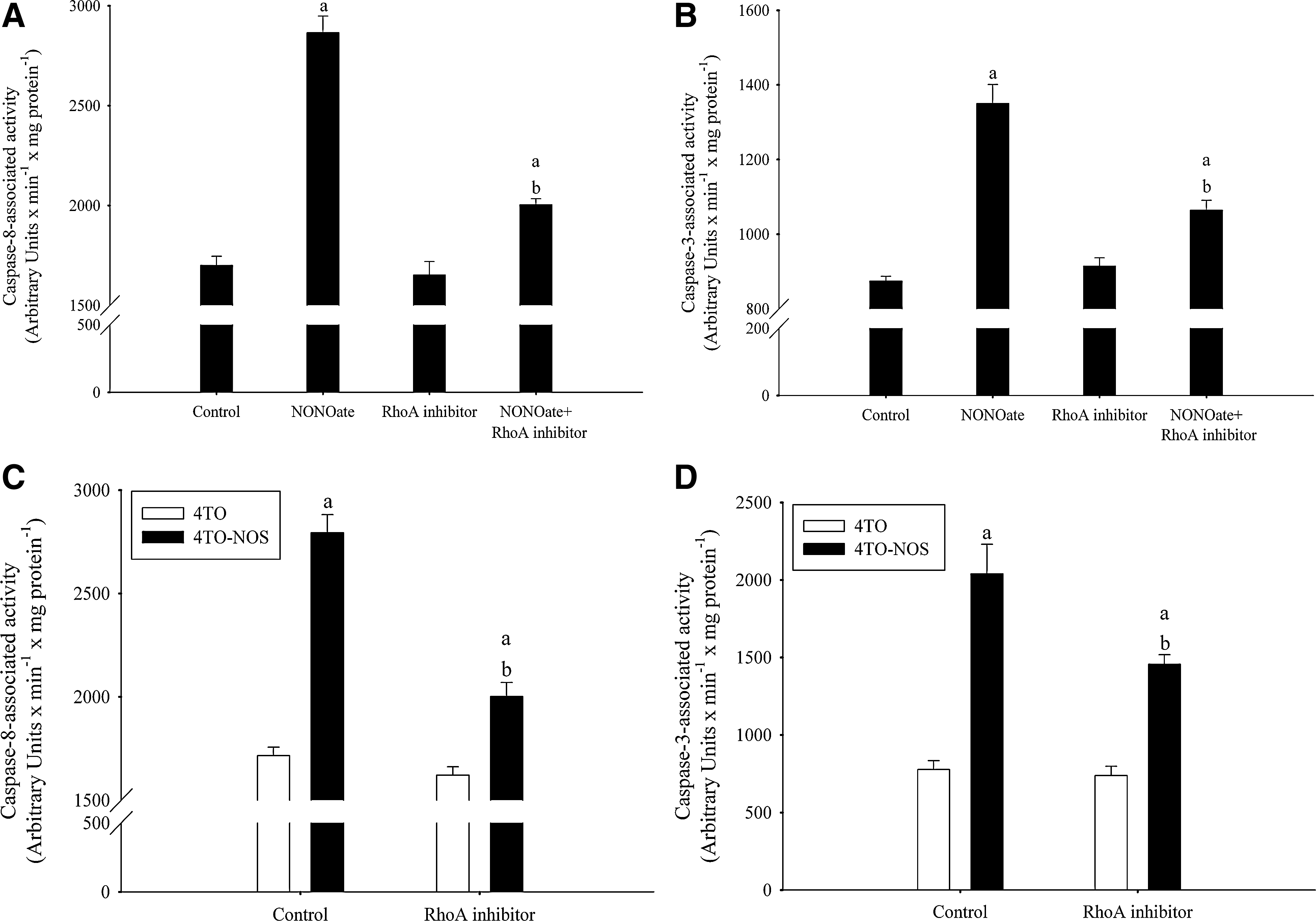
The involvement of RhoA kinase has been evaluated using Y-27632 as a highly potent, cell-permeable, selective, and ATP-competitive inhibitor of ROCK1 and ROCK2. The administration of Y-27632 reduced caspase-8 (Fig. 5A) and caspase-3 (Fig. 5B) activities induced by NONOate, as well as reduced caspase-8 (Fig. 5C) and caspase-3 (Fig. 5D) activities induced by NOS-3 overexpression in 4TO-NOS cells (p≤0.05).
The relationship between NO and p53 accumulation has been previously shown (13, 14). The involvement of p53 in our experimental conditions has been evaluated using siRNA interference technology. In this sense, the knockdown of p53 prevented the increase of CD95 expression (Fig. 6A) and caspase-8 (Fig. 6B) and caspase-3 (Fig. 6C) activities in 4TO-NOS cells (p≤0.05).

Role of oxidative stress during NOS-3 overexpression in HepG2 cells
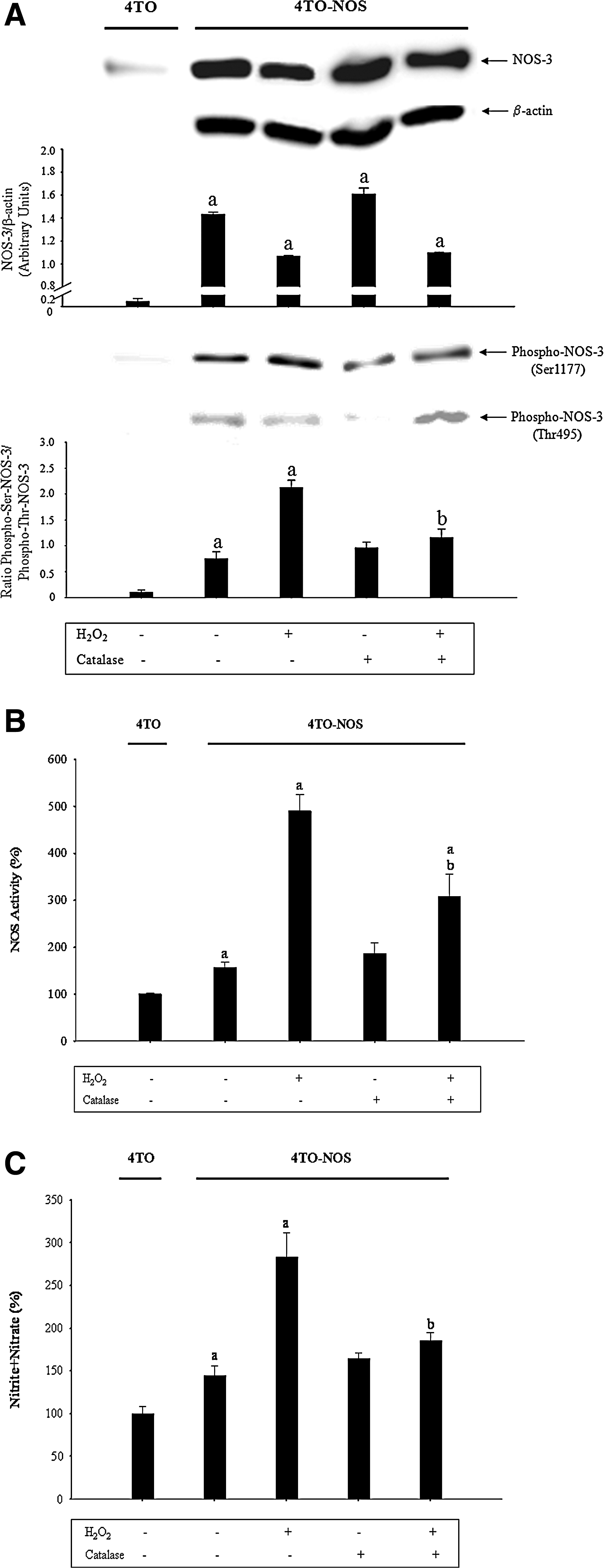
The production reactive oxygen species (ROS) was increased in the 4TO-NOS cell line (Fig. 7A) (p≤0.05). The addition of H2O2 increased caspase-8 (Fig. 7B) and caspase-3 (Fig. 7C) activities in 4TO-NOS. The activity of NOS-3 is tightly modulated by its phosphorylation state. The phosphorylation in Ser1177 and Thr495 residues increases or inhibits NOS-3 activity, respectively (12). H2O2 reduced significantly NOS-3 expression (Fig. 8A, upper panel) (p≤0.05). However, it also increased significantly Ser1177NOS-3 and reduced Thr495NOS-3 expression in 4TO-NOS, which resulted in an increase of the NOS-3 serine/threonine phosphorylation ratio (Fig. 8A, low panel) (p≤0.05). Consequently, H2O2 increased significantly NOS-3 activity (Fig. 8B) and nitrite+nitrate (Fig. 8C) concentration in the culture medium (p≤0.05). Catalase prevented the changes in NOS-3 phosphorylation and activity induced by H2O2 supplementation in 4TO-NOS cells (Fig. 8).

NOS-3-overexpressed HepG2 cells or intratumoral NO donor administration reduced tumor cell growth and increased the expression of p53 and cell death receptors in tumors developed in xenograft mouse model
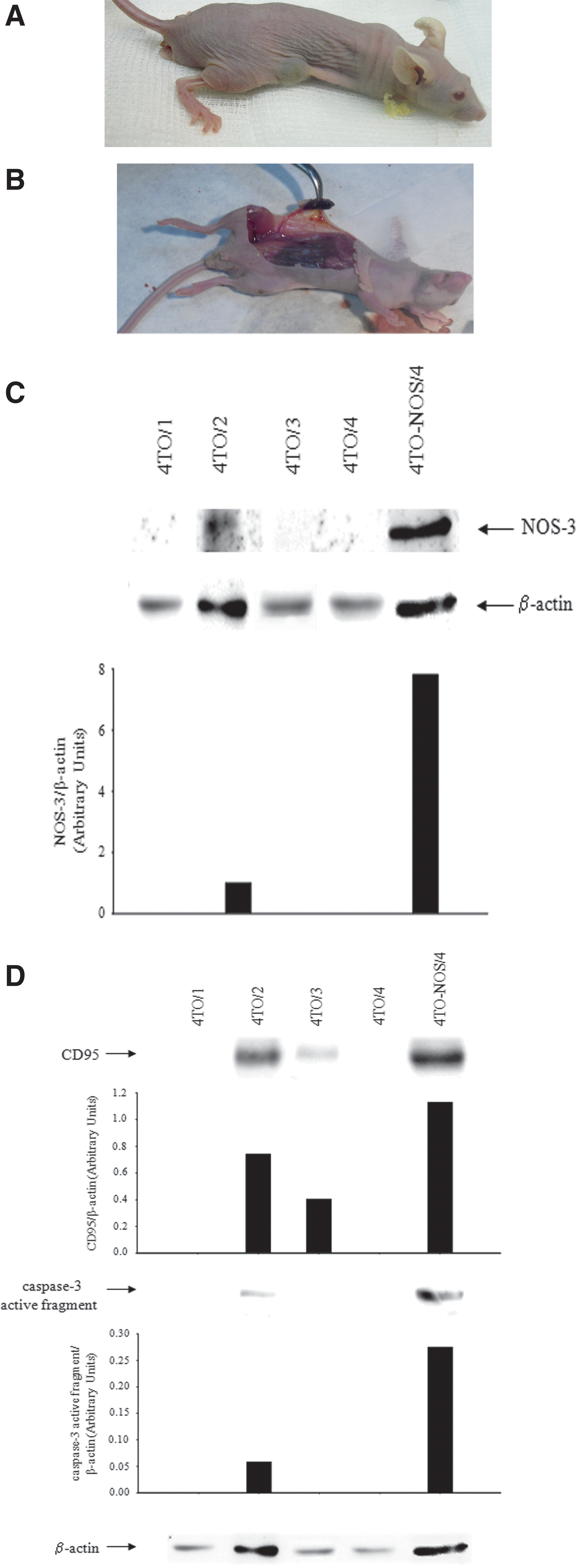
The subcutaneous implantation of 4TO cells generated tumor nodules in all nude mice (Fig. 9A, B and Table 1). Differently, the implantation of 4TO-NOS cells only induced tumor development in one animal that showed increased protein NOS-3 expression (Fig. 9C and Table 1). 4TO-NOS-3-derived tumor showed increased expression of CD95 and caspase-3 active fragment (Fig. 9D) (p≤0.05). The intratumoral administration of NO donor (NONOate) reduced the volume of tumors (80%) generated after subcutaneous 4TO cell implantation in nude mice (Fig. 10A and Table 2). In addition, NONOate increased tumor necrosis factor receptor type I (TNF-R1), CD95, and TNF-related apoptosis-inducing ligand (TRAIL) receptor −1 (TRAIL-R1) protein expression in tumor lysates (Fig. 10B) and in tumor sections (Fig. 10D, F, and H, respectively). Interestingly, the increased levels of cell death receptor protein expression induced by NO donor was associated with an increase of p53 and caspase-3 active fragment in tumors (Fig. 10B)

Cells (10×106) (4TO or 4TO-NOS) diluted in PBS (200 μL) were subcutaneously implanted in the right flank of four BALB/cOlaHsd-Foxn1nu nude mice. The animals were sacrificed at 30 days after cell implant. The volume was calculated as V=[(length)×(width)2×π]/6.
Cells (10×106) (4TO) diluted in PBS (200 μL) were subcutaneously implanted in the right flank of four BALB/cOlaHsd-Foxn1nu nude mice. When tumors reached 5–7 mm (day 20), the half of animals received an intratumoral administration of 2,2′-(hydroxynitrosohydrazino)bis-ethanamide (NONOate, 0.4 mg/kg b.w.) at days 20, 22, 24, and 26, and being sacrificed at day 27. The volume was calculated as V=[(length)×(width)2×π]/6.
Discussion
NO has demonstrated antitumoral activity in different experimental models. The aim of the present study was the identification of potential intracellular mechanisms leading to an increase of cell death by NO in hepatoma cells using different in vitro and in vivo experimental approaches. The increased intracellular concentration of NO using NO donors or stable NOS-3 overexpression promoted oxidative and nitrosative stress, p53 and CD95 expression, and cell death in cultured hepatoma cells. The administration of H2O2 increased the NOS-3 serine/threonine phosphorylation ratio and activity, and cell death in 4TO-NOS cells. 4TO-NOS-implanted cells or NONOate intratumoral administration to 4TO-derived tumors reduced tumor cell growth and increased the expression of p53 and cell death receptors (TNF-R1, CD95, and TRAIL-R1) in a xenograft mouse model.
NO exerts a pro- and antiapoptotic role depending on its local concentration, target cell, and the presence of other radical species such as the superoxide anion (4). The proapoptotic activity of the NO has been associated with the alteration of the mitochondrial potential and free-radical generation, activation of p53, alteration of the bcl-2 expression, and neutral sphingomyelinase-dependent production of ceramide (37, 57). The antiapoptotic properties of NO have been related to nitrosylation of different proteins such as caspases (28), PKCɛ, which reduces cFLIP release from the DISC (8), and cFLIP, avoiding its degradation by ubiquitination (7). NO increases CD95 expression and CD95-induced cell death in pulmonary artery smooth muscle cells (21) and neurons (38). The exogenous administration of NO donor increased CD95 expression and cell death in HepG2, Huh7, and Hep3B cells (Fig. 1). Interestingly, the expression of p53 is native, mutated, and absent in HepG2, Huh7 and Hep3B cells, respectively (23). NOS-3 overexpression increased p53 and CD95 expression, protein Tyr-nitration, and cell death in HepG2 cells (Figs. 2 and 4). The knockdown p53 experiments showed that p53 is involved in the increase of CD95 expression and cell death induced by NONOate and NOS-3 overexpression in HepG2 cells (Fig. 6). The alteration of the p53 gene in Huh7 and Hep3B cells suggests that other p53 gene family members may be involved during the induction by NO donor of CD95 expression and cell death in those hepatoma cell lines. In fact, p63 and p73 have also been associated with the increase of CD95, TNF-R1, and TRAIL-R1 expression and cell death sensitization in different hepatoma cell lines (53).
cFLIP is the homolog mammalian form of the previously described family of viral inhibitors (v-FLIPs) present in several γ-herpes viruses and molluscipoxvirus (54). cFLIP blocks the early signaling events of CD95 by interfering with the recruitment and activation of caspase-8 by FADD (24). The overexpression of cFLIP is involved in rendering cells resistant to death receptor-mediated apoptosis in ovarian cancer cells (1), and its increased expression has been associated with tumor resistance and avoiding immune surveillance (39). The reduction of the CD95/CD95L system with failure of transformed cells to undergo apoptosis has been frequently observed in HCC (22, 36). The regulation of the CD95/CD95L system represents a potential crucial step in tumorigenesis and a promising target for drug development against hepatocarcinogenesis (44). To date, several small molecules have been known to lower cFLIP expression and to sensitize tumor-resistant cells to death-receptor-mediated apoptosis (27). The downregulation of cFLIP by cytotoxic agents has also been shown to sensitize cells to death receptor-induced apoptosis (1, 29, 44). The study evaluated the alteration of cFLIPL/S bound to CD95 in 4TO-NOS cells. NOS-3 overexpression was associated with an increase of cFLIPL and reduction of cFLIPS bound to the CD95 receptor (Fig. 3D). The relative high expression of cFLIPS in cell lysate suggests the active degradation of cFLIPS in the DISC obtained from 4TO-NOS cells (Fig. 3D). The administration of NO donors altered the susceptibility of cFLIPL to ubiquitin–proteasome-mediated degradation and CD95-dependent cell death in a human bronchial epithelial cell line (6). Lavrik et al. (35) have observed that CD95 stimulation involves DISC formation at the cell membrane, and further formation of the cytosolic death effector domain protein-containing complex II composed of procaspase-8a/b, three isoforms of cFLIP (cFLIPL, cFLIPS, and cFLIPR), and FADD. Recently, it has been proposed that cFLIPL, but not cFLIPS, can be either anti- or proapoptotic depending on its expression level (6, 25). The presence of cFLIPL/S in cell lysate supports the existence of the cytosolic death complex during cell death in 4TO-NOS cells.
The induction of oxidative stress is identified as an early signaling event that promotes CD95 membrane trafficking and CD95-dependent cell death stimulation (48). ROS induce cFLIP degradation in human lung epithelial cells (32). NOS-3 overexpression increased oxidative/nitrosative stress, protein Tyr-nitration, CD95 expression, and cell death in 4TO-NOS-3 cells (Figs. 3, 4, and 7). These data are in agreement with previous studies in which induction of cell death by oxidative stress paralleled the increase of CD95 expression (16, 42, 58). The interaction of NO with superoxide anion (O2 •−) gives raise other more powerful radical species such as peroxinitrite involved in protein Tyr nitration (46, 51), which is relevant in cell death according to cell target, temporal interaction, and subcellular localization (10).
Rho regulates major cellular functions, such as membrane trafficking, phospholipid metabolism, cell cycle progression, cell transformation, apoptosis, and transcriptional activation (11, 19, 56). It has been shown that the NO-PKG pathway induces cell migration and adhesion via Rac1 activation in human artery smooth muscle cells (49). Our data showed that RhoA is involved in the induction of cell death by NO donor and NOS-3 overexpression in HepG2 cells (Fig. 5). It is feasible that cell death may results upon NOS/ROS-sensitive activation of Rho kinase.
NOS-3 activity is modulated by Ca2+/calmodulin, protein phosphorylation, protein–protein interactions, and subcellular localization. NOS-3 has been found to be phosphorylated at several consensus motifs by a number of kinases such as protein kinase (PK) type A (PKA), PKB/Akt, 5' adenosine monophosphate-activated protein kinase (AMPK), PKC, and Ca2+/calmodulin-dependent kinase II. (24). In particular, a serine residue in the reductase domain (Ser1177) and a threonine residue (Thr495) located within the CaM-binding domain are particularly important in promoting and inhibiting NOS-3 activity, respectively (12). The activation of phosphatidylinositol 3-kinase, serine kinases Akt, and PKA phosphorylates NOS-3 on Ser1177 and increases NOS-3 activity (9). The constitutive PKC probably phosphorylates NOS-3 on Thr495 (41). The protein phosphatase type I is responsible for dephosphorylation of NOS-3 on Thr495 based on its specificity for this site in NOS-3, whereas the protein phosphatase type 2 is responsible for dephosphorylation of NOS-3 on Ser1177 (41). NOS-3 overexpression induces oxidative stress in HepG2 cells (Fig. 7A). The lack of effectiveness of catalase in reducing caspase-8 and 3 activities in 4TO-NOS suggested that H2O2 is not critical during the induction of cell death in 4TO-NOS cells (Fig. 7B, C). However, H2O2 (100 μM) supplementation increased the NOS-3 Ser1177/Thr495 phosphorylation ratio, NOS activity, NO production, and cell death in 4TO-NOS cells (Figs. 7 and 8). The upregulation of superoxide dismutase has also been observed to stimulate NOS-3 activity induced by shear stress on endothelial cells (10).
The overexpression of NOS-3 induced oxidative/nitrosative stress and protein Tyr nitration, which was related to a reduction of the total cell number compared to control cells (Figs. 3E, F, 7, and 8). The effect of NOS-3 overexpression in cumulative proliferation doubling time has been addressed in a previous study (2). The proapoptotic properties of NOS-3 overexpression in HepG2 cells and the intratumoral administration of NO donor have been evaluated in an experimental xenograft mouse model. The subcutaneous implantation of 4TO cells induced the presence of tumors in all animals (4/4), but implantation of 4TO-NOS cells only induced tumor development in one animal (1/4) (Fig. 9 and Table 1). The reduced potential of 4TO-NOS to develop tumors was associated with increased expression of CD95 and caspase-3 active fragment (Fig. 9D). The intratumor delivery of NOS-2-generating cells has also been shown to reduce the tumor size induced by an ovarian and colon cancer cell implant in a xenograft mouse model (59). The administration of NO donor in 4TO-derived tumors increased the expression of p53, cell death receptors (TNF-R1, CD95, and TRAIL-R1), and caspase-3 active fragment in tumors, and reduced the tumor volume (Fig. 10).
Materials and Methods
Materials
All reagents were from Sigma-Aldrich Chemical Co., unless otherwise stated. NONOate (NOC-18) was purchased from Calbiochem. NO and RhoA were inhibited using L-NAME (Sigma-Aldrich Chemical Co.) and Y-27632 (ALX-270-333; Enzo Life Sciences AG). The study protocol has been approved by the Ethics Committee of the Institution.
Cell lines and culture conditions
HepG2 and Hep3B cell lines were obtained from ATCC-LGC. The Huh7 cell line was obtained from Apath L.L.C. HepG2 cells were transfected with the pcDNA4/TO (5100 pb; Invitrogen, Molecular Probes, Inc.) expression vector containing NOS-3 cDNA (3462 pb; NCBI, ImaGenes, full-length cDNA clone sequence BC063294) sequence under the control of the cytomegalovirus promoter (4TO-NOS). Twenty-one clones of HepG2 cells transfected with a NOS-3-overexpressing vector were obtained. This number was comparatively low in relation to that obtained in cells transfected with an empty vector. The selected clone showed the highest NOS-3 protein expression assessed by Western blot analysis. The expression of NOS-3 in 4TO cells was low in all clones. The transfected cells were selected with zeocine (15 mg/L; Invitrogen). The experiments measuring the expression of cFLIP isoforms by Western blot analysis in the cell lysate and CD95 immunoprecitation fractions required adequate positive controls obtained from 4TO-NOS cells stably transfected with the pcDNA 3.1 (+) (5428 pb; Invitrogen) vector, including either cFLIPL (1443 pb; NCBI, ImaGenes, full-length cDNA clone sequence NM_001127183) or cFLIPS (666 pb; NCBI, ImaGenes, full-length cDNA clone sequence NM_001127184), generating the stable 4TO-NOS-cFLIPL and 4TO-NOS-cFLIPS cell lines, respectively. In this case, cotransfected cell lines were additionally selected with geneticine (200 mg/L; Sigma-Aldrich Chemical Co.). Cells were routinely maintained in an EMEM (Eagle's Minimum Essential Medium; Sigma-Aldrich Chemical Co.) (pH 7.4) supplemented with 10% fetal bovine serum, 2.2 g/L HCO3Na, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin, and the corresponding selection antibiotic in 5% CO2 in air at 37°C.
Cells were maintained for 48 h before induction of cell death by agonistic anti-human CD95 antibodies (0.5 μg/ml; MBL Medical & Biological Laboratories CO., Ltd.). NONOate (0.1 mM) or L-NAME (5 mM) was administered 48 or 24 h before administration of cell death inducer, respectively. RhoA inhibitor (10 μM) was administered 1 h before NONOate administration, collecting samples 2 h later in wild-type HepG2 cells, or 12 h in 4TO and 4TO-NOS cells. The role of oxidative stress was carried out by H2O2 (100 μM; Merck KGaA) supplementation in presence of or not of catalase (1000 U/ml; Sigma-Aldrich Chemical Co.) administered 30 min before the oxidant. Samples were collected at 1, 6, or 12 h according to the experiments. The total cell number was determined by cell counts at 24, 48, 72, and 96 h after 24 h of cell stabilization.
Knockdown of p53
p53 was knocked down in 4TO and 4TO-NOS cells (20,000 cells/cm2). The transfection reagent oligofectamine (12252-011; Invitrogen) and the siRNA transfection medium (sc-36868; Santa Cruz Biotechnology, Inc.) were preincubated for 5 min at room temperature. Either p53 siRNA (160 pmol, sc-29435; Santa Cruz Biotechnology, Inc.) or control siRNA-A (80 pmol, sc-37007; Santa Cruz Biotechnology, Inc.) was mixed with the above-obtained mixture and incubated for 20 min at room temperature. Afterward, the interference solution was added to cultured cells for 7 h in absence of fetal bovine serum and antibiotic/antimycotic solution. Cell lysates were collected 24 h after cell transfection.
Measurement of cell death
Caspase-3-, 8-, and 9-associated activities, caspase-3 processing, and DNA fragmentation were used as parameters of apoptosis. Cells were treated with 20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 2 mM EDTA, 1% Triton X-100, 1 mM Na3VO4, 50 mM NaF, 1 mM PMSF, and a protease inhibitor cocktail (04-693-159-001; Roche Diagnostics GmbH) for the measurement of caspase-3, 8-, and 9-associated activity using Ac-DEVD-AFC (100 μM; Alexis Biochemicals, Enzo Life Sciences), Ac-LETD-AFC (100 μM; Alexis Biochemicals), and Ac-LEHD-AFC (100 μM; Alexis Biochemicals), respectively (17). The AFC-associated fluorescence (Ex 400; Em 505) was recorded using a GENios Microplate Reader (TECAN).
Caspase-3-active fragment (p17) was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) coupled to Western blot analysis. Proteins (100 μg) were separated by 14% SDS-PAGE and transferred to nitrocellulose. The membranes were incubated with the corresponding commercial primary antibodies against the caspase-3 active fragment (p17) (1/1000) (sc-7148; Santa Cruz Biotechnology, Inc.). The corresponding anti-rabbit (1/50,000) (sc-2301; Santa Cruz Biotechnology, Inc.) secondary antibodies were coupled to horseradish peroxidase revealing protein content by ECL (RPN2135; GE Healthcare). β-actin (1/5000) (ab8227; Abcam) was used as a cell protein-loading control. DNA fragmentation was measured by a commercial Cell Death Detection ELISAPlus (Roche) assay.
Measurement of NOS activity
NOS-3 activity was assessed following the procedure described by Knowles and Salter (30). Briefly, cells were treated with an extraction buffer (320 mM sucrose, 20 mM HEPES, 1 mM EDTA, 1 mM DTT, 10 μg/ml leupeptin, 2 μg/ml aprotinin, 10 mg/ml PMSF) at pH 7.2, collected, centrifuged at 400 g 5 min, and the resulting pellet was resuspended in a small volume of ice-cold lysis solution, homogenized with a micropistile, and sonicated three cycles of 10 s at 10-μm amplitude with 30 s cooling in ice/water in between. Samples were centrifuged at 18,000 g 5 min, precipitated, (250 μg protein) resuspended in an extraction buffer, and incubated in 50 mM KH2PO4, 1 mM CaCl2, 0.1 mM
Measurement of NO
The production of NO was measured by quantification of its related end products, nitrite/nitrate. In the assay, nitrate was converted to nitrite by nitrate reductase (EC 1.6.6.2), and the total nitrite was measured using the Griess reaction (18). Briefly, the samples were incubated with nitrate reductase (0.2 U/ml), FAD (5 mM), and NADPH (50 mM) for 20 min at 37°C. The reaction was stopped by the addition of sodium pyruvate (10 mM) and lactate dehydrogenase (24 mg/ml) for 5 min at 37°C, and precipitated with 1.4% ZnSO4. The total nitrite was allowed to react with the Griess reagent (1% sulfanilamide, 2.5% PO4H3, and 0.1% n-naphthylethylenediamine) for 10 min at 37°C, and was read at 540 nm using a GENios Microplate Reader (TECAN).
Determination of mitochondrial ROS
The production of ROS in mitochondria was monitored using a fluorescent probe, such as 2',7'-dichlorodihydrofluorescein diacetate (DCFH2-DA; Molecular Probes Europe BV), hydrolyzed by intracellular esterases to form DCFH2 in the cytoplasm, and oxidized by ROS to yield the highly fluorescent 2′,7′-dichlorofluorescein (DCF). The stock solution of DCFH2-DA was maintained in dimethyl sulfoxide and stored at −20°C until use. Cells were incubated with H2DCFDA (2 μM) for 30 min in a culture medium. Cells were washed with phosphate-buffered saline (PBS), and incubated with digitonin (10 μM) for 5 min to permeabilize cells to eliminate probe not retained in the mitochondria. Cells were maintained in PBS to avoid any interference with the measurement of cell fluorescence. The production of free radicals was in situ assessed as the enhancement on the fluorescence at Ex 500 nm/Em 520 nm measured in a GENios Reader (TECAN), respectively. The corresponding controls were carried out in cells incubated in presence/absence of solvent, dye, and cells.
Expression of TNF-R1, CD95, TRAIL-R1, NOS-3, and p53
The expression of TNF-R1, CD95, TRAIL-R1, phosphorylated and unphosphorylated NOS-3, and p53 protein expression were determined by SDS-PAGE coupled to Western blot analysis. Proteins (100 μg) were separated by 6% (NOS-3), 10% (p53), and 12% (TNF-R1, CD95, and TRAIL-R1) SDS-PAGE and transferred to nitrocellulose. The membranes were incubated with the corresponding commercial primary antibodies against TNF-R1 (1/1000) (sc-7895; Santa Cruz Biotechnology, Inc.), CD95 (1/1000) (sc-715; Santa Cruz Biotechnology, Inc.), TRAIL-R1 (1/1000) (sc-6823; Santa Cruz Biotechnology, Inc.), NOS-3 (1/1000) (sc-654; Santa Cruz Biotechnology, Inc.), Ser1177 phospho-NOS-3 (1/1000) (ref 9571; Cell Signaling Technology, Inc.), Thr495 phospho-NOS-3 (1/1000) (ref 9574; Cell Signaling Technology, Inc.), and p53 (1/1000) (sc-6243; Santa Cruz Biotechnology, Inc.). The corresponding anti-rabbit (1/50,000) (sc-2301; Santa Cruz Biotechnology, Inc.) or anti-mouse (1/50,000) (sc-2031; Santa Cruz Biotechnology, Inc.) secondary antibodies were coupled to horseradish peroxidase revealing protein content by ECL (RPN2135; GE Healthcare). β-actin (1/5000) (ab8227; Abcam) was used as a cell protein-loading control.
TNF-R1, CD95, and TRAIL-R1 expression in 4% paraformaldehyde-fixed tumor tissue sections (5 μm) mounted on glass slides coated with polylysine was also assessed by confocal microscopy. Samples were incubated overnight at 4°C with primary antibodies (anti-TNF-R1, anti-CD95, and anti-TRAIL-R1) diluted in 100 mM PBS, pH 7.4, containing 2% normal mouse serum and 0.05% Triton X-100. Sections were washed three times with PBS and incubated for 1 h at room temperature with anti-goat Alexa Fluor 488- (1/200) (A11055; Invitrogen, Molecular Probes, Inc.) and anti-rabbit Alexa Fluor 594- (A11058; Invitrogen, Molecular Probes, Inc.) labeled antibodies. Sections were washed several times with PBS, embedded in 70% glycerol, and mounted with cover slide. The specificity of the immunoreactivity was verified by incubating the sections with nonspecific primary antibodies. Six replicated immunostained sections were analyzed by an LSM 5 Exciter confocal microscope (Carl Zeiss) using a confocal imaging system (ZEN 2008; Microimaging Carl Zeiss).
Expression of nitrated proteins
Proteins were obtained by cell lysis in 20 mM Tris–HCl (pH 7.5), 0.1% NP-40, 0.1% SDS, 10 μg/ml leupeptin, 2 μg/ml aprotinin, and 10 mg/ml PMSF supplemented with a protease inhibitor cocktail (Sigma). Samples (3 μg protein) were separated by 12% SDS-PAGE in nonreducing conditions and transferred to nitrocellulose. The membranes were incubated with anti-3-nitrotyrosine (1/10,000) (N5538; Sigma-Aldrich Chemical Co.) primary antibodies, and anti-mouse (1/50,000) (sc-2031; Santa Cruz Biotechnology, Inc.) as secondary antibodies, which were coupled to horseradish peroxidase revealing protein content by ECL (GE Healthcare). β-actin (1/5000) (ab8227; Abcam) was used as a cell protein-loading control.
Expression of cFLIPL and cFLIPS
The expression of cFLIPL and cFLIPS bound to CD95 receptor was assessed by CD95 immunoprecipitation coupled to SDS-PAGE and Western blot analysis. A volume of cell lysate (10 mg protein) was mixed with an immunoprecipitation buffer (20 mM Tris-HCl, pH 7.5, 1% Triton X-100, 150 mM NaCl, 10% glycerol, 1 mM Na3VO4, 50 mM NaF, 2 mM EDTA, 1 mM PMSF, and cocktail protease inhibitors) and 2.4 μg anti-human CD95 (sc-715; Santa Cruz Biotechnology, Inc.) antibodies chemically (dimethyl pimelimidate; Sigma-Aldrich Chemical Co.) cross-linked with protein G Sepharose (40 μL; GE Healthcare) at 4°C overnight with rotation. Afterward, the sample was washed with the immunoprecipitation buffer, resuspended in a loading buffer without β-mercaptoethanol, and run into 12.5% SDS-PAGE. The expression of cFLIPL and cFLIPS was assessed by Western blot analysis using anti-cFLIP (1/1000) (Alx-804-428; Enzo Life Sciences) as primary antibodies, and anti-mouse (1/50,000) (sc-2031; Santa Cruz Biotechnology, Inc.) as secondary antibodies. Different controls such as 4TO-NOS-cFLIPS- and 4TO-NOS-cFLIPL-overexpressed cells, cross-linked anti-human CD95 without sample, and cell lysates from each nonimmunoprecipitated samples were run in parallel with the immunoprecipitated samples.
Effect of NOS-3 overexpression and NO donor intratumoral administration in tumor growth in xenograft mouse model
All animal care and experimentation procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the National Academy of Sciences. Two different experimental strategies were designed in male BALB/cOlaHsd-Foxn1nu nude mice (Harlan Laboratories) aged 4 weeks (weighing 18–22 g). Twelve male animals were randomly divided into three groups according to subcutaneous administration of solvent, 4TO and 4TO-NOS cells. Cells (10×106) were diluted in PBS (200 μl) and administered subcutaneously into the right flank of mice. The animals were sacrificed 30 days after cell implantation. The tumour volume was calculated using the formula: V=[(length)×(width)2×π]/6. The second experimental strategy was developed in ten nude mice that received the subcutaneous administration of 4TO (10×106) cells diluted in PBS (200 μl) into the right flank of animals. When tumors reached 5–7 mm (day 20), the half of animals received an intratumoral administration of NONOate (0.4 mg/kg b.w.) at days 20, 22, 24, and 26, and were sacrificed the day after.
Statistical analysis
Results are expressed as mean±SEM of five independent experiments. Data were compared using ANOVA with the least significant difference test as post hoc multiple comparison analysis. The statistical differences were set at p≤0.05. The groups with “a” were significantly different versus the corresponding control group. The groups with “b” were significantly different versus the corresponding group without anti-CD95 antibodies.
Footnotes
Acknowledgments
This study has been supported by the Instituto de Salud Carlos III (FIS 09/00185), Consejería de Innovación, Ciencia y Empresa (“Estancia Excelente,” call 2007), and CIBERehd (Foreign Research Stage, call 2008). CIBERehd was funded by the Instituto de Salud Carlos III.
Author Disclosure Statement
Raúl González, Gustavo Ferrín, Patricia Aguilar-Melero, Isidora Ranchal, Clara I. Linares, Rosario I. Bello, Manuel De la Mata, Vladimir Gogvadze, José A. Bárcena, José M. Álamo, Sten Orrenius, Francisco J. Padillo, Boris Zhivotovsky, and Jordi Muntané have no actual or future commercial associations with the study that might create a conflict of interest in connection with the submitted manuscript.