
Abstract
Introduction
DNA Methylation
The inheritance of information based on gene expression levels is known as epigenetics, as opposed to genetics, which refers to the information transmitted on the basis of gene sequence (40). Epigenetic mechanisms include DNA methylation, covalent modifications of histones, and RNA interference, all of which alter gene expression and function (38). In contrast to genetic changes in human diseases, epigenetic changes are gradual in onset and progressive, their effects are dose dependent, and are potentially reversible by dietary and pharmacologic manipulations (11, 86).
Of the various epigenetic mechanisms, DNA methylation of the cytosines located within the cytosine-guanine (CpG) sequences is the most widely studied and well characterized. DNA methylation is heritable, tissue- and, species specific, and an important epigenetic inverse determinant in gene expression (11, 72). Methylation also plays a pivotal role in the maintenance of DNA integrity and stability, and in chromatin modifications (41, 72). In normal cells, up to 80% of all CpG sites in human DNA are methylated (41, 72). However, this global methylation occurs primarily in the bulk of the genome where CpG density is low, including exons, noncoding regions, and repeat DNA sites, and allows correct organization of chromatin in active and inactive states (Fig. 1) (61). By contrast, most CpG rich areas clustered in small stretches of DNA termed “CpG islands,” which span the 5′ end of approximately half of all transcribed human genes including the promoter, untranslated region, and exon 1, are unmethylated in normal cells, thereby allowing transcription to occur (Fig. 1) (41, 72, 135). The methylation of promoter region CpG islands, termed gene-specific methylation, causes stable, heritable transcriptional silencing (41, 72). This is mediated by the transcriptional repressor, methyl-CpG binding protein-2 (MeCP2), which binds methylated CpG islands and recruits a complex containing a transcriptional co-repressor and a histone deacetylase (16, 73). The deacetylation of histones suppresses transcription by allowing tighter nucleosomal packaging, thus rendering an inactive chromatin conformation (15, 23).

DNA methylation is a dynamic process between active methylation, mediated by CpG DNA methyltransferases (DNMT1, 3a, 3b) using S-adenosylmethionine (SAM) as the methyl donor, and active and passive removal of methyl groups from 5-methylcytosine residues by a purported demethylase (MBD2) (99). During embryogenesis, active and passive demethylation of the paternal and maternal methylation patterns, respectively, occurs, which erases significant parts of the parental DNA methylation. This is followed by de novo methylation, which establishes a new DNA methylation pattern soon after implantation, with methylation limited to non-CpG island areas, except for the rare genes silenced in normal cells (Fig. 2) (99, 133). Themaintenance of CpG DNA methyltransferase (DNMT1) uses hemimethylated sites that ensure DNA methylation patterns, whereas de novo CpG DNA methyltransferases (DNMT3a, 3b) do not require pre-existing methylation and, therefore, establish a new DNA methylation pattern (99).

DNA methylation is critically involved in regulating many developmental and cellular processes, including embryonic development, transcription, chromatin structure, X chromosome inactivation, genomic imprinting, and chromosome stability (134). Aberrant patterns and dysregulation of DNA methylation are mechanistically related to the development of several human diseases, including cancer (41, 72).
Folate
Folate is a water-soluble B vitamin that is naturally present in many foods, including green leafy vegetables, asparagus, broccoli, Brussels sprouts, citrus fruit, legumes, dry cereals, whole grain, yeast, lima beans, liver, and other organ meats. FA is the fully oxidized monoglutamyl synthetic form of folate that is commercially used in supplements and in fortified foods (Fig. 3). Naturally occurring folates are very unstable, rapidly lose their activity in foods, and are easily oxidized under low pH (114). Folate bioavailability varies widely depending on the food source and preparation method (114). Approximately 50%–75% of the original folate values are lost through food harvesting, storage, processing, and preparation (114). In contrast, FA is highly stable for months or even years and has a higher bioavailability compared with naturally occurring folates (114). FA is neither found in nature nor is it a normal metabolite. It should be reduced, first to dihydrofolate (DHF) and then to tetrahydrofolate (THF) by DHF reductase (DHFR) and methylated to 5-methylTHF (the predominant folate found in blood) in the liver and, to a lesser degree, in the intestine (179), before it can enter the folate cycle (Fig. 4). Since the capacity to reduce folate is limited, high intakes of FA result in its appearance of being unaltered in circulation (76).


Biochemical Functions of Folate
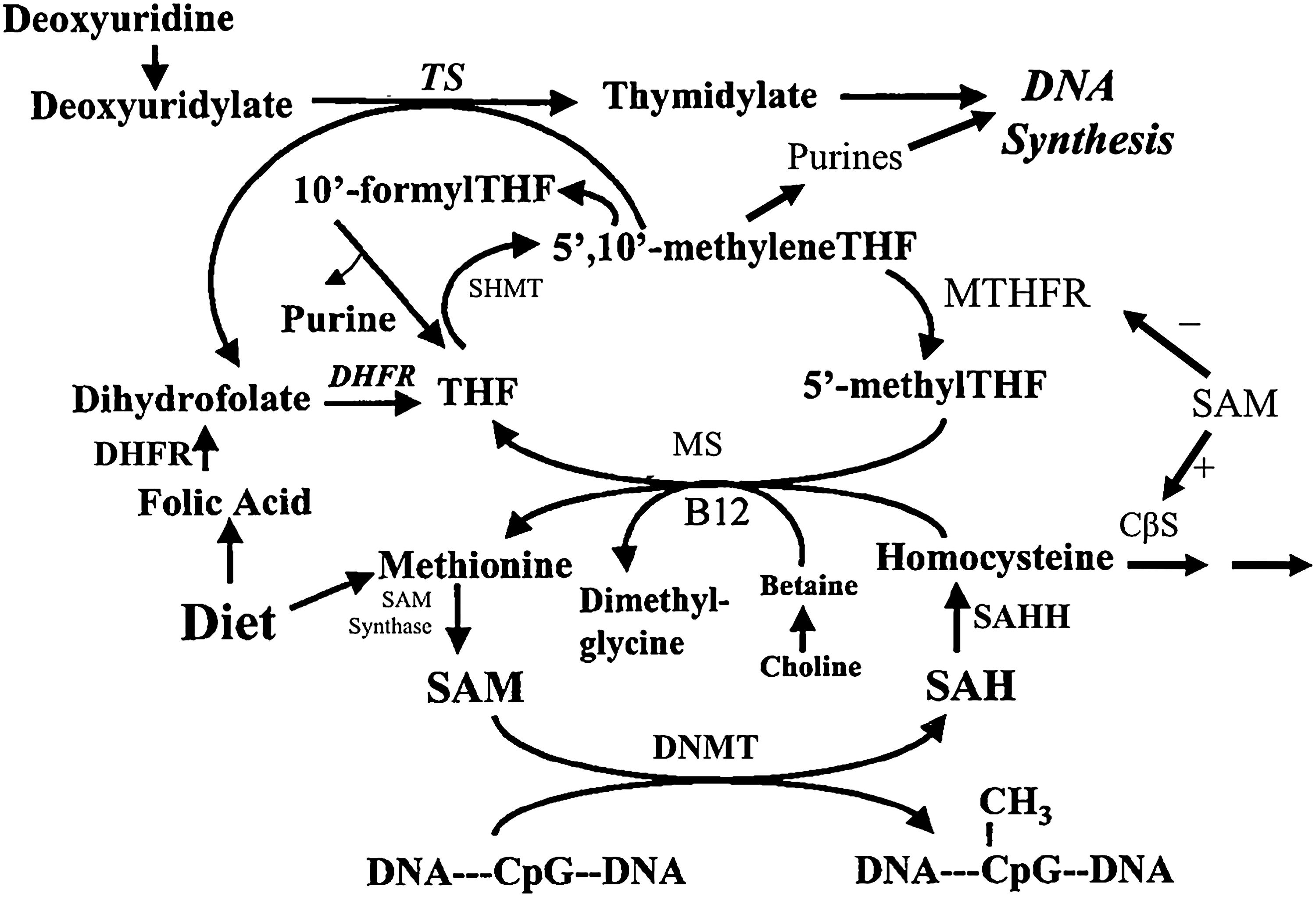
Folate participates in the transfer of one-carbon units involved in nucleotide biosynthesis, methionine cycle, and biological methylation reactions (Figs. 4 and 5) (141). As an essential cofactor for the de novo biosynthesis of nucleotides (106, 141, 169), folate is important in DNA synthesis, stability and integrity, and repair. In the methionine cycle, 5-methylTHF transfers single methyl groups to homocysteine, catalyzed by methionine synthase (20, 98), to synthesize methionine. This ensures the provision of SAM, the primary methyl group donor for most biological methylation reactions (84, 86). After donating the methyl group, 5-methylTHF is converted to THF and then to 5,10-methyleneTHF by serine hydroxymethyltransferase (58, 155). 5,10-methyleneTHF is a key substrate in folate metabolism, which can be directed toward nucleotide biosynthesis or toward methionine regeneration (59). Methylenetetrahydrofolate reductase (MTHFR) catalyzes the irreversible conversion of 5,10-methyleneTHF to 5-methylTHF (161).
Folate in Health and Disease
Folate plays an important role in human health and disease. Folate deficiency has been linked to the development of anemia, cardiovascular disease, neural tube defects (NTDs) and congenital disorders, adverse pregnancy outcomes, neuropsychiatric disorders, and cognitive impairments (114). In addition, a substantial amount of epidemiologic evidence suggests an inverse association between folate status (assessed by dietary folate intake or by the measurement of blood folate levels) and the risk of several malignancies, including cancer of the lungs, oropharynx, esophagus, stomach, colorectum, pancreas, cervix, ovary, prostate, and breast and the risk of neuroblastoma and leukemia (82, 83, 87, 88). More recently, an emerging body of evidence has warned of the potentially harmful effects of FA supplementation, including resistance or tolerance to antifolate drugs used against arthritis (34, 79) and cancer (19, 136, 182); decreased natural killer cell cytotoxicity (160); genetic selection of disease alleles (147); an increased risk of insulin resistance and obesity (181), and asthma (175) in children born to mothers supplemented with high levels of FA; and an increased risk of cognitive impairment in the elderly in combination with low vitamin B12 status (116, 137, 138, 140). Except for anemia and NTDs, the precise nature and magnitude of the relationship between folate status and the risk of these diseases have not been uniformly consistent and remain to be clearly elucidated (114, 147).
Folate and CRC
The highly controversial relationship between folate and carcinogenesis has been most widely studied for CRC. In general, the portfolio of epidemiologic and clinical evidence indicates ∼20%–40% reduction in the risk of CRC or adenoma in subjects with the highest dietary intake or blood levels of folate compared with those with the lowest intake or blood levels (77, 82, 87, 88). The role of folate in colorectal carcinogenesis has been further strengthened by the observations that genetic polymorphisms in the folate metabolic pathway (e.g., MTHFR C677T polymorphism) modify CRC risk (8, 89, 126).
Although there is no definitive evidence supporting the protective effect of folate supplementation on colorectal carcinogenesis from human experiments at present, several small intervention studies have demonstrated that folate supplementation can improve or reverse surrogate endpoint biomarkers of CRC (14, 28, 30, 80, 90, 97, 121), and some epidemiologic studies have shown a beneficial effect of multivitamin supplements containing ≥0.4 mg FA on CRC risk and mortality (56, 57, 69). However, in the Aspirin/Folate Polyp Prevention Study, FA supplementation was associated with a 67% increased risk of advanced lesions with a high malignant potential in men and women with a history of colorectal adenomas (27). Furthermore, a positive association between FA supplementation and prostate cancer risk was reported as a secondary observation in this trial (43). In contrast, some human FA CRC chemoprevention trials have failed to demonstrate the tumor-promoting effect of FA supplementation (70, 104, 121, 180). In a study from Norway in which the primary endpoint was the cardiovascular outcome in response to FA and other B vitamin supplementation, treatment with FA (0.8 mg/day) and vitamin B12 (0.4 mg/day) for a median period of 36 months significantly increased overall cancer incidence and mortality by 21% and 38%, respectively (37). However, in a meta-analysis of 8 randomized trials of FA supplementation involving 37,485 individuals at increased risk of cardiovascular disease, FA supplementation of a median duration of 5 years had no significant effects on vascular outcomes (the primary endpoint), overall cancer incidence, cancer mortality, or all-cause mortality (25). Two recent ecologic studies that examined a temporal postfortification trend of CRC incidence in the United States, Canada, and Chile reported increased CRC rates in these countries following fortification, suggesting that FA fortification may have been wholly or partly responsible for this disturbing trend (63, 111). However, two large prospective epidemiologic studies conducted after FA fortification in the United States have suggested a CRC protective of consuming adequate amounts of folate and have not demonstrated a CRC-promoting effect of FA supplementation (55, 153).
The data from animal studies generally support a causal relationship between folate depletion and CRC risk and an inhibitory effect of modest levels of folate supplementation on colorectal carcinogenesis (83). However, animals studies have also shown that folate supplementation may increase CRC risk and accelerate CRC progression if physiological levels are exceeded or if it is provided after neoplastic foci are established in the colorectum (83, 84).
Mechanisms of action
Several biologically plausible mechanisms exist that explain this seemingly dual role of folate in CRC cancer. As an essential cofactor for nucleotide biosynthesis, folate plays an important role in DNA synthesis, integrity, and repair, aberrancies of which are mechanistically related to cancer development (87). In normal tissues, FA supplementation provides nucleotide precursors for DNA synthesis and replication. This ensures DNA fidelity, maintenance of DNA integrity, and optimal DNA repair, therefore reducing the risk of neoplastic transformation (Fig. 6) (87). In contrast, FA supplementation promotes the progression of (pre)neoplastic lesions by providing nucleotide precursors to the rapidly replicating transformed cells, allowing accelerated proliferation (Fig. 6) (87). Folate also modulates DNA methylation of cytosine within the cytosine-guanine (CpG) sequences because of its role in the provision of SAM (86). Neoplastic cells simultaneously harbor widespread global DNA hypomethylation and gene-specific promoter CpG island hypermethylation (86). Global DNA hypomethylation is an early and consistent event in colorectal carcinogenesis and contributes to CRC development through several mechanisms, including chromosomal instability (86). DNA methylation at promoter CpG islands silences transcription and, hence, inactivates the function of a wide array of tumor suppressor and cancer-related genes (86). FA supplementation is able to reverse pre-existing global DNA hypomethylation and increase the extent of global DNA methylation above the pre-existing level (Fig. 6) (86), thereby reducing the risk of neoplastic transformation. In contrast, FA supplementation may cause de novo methylation of CpG islands of tumor suppressor genes, with consequent gene inactivation leading to tumor development and progression (Fig. 6) (87).

Folate and DNA methylation
Due to the folate's involvement in converting homocysteine to methionine, leading to the provision of SAM (Fig. 4) (141), we and others have been investigating whether folate deficiency or supplementation can modulate DNA methylation. In addition, there are certain periods of growth and development that are particularly vulnerable to environmental dietary exposures, and this may modify DNA methylation patterns with consequent permanent changes in phenotype and other functional ramifications.
Effects of folate status on DNA methylation in animal studies
Folate deficiency
The effect of folate deficiency and supplementation on DNA methylation in rodents has been previously reviewed (31, 85, 86). Briefly, it appears that prolonged and severe folate deficiency induces global DNA hypomethylation in the rodent liver (10, 22), while moderate deficiency does not have a consistent effect. Despite altering hepatic levels of one-carbon metabolism intermediates and reducing SAM to S-adenosylhomocysteine (SAH) concentration ratios, moderate folate deficiency did not induce hepatic global hypomethylation (36, 91, 162). In contrast, there are some reports of folate deficiency increasing genomic methylation. Moderate folate deficiency for 5 weeks in mice was shown to induce a significant 56% increase in genomic DNA methylation in the liver followed by the return of genomic DNA methylation value to that of the baseline by 8 weeks (149). In a more recent study, dietary folate deficiency provided during the postweaning period through childhood to puberty significantly increased genomic DNA methylation by 34%–48% in rat liver that persisted into adulthood after a return to the control diet at puberty (93).
In the colorectum, the majority of animal studies demonstrate a resistance to altered SAM and SAH levels and do not support an association between folate deficiency and genomic DNA methylation (31, 85, 86). However, a recent study reported that chronic, severe folate deficiency in older adult mice induced significant genomic DNA hypomethylation in the colon, and a nonsignificant degree of genomic DNA hypomethylation in the small intestine and spleen (103). Contrary to this observation, a study by Sohn et al. (148) demonstrated that folate deficiency of a short duration and severe degree may induce genomic DNA hypermethylation in the colon (148). The paradoxical effect of folate deficiency on increasing genomic methylation in hepatic and colonic tissue is likely due to the compensatory up-regulation of Dnmt and of the choline and betaine-dependent transmethylation pathway. If adequate levels of dietary folate and other methyl group donors are provided in adolescence and continued into adulthood, then this pattern of genomic DNA hypermethylation is maintained. On the other hand, if continual dietary folate deficiency is imposed, then this compensatory hypermethylation pattern will not be maintained.
Folate supplementation
The effect of folate supplementation alone on DNA methylation in rodent liver has not yet been clearly elucidated. A recent study has found that dietary folate supplementation at four times the basal dietary requirement of the rat (8 mg/kg) did not affect genomic DNA methylation in adult rat liver, regardless of the timing or duration of supplementation (93). Another study found that dietary folate supplementation at 20 times the basal dietary requirement of the rat (40 mg/kg) provided for 4 weeks in weanling rats did not alter SAM and SAH, SAM to SAH concentration ratios, and genomic DNA methylation in the liver (1). Interestingly, in older rats fed a folate deficient (0 μmol/kg), replete (4.5 μmol/kg), or supplemented (18 μmol/kg) diet for 8 and 20 weeks, hepatic SAH levels decreased, while genomic DNA methylation in liver increased incrementally with increasing levels of dietary folate, and folate-supplemented rats demonstrated a significantly greater degree of genomic DNA methylation compared with folate-deplete rats at both 8 and 20 weeks (22). In the colon, one study found that dimethylhydrazine administration in conjunction with folate supplementation of 8 and 40 mg/kg for 20 weeks in weanling rats did not alter concentrations of SAM, SAH, SAM to SAH concentration ratios, and DNA methylation (92). At the same time, weanling rats fed a folate-supplemented diet (8 mg/kg) for 5 weeks showed no change in colonic concentrations of SAM, SAH, SAM to SAH concentration ratios, and colonic DNA methylation, and, in addition, p53-specific DNA methylation was not altered (148). Interestingly, dietary folate supplementation (18 μmol/kg) in both young and older rats for 8 and 20 weeks resulted in a decrease in colonic SAH concentrations, although genomic DNA methylation in the colon was not altered (21).
The results from these studies suggest that in rodents, the liver and colon are generally resistant to changes in the SAM and SAH levels from folate supplementation, and, consequently, are resistant to changes in DNA methylation. Taken together, the results from animal studies suggest that DNA methylation patterns are gene and site specific and depend on cell type, target organ, and stage of transformation as well as on the timing, degree, and duration of folate intervention.
Effects of folate status on global DNA methylation in human studies
Folate deficiency
There are several observations in humans suggesting that an altered folate status can affect genomic DNA methylation (see Table 1). In a metabolic unit setting, folate depletion in older female volunteers (49–63 years of age) for 7–9 weeks has been demonstrated as reducing genomic DNA methylation in leukocytes (67, 132). Furthermore, FA supplementation, even at modest levels, was shown to reverse the extent of genomic hypomethylation in one study (67). However, in an earlier study by the same group, changes in in vivo methylation capacity (as measured by the ability to methylate orally administered nicotinamide as detected in the urine as methylated metabolites) in response to dietary folate (25 μg folate/day) and methyl group restriction for 30 days was not observed in healthy male volunteers (33–46 years of age) (68).
Folate supplementation
In some human intervention studies, folate supplementation at 12.5–25 times the daily requirement for 3–12 months significantly increased the extent of colonic genomic DNA methylation in subjects with resected colorectal adenoma or cancer (28, 30, 90), whereas no such effect was observed in patients with chronic ulcerative colitis who were given folate supplementation at 12.5 times the daily requirement for 6 months (29) or in participants from the Aspirin/Folate Polyp Prevention Study, as determined by the methylation of long interspersed nucleotide elements (LINE-1) (44). Folate supplementation at three and five times the daily requirement, which was sufficient to improve and correct a marker of DNA damage, also failed to modulate genomic DNA methylation in the lymphocytes of healthy volunteers (12, 42). In another study, daily folate supplementation with 15 mg 5-methyltetrahydrofolate for 8 weeks has been observed as restoring genomic DNA methylation in lymphocytes to normal levels in 32 men with uremia, hyperhomocysteinemia, and pre-existing genomic DNA hypomethylation (65). In patients with colorectal adenomas, a physiological dose of FA (0.4 mg/day) for 10 weeks has been demonstrated as significantly increasing genomic DNA methylation in both lymphocytes (by 31%) and colonic mucosa (by 25%) compared with placebo (127).
Observational studies
Several human observational studies have investigated correlations between DNA methylation and folate status (see Table 2). In subjects with adequate folate, no significant correlations between genomic lymphocyte DNA methylation and red blood cell (RBC) folate and plasma homocysteine concentrations have been reported (42, 118), whereas a positive relationship has been found in adults chronically exposed to arsenic (123). Studies evaluating the relationship between serum folate status and methylation-related intermediates have also generally failed to demonstrate an association in healthy adults (62) and the elderly (13). On the other hand, colonic global DNA methylation is positively correlated with serum and RBC folate concentrations and negatively correlated with plasma homocysteine concentrations in individuals with colonic adenomas and adenocarcinomas (3, 128) and in those without these lesions (129). Folate levels and SAM to SAH concentration ratios have also been reported as being lower (by 28%) in malignant tissue compared with normal-appearing adjacent colon mucosa in subjects with CRC (4). Additional evidence lends support to a positive relationship between folate status and global DNA methylation. In a combined analysis of CRCs from participants from the Nurse's Health Study and the Health Professionals Follow-up Study, the risk of genomic hypomethylation (determined by <55% LINE-1 methylation) was 43% lower in subjects with a high (≥0.4 mg) total daily folate intake compared with those with a low (<0.2 mg) total daily folate intake (139). In a study that stratified folate intake according to pre- and postfortification levels, the observed inverse association between leukocyte genomic DNA methylation and adenoma was stronger among subjects with a low (<0.317 mg pre- and <0.413 mg postfortification) total folate intake as compared with those with a high (≥0.317 mg pre- and ≥0.413 mg postfortification) total folate intake in either of the fortification periods (102). Furthermore, in women positive for the human papillomavirus, supraphysiological concentrations of plasma folate (>19 ng/ml) in combination with sufficient levels of vitamin B12 (≥200.6 ng/ml) were significantly associated with greater genomic methylation of peripheral blood mononuclear cells compared with women with lower folate and lower vitamin B12 (124). One human study reported that serum and cervical tissue folate concentrations correlated inversely, albeit weakly, with cervical genomic DNA methylation (47); however, such a relationship was not observed for RBC (46) or circulating plasma folate (124) in exfoliated cervical cells.
RBC, red blood cell; DFE, dietary folate equivalent.
In summary, the data from human clinical studies suggest that controlled folate depletion in a metabolic unit appears to reduce genomic DNA methylation in leukocytes of older individuals. However, there are no conclusive data suggesting that folate deficiency of a physiologically and clinical relevant degree induces significant genomic DNA hypomethylation in the colorectum. In contrast, folate supplementation, even at modest supplemental levels, appears to be able to increase genomic DNA methylation in the colorectum in certain situations. The majority of observational studies have described a direct relationship between dietary and blood levels of folate and genomic DNA methylation in both lymphocytes and colonic tissues such that a low folate status is associated with genomic hypomethylation. This positive association is more consistent in individuals with colorectal adenomas, adenocarcinomas, or previously resected neoplastic tumors as well as in those at a greater risk of health complications compared with normal subjects.
Effects of folate status on gene-specific methylation in human studies
Observational studies investigating CRC
For gene-specific DNA methylation, the majority of human observational studies have investigated the role of folate status in CRC (see Table 3). Aberrant CpG island methylation is characteristic of tumor development, and specific promoter CpG islands are frequently and simultaneously methylated in sporadic CRC, leading to transcriptional silencing (32, 75, 110, 158). In the Netherlands Cohort Study on Diet and Cancer, the prevalence of CpG island promoter hypermethylation was higher, albeit nonsignificantly, in CRCs derived from patients with low folate/high alcohol intake compared with CRCs from patients with high folate/low alcohol intake for each of the six tested genes (APC, p14, p16, hMLH1, O6-MGMT, and RASSF1A) (165). The number of CRCs with at least one gene methylated was higher (84%) in the low folate intake/high alcohol intake group compared with the high folate intake/low alcohol intake group (165). A later follow-up analysis in a subcohort of this population did not report any effect of isolated dietary folate intake on the risk of CRCs specifically presenting with MLH1 hypermethylation (33). Al-Ghnaniem et al. also examined gene-specific methylation in biopsies of normal-appearing colorectal mucosa from subjects with and without colorectal neoplasia (3). In general, patients with neoplasia were reported to have lower serum folate and promoter CpG hypermethylation of the ERα and MLH1 genes compared with disease-free patients. Estrogen receptor alpha (ERα) methylation was also positively correlated with plasma homocysteine in all subjects, but significant inverse correlations between promoter CpG methylation and folate status were not observed (3). In contrast, a modest degree of colorectal CpG hypermethylation of the ERα and SFRP1 genes was significantly associated with higher RBC folate levels in participants from the Aspirin/Folate Polyp Prevention Study (170). The odds of promoter methylation of CDKN2A, MLH1, CACNA1G, NEUROG1, RUNX3, SOCS1, IGF2, and CRABP1 in colorectal tumors are also greater in patients with high circulating levels of plasma folate (166). Colorectal carcinomas with frequent promoter methylation have been shown as having higher tumor concentrations of different folate metabolites, including 5,10-methyleneTHF and THF (75).
ESCC, esophageal squamous cell carcinoma; NSCLC, nonsmall cell lung cancer.
Observational studies investigating other cancers
The effect of folate status on gene-specific methylation patterns at other tissue sites has also been examined. In women with cervical dysplasia, although lower RBC folate and cervical promoter hypermethylation of several tumor suppressor genes were both independently associated with increasing severity of cervical cancer, inverse correlations between folate and methylation were not observed (46). Similarly, no associations were found between dietary folate intake and promoter methylation of E-cadherin, p16, and RAR-β2 in breast tumors (157). For the ERα gene, however, lower dietary folate intake was associated with twofold greater odds, albeit nonsignificantly, of breast cancer with ERα promoter methylation. In addition, high folate intake has been shown to offer protection against methylation of several genes in subjects with a long history of smoking (154). The dietary folate intake derived from fruits was positively associated with a 34% increase in methylation frequency of the MGMT gene in esophageal squamous cell carcinoma (171) and tissue folate levels, measured as the sum of 5,10-methyleneTHF and THF, whereas it positively correlated with LINE-1, CDH13, and RUNX3 methylation in nonsmall cell lung cancer (71).
In summary, the direction and magnitude of effect due to dietary and blood folate concentrations on gene-specific methylation remain unclear. Some studies demonstrate a greater prevalence or risk of aberrant hypermethylation of certain genes involved in colorectal carcinogenesis in subjects with low folate, while others have reported this in subjects with high folate. The discrepancies in identifying a clear association between folate status and gene-specific methylation may be explained, in part, by the different methods of stratifying folate levels for comparison and the use of different markers to evaluate folate status. Dietary intake and serum levels of folate may not necessarily be reflective of folate concentrations in the target organ. Moreover, blood as a surrogate marker of methylation is not always representative of tissue-specific methylation (113). These studies are also complicated by the lack of consistency in the specific genes investigated and the sampling of different CpG sites in different tissues.
Recently, the possibility of an inverse relationship between folate supplementation and DNA methylation status has been raised. Among reproductive-age women not previously exposed to FA in China, FA supplementation (0.1, 0.4, and 4 mg/day) for 1 month significantly decreased genomic methylation by 13% and continued to remain significantly lower with further intervention (130). An interesting observation in this study is that after a 3 month washout period after FA treatment, genomic methylation decreased even further (by 23%) relative to baseline values. This seemingly paradoxical effect of FA supplementation on global DNA methylation may be partly explained by the preferential shuttling of the flux of one-carbon units to the nucleotide synthesis pathway over the methionine cycle necessary for biological methylation reactions in response to FA supplementation. Although FA is an inhibitor of DHFR (9), and an enzyme important in the maintenance of the intracellular folate pool, in certain situations, it may upregulate DHFR (74), and this upregulation may increase thymidylate synthase activity, because the transcription of these genes is co-regulated by several transcription factors (Fig. 4) (145, 150). A mathematical modeling framework has indicated that this would increase thymidylate production, thereby increasing cellular proliferation, at the expense of biological methylation reactions (119).
MTHFR Polymorphisms and DNA Methylation
There is evidence that folate status influences DNA methylation through an interaction with the MTHFR C677T polymorphism (see Table 4). MTHFR is a critical enzyme in folate metabolism that catalyzes the irreversible conversion of 5,10-methyleneTHF to 5-methyltetrahydrofolate, thereby playing an important role in DNA synthesis, maintenance of nucleotide pool balance, and DNA methylation (Fig. 4) (49, 82). The MTHFR C677T polymorphism causes thermolability and reduced MTHFR activity, leading to lower levels of 5-methyltetrahydrofolate, an accumulation of 5,10-methyleneTHF, increased plasma homocysteine levels (a sensitive inverse indicator of folate status), and changes in the cellular composition of one-carbon folate derivatives (Fig. 4) (49, 82). Studies have also indicated that another polymorphism in the MTHFR gene (A1298C) may modulate the genomic DNA methylation in human lymphocytes, although the degree and direction of change have not been clearly established (17, 50). More recent investigations of folate status and DNA methylation in humans include analysis of common MTHFR polymorphisms.
MTHFR, methylenetetrahydrofolate reductase; CRC, colorectal cancer; NA, not applicable; HNSCC, head and neck squamous cell carcinoma.
In human intervention trials, daily folate restriction with 135 μg of dietary folate equivalents (DFEs) for 7 weeks followed by a repletion with 400 or 800 μg DFE for 7 weeks did not influence leukocyte genomic DNA methylation in a group of young and healthy women wild type for MTHFR C677T (5). However, another study by the same group observed significantly lower leukocyte genomic DNA methylation in women homozygous for the polymorphism relative to women with the CC or CT genotype at the end of the 14-week period of folate treatment (6). Similarly, Shelnutt et al. reported a nonsignificant decrease in leukocyte genomic DNA methylation in women depleted of folate (115 μg DFE/day for 7 weeks) (142). This was corrected by the folate repletion of 400 μg DFE/day for 7 weeks but only in women with the MTHFR 677TT genotype. However, an in vivo analysis of genomic monocyte DNA methylation in a subgroup of women from this same population, as determined by methyldeoxycytidine enrichment after radiolabeled infusions of [13C5]methionine, indicated that folate-dependent intracellular one-carbon metabolism was suppressed after 7 weeks of folate restriction (115±20 μg DFE/day), but this effect was independent of the MTHFR genotype (131). Among a small group of hyperhomocysteinemic men with normal renal function and homozygous for the MTHFR C677T polymorphism, supplementation with 5 mg FA/day for 8 weeks also did not induce any genomic methylation changes in peripheral blood mononuclear cells (125).
In observational studies, genomic DNA hypomethylation in peripheral blood monouclear cells has been observed in subjects homozygous for the MTHFR 677TT genotype when compared with CC wild-type individuals (49). When analyzed according to folate status, however, only TT subjects with low levels of folate had hypomethylated DNA (49). In healthy human volunteers, leukocyte genomic DNA methylation was positively and significantly related to RBC folate concentrations in subjects with the MTHFR 677TT genotype, but not in those with wild-type MTHFR (152). Furthermore, a strong trend toward diminished DNA methylation was also observed in subjects with the TT variant with lower plasma folate levels (152). However, in a population of pregnant women, where the majority were deficient in folate (<0.6 mg/day according to World Health Organization and Institute of Medicine guidelines), dietary folate consumption did not influence global methylation in maternal blood, while women carrying the MTHFR 677 T allele had a greater risk of global DNA hypomethylation if they were vitamin B6 deficient (96).
In a study that investigated the combined effects of FA (12.5 times the daily requirement) and vitamin B12 (1.25 mg/day) supplementation for 6 months on the promoter methylation of six tumor suppressor and DNA repair genes frequently reported to be aberrantly methylated in CRC, a trend toward a 67% increase in promoter hypermethylation was reported in the rectal mucosa of patients with resected colorectal adenomas, although this did not reach statistical significance (163). However, further investigation of the six genes revealed that folate intake interacted with the MTHFR C677T polymorphism to influence CpG promoter methylation in colorectal adenomas such that among individuals homozygous for this variant, the risk of promoter methylation was inversely related to dietary folate intake, but statistical significance was only observed for the O6-MGMT DNA-methyltransferase gene (164). The results from this research group suggest that higher folate intakes may increase methyltransferase expression and subsequent methylation activity, particularly in individuals with adenomas and reduced MTHFR enzyme activity (164). Furthermore, Slattery et al. initially failed to identify a significant association between dietary folate and colon tumor CpG island methylation of p16, MLH1 and MINT-1, −2, and −3 loci (146), but in their follow-up analysis, subjects heterozygous or homozygous for the MTHFR A1298C genotype with low folate/low methionine/high alcohol intake had more than twofold greater odds of developing tumors presenting CpG island hypermethylation compared with subjects with the wild-type genotype and high folate/high methionine/low alcohol intake (32). A greater risk of p16 hypermethylation in head and neck squamous cell carcinomas was also observed in subjects with low dietary folate compared with those with high dietary folate, which was further exacerbated in subjects with the MTHFR 677TT genotype (94). In contrast, one study reported that the prevalence of promoter methylation of p16, but not hMLH1 or hMSH2, was significantly higher in CRCs from patients with high serum folate concentrations, that the odds of tumor promoter methylation were significantly higher in patients with high circulating folate levels, and that this positive association was further modified by the MTHFR C677T polymorphism, reaching significance only in subjects heterozygous or homozygous for the MTHFR C677T polymorphism (115).
Taken together, these studies suggest that common MTHFR polymorphisms associated with impaired enzyme activity interact with folate in a manner that modulates both genomic and gene-specific DNA methylation. Human observational studies provide evidence that the MTHFR C677T polymorphism is associated with genomic hypomethylation in leukocytes, which may be mediated, in part, by a low status in folate or other methyl donors. For the colorectum and other tissue sites, whether or not the MTHFR C677T and A1298C polymorphism in conjunction with marginal folate status affects DNA methylation needs to be further characterized. These studies emphasize the importance of taking into consideration the interactions between folate status and critical genes in the folate and one-carbon metabolic pathway when investigating the effect of folate nutrition on DNA methylation.
Periods of Increased Susceptibility to Folate and DNA Methylation Changes
Pregnancy and early postnatal life
The importance of the immediately surrounding environment during critical periods of prenatal and early postnatal development has been known to affect lifelong health and the risk of disease. This field of study, known as the developmental origins of health and disease (DOHaD), has been in existence for nearly 50 years, but recent developments have generated tremendous interest in this area of research. Since epigenetic patterns are established in utero, aberrations in this dynamic process have been proposed to be an underpinning mechanism in the DOHaD hypothesis. One possibility is such that maternal exposures, including nutrition, can alter the intrauterine one-carbon precursor milieu and, as a result, disrupt one-carbon metabolism in the developing offspring. At present, the study of maternal nutrition and its effects on DNA methylation patterns as a predictor of health and risk of disease in the offspring has become a rapidly emerging field in the research community. Several preliminary studies have shown that maternal FA supplementation during pregnancy can modify the offspring's methylome with subsequent changes in phenotype.
Effects of maternal environment on DNA methylation in the offspring
Studies using viable yellow agouti mice have unequivocally demonstrated that maternal dietary methyl group supplementation containing FA can permanently alter the phenotypic coat color of the offspring via increased CpG methylation in the promoter region of the agouti gene (174, 178). Similarly, a methyl group-rich diet has been shown as significantly reducing the proportion of progeny with a kinked tail phenotype, and this was paralleled to a higher degree of CpG methylation in the promoter region of the AxinFused gene (173). In the agouti mouse model, folate has also been shown as interacting with other environmental exposures during the intrauterine period to modulate methylation patterns in the developing offspring. Bisphenol A is an estrogenic xenobiotic chemical used in the manufacturing of polycarbonate plastics and is associated with higher body weight, increased risk of cancer, and other chronic health conditions (108). Exposure to this chemical has been shown to shift the coat color of agouti mice by decreasing CpG methylation of the agouti gene when provided in utero. Moreover, maternal methyl donor supplementation including FA successfully reversed the epigenetic and phenotypic effects of bisphenol A (35). In rats, promoter methylation of the Pparγ and Gr genes has been observed as being significantly lower, by 20% and 22.8%, respectively, in offspring from dams fed a protein-restricted diet compared with those fed a control diet during pregnancy (101). Accompanying increases in protein expression of the Pparγ and Gr genes were also reported, and maternal supplementation with FA prevented these changes (101).
Effects of maternal folate status on DNA methylation in the animal offspring
The effect of FA supplementation alone in utero on epigenetic modulation in the offspring has been displayed in other animal studies (see Table 5). In mice heterozygous for the folate binding protein gene (Folbp1+/−), daily administration of folinic acid by gavage during the periconceptional period until day 15.5 of gestation significantly decreased genomic DNA methylation in both the liver and brain tissues of the offspring (45). In other rodent studies, maternal folate deficiency and supplementation with other nutritional factors were not observed as affecting offspring liver genomic methylation (39, 109). However, in hyperhomocysteinemic rats, maternal dietary folate deficiency was significantly associated with placenta genomic DNA hypomethylation (81). In addition, positive significant correlations between placental genomic DNA methylation and folate levels in the placenta, plasma, and liver were reported (81). Kulkarni et al. also investigated the effects of altered maternal FA in the presence or absence of vitamin B12 on placental global DNA methylation in rats and reported that FA supplementation (8 mg/kg) in the absence of vitamin B12 results in genomic DNA hypomethylation, suggesting that the ratio of FA and vitamin B12 that may have an important role in determining genomic methylation patterns (95). More recently, a study examining both maternal and postweaning folate supply on global methylation in the small intestine of adult mice offspring observed that maternal folate depletion (0.4 mg/kg) during pregnancy and lactation is associated with DNA hypomethylation compared with control (2 mg/kg) and supplemented (8 mg/kg) groups, regardless of postweaning folate diet (112). In an animal model involving sheep, maternal periconceptional folate and vitamin B12 restriction led to aberrant methylation patterns in 4% of the 1400 CpG islands examined (144). Furthermore, the adult male offspring displayed increased adiposity, insulin resistance, altered immune function, and high blood pressure (144).
FA, folic acid; IGF2, insulin-like growth factor-2; DMR, differentially methylated region; NS, not significant; NA, not applicable.
Experimental animal tumorigenesis models have shown that maternal dietary folate manipulation can modify both DNA methylation patterns and the risk of cancer in the developing offspring. Maternal FA supplementation, equivalent to ∼1 mg FA/day in humans, provided in utero and during lactation significantly decreased mammary genomic methylation by 7%, and increased the risk of mammary tumors in the offspring (107). In contrast, maternal FA supplementation at the same level and duration significantly increased colorectal genomic methylation by 3%, and reduced the odds of developing colorectal tumors in the offspring (143). Similarly, in the Apc1638N spontaneous mouse model of CRC, the offspring of B-vitamin (including FA) supplemented mothers displayed a mild degree of genomic hypomethylation in the small intestine mucosa and decreased tumor occurrence (24). This suggests that the effect of maternal FA supplementation on cancer risk in the offspring may be organ specific, and the outcome may be mediated by changes in global DNA methylation.
Effects of maternal folate status on DNA methylation in the human offspring
Recent human studies provide further support of the effect of folate supplementation in utero on epigenetic consequences in the offspring (see Table 5). A preliminary prospective study in the United Kingdom found an inverse correlation between cord plasma homocysteine concentrations and global DNA methylation using methylation of LINE-1 sequences as a surrogate in cord lymphocytes in the offspring of 24 pregnant women (52). Although the results of this study are consistent with the biological functions of folate, significant associations between maternal FA use and cord blood folate and genomic lymphocyte methylation were not reported. Interestingly, an association between genomic lymphocyte methylation and fetal birth weight was identified (52). Further interrogation of CpG loci associated with specific genes in a subset of the same cohort by Fryer et al. found that CpG dinucleotide methylation patterns were also significantly correlated with plasma homocysteine, LINE-1 methylation, and birth weight centile (51).
A limited number of studies have investigated the effect of maternal folate exposure on CpG methylation of the imprinted insulin-like growth factor-2 gene (IGF2), which is important for growth and development. A cross-sectional examination of a population unexposed to FA supplements before and during pregnancy revealed no significant associations between cord blood promoter methylation of the IGF2 gene and serum folate levels in either mother's or umbilical cord blood (7). However, in an observational study conducted in the Netherlands, periconceptional maternal FA use of 400 μg/day significantly increased, by 4.5%, the methylation of the IGF2 differentially methylated region (IGF2 DMR) in whole blood derived from children at 17 months of age (151). An independent inverse association was also observed between IGF2 DMR methylation and birth weight. More recently, the North American Newborn Epigenetic Study also evaluated the effect of the exposure of maternal FA supplementation before and during pregnancy on the methylation profile of two DMRs known to regulate IGF2 expression (64). Using cord blood leukocytes, no evidence for methylation differences at the IGF2 DMR was found while methylation levels at the second H19 DMR decreased significantly among FA users compared with nonusers (64). Both IGF2 DMR hypomethylation and H19 DMR hypermethylation have been independently associated with increased IGF2 transcriptional activity and loss of imprinting, and have been observed in several malignancies (64). While both studies report favorable shifts in methylation changes of the IGF2 and H19 DMRs with FA supplementation, the functional outcomes of these methylation changes require further investigation. More recently, the question of whether mothers themselves may be susceptible to changes in DNA methylation as a result of modified folate nutrition during pregnancy has been raised. In mice fed folate-adequate (2 mg/kg) and folate-deficient (0.4 mg/kg) diets before mating and during pregnancy and lactation, postpartum methylation of Igf2 DMR1 and Slc39a4 at specific CpG sites was significantly lower in folate-deficient rats (DNA methylation measured as an average across blood, liver, and kidney) (112). Interestingly, for Igf2 DMR2, a significant interaction between dietary folate and tissue type for overall methylation was observed where CpG methylation was lower in blood and higher in liver in low-folate dams (112).
Given the association between folate deficiency and the risk of NTDs, a limited number of human studies have investigated whether aberrant DNA methylation patterns can be an underlying mechanism of congenital malformations. Two case-control studies have examined the relationship between maternal folate status and genomic methylation of neural tissue from NTD-affected human fetuses (18, 172). While both studies reported lower maternal circulating folate concentrations and genomic brain hypomethylation in NTD fetuses compared with controls, only the study by Chang et al. observed a significant correlation between the methylation changes and folate status (18). Both studies suggest that folate inadequacy may interfere with one-carbon metabolism and affect normal fetal development.
Although it is difficult to directly compare the outcomes of the studies just reviewed due to inherent differences in study design and research models utilized, several lines of evidence support the theory that DNA methylation patterns of the developing offspring can be significantly modulated by environmental intrauterine exposures, including varying levels of FA alone or in combination with other methyl donors. Furthermore, the changes in methylation status may be associated with developmental changes and permanent alterations in phenotype with potential health consequences in later adult life. Whether or not folate-mediated epigenetic changes in utero can affect the risk of chronic disease development in adulthood remains unclear. Several studies indicate that maternal nutrition during pregnancy accurately predicts early life nutrition of the developing offspring (117, 120). Maternal biomarkers of B vitamins, including folate, have been shown as significantly correlating with cord blood folate (120) and infant folate at 6 months (60). Furthermore, folate levels in umbilical cord blood are thrice the level found in the mother (156). Thus, in this specific population of pregnant women, the possibility exists that an increased exposure to dietary and circulating folate translates to a heightened intrauterine folate environment, and this may have significant implications in the growth, development, and subsequent risk for disease in the offspring.
Aging
The study of age-related epigenetic changes has recently become a growing topic in research. As we age, a variety of internal and external factors play a role in disease susceptibility. Exposure to numerous environmental factors throughout life alters epigenetic patterns previously established in utero. Studies involving monozygotic twins clearly illustrate how environmental factors, including diet, can lead to the divergence of epigenetic modifications with age (48). Therefore, research on modifying or limiting exposures, which lead to epigenetic changes, is of significant interest.
Mechanism of action
Aging has been shown as modifying patterns of DNA methylation in a variety of species and tissues and is generally associated with genomic DNA hypomethylation and gene-specific promoter hypermethylation in a tissue-specific manner (53, 66, 167, 176). Genomic DNA hypomethylation is thought to occur with decreased activity of DNMT1, the methyltransferase responsible for maintaining and translating methylation patterns to subsequent DNA strands (168). Although not yet clear, gene-specific hypermethylation is thought to be due to upregulation of methyltransferases DNMT3a and 3b, which are responsible for de novo methylation (105).
Age-related methylation is a common event in human tissues and an important contributor to promoter CpG island hypermethylation of several genes with consequent gene inactivation of tumor suppressor genes observed in colorectal and other cancers (159). Furthermore, aging is an independent determinant of CRC risk (177). It is, therefore, possible that age-related DNA methylation of certain genes serves as a functional link between aging and CRC by providing a selective advantage for normal colon cells through deregulating growth and differentiation (159). These cells may then be at a higher susceptibility for acquiring genetic lesions such as mutations.
Effects of folate status and aging on DNA methylation in animal studies
It has been proposed that environmental exposures or modifier genes may play a role in age-related methylation changes (2). FA exposure plays a significant role in this phenomenon, as folate participates in methylation reactions. A number of studies have shown that folate and aging may act synergistically to alter DNA methylation patterns (see Table 6). A recent animal study reported diminished genomic and increased p16 promoter DNA methylation in the colon of aged mice compared with those of young mice (78). Interestingly, both genomic and p16 promoter DNA methylation increased in a manner that was directly related to dietary folate in old mice, whereas this pattern was not evident in young mice (78). In older rat liver, dietary folate over a wide range of intakes was also shown as modulating genomic DNA methylation, as genomic DNA methylation increased with increasing levels of dietary folate (22). Another rodent study has shown the elder rat colon to be highly susceptible to folate depletion, with consequent changes in SAM and SAH, compared with the young colon (21). Therefore, folate deficiency in the aging colon may predispose it to changes in SAM and SAH and consequent DNA methylation changes more readily than that in the young colon. Therefore, folate status and DNA methylation changes may serve as a functional link between aging and CRC.
NS, not significant.
Effects of folate status and aging on DNA methylation in human studies
In human studies, moderate folate depletion has been shown as being associated with diminished genomic DNA methylation in lymphocytes (67) and leukocytes (132) in healthy, postmenopausal women. This direct relationship between folate status and genomic DNA methylation is similar to the trends demonstrated in previous animal studies. Interestingly, however, genomic hypomethylation in lymphocytes was reversed after 3 weeks of folate repletion (286–516 μg/day) (67), but no such reversal was observed in leukocyte genomic methylation after folate repletion (415 μg/day) for 7 weeks (132). It has been suggested that older age and lower levels of folate may increase the response time of returning methylation levels to normal levels (132).
In terms of gene-specific methylation, a recent multi-center chemoprevention trial found a 1.7% increase (p<0.001) in methylation levels in normal colorectal mucosa of the ERα and a 2.9% methylation increase (p<0.001) in secreted frizzled related protein-1 (SFRP1) for each 10 years of age. ERα and SFRP1 are thought to regulate cell growth and differentiation and are, therefore, two genes important for cancer development. There was a positive, nonsignificant association between year 3 RBC folate levels and methylation levels in ERα and SFRP1. However, there were no associations indicating percentage ERα or SFRP1 methylation levels leading to an increased risk of adenoma or hyperplastic polyps (170), indicating that the epigenetic changes may not lead to detrimental phenotypic outcomes.
Li et al. found that human cells from older subjects (>50 years old) cultured in low folate (10 nM) resulted in decreased killer cell immunoglobulin-like receptor 2DL2 (KIR2DL2) promoter region DNA methylation levels in T cells than in cells from younger subjects (100). Interestingly, the cells from older subjects exposed to higher folate levels (40 nM) had less methylation in the KIR2DL2 promoter region than those in younger subjects exposed to either folate concentration, indicating that adequate folate levels did not return methylation levels to those found in younger subjects. The partial demethylation in KIR2DL2 resulted in increased gene expression of KIR2DL2 but only in the older subjects, and homocysteine exacerbated this relationship (100). However, these data are based on a small subset (three older, three younger) of study subjects. The resulting promoter region hypomethylation of KIR2DL2 is the opposite of what is usually associated with aging. Nonetheless, the study indicates low folate and age lead to changes in methylation patterns that result in the differential expression of a gene involved in autoimmunity and acute coronary syndromes.
Since folate and increased age have been shown as having a synergistic effect on altered DNA methylation patterns, it is important to consider folate exposure in this population. Recent data indicate folate exposure in the elderly and are adequate across all age groups with <1% of the Canadian population being folate deficient (26). Furthermore, median RBC folate levels were the highest among 60–79 year olds at 1409 nM. This is above the high RBC folate concentration cutoff of 1360 nM, which reflects the 97th percentile from the National Health and Nutrition Examination Survey (1999–2004) (122). The effects of supraphysiological FA levels in a population who is at a higher risk for cancer development are currently unknown; however, the potential for increased aberrant DNA methylation patterns and subsequent altered gene expression cannot be ignored. Concerns for increased FA levels in this vulnerable population have recently been raised (54).
The results from these studies suggest that the aging process can alter DNA methylation, and folate may further modify DNA methylation in the elderly in a tissue-specific manner. At present, the precise mechanism of how folate and aging interact to modulate DNA methylation has not been clearly elucidated. Folate intakes in the elderly seem to be adequate, and there is some concern that this population is reaching supraphysiological levels. However, data on epigenetic changes in the elderly suggest that it is important to consider environmental exposures over a lifetime and short-term folate exposure, in this case, may not be enough to return methylation levels to those found in younger individuals. However, therapeutic measures targeted at epigenetic modifications are desirable and worth researching while considering that epigenetic pathways are potentially reversible. Further studies are required to investigate the interaction between folate and aging on genomic and gene-specific DNA methylation and subsequent disease susceptibility.
Conclusions
Genetic changes in cancer are abrupt in onset, their effects are often all-or-nothing, the loss of function occurs at a fixed level, and they are not reversible in most cases. In contrast, epigenetic changes are gradual in onset and progressive, their effects are dose dependent, and are potentially reversible. These observations present new opportunities in cancer risk modification and prevention using dietary and lifestyle factors and potential chemopreventive drugs. In this regard, folate has been a focus of intense interest because of an inverse association between folate status and the risk of several malignancies and due to its potential ability to modulate DNA methylation. The portfolio of evidence from animal, in vitro, and human studies collectively suggests that the effects of folate deficiency and supplementation on DNA methylation are highly complex and appear to depend on cell type, target organ, and stage of transformation and are gene and site specific. These studies also suggest that changes in DNA methylation depend on the magnitude and duration of folate manipulations, on interactions with other methyl group donors and dietary factors, and on genetic variants in the folate metabolic and one-carbon transfer pathways. Although some similarities exist, animal models differ from humans in several important physiological aspects, including bioavailability, metabolism, and excretion of folate. Therefore, any extrapolation of the observations from these models to human situations should be very cautiously made. Furthermore, animal models may produce variable results owing to species differences, different diet compositions, and variable dose, time, and duration of folate manipulations. Recent studies have demonstrated the exposure to folate deficiency, and supplementation in the intrauterine environment and early life and during the aging process may have profound effects on DNA methylation with significant functional ramifications. Although the jury is still out, the potential for folate to modulate DNA methylation and, thus, modify the risk of chronic diseases including cancer in humans remains provocative and is worthy of further studies.