
Abstract
Introduction
It has been shown that the kidney expresses all the RAS components such as angiotensinogen (AGT), ACE, and AT1, particularly in proximal tubular epithelial cells (PTCs) (19, 23). Ang II content in the proximal tubular compartments is much higher than that can be explained on the basis of equilibration with the circulation concentration (37), suggesting that intrarenal RAS activity is regulated in a manner distinct from circulating RAS. However, the independent regulation of intrarenal RAS remains unclear.
Innovation
Activation of intrarenal renin–angiotensin system (RAS) is critical in the progression of chronic kidney diseases (CKDs). However, the regulation of intrarenal RAS remains unclear. The present study for the first time demonstrated that advanced oxidation protein product (AOPP)-albumin activated intrarenal RAS by a CD36-dependent, redox-sensitive signaling. The triggering effect of AOPP-albumin was 100-times stronger than that of unmodified albumin. Intrarenal AOPP accumulation was correlated with angiotensin II production in renal biopsies from patients with IgA nephropathy. These results suggest that AOPPs might be a potential target for the therapeutic intervention of CKD.
Several factors have been implicated in activation of the intrarenal RAS, including hyperglycemia (18), reduced availability of nitric oxide (9), high-salt diet (24), and albuminuria (25). Our previous study indicated that albuminuria activates RAS in PTCs via interacting with the endocytic receptor megalin/cubulin (2). Noteworthy, albumin would be modified by oxidants under the condition of oxidative stress. Accumulation of oxidized protein products, such as advanced oxidation protein products (AOPPs), has been well recognized in diseases with oxidative stress, including CKDs and diabetes (30, 33). AOPPs are a family of dityrosine-containing protein products generated by reaction of proteins with hypochlorous acid (HOCl) and mainly carried by albumin in vivo (44, 45). Chronic accumulation of AOPPs is associated with aggravated renal fibrosis in remnant kidney and experimental diabetic nephropathy (26, 39). More importantly, clinical studies indicated that the AOPP level is a strong predictor for the progression of IgA nephropathy (1, 4), suggesting that AOPPs might be a class of renal pathogenic mediators involved in the progression of CKDs. However, the mechanism underlying the pathogenic effect of AOPPs remains to be further investigated.
The present study was designed to test the hypothesis that AOPP-modified albumin (AOPP-albumin) may trigger the activation of intrarenal RAS. We demonstrated that, both in vitro and in vivo, AOPP-albumin upregulated the expression of almost all the components of RAS and increased the activity of ACE in PTCs. The effect of AOPP-albumin is 100-times stronger than that of unmodified albumin. Unlike native albumin, AOPP-induced activation of intrarenal RAS was mediated mainly by a CD36-dependent redox pathway. In patients with CKD, deposition of AOPPs was found in renal tubular epithelial cells and was associated with Ang II formation. These results revealed a new mechanism underlying activation of intrarenal RAS and provided a link between protein oxidative damage and the progression of CKDs.
Results
AOPPs activated RAS in cultured PTCs
First of all, we examined the effect of AOPPs on the expression of the major components of RAS that have been shown to be expressed on tubular cells (19, 23). As shown in Figure 1, exposure of rat PTCs (NRK52E) to an indicated amount of AOPP–rat serum albumin (AOPP-RSA) upregulated the expression of AGT, ACE, and AT1 in a dose- (Fig. 1A, C) and time-dependent (Fig. 1B, D) manner at both mRNA (Fig. 1A, B) and protein (Fig. 1C, D) levels. About 50 μg/ml of AOPPs resulted in a significant increase of the expression of AGT, ACE, and AT1. We previously showed that native albumin activated RAS in PTCs (2). Therefore, we compared the effect of AOPP-RSA and unmodified RSA. As shown in Figure 1A, RSA, at the concentration of 100 μg/ml, did not affect the expression of AGT, ACE, and AT1 compared with untreated cells. About 10 mg/ml of native RSA showed a comparable effect to that of 100 μg/ml of AOPP-RSA.
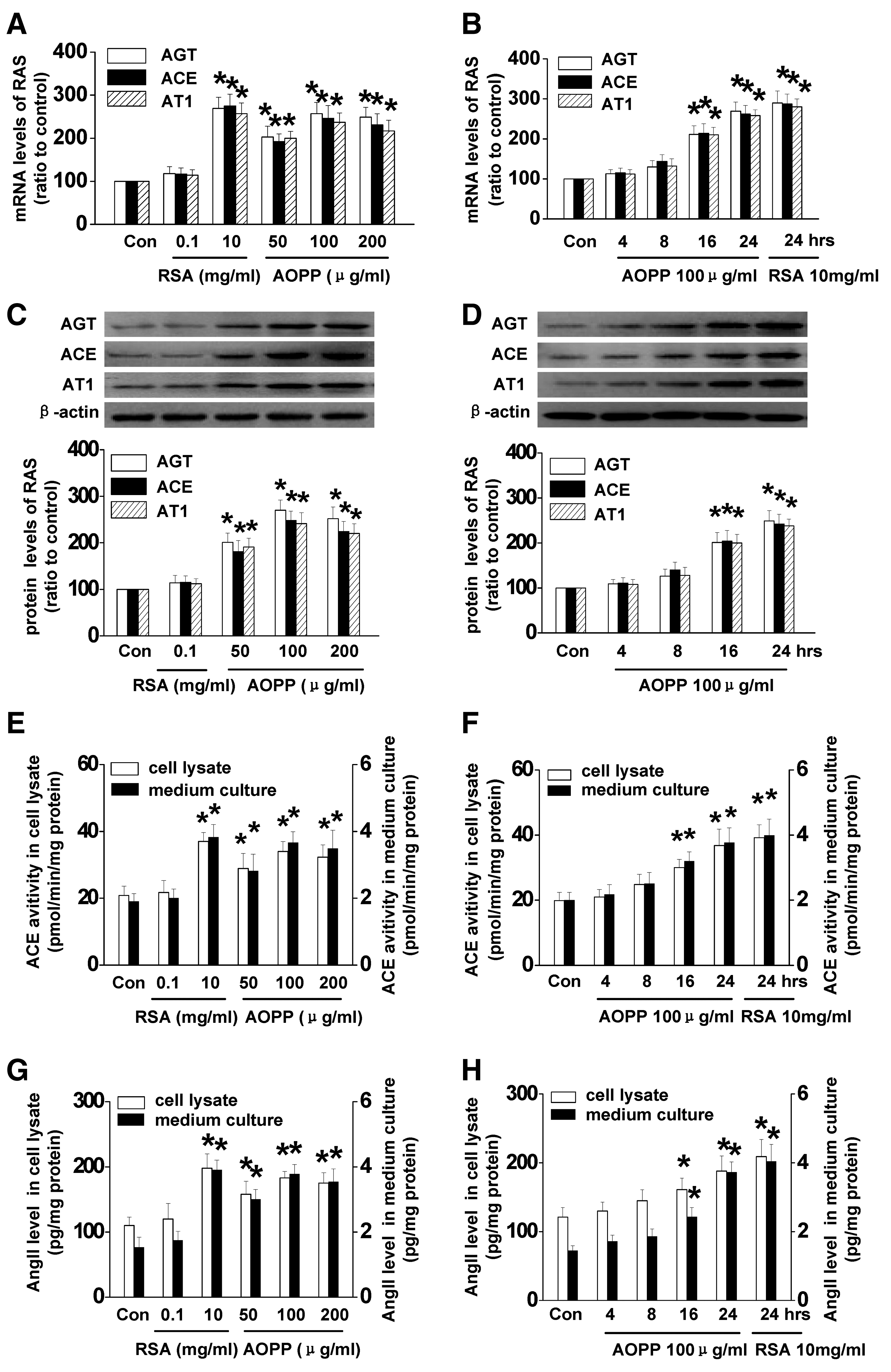
To further determine whether AOPP-RSA increases ACE function, the activity of ACE in the cell lysates and supernatants was examined. As shown in Figure 1E and F, stimulation of AOPPs enhanced ACE activity in a dose- (Fig. 1E) and time-dependent (Fig. 1F) manner. In addition, AOPP-RSA treatment increased Ang II production in a dose- (Fig. 1G) and time-dependent (Fig. 1H) manner. The effect of 100 μg/ml of AOPP-RSA was comparable with that of 10 mg/ml of RSA (Fig. 1E, G). Altogether, these results indicated that AOPP-RSA stimulation resulted in RAS activation in PTCs, and its effect was 100-times stronger than that of unmodified albumin.
AOPP-induced activation of RAS was mainly mediated by CD36
The cells used in the present study expressed the endocytic receptor megalin/ cubilin and its complement chloride channel (Clc) 5, and scavenge receptor (SR), including CD36, receptor for advanced glycation end products (RAGE), SR class B type 1 (SR-B1), SR-A, lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), and AGE-R3 (Galectin-3) (Fig. 2A). To determine the receptor-mediating AOPP-induced RAS activation, we silenced these receptors using small interfering RNA (siRNA) transfection before exposure to AOPP-RSA. The efficiency of each siRNA was demonstrated by western blot (Fig. 2A). Transfection of siRNA resulted in ∼70% decrease of the protein expression in PTCs (Supplementary Fig. S1; Supplementary Data are available online at

AOPPs triggered RAS activation via PKCα-NADPH oxidase-dependent reactive oxygen species production
We previously showed that AOPP-RSA induced reactive oxygen species (ROS) production in podocytes. Therefore, we examined the intracellular ROS level in NRK52E cells. As shown in Figure 3A, AOPP-RSA increased ROS generation in a dose- and time-dependent manner. Noteworthy, stimulation of cells with 100 μg/ml of AOPP-RSA resulted in a significant increase of intracellular ROS, whereas native RSA at the same concentration had no effect. To generate the amount of ROS as that of 100 μg/ml of AOPPs-RSA, 10 mg/ml of RSA, which is 100-times stronger as much as AOPP-RSA, was required.

To verify the enzymatic source of ROS generation, PTCs were preincubated with inhibitors of various enzymatic systems involved in ROS production before AOPP-RSA stimulation. As shown in Figure 3B, the NADPH oxidase inhibitor diphenylene iodonium (DPI) or apocynin significantly inhibited AOPP-induced ROS production, whereas the inhibitors of the mitochondrial respiratory chain complexes and the inhibitors of nitric oxide synthase, cyclooxygenase, or xanthine oxidase had no effect. To further confirm the role of NADPH oxidase, we knocked down p47phox, a subunit of NADPH oxidase, by siRNA. The efficiency of siRNA was examined by western blot (Supplementary Fig. S2). As shown in Figure 3B, silencing p47phox significantly attenuated AOPP-induced ROS production. Taken together, these data suggest that AOPP-induced ROS production was specifically mediated by NADPH oxidase.
To further confirm the involvement of NADPH oxidase, we analyzed NADPH oxidase activity in AOPP-RSA-stimulated PTCs. As shown in Figure 3C and D, treatment of AOPPs led to phosphorylation of p47phox (Fig. 3C) and increased expression of NADPH oxidase subunits p47phox, p22phox, Nox4, and Nox2 (Fig. 3D). Coimmunoprecipitation assay revealed that the interaction of p22phox with p47phox, Nox4, and Nox2 was significantly increased in AOPP-RSA-treated cells (Fig. 3C), suggesting that AOPP-RSA induced the activation of NADPH oxidase.
To clarify the upstream event responsible for AOPP-induced NADPH oxidase activation, we examined the phosphorylation of all subunits of protein kinase C (PKC) (α, ζ, δ, θ, and ɛ) that have been shown to be present in PTCs (5, 47). As shown in Figure 4A, treatment of AOPPs significantly enhanced the phosphorylation of PKCα (p<0.01), but had no effect on the phosphorylation of PKCζ, PKCδ, PKCθ, and PKCɛ. To evaluate the role of PKCα on AOPP-induced NADPH oxidase activation, PTCs were incubated with a pan-PKC inhibitor Gö6983 (0.1 μM) (10) or a PKCα inhibitor Gö6976 (10 nM) (32). As shown in Figure 4B and C, Gö6976 had similar effect as Gö6983 on inhibiting AOPP-induced phosphorylation of p47phox (Fig. 4B) and ROS production (Fig. 4C), suggesting PKCα activation is the upstream event of NADPH oxidase-dependent ROS production.
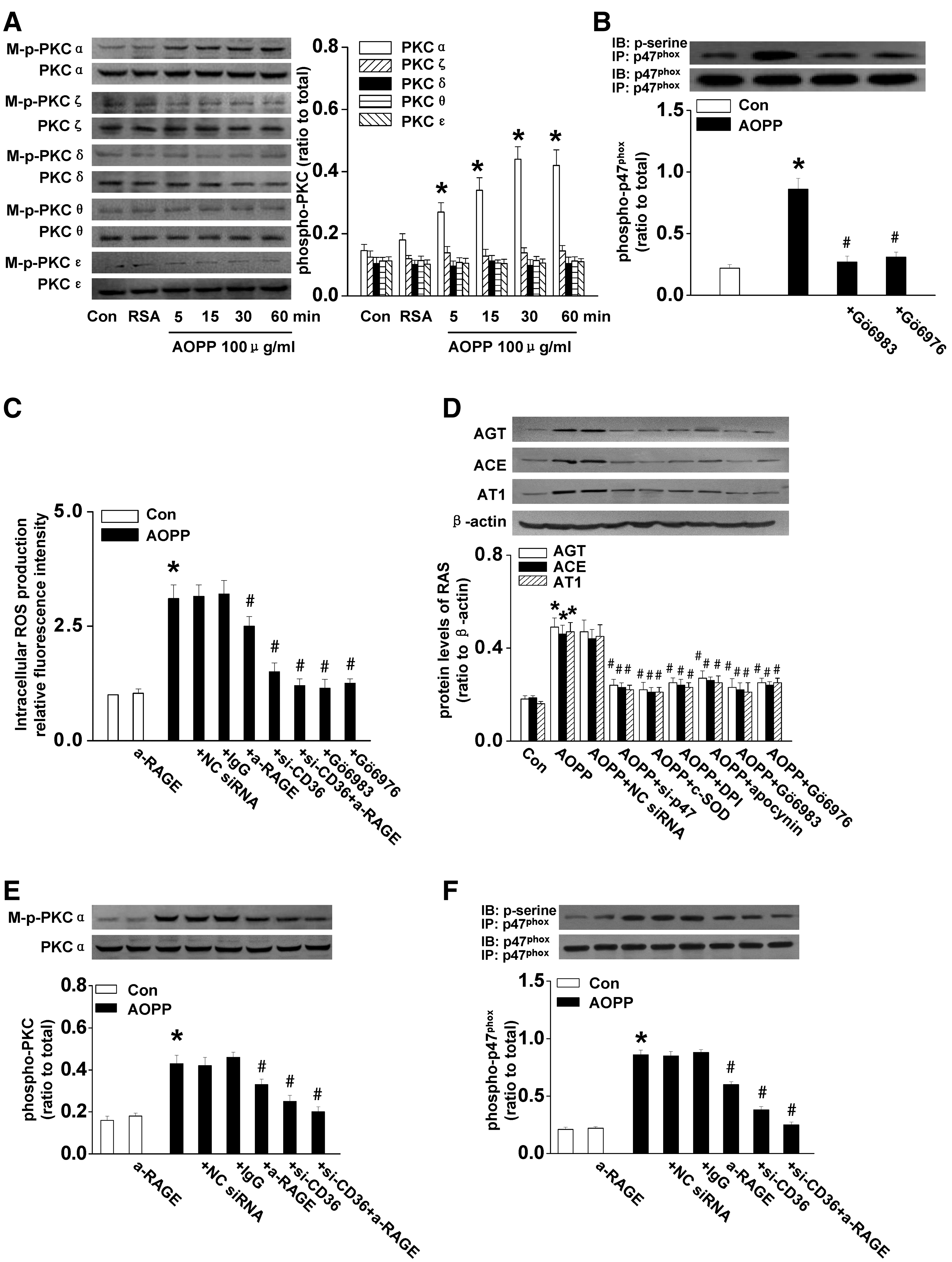
We next examined the role of PKCα and NADPH oxidase in RAS activation. As shown in Figure 4D, DPI, apocynin, Gö6983, Gö6976, or p47phox siRNA significantly inhibited AOPP-induced expression of AGT, ACE, and AT1, indicating that the PKCα-NADPH oxidase pathway was responsible for AOPP-induced RAS activation.
To clarify the role of CD36 in AOPP-induced activation of the PKCα-NADPH oxidase-ROS axis, we blocked CD36 and/or RAGE before AOPP-RSA treatment. Knocking down CD36 significantly inhibited AOPP-induced PKCα phosphorylation (Fig. 4E), NADPH oxidase activation (Fig. 4F), and ROS production (Fig. 4C). Blocking CD36 and RAGE together showed a more dramatic effect (Fig. 4C, E, F).
Altogether, these data indicated that the CD36/RAGE-PKCα-NADPH oxidase-ROS axis is responsible for AOPP-induced RAS activation in cultured PTCs.
AOPPs activated nuclear factor-κB and activation protein 1 via the CD36-PKCα-NADPH oxidase pathway
Activation of Nuclear factor-κB (NF-κB) and activation protein 1 (AP-1) has been shown to regulate the expression of major components of RAS (40, 41). Therefore, we examined the effect of AOPP-RSA on the activation of NF-κB and AP-1. As shown in Figure 5A and B, AOPP-RSA upregulated the expression of c-jun and c-fos and enhanced the phosphorylation of the c-jun and p65 subunits of NF-κB in a time-dependent manner. We next evaluated the involvement of the CD36/RAGE-PKCα-NADPH oxidase axis in AOPP-induced activation of NF-κB and AP-1. As shown in Figure 5C and D, knockdown CD36 significantly inhibited AOPP-induced upregulation of c-jun and c-fos and phosphorylation of c-jun and p65. Blocking CD36 and RAGE together completely abolished AOPP-augmented activation of AP-1 and NF-κB. Furthermore, AOPP-triggered activation of AP-1 and NF-κB was attenuated by DPI, apocynin, Gö6983, Gö6976, or p47phox siRNA (Fig. 5E, F), suggesting that AOPPs activated NF-κB and AP-1 through the CD36/RAGE-PKCα-NADPH oxidase pathway.
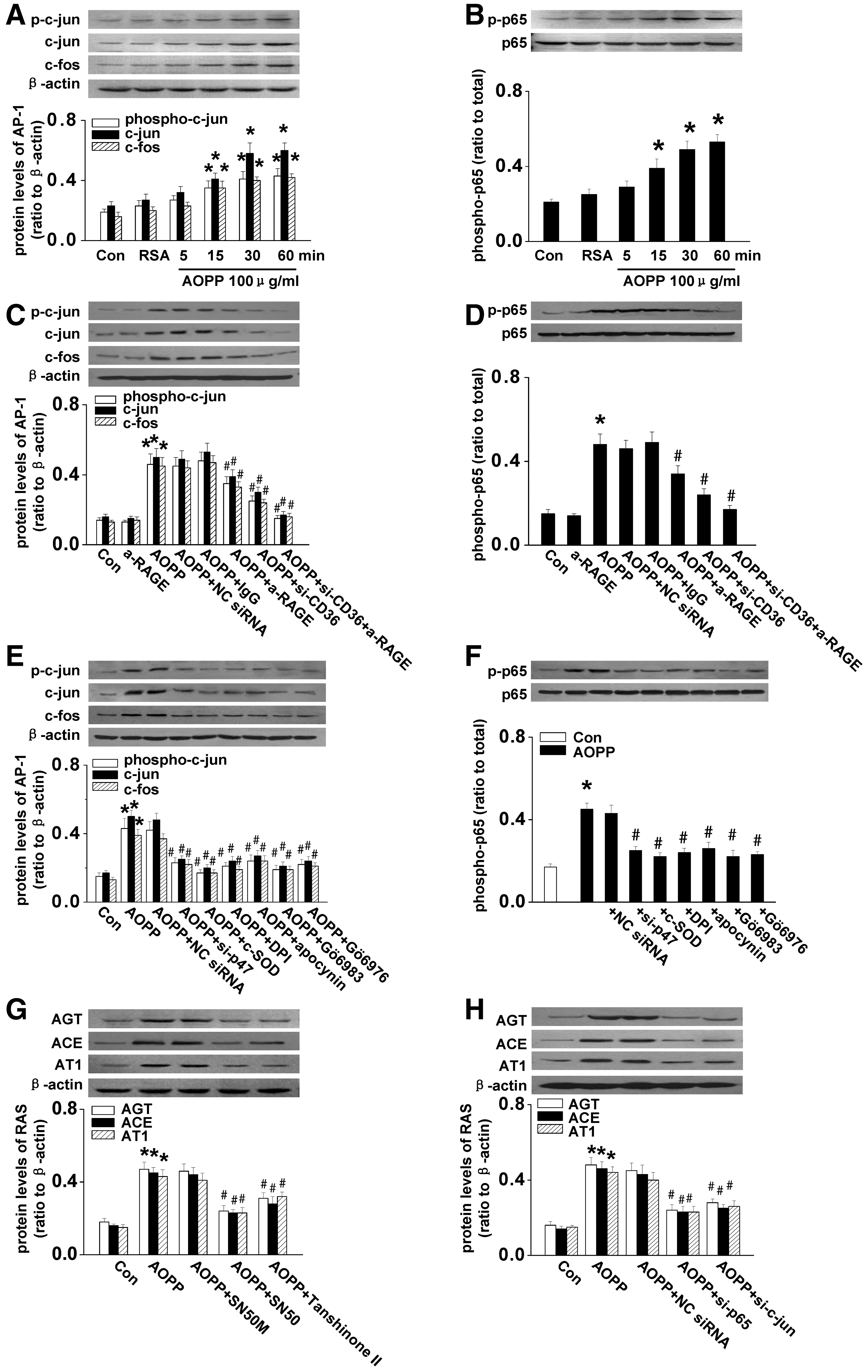
To determine the role of AP-1 and NF-κB in the expression of RAS components, NRK52E cells were incubated with an inhibitor of AP-1 (Tanshinone II) or NF-κB (SN50) before AOPP-RSA stimulation. As shown in Figure 5G, Tanshinone II or SN50 significantly attenuated AOPP-induced expression of AGT, ACE, and AT1. This result was further confirmed by siRNA, specifically knockdown p65 subunit of NF-κB or c-jun (Fig. 5H and Supplementary Fig. S3), indicating that AGT, ACE, and AT1 are the target genes regulated by AP-1 or NF-κB upon stimulation of AOPPs.
Chronic AOPP-loading activated intrarenal RAS
To examine whether the effects observed in vitro occurred in vivo, we treated Sprague-Dawley rats with unilateral nephrectomy (UNX) together with daily intravenous injections of 50 mg/kg/day AOPP-RSA for 3 weeks. Immunohistochemistry staining showed that the strong positive staining for AGT, ACE, AT1, or AOPPs was mainly detected in PTCs in AOPP-RSA-challenged rats (Fig. 6A). Increased activation of intrarenal RAS in AOPP-RSA-treated rats was further confirmed by the upregulation of the mRNA and protein level of AGT, ACE, and AT1 (Fig. 6B, C), enhanced activity of ACE (Fig. 6D), increased concentration of Ang II in the renal cortex (Fig. 6E), and increased concentration of AGT in urine (Fig. 6F). The level of intrarenal RAS activation in rats injected with 50 mg/kg/day AOPP-RSA was comparable with that in rats treated with 5 g/kg/day RSA (Fig. 6A–F), indicating that the effect of AOPP-RSA was 100-times that of unmodified RSA. However, loading of AOPPs did not affect the plasma levels of AGT, Ang II, and the activity of serum ACE (Supplementary Table S1).
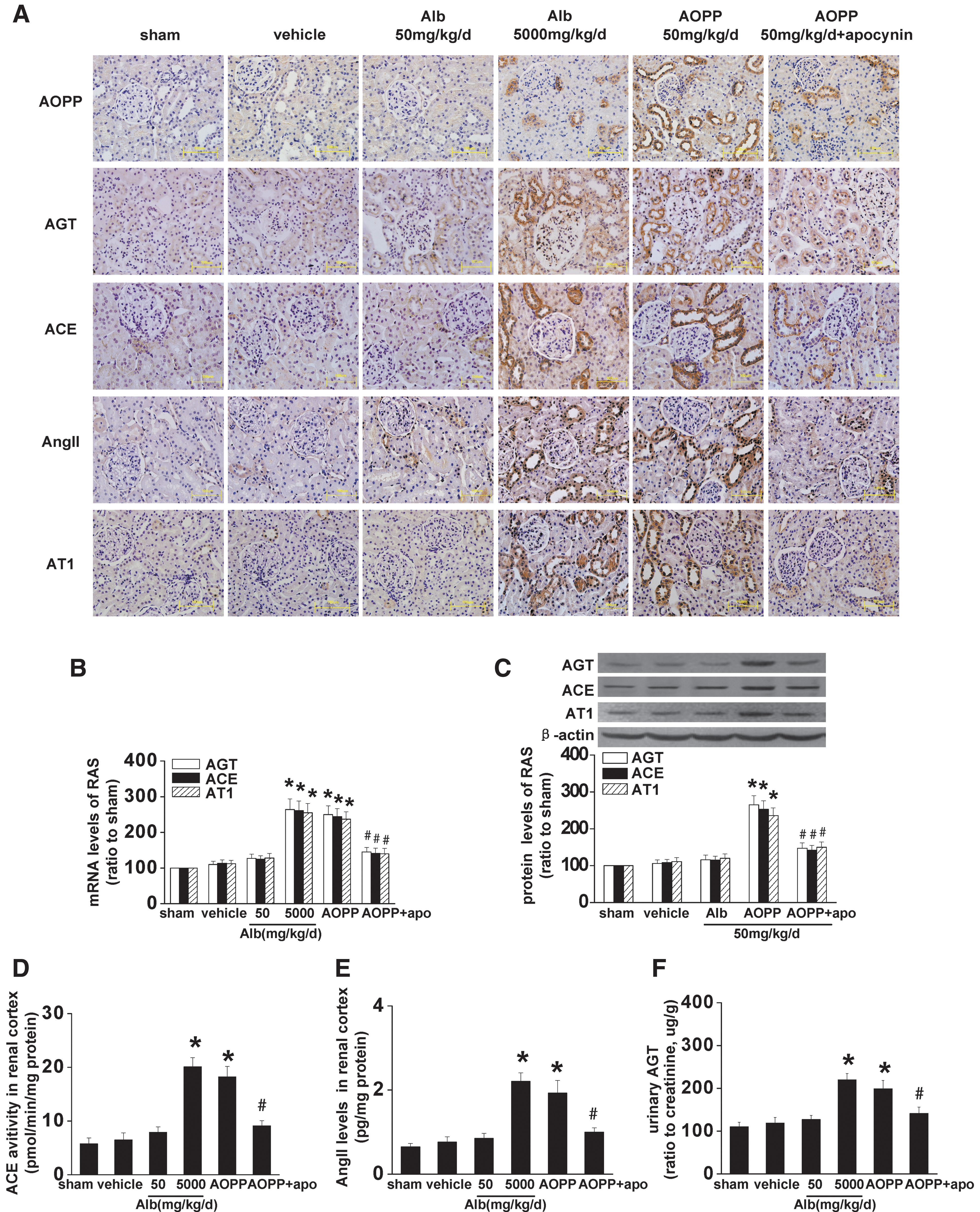
Moreover, intravenous injections of 50 mg/kg/day of AOPPs resulted in an approximately onefold increase, which is the level detected in patients with diabetes and CKD (22, 45), in the plasma level of AOPPs. Noteworthy, this level of plasma AOPPs caused glomerular hyperfiltration as manifested by an increased renal mass and enhanced urinary albumin excretion (Table 1), a phenomenon similar to that of early diabetes (14).
p<0.05 versus sham; b p<0.05 versus vehicle; c p<0.05 versus AOPP 50 mg/kg/day.
Data are expressed as mean±SD, n=6.
0 w, 0 week; 3 w, 3 week; BP, blood pressure; KW/BW, kidney weight/body weight; UAE/creatinine, urinary albumin excretion measured by using three consecutive 24-h urine collection/creatinine; RSA, rat serum albumin; AOPP, advanced oxidative protein product; UNX, unilateral nephrectomy.
AOPP-induced intrarenal RAS activation was dependent on the PKCα-NADPH oxidase-AP-1/NF-κB pathway
We next examined the intracellular pathway mediating AOPP-induced activation of intrarenal RAS. Compared with UNX control rats, AOPP-challenged rats had a significant increase in phosphorylated PKCα and p47phox on the cell membrane (Fig. 7A, B), and an enhanced level of superoxide in the homogenates of renal cortex (Fig. 7C). Furthermore, AOPP challenge resulted in significant upregulation of c-jun and c-fos and enhanced phosphorylation of c-jun (Fig. 7D) and p65 (Fig. 7E). These data suggested that AOPP loading activated the intrarenal PKCα-NADPH oxidase-NF-κB/AP-1 pathway.
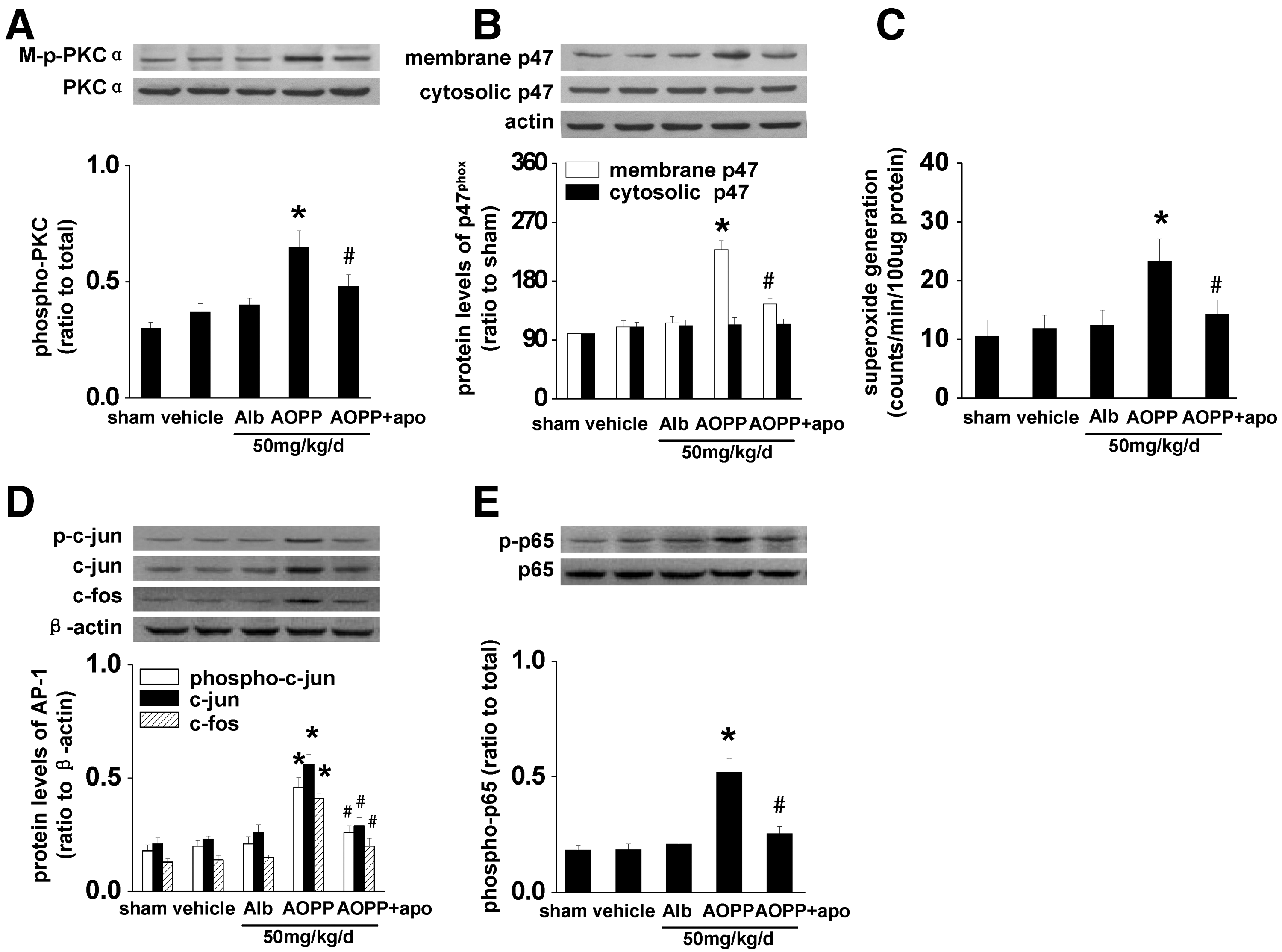
To confirm the role of NADPH oxidase in AOPP-induced RAS activation in vivo, AOPP-challenged rats were intragastrically administrated with apocynin at 100 mg/kg/day. Inhibition of NADPH oxidase by apocynin significantly attenuated AOPP-triggered activation of the PKCα-NADPH oxidase-NF-κB/AP-1 pathway (Fig. 7), as well as activation of intrarenal RAS (Fig. 6).
AOPP-induced intrarenal RAS activation was mediated mainly by CD36
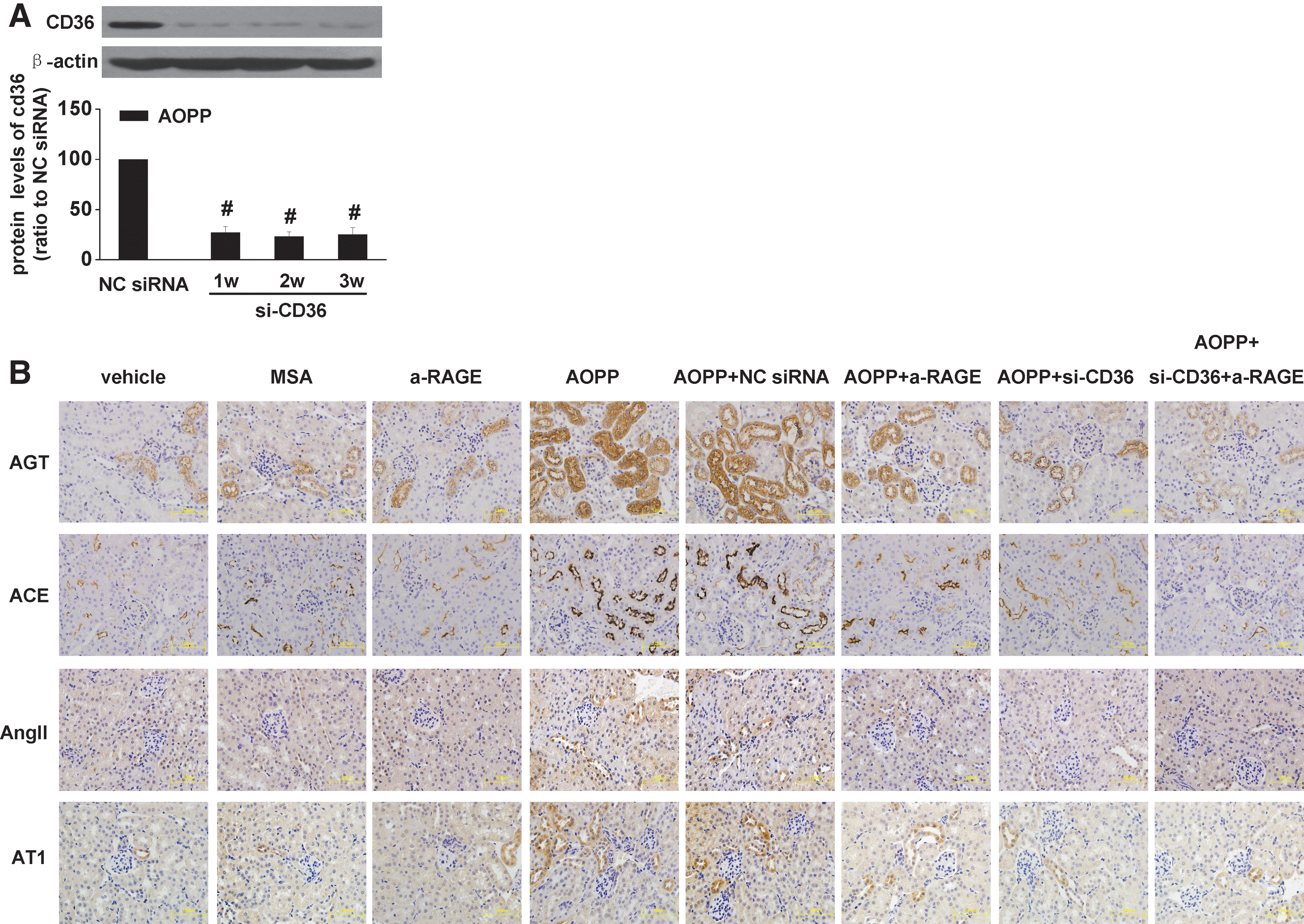
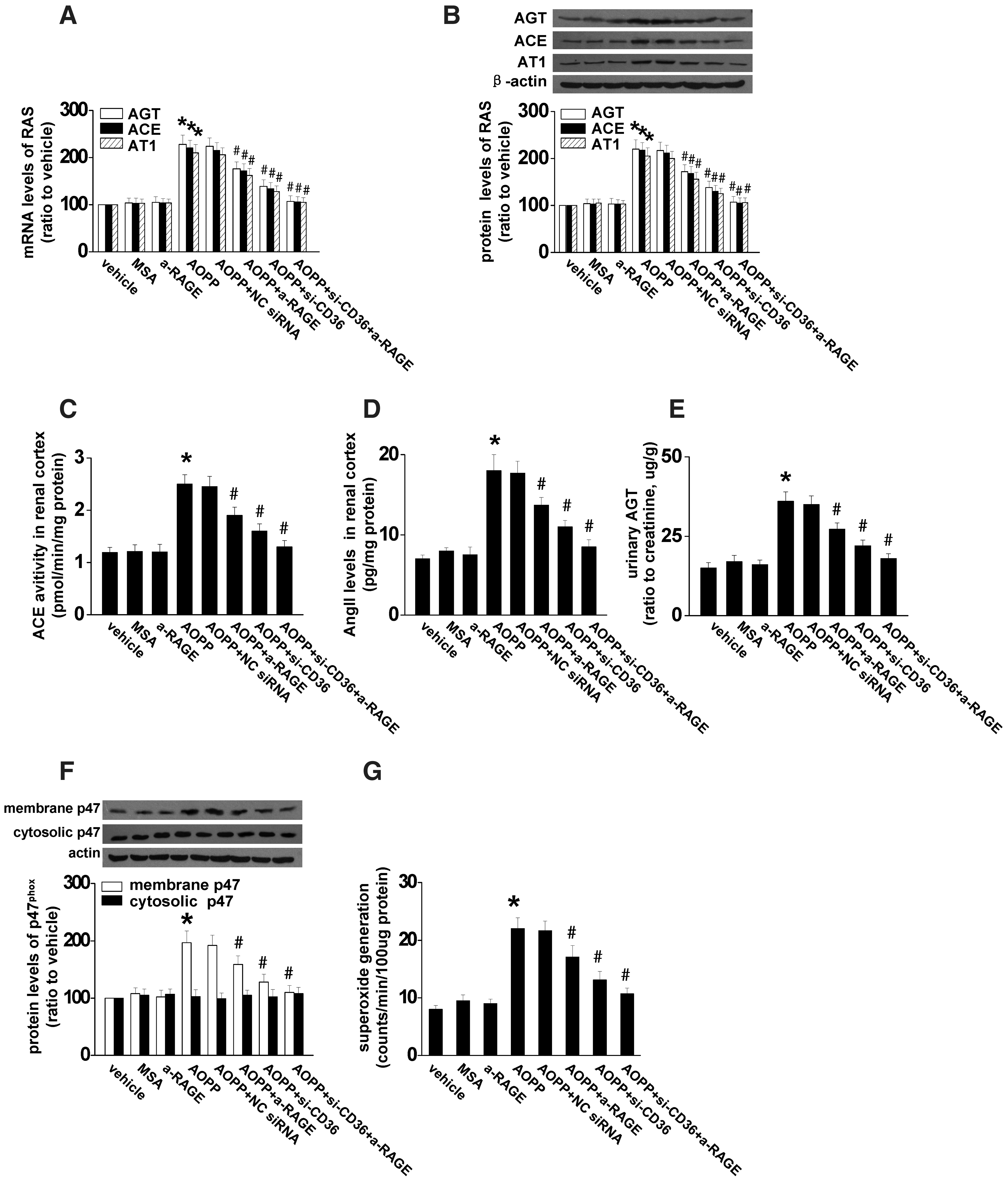
To determine the receptor-mediating AOPP-induced RAS activation in vivo, BALB/c mice were treated with AOPP-modified mouse serum albumin (AOPP-MSA) together with CD36 siRNA, anti-RAGE, or CD36 siRNA plus anti-RAGE for 3 weeks. The efficiency of CD36 siRNA on knockdown CD36 expression was demonstrated by western blot (Fig. 8A). Immunohistochemistry staining indicated that blockade of CD36, and to a less extent, blocking RAGE, significantly decreased the expression of AGT, ACE, and AT1 in renal tubular cells (Fig. 8B). Combination of CD36 siRNA with anti-RAGE completely abolished AOPP-triggered intrarenal RAS activation. Furthermore, CD36 siRNA remarkably attenuated AOPP-induced expression of intrarenal AGT, ACE, and AT1 at both mRNA and protein levels (Fig. 9A, B), decreased the activity of ACE (Fig. 9C), attenuated the production of Ang II (Fig. 9D), and urinary excretion of AGT (Fig. 9E). In addition, blocking CD36 significantly inhibited AOPP-induced NADPH oxidase activation (Fig. 9G) and ROS production (Fig. 9H). Likewise, AOPP-induced albuminuria was significantly attenuated by CD36 blockade (Table 2). Blocking CD36 together with RAGE almost completely abolished the triggering effect of AOPPs (Fig. 9 and Table 2). These data suggested that AOPP-induced activation of intrarenal RAS was mediated mainly by CD36.
p<0.05 versus MSA 50 mg/kg/day; b p<0.05 versus AOPP 50 mg/kg/day; c p<0.05 versus vehicle.
Data are expressed as mean±SD, n=6.
MSA, mouse serum albumin; RAGE, receptor for advanced glycation end products; siRNA, small interfering RNA.
Renal accumulation of AOPPs was correlated with RAS activation in human CKDs
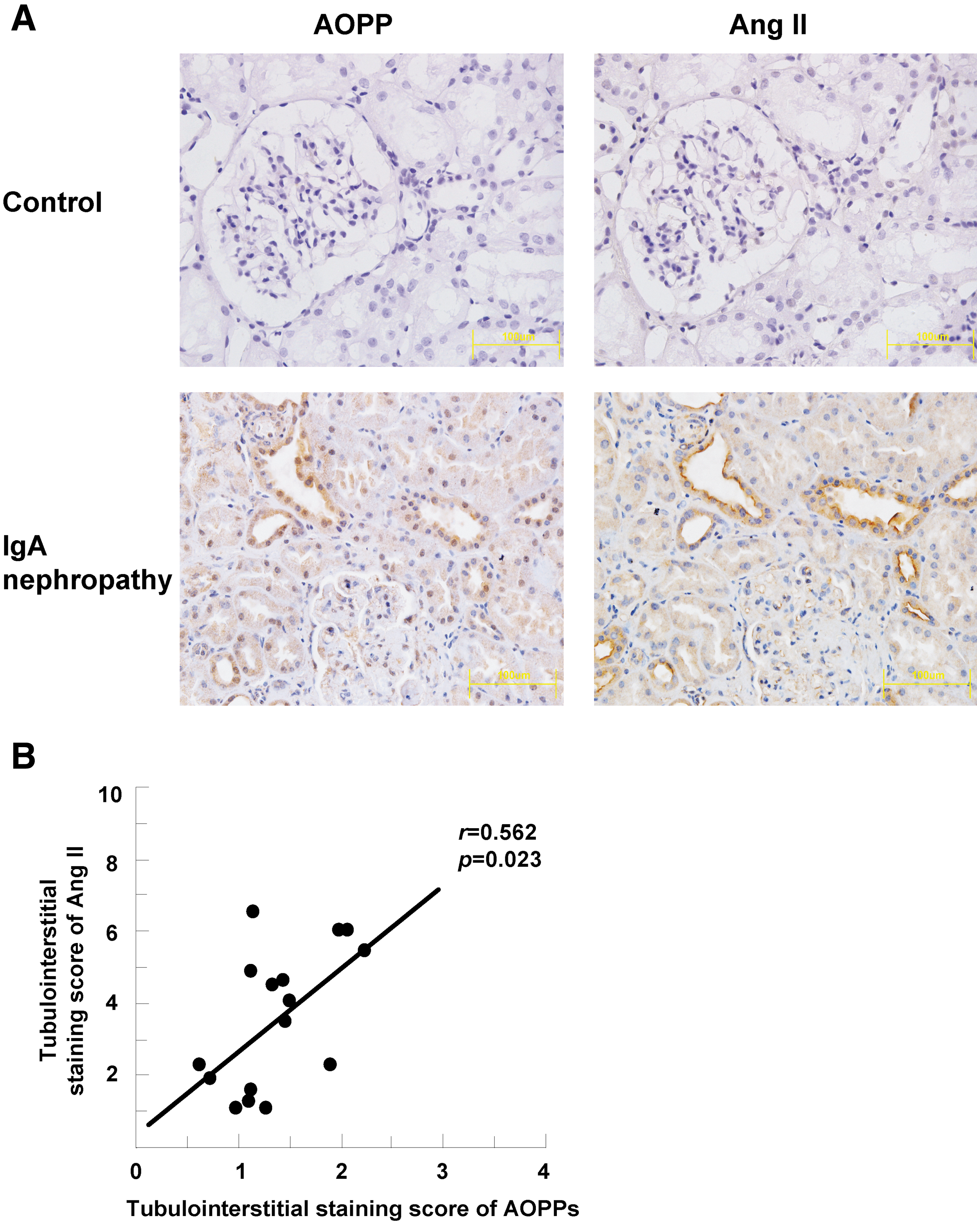
To evaluate the clinical relevance of AOPP-activated intrarenal RAS, we examined the expression of AOPPs and Ang II by immunohistochemistry in sequential sections of renal biopsies from 19 patients with IgA nephropathy. The normal renal tissue adjacent to renal tumor was used as normal control. As shown in Figure 10A, AOPPs and Ang II were predominantly co-localized in renal tubular cells in renal tissues from patients with IgA nephropathy, whereas no positive signal for AOPPs and Ang II was detected in the normal renal tissue. Semiquantitative analysis revealed that the expression of AOPPs was significantly correlated with that of Ang II (Fig. 10B), suggesting that accumulation of AOPPs was associated with intrarenal RAS activation in human IgA nephropathy.
Discussion
Inappropriate activation of intrarenal RAS plays a critical role in the progression of CKD. However, little is known about the independent regulation of local RAS activation. In the present study, we provided both in vitro and in vivo evidence that AOPP-albumin activated RAS in renal tubular epithelial cells. The triggering effect of AOPP-albumin was much stronger than that of unmodified albumin and was mainly mediated by a CD36-dependent pathway involving activation of PKCα, NADPH oxidase, and NF-κB/AP-1. Chronic AOPP–albumin loading in UNX rats resulted in AOPP deposition in PTCs accompanied with RAS activation and functional perturbations such as increased urinary albumin excretion. Although the AOPPs used in the study were prepared in vitro by exposure of albumin to high concentration of HOCl, our previous study has demonstrated that the biological effect of the AOPPs prepared by this method is similar with that isolated from the patients (13). In addition, accumulation of AOPPs was also detected in renal tubular cells and correlated with RAS activation in renal biopsies from patients with IgA nephropathy. To the best of our knowledge, this is the first study demonstrating that AOPPs activated intrarenal RAS through a CD36-dependent, redox-sensitive pathway.
Our in vitro study found that AOPP-albumin upregulated the expression of almost all components of RAS and increased the activity of ACE in PTCs. Data from our in vivo study demonstrated that deposition of AOPPs in renal tissues, particularly in renal tubular epithelial cells, was associated with activation of intrarenal RAS when its plasma concentration was increased to the level detected in patients with diabetes and CKDs (onefold higher than the mean of normal level) (22, 45), indicating that accumulation of AOPPs might be a trigger for intrarenal RAS activation. The effect of AOPPs on intrarenal RAS activation was similar in both rat and mouse models, which is consistent with our previous studies that the renal histological and functional changes responsible for an AOPP challenge are very similar between the two species (48). However, AOPP loading at this level seemed not to trigger the circulating RAS. The reason for the differential effect of AOPPs on intrarenal and circulating RAS is not clear, but might relate to the fact that intrarenal RAS activity is regulated in a manner distinct from the circulating RAS concentration (23). Previous studies have demonstrated that the Ang II contents in renal tissues are much higher than can be explained on the basis of equilibration with the circulating concentrations (23). Furthermore, the demonstration of much higher concentrations of Ang II in specific regions within the kidney indicated selective local regulation of intrarenal RAS (31).
Exposure of PTCs to excessive albumin also increases the activation of RAS (2). However, our in vitro and in vivo data demonstrated that AOPP-modified albumin is a much stronger modulator for intrarenal RAS compared to native albumin: AOPP-RSA, at 1 percent of the concentration of native RSA, upregulated the expression of RAS in cultured PTCs. Furthermore, chronic loading of AOPP-RSA at 50 mg/kg/day resulted in a similar increase in activation of intrarenal RAS as that of RSA at 5 g/kg/day. These results suggest that, in conditions with increased protein oxidative damage such as CKDs and diabetes, increased AOPP-albumin in the lumen of renal tubules might cause a more dramatic activation of local RAS than native albumin.
Oxidative stress is a common feature in diabetes and CKDs (15, 38). Increased plasma concentration of AOPPs has been reported in patients with IgA nephropathy and diabetes (4, 34). We found in the present study that AOPPs were mainly deposited in tubular epithelial cells in patients with IgA nephropathy. Renal AOPP accumulation was co-localized with Ang II and correlated with Ang II formation in renal tubules, suggesting that AOPPs may be involved in progression of CKDs through activation of intrarenal RAS. Supporting this hypothesis, previous clinical studies have shown that AOPP deposition is found in biopsies from patients with various CKDs (11, 27, 29), and the plasma level of AOPPs significantly correlated with renal progression in patients with IgA nephropathy and diabetic nephropathy (1, 35). Blocking RAS using RAS inhibitors reduces albuminuria and improves the outcome of CKDs (3, 46).
The underlying mechanism responsible for the differential effect between AOPP-modified albumin and native albumin remains to be investigated. Unlike unmodified albumin, which functions through an endocytic receptor megalin/cubulin (2), AOPP-albumin exerts its biological activity mainly through the class B scavenger receptor CD36. Knockdown or blocking CD36, and to a much less extent, blocking RAGE, dramatically attenuated AOPP-albumin-induced intrarenal RAS activation both in vitro and in vivo. Blocking CD36 and RAGE together completely abolished the triggering effect of AOPP-albumin. However, knockdown megalin/cubilin or other scavenger receptors had no effect on AOPP-albumin-induced RAS activation. Consistent with our finding, previous study has shown that AOPPs promoted TGF-β production via CD36 on PTCs (20).
In the present study, we deciphered the intracellular signaling responsible for the triggering effect of AOPPs on RAS. AOPP challenge induced phosphorylation of PKCα, promoted NADPH oxidase-dependent ROS generation, and resulted in activation of NF-κB/AP-1. Interestingly, all the three RAS genes tested in the current study (ACE, AGT, and AT1) have binding sites for NF-κB and AP-1 (6, 7, 21, 28, 42). Blocking this pathway significantly abolished AOPP-induced intrarenal RAS activation. Noteworthy, AOPPs might exert a different biological activity in various renal cells via different receptors. Our previous studies indicated that AOPP-albumin triggers apoptosis of podocytes (48) and inflammation in endothelial cells via interacting with RAGE (13). AOPP stimulation increases extracellular matrix production in cultured mesangial cells (43). Iwao et al. reported that AOPPs promote TGF-β expression in PTCs via binding with CD36 (20). Although AOPPs interact with various receptors, the postreceptor signaling appears quite similar. NADPH oxidase activation is the common intracellular signaling event elicited by AOPPs in various cell types (13, 43, 48). The present study demonstrated that inhibition of this enzyme significantly diminished AOPP-induced intrarenal RAS activation in cultured PTCs and in AOPP-challenged animals. These data provided important information for understanding the pathogenic effect of AOPPs.
Taken together, our findings delineate an important role for AOPPs in activating intrarenal RAS. AOPPs deposited in renal tissues, particularly in renal tubular epithelial cells, when the plasma concentration increased to the level detected in diabetes and CKDs. AOPP-albumin activated RAS in PTCs through a CD36-mediated, redox sensitive pathway. Given the fact that accumulation of AOPPs is prevalent in diabetes and CKD, we propose that AOPPs accumulated in renal tubules may promote formation of the vicious feedback loop between oxidative stress and RAS activation, and thus may maintain or amplify the renal injury. Strategies aimed at reducing AOPP accumulation could slow the progression of CKD.
Materials and Methods
AOPP–albumin preparation
AOPP-rat/MSA was prepared in vitro as described previously (13, 48). Briefly, fatty acid-free rat/MSA (Sigma, St. Louis, MO) was exposed to 200 mM HOCl (Fluke, Buchs, Switzerland) for 30 min in the absence of free amino acid/carbohydrate/lipids. The preparation was dialyzed overnight against phosphate-buffered saline (PBS) to remove free HOCl.
AOPPs and RAGE monoclonal antibody preparation
Monoclonal antibodies against AOPPs (Clone 3F2) and RAGE (Clone B2-2) were generated as described previously (8, 12, 27, 49).
Animal model
All animal procedures were approved by the Animal Experiment Committee of the Southern Medical University. Details are described in the Supplementary Materials and Methods. Briefly, male Sprague-Dawley rats were subjected to UNX. One week later, the UNX rats were randomized into four groups (n=6 in each group) and received daily intravenous injection of vehicle (endotoxin-free PBS, pH 7.4), native RSA (50 mg/kg per day), AOPP-RSA (50 mg/kg per day), or AOPP-RSA (50 mg/kg per day) plus intragastric administration of apocynin, respectively.
To block CD36 and/or RAGE, the UNX male BALB/c mice were administrated with AOPP-MSA (50 mg/kg per day) together with mouse CD36 siRNA by intravenous injection, anti-RAGE monoclonal antibody by intraperitoneal injection, or CD36 siRNA plus anti-RAGE, respectively.
Patients
The renal biopsy sections were taken from 19 adult patients with biopsy-proven primary IgA nephropathy. Normal renal tissues adjacent to the tumor (renal cell carcinoma) collected from nephrectomies (n=3) were served as controls. The demographic, clinical, and laboratory data of the patients are presented in Table 3.
All the patients are freshly diagnosed without any therapy. Those with eGFR<80 ml/min/1.73 m2 have been excluded.
Values are expressed as mean±SD, bMale/Female or cYes/No;
eGFR was calculated as the Modification of Diet in Renal Disease (MDRD) formula.
BMI, body mass index; MAP, mean arterial blood pressure; eGFR, estimated glomerular filtration rate.
Statistical analysis
All data were expressed as mean±SD. Continuous variables between groups at each time point were compared using a one-way analysis of variance, followed by a least significant difference (LSD) method when p<0.05. Differences of the variables between two time points were determined by independent samples t-test. The relationship between variables was assessed by the Pearson correlation analysis. Statistical analyses were conducted with SPSS 13.0 for Windows (SPSS, Chicago, IL). p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
This study is supported by the National 973 program (No. 2012CB517700 and 2011CB504005), National Nature and Science Grants (No. 30830056 and U0932002) to Dr. Fan Fan Hou, and National Nature and Science Young Investigator Grant (81200502) to Dr. Wei Cao.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.