
Abstract
Introduction
Innovation
We have shown earlier that aldose reductase (AR) inhibition is antioxidative, anti-inflammatory, and chemopreventive. However, the molecular mechanisms by which AR promotes colon cancer cell proliferation are not known. Herein, we provided a novel role of AR in the regulation of microribonucleic acid (miR)-21 and its target phosphatase and tensin homolog (PTEN) in oxidative stress-induced colon cancer cells. Our findings show that AR promotes colon cancer growth by regulating the reactive oxygen species/phosphatidylinositol 3-kinase/serine/threonine kinase/forkhead box O3A (FOXO3a)/activator protein-1(AP-1)/miR-21/PTEN pathways. Our results for the first time indicate that AR inhibition by decreasing the miR-21 expression and increasing the levels of PTEN and FOXO3a prevents colon cancer growth. Our studies also indicate a novel role of FOXO3a in preventing miR-21 expression by attenuating AP-1 activation.
Recent studies have demonstrated that microribonucleic acids (miRs), a class of small noncoding 18- to 25-nucleotide-long RNAs, negatively regulate gene expression by binding to the 3′-untranslated region of target messenger RNAs (mRNAs), which cause translational repression or degradation of target proteins (35, 28). miRs have indeed important regulatory functions in processes such as differentiation, proliferation, and inhibition of apoptosis (28). Deregulation of miRs affects normal cell growth that leads to a variety of disorders, including malignancies (15, 28). miR-21 is overexpressed in various types of solid tumors as well as other malignancies (5, 6, 8, 13, 18, 21, 24, 40, 44). Further, increased expression of miR-21 has been implicated in various processes involved in carcinogenesis, including inhibition of apoptosis (6), promotion of cell proliferation (24), and stimulation of tumor growth (27). Regulation of cell proliferation by miR-21 could be by attenuating the transcription of the target gene/protein. Phosphatase and tensin homolog deleted on chromosome 10 (phosphatase and tensin homolog [PTEN]) has been shown to be a validated target of miR-21 (21). Recent studies have shown that inactivation as well as downregulation of PTEN play an important role in tumor progression (29, 41). However, the mechanisms of PTEN inactivation/degradation are poorly understood. Degradation of PTEN by miRs, particularly by miR-21, and blocking the transcription/translation of PTEN by the inactivation of its transcription factors such as FOXO3a, a member of forkhead transcription factors, through the PI3K/serine/threonine kinase (AKT) pathway, play a major role in tumorigenesis. The forkhead family of transcription factors is characterized by the presence of a conserved 100-amino-acid deoxyribonucleic acid (DNA)-binding domain that participates in regulating diverse cellular functions such as apoptosis, differentiation, metabolism, proliferation, and survival (1). FOXO3a that transcribes various genes becomes inactive when phosphorylated by protein kinase AKT. Upon phosphorylation by AKT at Thr-32, Ser-253, and Ser-315, FOXO3a binds to 14-3-3 proteins and loses the transcriptional functions (4). To investigate if the antioxidative property of AR inhibition prevents colon cancer growth by regulating the expression of oxidative stress-induced miRNAs and their target proteins, we examined the molecular mechanisms by which AR regulates miR-21 and PTEN expression in human colon cancer cells. Our results demonstrate that inhibition of AR represses the expression of miR-21 through regulating ROS/PI3K/AKT/FOXO3a/AP-1 signaling, which in turn increases PTEN, and thereby prevents colon cancer growth.
Results
Regulation of growth factor-induced miRs by aldose reductase in human colon cancer cells
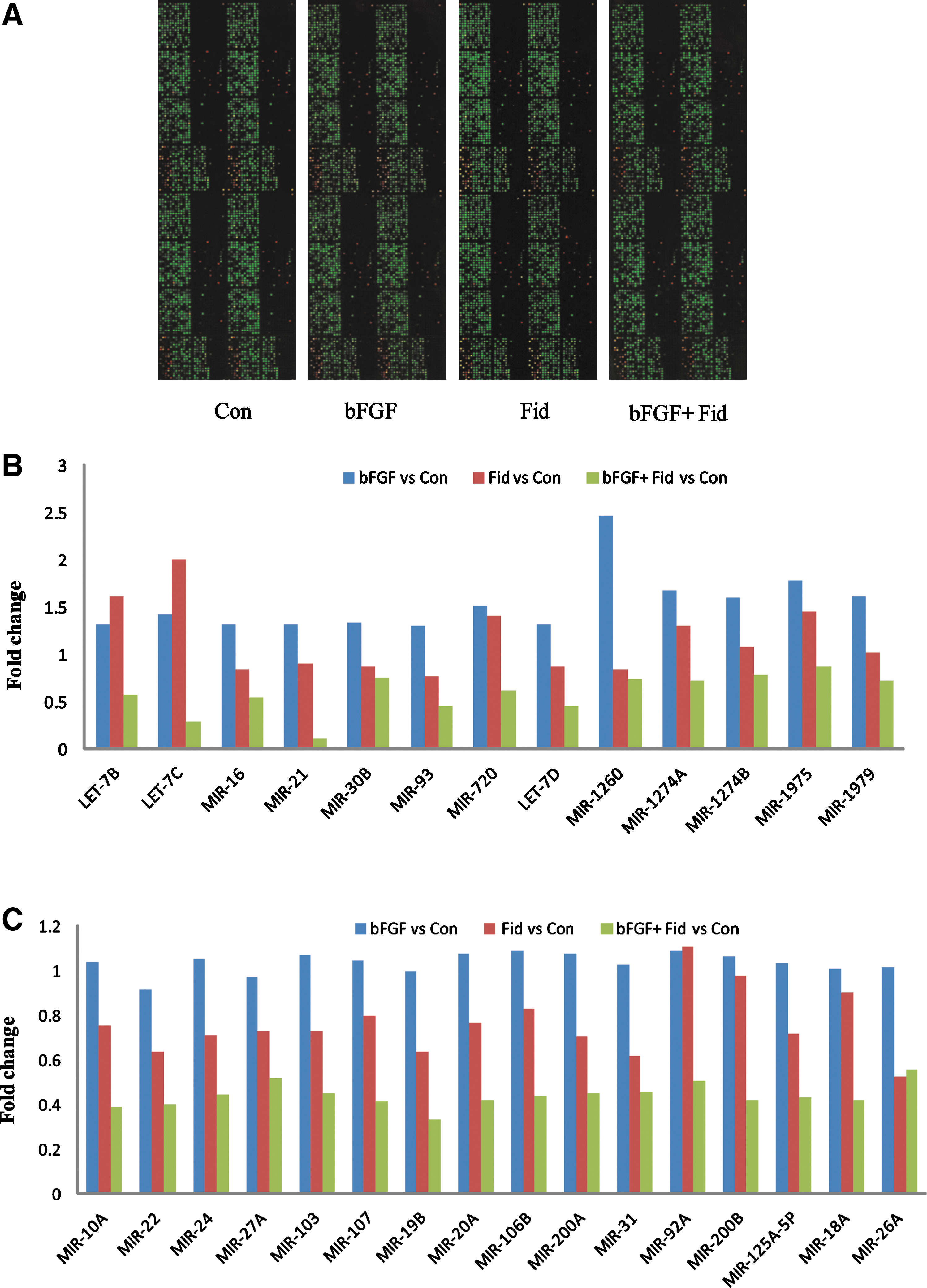
Aberrant expression of miRs that regulate the expression of various genes is associated with numerous diseases, including tumor initiation and progression (13, 15). Therefore, we first examined how oxidative stress-response protein, AR, regulates miR expression in growth factor-treated human colon cancer cells (HT29) by performing miR microarray (Fig. 1A). Approximately 13 miRs were upregulated (1.3–2.4-fold) in growth factor-treated cells. They were let-7b, let-7c, let-7d, miR-16, miR-21, miR-30b, miR-93, miR-720, miR-1260, miR-1274a, miR-1274b, miR-1975, and miR-1979. Incubation of HT29 cells with AR inhibitor significantly suppressed the expression of these miRs (Fig. 1B). In addition, expression of 16 miRs (miR-10a, miR-22, miR-24, miR-27a, miR-103, miR-107, miR-19b, miR-20a, miR-106b, miR-200a, miR-31, miR-92a, miR-200b, miR-125-5p, miR-18a, and miR-26a) was not changed when stimulated with bFGF, whereas incubation with AR inhibitor significantly suppressed (∼2-fold) the expression of these miRs (Fig. 1C). This suggests that AR regulates the expression of various miRs in colon cancer.
Regulation of miR-21 and its target protein, PTEN, by AR in human colon cancer cells
To further confirm the microarray data, we next investigated the role of AR in bFGF

Effect of AR inhibition on the activation of PTEN in growth factor-induced HT29 colon cancer cells
Since, PTEN levels in cancer cells are regulated by specific protein kinases such as PI3K/AKT and also by miR-21 (21, 29, 41), and our results suggest that AR regulates PTEN protein expression, therefore we examined how AR inhibition restores growth factor-induced activation and expression of PTEN. As shown in Figure 2E, stimulation of HT29 cells with EGF in a time-dependent manner increased the phosphorylation of PTEN, and inhibition of AR significantly decreased it. Further, as shown in Figure 2C and D, stimulation of cells with EGF and bFGF decreased PTEN protein expression, and AR inhibitor significantly increased the expression of PTEN compared to that by growth factors alone, suggesting that inhibition of AR not only activates PTEN but also increases the expression of PTEN in colon cancer cells. To further confirm the role of PTEN in HT29 cell proliferation, we ablated the cells with PTEN siRNA and incubated with EGF. This resulted in increased cell proliferation in PTEN-ablated cells compared to EGF alone (Fig. 2F). Increased cell proliferation was further confirmed by proliferating cell nuclear antigen (PCNA) expression. HT29 cells ablated with PTEN siRNA showed increased PCNA expression compared to EGF alone (Fig. 2G). Further, AR inhibition prevented the EGF-induced cell growth in control siRNA-treated cells, but not in the PTEN-siRNA treated cells (Fig. 2F). AR inhibitor, fidarestat, added in the culture medium did not cause significant floating of cells.
Effect of miR-21 mimic and inhibitor on PTEN expression and HT29 cell proliferation
Since miR-21 is well known to regulate the expression of PTEN, we next examined the role of AR in mediation of miR-21-regulated PTEN expression as well as cancer cell proliferation. For these studies, HT29 cells were transfected with miR-21 mimic, followed by stimulation with EGF for 24 h. Cell proliferation was higher in the cells treated with EGF+miR-21 mimic compared to the cells treated with EGF alone (Fig. 3A). The increased growth of cells was significantly attenuated by AR inhibitor as well as miR-21 inhibitor. However, AR inhibitor, miR-21 mimic, and miR-21 inhibitor individually did not affect HT29 cell proliferation (Fig. 3A). Further, as shown in Figure 3B, treatment of HT29 cells with EGF alone as well as EGF+miR-21 mimic downregulated the PTEN expression, and miR-21 inhibitor significantly prevented it. Thus, our results indicate that inhibition of AR prevents colon cancer growth by regulating the miR-21-mediated expression of PTEN in colon cancer cells.

Effect of AR inhibition on the activation of FOXO3a in growth factor-induced HT29 colon cancer cells
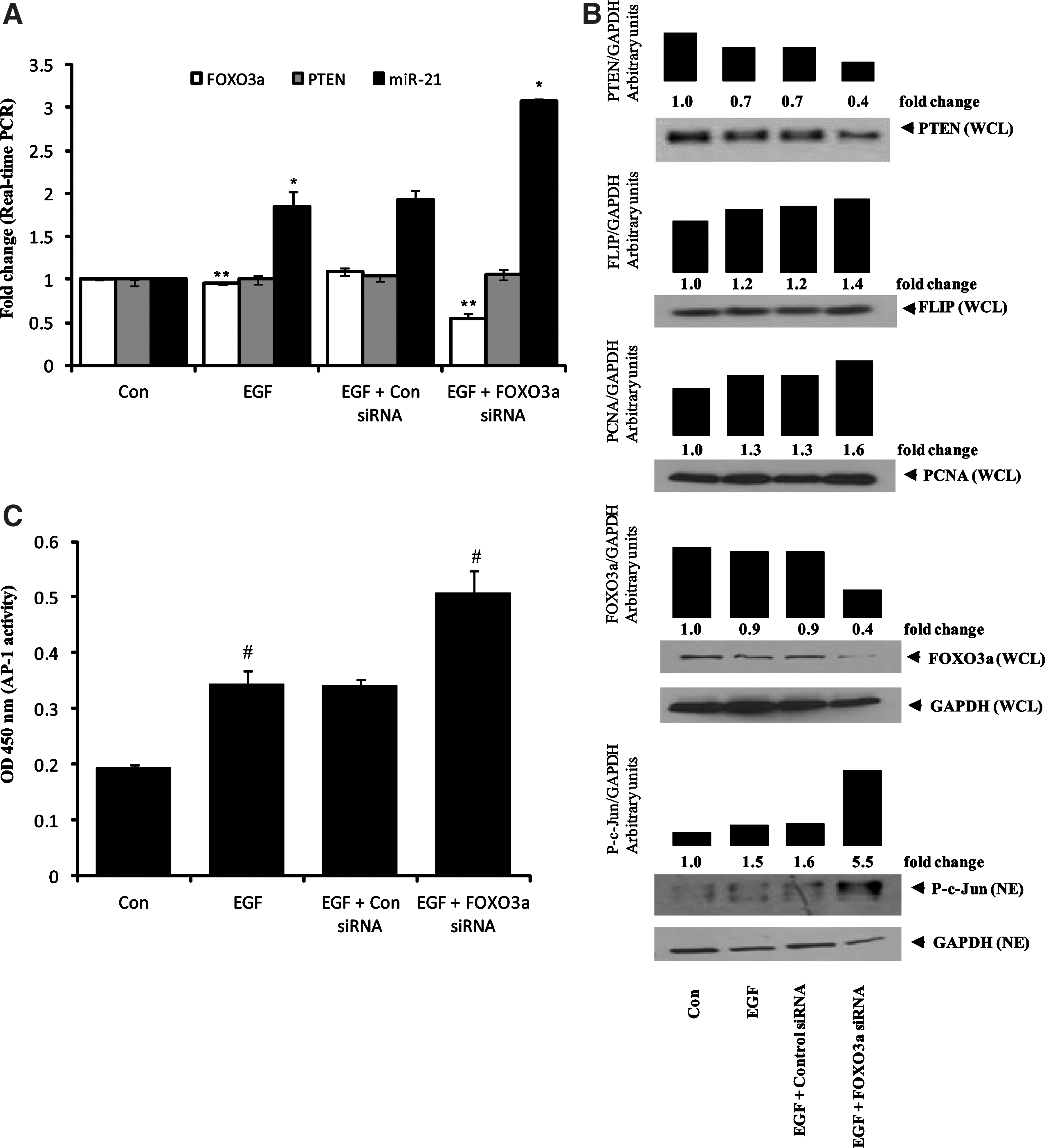
Recently, FOXO3a has been shown to be one of the transcription factors for PTEN and a repressor of miR-21 expression (7, 42). To test our hypothesis that AR regulates PTEN and miR-21 expression in colon cancer cells through FOXO3a, we investigated the effect of AR inhibitor on the activation and expression of FOXO3a. As shown in Figure 3C, EGF inactivated FOXO3a in a time-dependent phosphorylation. In contrast, preincubation of HT29 cells with AR inhibitor significantly decreased the phosphorylation of FOXO3a. To further understand the role of AR inhibition in FOXO3a expression in HT29 and Caco-2 colon cancer cells, we treated the cells with or without AR inhibitor for 24 h, followed by stimulation by the growth factors, EGF and bFGF. As shown in Figure 3D and E, stimulation of HT29 and Caco-2 cells with growth factors and preincubation with AR inhibitor significantly increased the expression of FOXO3a. Since we did not observe any change in the PTEN mRNA level in HT29 cells with or without AR inhibitor, we concluded that FOXO3a may not be solely responsible for PTEN transcription. Although FOXO3a is known to repress miR-21 expression, the exact mechanism is not known. Since AP-1 is a transcription factor for miR-21 expression, it is likely that FOXO3a may repress miR-21 expression through AP-1 inactivation. Therefore, we ablated HT29 cells with FOXO3a siRNA and stimulated with EGF for 48 h. Real-time PCR results showed that HT29 cells ablated with FOXO3a siRNA did not affect PTEN mRNA levels, whereas miR-21 expression was significantly upregulated compared to EGF alone (Fig. 4A). These results demonstrate that FOXO3a represses miR-21 expression. Further, HT29 cells ablated with FOXO3a siRNA and stimulated with EGF showed increased AP-1 activation (c-Jun phosphorylation) compared to EGF alone (Fig. 4B). Also, HT29 cells ablated with FOXO3a siRNA showed increased PCNA and FLIP expression compared to EGF alone (Fig. 4B). We also quantified the DNA-binding activity of AP-1 in HT29 cells ablated with FOXO3a siRNA using a transcription factor enzyme-linked immunosorbent assay kit (Panomics). Ablation with FOXO3a siRNA in HT29 cells resulted in increased DNA-binding activity of AP-1 compared to EGF alone (Fig. 4C). This novel finding suggests that FOXO3a represses miR-21 expression through inhibition of AP-1 activation in colon cancer cells.
Inhibition of AR prevents EGF-induced miR-21 expression by regulating AP-1 and its upstream targets
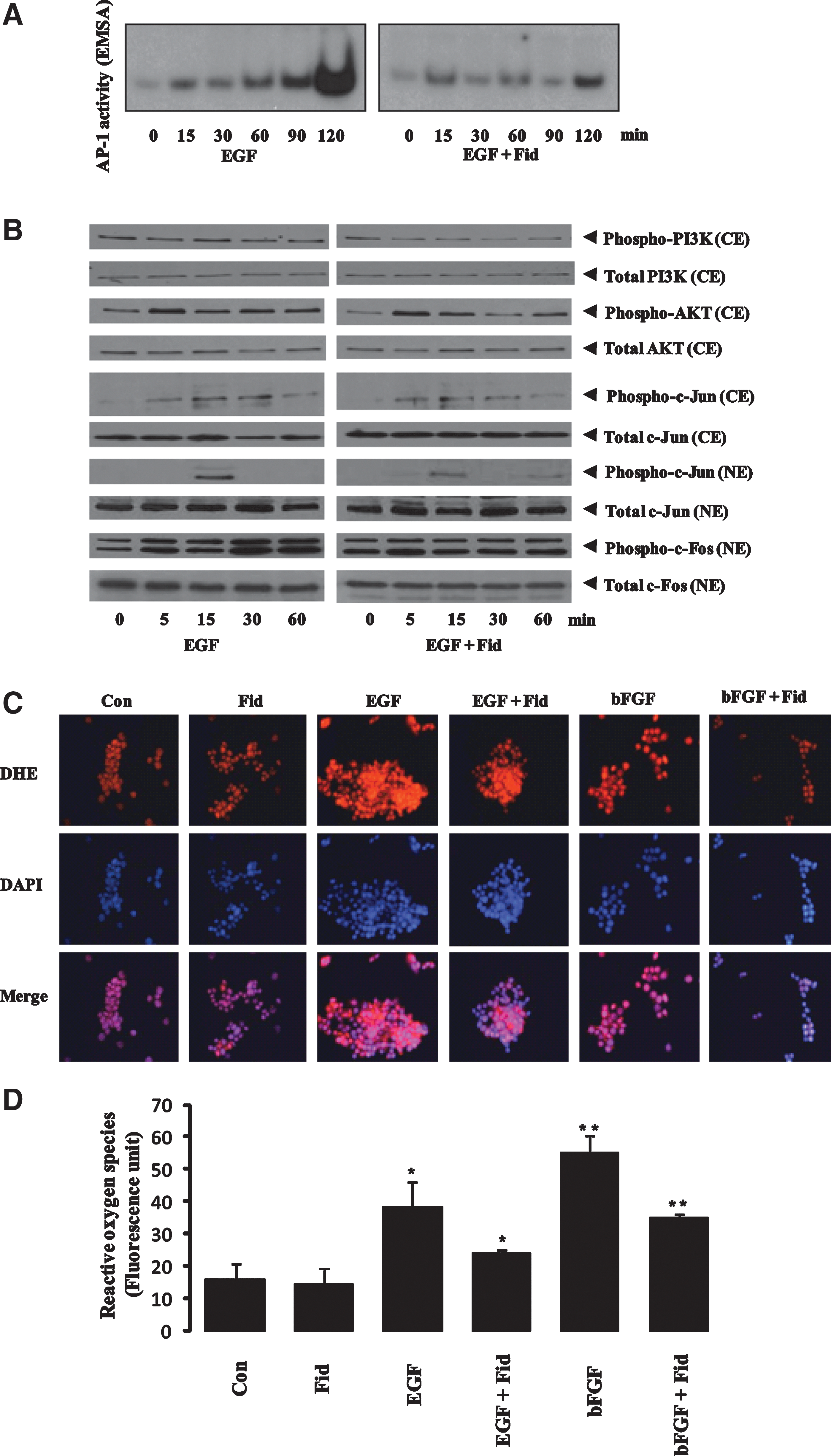
Since miR-21 expression is regulated by AP-1, which in turn is activated by the PI3K and AKT pathway, we determined the effect of AR inhibitor on EGF-induced AP-1 activation. As shown in Figure 5A, stimulation of HT29 cells with EGF increased DNA-binding activity of AP-1, which was significantly prevented by preincubation of HT29 cells with AR inhibitor. To further understand how AR inhibition prevents miR-21 expression, we examined the activation of PI3K and AKT in colon cancer cells. As shown in Figure 5B, stimulation of HT29 cells with EGF activated PI3K and AKT, and the AR inhibitor, fidarestat, inhibited it. Activation of AP1 was inhibited by inhibition of c-Jun and c-Fos phosphorylation in the presence of AR inhibitor (Fig. 5B). Since AP-1 is a redox-sensitive transcription factor, we next examined the effect of AR inhibition on growth factor-induced ROS. Treatment of HT29 cells with growth factor for 1 h caused a marked increase in ROS formation, and preincubation of cells with AR inhibitor prevented it (Fig. 5C, D). These results suggest that inhibition of AR decreases miR-21 expression by attenuating the ROS/PI3K/AKT/FOXO3a/AP-1 pathway.
Inhibition of AR prevents the progression of colon cancer in nude mice by modulating miR-21, PTEN, and FOXO3a expression
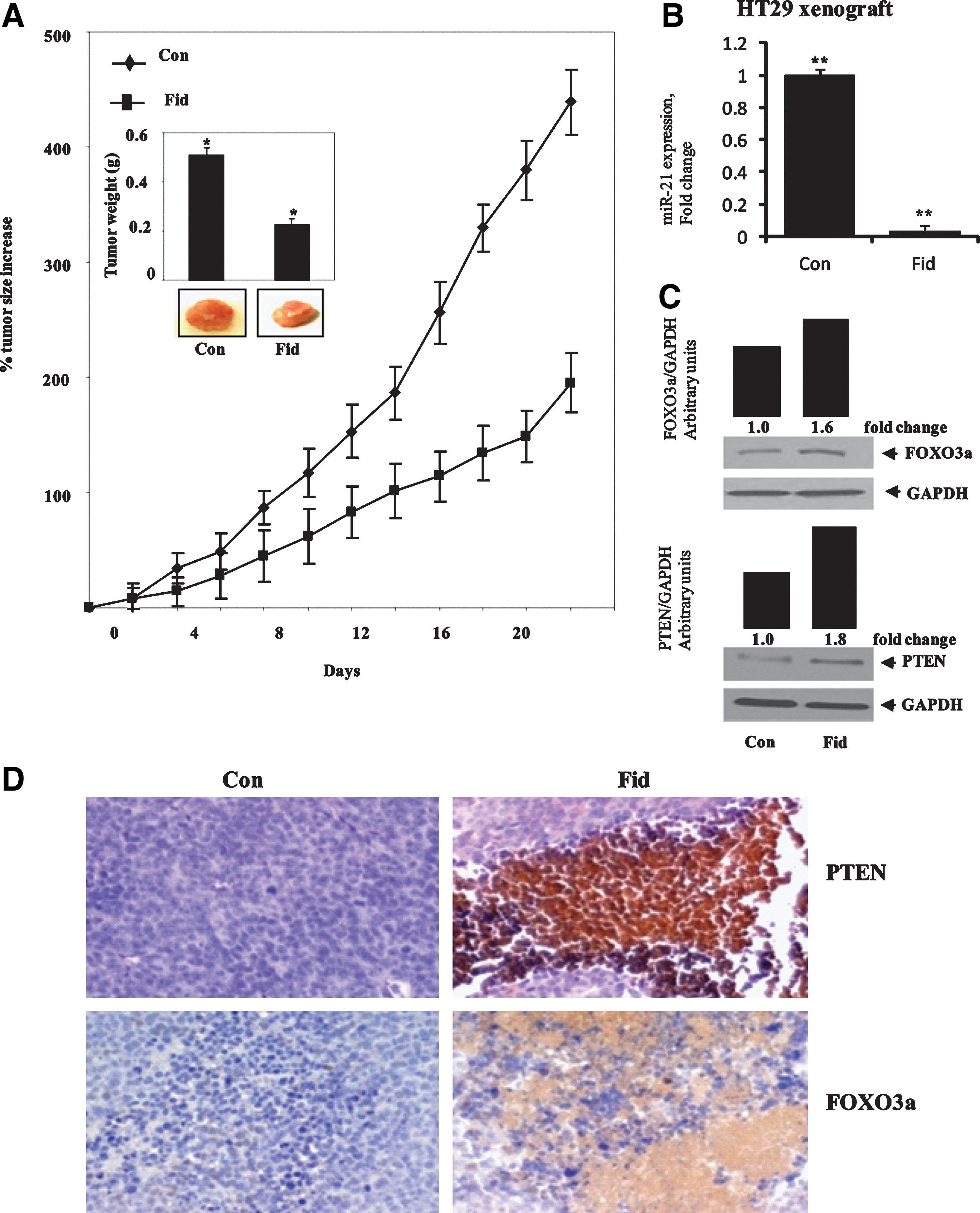
HT29 cells were injected subcutaneously in nu/nu nude mice, and the tumor was allowed to grow to ∼30 mm2 over a period of ∼3 weeks as described earlier (33). Subsequently, the animals were treated with AR inhibitor, fidarestat (50 mg/kg body weight/day), in drinking water. At different days, tumor volumes were measured by using a caliper, and the percentage increase in tumor size was calculated by taking 30 mm2 as starting zero percent (Fig. 6A). When the tumor size in the control mice (without AR inhibitor) reached almost 160 mm2, all the mice were killed, and the tumors were excised. The weight as well as the size of the tumors in AR inhibitor-treated group were significantly less compared to control mice (Fig. 6A). Since inhibition of AR prevented tumor growth in nude mouse xenografts, we next determined the effect of AR inhibition on the expression of miR-21 in xenograft tissues. Our results shown in the Figure 6B indicate that miR-21 expression was almost completely inhibited in fidarestat-treated mice compared to control mice. We next determined the effect of AR inhibition on the expression of PTEN and FOXO3a in xenograft tissues. As shown in Figure 6C and D, increased expression of PTEN and FOXO3a was observed in fidarestat-treated mice compared to control mice. Overall, our results indicate that AR inhibition could prevent colon cancer growth by downregulating the expression of miR-21 and upregulating the expression of PTEN and FOXO3a.
Discussion
ROS are known to play a crucial role in various cellular processes such as proliferation, cell migration (metastasis), differentiation, DNA modification, and apoptosis (36, 20, 37, 43, 17, 14, 11). The cellular processes mediated by ROS depend on the source and the extent of ROS that can cause cell proliferation as well as cell death (36, 20, 37, 43, 17, 14, 11). ROS are critical for both inflammation and tumor growth, as they control a myriad of signaling pathways by activating various transcription factors, AP-1, NF-kB, STAT3, etc (25, 12, 3). Herein, we have examined how AR modulates the growth factor-induced colon cancer growth via ROS-mediated activation of miR21 and its target proteins such as PTEN.
Dysregulation of cell signaling that promotes tumor proliferation is controlled by miRs, which function as both oncogenes and tumor suppressors (15). In most of the cancers, upregulated oncogenic miRs could downregulate their target tumor suppressor genes and facilitate proliferation of cancer cells. By using the miR microarray, we have found ∼13 miRs, including widely studied miR-21, upregulated in bFGF-treated human colon cancer HT29 cells. miR-21 is one of the oncogenic miRs that is highly expressed in a variety of malignant tumors. Because miR-21 targets various tumor suppressors (6), the upregulation of this miR may result in increased inflammatory markers such as proapoptotic proteins. Since miR-21 is upregulated under oxidative stress (6, 21, 27), and AR mediates the oxidative stress-mediated cell proliferation and inflammatory signaling in various diseases, including angiogenesis and cancer (22, 23, 30, 31, 33, 34), we determined the role of AR in the modulation of miR-21 expression in human colon cancer cells. Our results demonstrate that AR inhibition suppresses oncogenic miR-21 expression and prevents the proliferation of cancer cells. We have further identified PTEN as a putative target of miR-21. In particular, we showed that AR inhibition upregulates the PTEN and FOXO3a levels and downregulates miR-21, and thereby facilitates the inhibition of tumorigenesis. We have thus demonstrated that AR regulates the expression of miR-21 and its target PTEN, which are the key regulators of numerous physiological cellular processes such as proliferation, metabolism, and apoptosis via PI3K/AKT/AP-1 signaling.
The functional significance of miR-21 largely depends on the target genes that it regulates. Recently, PTEN has been validated as a target for miR-21 (21). It has been reported that PTEN can induce apoptosis and control cell growth, invasion, and angiogenesis through interference with several signaling pathways (19). PTEN is a dual protein–lipid phosphatase that dephosphorylates the secondary messenger produced by PI3K and interrupts the downstream activation of AKT, which is known to be involved in tumorigenesis (19). Further, PTEN is one of the most frequently inactivated/downregulated tumor suppressors in different types of cancer (29, 41). Several mechanisms for PTEN downregulation in various cancers such as genetic mutation, promoter methylation, and phosphorylation by the upstream kinase, inactivation/downregulation of transcription factor, and targeting by oncogenic miRs, especially miR-21, have been proposed (10, 29, 41). Since we have shown that AR inhibition decreases miR-21 expression in colon cancer cells, we envisioned that AR inhibition could restore PTEN levels, thereby preventing the cell proliferation. Indeed, inhibition of AR in growth factor-induced colon cancer cells resulted in upregulation of PTEN protein levels, indicating a major role of AR in PTEN expression. Since phosphorylation of PTEN by various upstream kinases also causes its inactivation (10), we investigated the role of AR inhibition in PTEN phosphorylation. Our results showed that AR inhibition prevents the phosphorylation of PTEN in growth factor-induced human colon cancer cells, thereby activating PTEN. Further, AR inhibition not only activates PTEN but also increases the expression of this protein in cancer cells, thereby preventing the proliferation of colon cancer cells. Therefore, AR could be a novel target to prevent colon cancer. Further, FOXO3a is one of the important transcription factors besides protein 53, early growth response factor 1, peroxisome proliferator-activated receptor-γ, and upstream stimulatory factor 1 for PTEN (7), and we investigated the effect of AR inhibition on FOXO3a activation/expression in human colon cancer cells. Our results showed that AR inhibition significantly increased the expression and activation of FOXO3a, indicating that FOXO3a could transcribe PTEN in colon cancer cells. Since we did not observe a significant difference in PTEN mRNA levels in growth factor-treated HT29 cells with or without AR inhibitor, we concluded that FOXO3a might not be solely responsible for PTEN transcription. Although FOXO3a is known to repress miR-21 expression (42), the exact mechanism has not been identified. Since AP-1 is a transcription factor for miR-21 (32), we hypothesized that FOXO3a may repress miR-21 expression through AP-1 inactivation. Our results showed a novel finding that FOXO3a represses miR-21 expression by inhibiting the activation and DNA-binding activity of AP-1 (Fig. 4). We found that AR inhibition downregulates miR-21 expression by inhibiting PI3K/AKT/AP-1 and upregulates FOXO3a and PTEN in colon cancer cells (Fig. 7). It is likely that decreased growth factor-induced activation of AP-1 by AR inhibition could be by increased expression of FOXO3a, which is known to repress miR-21 expression. To investigate how miR-21 promotes cell proliferation and downregulates PTEN, we transfected HT29 cells with miR-21 mimic and inhibitor. HT29 cells transfected with the miR-21 mimic and stimulated with EGF showed increased cell proliferation and downregulation of PTEN compared to EGF alone. These results inversely correlated with miR-21 inhibition and demonstrated that miR-21 promotes cell proliferation via downregulation of PTEN.
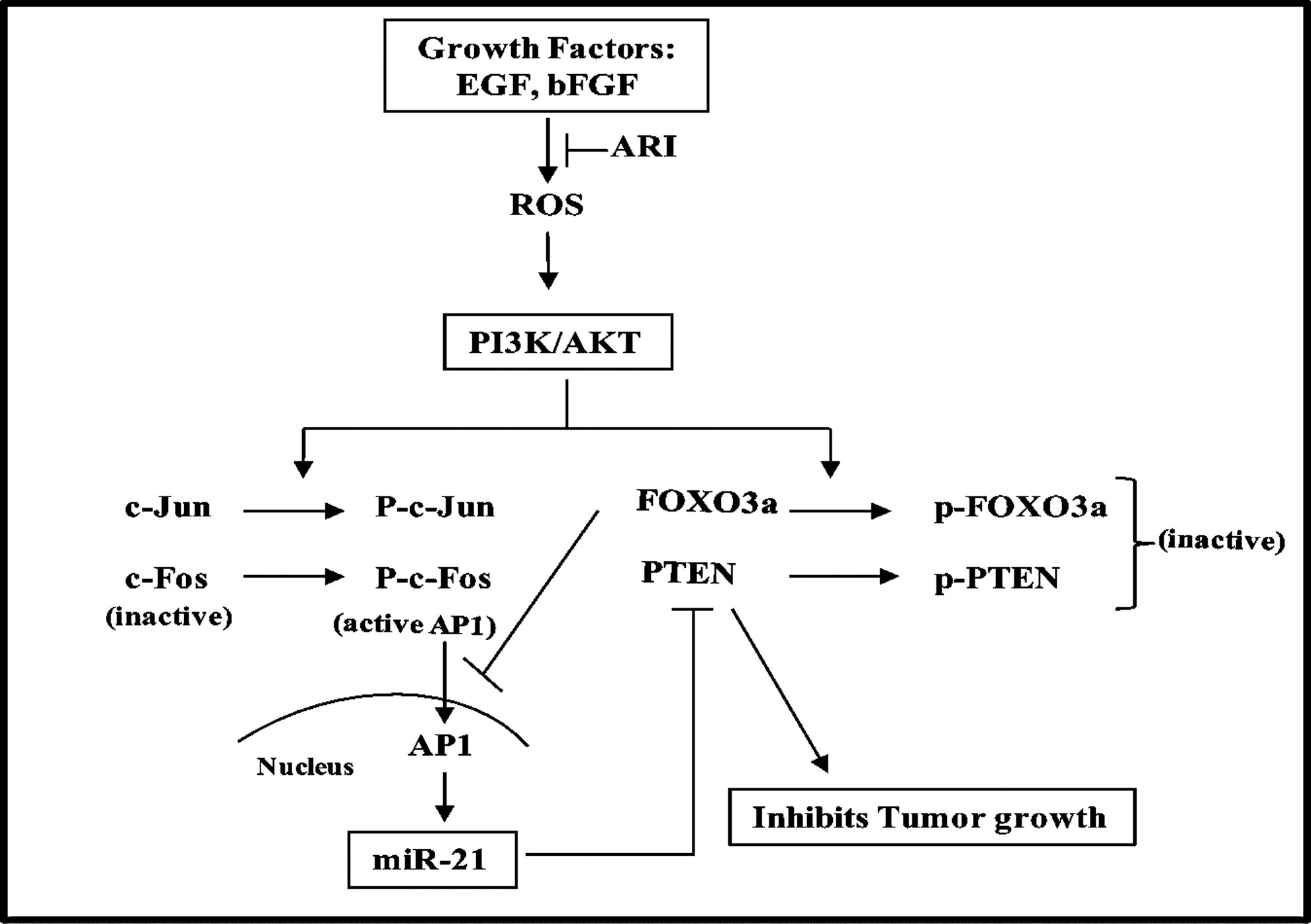
In summary, we have shown that by downregulation of miR-21 expression, AR inhibition prevents growth factor-induced colon cancer cell proliferation. AR inhibition also prevents the activation (phosphorylation) of PI3K/AKT and its mediated inactivation (phosphorylation) of PTEN and FOXO3a. Further, inhibition of AR increased the growth factor-mediated expression of PTEN and FOXO3a, which are important negative regulators of the PI3K/AKT/AP-1 pathway that regulates tumor cell proliferation.
Materials and Methods
Materials
McCoy's 5A medium, RPMI 1640, phosphate-buffered saline (PBS), penicillin/streptomycin solution, trypsin, and fetal bovine serum (FBS) were purchased from Invitrogen. Antibodies against PTEN, phospho-PTEN (S380/Thr382/383), FOXO3a, phospho-FOXO3a (S253), PI3K, phospho-PI3K (p85-Tyr458/p55-Tyr199), AKT, phospho-AKT (S473), c-FOS, phospho-c-Fos (S32), c-Jun, phospho-c-Jun (S73), PCNA, FLIP, and GAPDH were purchased from Cell Signal, Inc., and Santa Cruz Biotechnology, Inc., respectively. Fidarestat was obtained as a gift chemical from Sanwa Kagaku Kenkyusho Co., Ltd., and Livwel Therapeutics, Inc. EGF, bFGF, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and other reagents used in Western blot analysis were obtained from Sigma. On-Target plus SMARTpool PTEN siRNA against PTEN was purchased from Dharmacon RNAi Technologies (Thermo Scientific). All other reagents used were of analytical grade.
Cell culture
The human colon cancer cell lines SW480, Caco-2, HCT116, and HT29 were obtained from the American Type Culture Collection. SW480, and Caco-2 cells were grown in RPMI 1640 and Dulbecco's modified Eagle medium supplemented with 10% FBS and 1% penicillin/streptomycin, respectively. HCT116 and HT29 cells were maintained and grown in McCoy's 5A medium supplemented with 10% FBS and 1% penicillin/streptomycin.
miR microarray analysis
HT29 cells were serum-starved in McCoy's 5A medium containing 0.1% FBS in the presence or absence of fidarestat (2 μM) for 24 h and then stimulated with EGF for another 12 h. After 12 h, cells were collected, quickly frozen in liquid nitrogen, and stored at −80°C until further analysis. Cells were sent to Miltenyi Biotec, CA, for miR microarray analysis using universal reference. Briefly, total RNA was isolated using standard RNA extraction protocols (Trizol). Samples were labeled according to the undisclosed miRXplore™ User manual. Fluorescent-labeled samples were hybridized overnight to miRXplore Microarrays using the a-Hyb™ Hybridization Station. In general, control samples [Universal Reference (UR)] were labeled with Hy3, and experimental samples were labeled with Hy5. The miRXplore Universal Reference (UR) represents a defined pool of synthetic miRs for comparison of multiple samples. Fluorescence signals of the hybridized miRXplore Microarrays were detected using a laser scanner (Agilent Technologies). Mean signal and mean local background intensities were obtained for each spot of the microarray images using ImaGeneâ software (Biodiscovery). Low-quality spots were flagged and excluded from data analysis. Unflagged spots were analyzed with PIQORTM Analyzer software that included background subtraction to obtain the net signal intensity, data normalization, and calculation of the Hy5/Hy3 ratios for the sample. As an additional quality-filtering step, only spots/genes were taken into account for the calculation of the Hy5/Hy3 ratio that had a signal that was equal or higher than the 50% of the background (Fig. 1A).
Quantitative real-time PCR analysis of miR-21 expression
HT29 cells were serum-starved in McCoy's 5A medium containing 0.1% FBS in the presence or absence of fidarestat (2 μM) for 24 h and then stimulated with bFGF for another 8 h. Subsequently, total RNA was isolated using standard RNA extraction protocols (Trizol) and quality checked via the Agilent 2100 Bioanalyzer platform. miR-enriched total RNA was extracted from HT29 cells using the mirVana miRNA isolation kit (Ambion). miR-21 was quantified using TaqMan miR Assays (Applied Biosystems). U6 RNA was used for normalization of miR expression. Analysis and fold changes were determined using the comparative threshold cycle (Ct) method.
Measurement of cytotoxicity
HT29 cells were cultured in McCoy's 5A medium in a 96-well plate at 2500 cells/well. Subconfluent cells were growth-arrested in 0.1% FBS with or without the AR inhibitor, fidarestat (2 μM), or transfected with miR-21 mimic and inhibitor, and PTEN siRNA or control siRNA using Hiperfect reagent (Qiagen). After 24 h, cells were treated with EGF (10 ng/ml) and incubated for 24 h. Cell viability was determined by MTT assay as described previously (34).
Transient transfection with miRNA mimic and miRNA inhibitor
miR-21 mimic and inhibitor were transfected into HT29 cells according to the manufacturer's protocol. Briefly, 2×105 HT-29 cells were transfected with 500 nM of inhibitor and 50 nM of mimic of miR-21 (Qiagen GmbH) for 24 h. Cells were also serum-starved in McCoy's 5A medium containing 0.1% FBS for 24 h, followed by stimulation with EGF (10 ng/ml) for another 24 h.
Western blots and electrophoretic mobility-shift assay
The expression of PTEN, phospho-PTEN, FOXO3A, phospho-FOXO3A, PI3K, phospho-PI3K, AKT, phospho-AKT, c-FOS, phospho-c-FOS, c-Jun, phospho-c-JUN, PCNA, FLIP, and GAPDH proteins was determined by Western blot analysis. The antigen–antibody complex was detected by enhanced chemiluminescence (Pierce). Electrophoretic mobility-shift assay was performed as described earlier (23). The radiolabeled bands were quantified by using Kodak Image Station 2000R.
Determinations of ROS
ROS were determined in HT29 cells treated with or without AR inhibitor using ROS-sensitive indicators dihydroethidium (DHE) and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescin diacetate, acetyl ester (CM-H2DCFDA) from Invitrogen. For ROS detection by DHE, ∼1×105 HT29 cells were seeded on chambered slides and incubated for 24 h at 37°C. The cells were serum-starved in 0.1% FBS medium containing 10 μM fidarestat for overnight. The cells were then stimulated with EGF(10 ng/ml) and bFGF (15 ng/ml) for 1 h. Subsequently, the cells were washed with cold PBS twice and incubated with 5 μM DHE in PBS at 37°C in a humidified chamber for 30 min. The cells were washed in PBS twice and mounted with a fluorsave mounting medium containing DAPI (Vector Laboratories, Inc.). The photomicrographs were obtained and analyzed with a Nikon epifluorescence microscope with a 585-nm long-pass filter. ROS were quantified by CM-H2DCFDA fluorescent dye, and ∼2.5×104 HT29 cells were seeded in a 24-well plate in triplicate and incubated for 24 h at 37°C. The cells were starved in a medium containing 0.1% serum without or with 10 μM fidarestat for overnight and subsequently washed with 1 × Hank's buffered salt solution (HBSS) buffer and incubated with 5 μM CM-H2DCFDA at 37°C for 30 min, washed again to remove excess CM-H2DCFDA, and treated with EGF and bFGF in presence or absence of 10 μM fidarestat for 1 h. At the end of incubation, the cells were washed twice with cold 1 × HBSS buffer, and fluorescence was measured with a CytoFluor II fluorescence plate reader (PerSeptive Biosystems, Inc.) at excitation 485 nm and emission 528 nm.
Nude mouse xenografts
The effect of AR inhibition on tumor growth of human colon cancer cells (HT29) in nude mouse xenografts was studied as described earlier (33). Briefly, 5–6-week-old athymic nude nu/nu mice (Charles River) were injected s.c. with 1×106 HT29 human colon adenocarcinoma cells in 100 μl PBS. Animals were treated with fidarestat (50 mg/kg body weight/day) in drinking water when the tumor surface area exceeded 30 mm2 (day 21). Tumors were measured in two dimensions using calipers. All animal experiments were carried out in accordance with a protocol approved by the Institutional Animal Care and Use Committee.
Statistical analysis
Data presented as mean±SE and p-values were determined by unpaired Student's t-test using Microsoft Office Excel 2007 software. p<0.05 was considered as statistically significant.
Footnotes
Acknowledgment
Supported by an NIH grant CA129383 to SKS
Author Disclosure Statement
The authors have no conflicts of interest to disclose.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.