
Abstract
Introduction
Innovation
Mitochondria are a major source of reactive oxygen species (ROS). Here we show that mutants lacking the mitochondrial proteins Cmc1 or Coa4 show an impaired assembly of cytochrome c oxidase, the terminal complex of the respiratory chain. Interestingly, the inability of these strains to grow under respiratory conditions is not due to their reduced respiratory competence per se, but rather due to their strongly increased production of hydrogen peroxide. Quenching of ROS completely suppresses their growth defect. We propose that assembly intermediates of respiratory chain complexes significantly contribute to mitochondrial ROS production, thereby preventing the propagation of cells that contain malfunctional mitochondria.
The assembly of respiratory chain complexes is an intricate process for several reasons: (i) The levels of mitochondrially encoded and nuclear encoded subunits need to be coordinated, which requires a regulatory cross talk from mitochondria to the nucleus that is only poorly understood (31); (ii) the complexes of the respiratory chain contain several reactive coenzymes, such as heme groups, iron-sulfur clusters, and copper or zinc ions, which need to be inserted during the assembly process; (iii) most mitochondrially encoded subunits consist of many transmembrane spans and belong to the most hydrophobic proteins of a eukaryotic cell; to prevent their unproductive aggregation, they need to be inserted into the inner membrane in a cotranslational fashion, which is achieved by physical tethering of the mitochondrial Oxa1 insertase to mitochondrial ribosomes (25, 27, 50); (iv) possibly the largest problem is the high reactivity of the reaction centers in respiratory chain complexes; assembly intermediates of these enzymes might lead to the direct transfer of single electrons to oxygen giving rise to the production of superoxide and other reactive oxygen species (ROS) (24, 29, 39). Although the potentially hazardous role of these intermediates was proposed a long time ago (46), experimental evidence for a physiological relevance of these intermediates was so far only described following exposure of cells to externally added hydrogen peroxide (26, 53).
Mainly by genetic studies carried out over the last three decades in the yeast Saccharomyces cerevisiae, a large number of assembly factors have been identified that coordinate the biogenesis of respiratory chain complexes (11, 52). Despite their early discovery and the relevance of respiratory chain assembly for human diseases, the function of many of these factors is still largely unclear. One ill-defined group of these assembly factors are the “twin Cx9C” proteins. They are characterized by two sequence motifs each containing two cysteine residues that are separated by nine amino acid residues. These proteins are located in the intermembrane space (IMS) of mitochondria. S. cerevisiae contains 14 members of this family, most of which are critical for the accumulation of normal amounts of cytochrome c oxidase and for growth on nonfermentable carbon sources (9, 32). The cysteine residues in these proteins are required for their targeting to mitochondria; during the import reaction, these proteins interact with the oxidoreductase Mia40 in the IMS, which converts the reduced thiols in the imported precursor into oxidized disulfides that are found in the mature forms of these proteins (3, 10, 36, 41).
In order to identify a role of redox regulation for the biogenesis of the respiratory chain, we performed an unbiased screen to identify yeast mutants in which the competence to respire is increased by the reductants dithiothreitol (DTT) and/or reduced glutathione (GSH). Thereby, we identified two mutants lacking “twin Cx9C” proteins whose respiratory deficiency was suppressed by DTT and GSH. Interestingly, the reductants did not cure the primary defects in respiratory chain assembly of these strains. However, they counteracted a toxic effect that is presumably due to increased oxidative stress resulting from the presence of assembly intermediates of cytochrome c oxidase. Our data suggest that defects in the function or biogenesis of mitochondrial enzymes lead to the respiration-induced production of hydrogen peroxide, which can impair cell growth. Based on the observations of this study we propose that an intracellular redox-dependent signaling pathway dampens the propagation of mutants that produce increased levels of mitochondria-derived oxidative stress.
Results
The growth defect on glycerol of many yeast mutants is suppressed by addition of reductants
To screen for yeast mutants that depend on externally added reductants in order to grow on nonfermentable carbon sources, we performed a genome-wide unbiased growth analysis. To this end we used a subset of the yeast Matα deletion library which contained 386 deletion mutants that showed a reduced growth on the nonfermentable carbon source glycerol (35). These strains and a wild type for control were grown overnight in the presence of the fermentable carbon source glucose and then spotted with a pinning tool onto plates containing the nonfermentable carbon source glycerol in the absence or presence of 5 mM GSH or DTT. The plates were incubated at either 25°C or 30°C and the growth of the colonies was compared. The growth of 44 mutants was stronger on at least two of the four reductant-containing plates than on the control plates (Table 1). Of these strains 18 (40%) lacked mitochondrial proteins, most of which are components required for the biogenesis or function of the respiratory chain, including many factors required for mitochondrial protein synthesis. The second largest group of proteins (nine strains, i.e., 20%) comprises nuclear factors that influence transcription. The potential growth stimulation by GSH and DTT for the mutants that showed the strongest stimulation by reductants was further confirmed by drop dilution assays (Fig. 1). The minor differences between the growth phenotypes presented in Table 1 and Figure 1 are presumably due to the different conditions of the precultures from which the plates were prepared (glucose-based, largely stationary cultures for Table 1 and galactose-based exponentially growing cells for Fig. 1). In summary, we conclude that addition of reductants stimulates respiratory growth of many yeast mutants, in particular of those showing defects in gene expression.

GSH, reduced glutathione.
Reductants suppress the growth defect of mutants lacking Cmc1 and Coa4 on nonfermentable carbon sources
Two strains that showed a strong respiratory growth stimulation by GSH and DTT caught our attention because they lacked genes for the “twin Cx9C” proteins Cmc1 and Coa4, which were shown before to play a role in the biogenesis of cytochrome c oxidase (7, 22, 23, 32). These strains showed no (Δcmc1) or poor (Δcoa4) growth on glycerol but were strongly stimulated by either GSH or DTT. Even growth of a double mutant in which both Cmc1 and Coa4 are absent can be rescued by GSH and DTT (Fig. 2A). Reductant-dependent growth on glycerol was not observed for a mutant lacking the structural Cox6 subunit of cytochrome c oxidase.
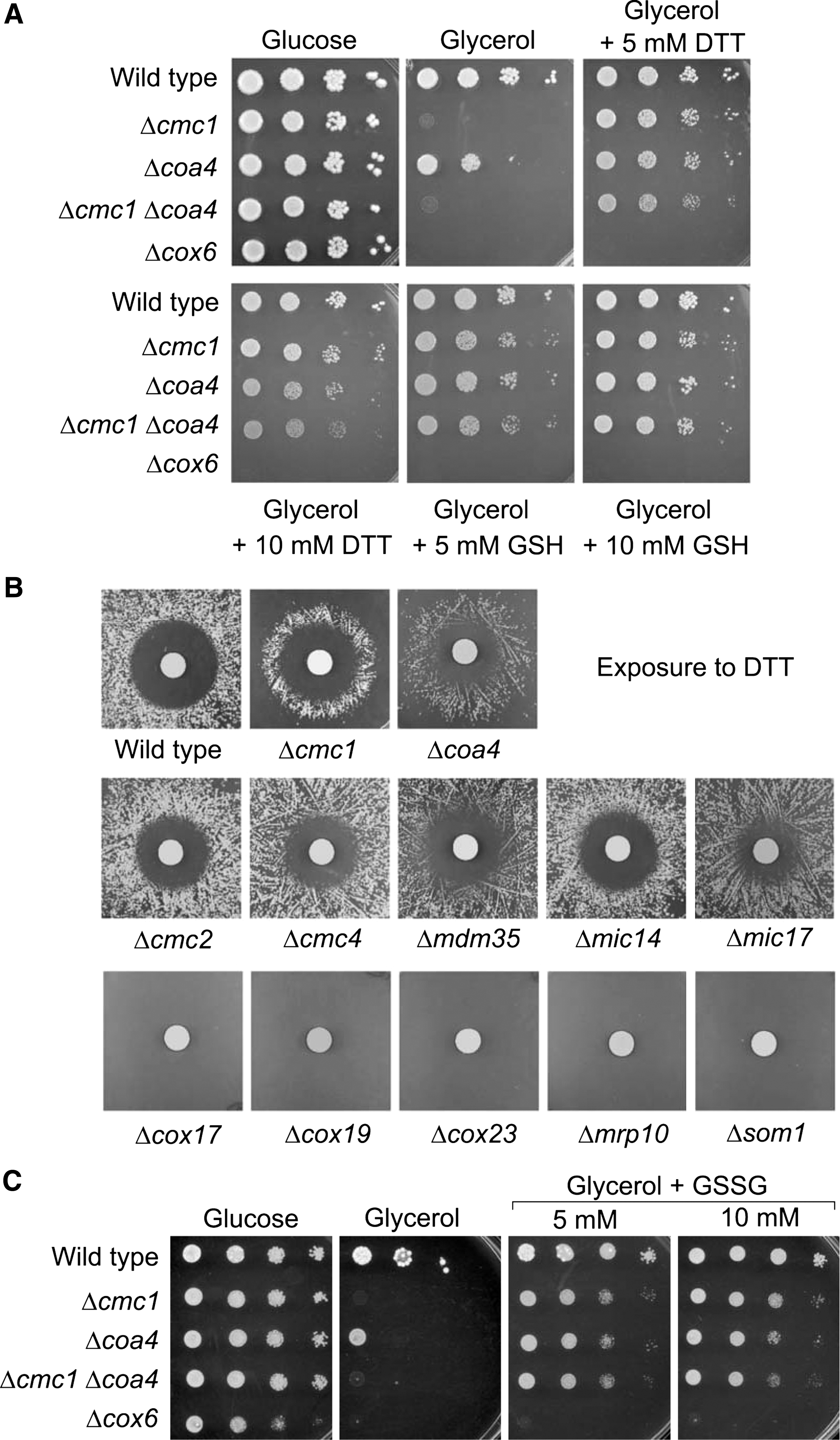
The positive effect of DTT on the growth of Δcmc1 and Δcoa4 cells was apparent when DTT-soaked filter papers were placed on a lawn of cells on YPG plates (Fig. 2B). The high DTT concentration directly around the filter prevented cell growth even of wild type leading to a colony-free halo. When Δcmc1 and Δcoa4 cells were used, a ring of colonies was observed that grew where the DTT concentration was neither too high nor too low. Ring-like halos were not observed with mutants lacking other “twin Cx9C” proteins (Fig. 2B).
The positive effect of the added reductants might be explained by a direct reduction of cellular thiols or, less directly, by an increase of the cellular capacity to buffer redox changes. To differentiate between both effects we next tested whether the addition of oxidized glutathione (GSSG) suppresses the growth defect of the mutants as well. As shown in Figure 2C, GSSG showed a strongly stimulating effect on respiratory growth of Δcmc1 and Δcoa4 cells. This suggests that it is not the direct reducing activity of the added reagent per se that is required as the uptake of GSSG, which can be reduced intracellularly to GSH by glutathione reductase in an NADPH-dependent process, obviously is sufficient to render the cells respiration-competent.
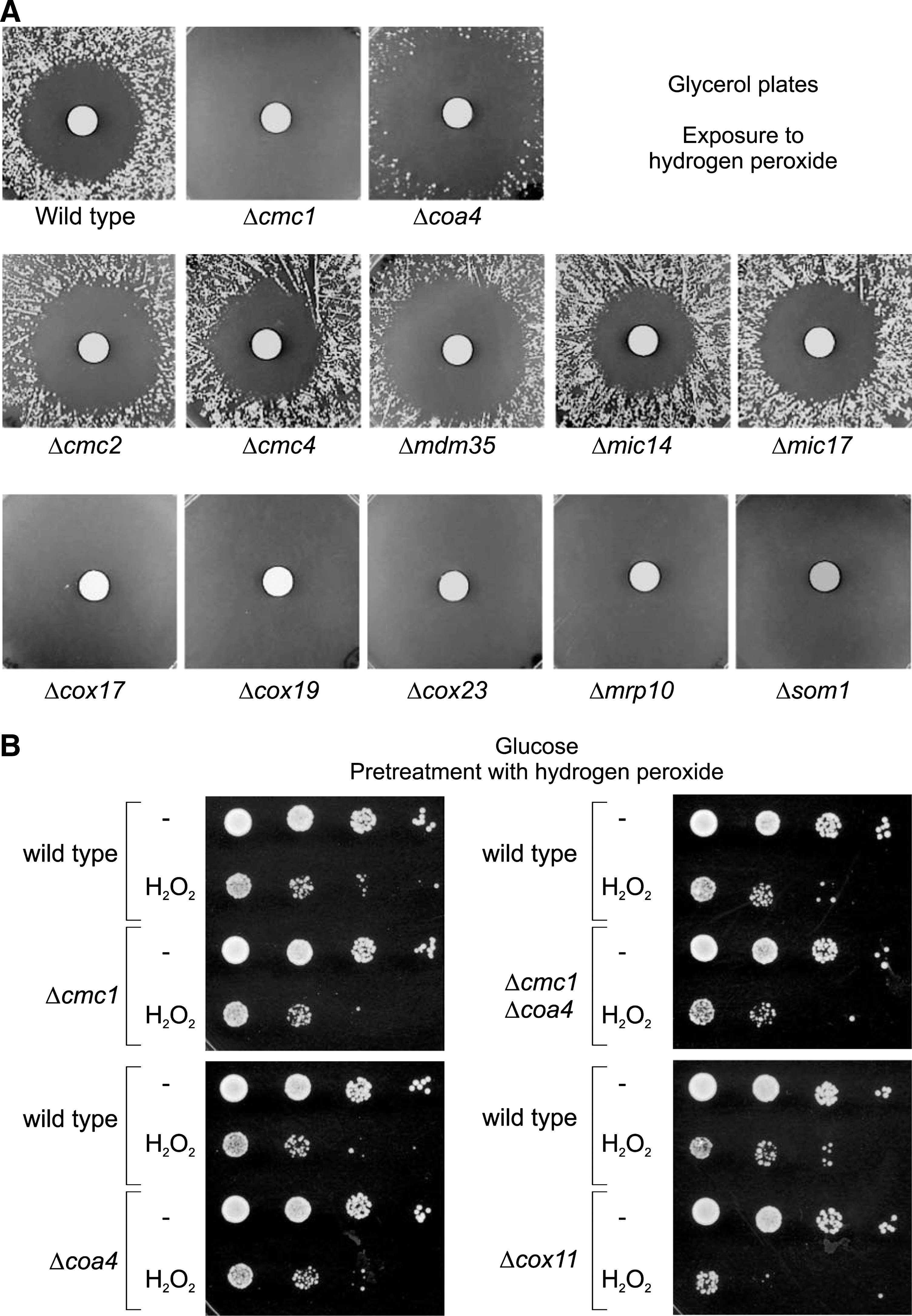
None of the yeast mutants lacking “twin Cx9C” proteins was suppressed upon exposure to the oxidizing reagent hydrogen peroxide (Fig. 3A). It was shown before that some mutants like Δcox11 that lead to defects in the assembly of cytochrome c oxidase show an increased toxicity upon incubation with hydrogen peroxide (26, 53). However, we did not find an increased hydrogen peroxide toxicity of Δcmc1 and Δcoa4 cells as long as they were cultured on glucose-containing medium (Fig. 3B).
Cmc1 or Coa4 exhibit specific nonredundant functions in mitochondria
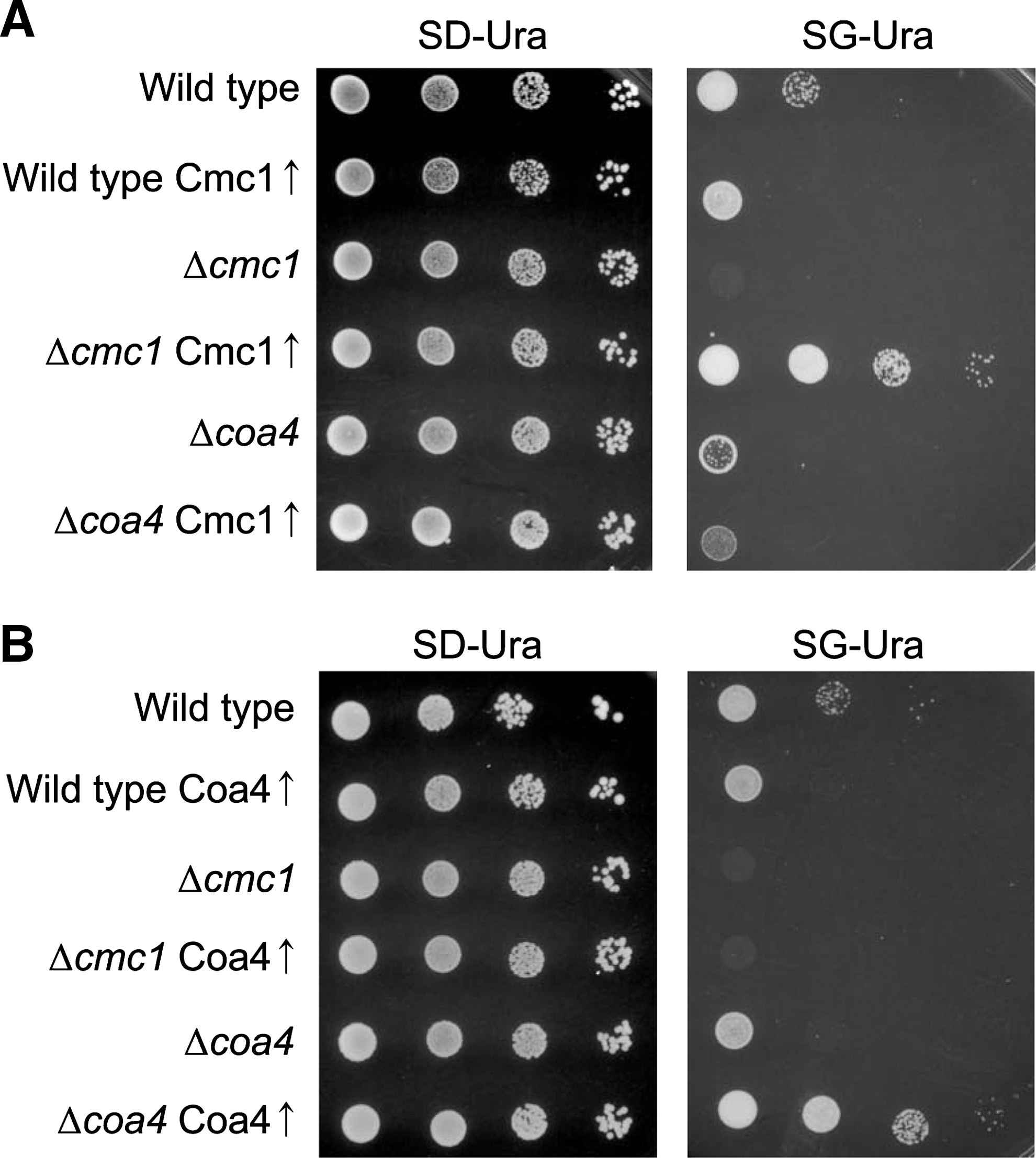
The similarities of Cmc1 and Coa4 in protein structure and in the growth phenotypes of the deletion mutants led us to test whether overexpression of one of the proteins can suppress the defects in the deletion mutants that lack the other protein (Fig. 4A, B). Whereas the overexpression plasmids carrying CMC1 and COA4 clearly suppressed the defects in Δcmc1 and Δcoa4 cells, respectively, they did not show any effect on the other mutant. Hence, we conclude that Cmc1 and Coa4 carry out nonredundant functions in mitochondria.
Mutants lacking Cmc1 or Coa4 show reduced levels of cytochrome c oxidase
The growth defect of Δcmc1 and Δcoa4 mutants on nonfermentable carbon sources pointed to a defect in mitochondrial respiration in these strains. Indeed, oxygen consumption measurements with isolated mitochondria confirmed strongly reduced respiration levels in the mutants, particularly in Δcoa4 mitochondria (Fig. 5A).

It was previously demonstrated that Cmc1 and Coa4 play a direct or indirect role in the assembly of cytochrome c oxidase (7, 22, 23). We therefore analyzed the activity of cytochrome c oxidase in Δcmc1 and Δcoa4 mitochondria (Fig. 5B). This revealed that in the absence of Cmc1 about 40% of the cytochrome c oxidase activity of wild-type mitochondria is found, and in the absence of Coa4 only about 25%. This correlates with Western blot signals of Cox2, one of the mitochondrially encoded core subunits of cytochrome c oxidase, which was also diminished, but not absent, in Δcmc1 and Δcoa4 mitochondria (Fig. 5C). Oxygen consumption—thus, respiration—was not stimulated in mitochondria of these strains by addition of GSH or the antioxidant ascorbic acid (Fig. 5D), indicating that reductants had no direct effect on the performance of the respiratory chain in any of these mitochondria.
In summary, we find reduced levels of cytochrome c oxidase in both mutants, although it should be noted that cytochrome c oxidase is still present in these strains and still exhibits residual activity. Interestingly, respiratory activity was less affected in Δcmc1 than in Δcoa4 mitochondria although its growth defect on glycerol was much more pronounced. This indicated that the reduced activity of cytochrome c oxidase might not be exclusively responsible for the growth arrest.
Reductants do not increase the levels of cytochrome c oxidase in Δcmc1 and Δcoa4 mutants
The suppression of the growth phenotype of the Δcmc1 and Δcoa4 mutants on nonfermentable media by DTT and GSH might suggest that reductants can restore normal cytochrome c oxidase levels in these strains. Diminished levels of cytochrome c oxidase might be due to a reduced production or an increased degradation of the enzyme. To monitor the synthesis of cytochrome c oxidase subunits as well as their stability, we radiolabeled mitochondrial translation products in whole yeast cells and followed their stability in subsequent chase reactions (Fig. 6A). In wild-type cells, all three mitochondria-encoded subunits of cytochrome c oxidase were efficiently synthesized and remained relatively stable during the 60 min of the postlabeling period. In contrast, Cox1 was synthesized only at reduced levels in the Δcmc1 and Δcoa4 cells (Fig. 6A, B). Diminished levels of Cox1 synthesis are characteristic for strains with defects in the assembly of cytochrome c oxidase (5, 21, 37, 44, 45). The levels of Cox1 were not increased in the presence of DTT, indicating that DTT did not suppress the assembly defect in these strains (Fig. 6B). Cox2 and Cox3, the two other subunits of cytochrome c oxidase, were synthesized at normal levels. However, their amounts rapidly declined during the chase reaction, most severely in the double mutant (Fig. 6A, C). The presence of DTT did not suppress the instability of Cox2 and Cox3, again indicating that reductants do not improve biogenesis of cytochrome c oxidase in these strains. This is further supported by Western blots (Fig. 6D), which revealed the presence of strongly reduced levels of Cox2 in the Δcmc1 and Δcoa4 mutants regardless of whether the cells were grown in the absence or the presence of DTT or ascorbic acid. In summary, although the presence of DTT suppressed the growth defects of Δcmc1 and Δcoa4 mutants, it did not alleviate the reduced biogenesis of cytochrome c oxidase in these strains. This suggests that the growth arrest is caused by side effects of these assembly mutants rather than directly by the reduction of the levels of cytochrome c oxidase.

Mitochondria of Δcmc1 and Δcoa4 mutants produce increased levels of hydrogen peroxide
We speculated whether the problems in respiratory chain assembly in the Δcmc1 and Δcoa4 mutants result in an increased accumulation of ROS, which reduces their viability. To test this, we incubated mitochondria in the presence of succinate and measured the amount of hydrogen peroxide produced using a fluorescence-based Amplex red assay (Fig. 7A). Indeed we found significantly increased levels of hydrogen peroxide produced in these mutants. Moreover, we observed an increased carbonylation of mitochondrial proteins, which is indicative for oxidation-driven damage in these strains (42) (Fig. 7B). However, we do not regard it as likely that the rapid degradation of mitochondrial translation products that was observed in the Δcmc1 and Δcoa4 mitochondria was due to their oxidative damage since immunoprecipitation experiments with dinitrophenyl-specific antibodies after dinitrophenylhydrazone-modification did not show any elevated levels of carbonylated translation products in these mutants (Fig. 7C). Thus, we suspect that the oxidative damage is rather a consequence of the cytochrome c oxidase defect rather than its cause.
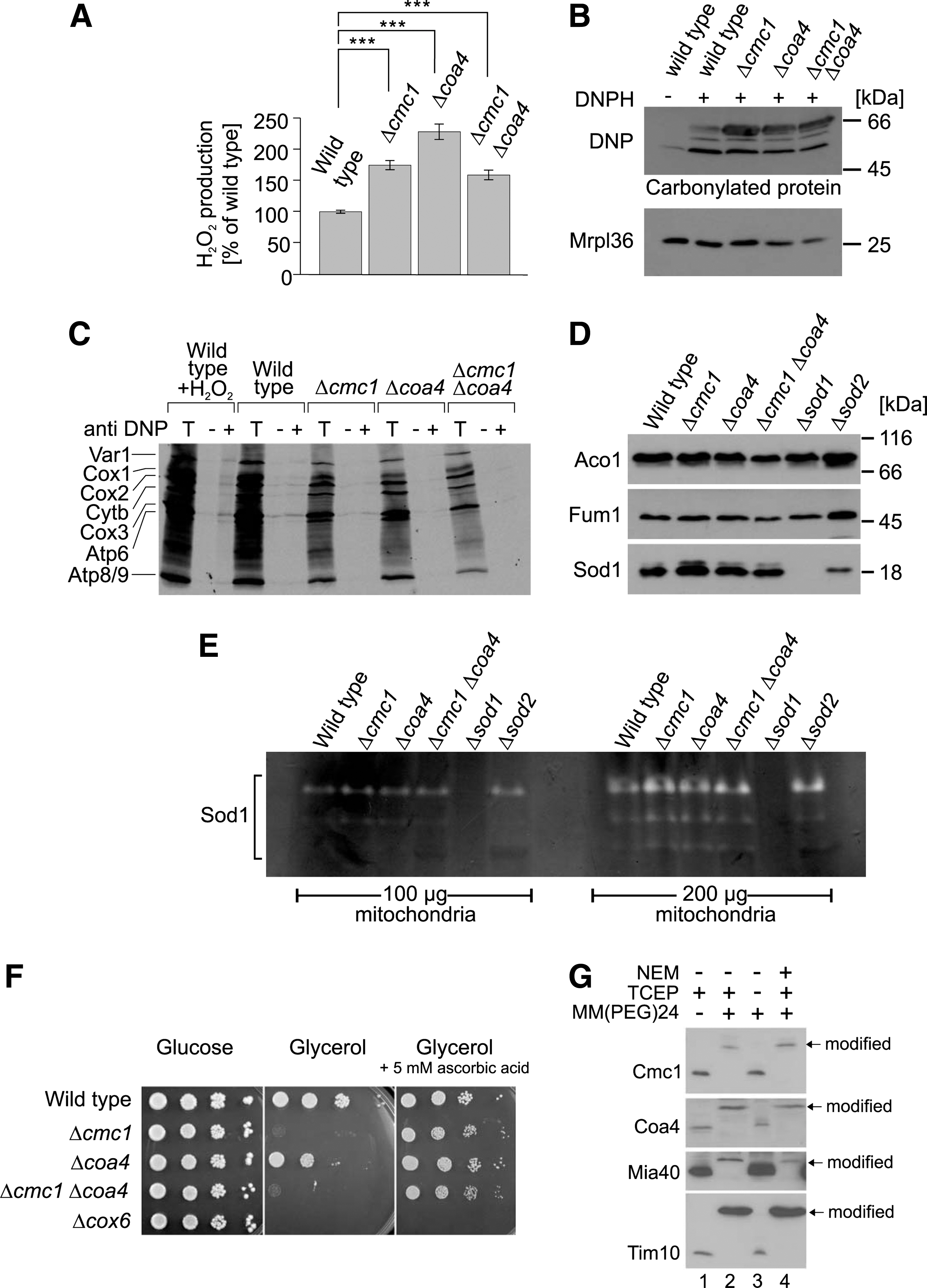
Mitochondria contain two superoxide dismutases that produce hydrogen peroxide from superoxide radicals: Sod1 in the IMS and Sod2 in the matrix. Increased levels of Sod1 in Δcmc1 mitochondria were reported before (22), but no changes in the protein levels of Sod1 or Sod2 were observed with the strains used in this study (Fig. 7D). Activity gels revealed comparable activities of superoxide dismutase activity in wild type, Δcmc1, and Δcoa4 mitochondria excluding that the different levels of hydrogen peroxide produced by the different mitochondria are due to different superoxide dismutase activities (Fig. 7E). Thus, it appears more likely that the increased ROS levels caused by the defects in the assembly of the respiratory chain complexes are responsible for the observed growth arrest. In order to test this hypothesis, we analyzed cell growth in the presence of the antioxidant ascorbic acid (Fig. 7F). Interestingly, ascorbic acid completely suppressed the growth defect of the mutants. Although ascorbic acid treatment can reduce sulfenylated thiols, which might prevent disulfide formation (38, 55), the strong suppression by ascorbic acid suggests that quenching of ROS rather than the reduction of disulfides allows growth of the Δcmc1 and Δcoa4 mutants on nonfermentable carbon sources.
It remained possible that Cmc1 and Coa4 serve as scavengers for hydrogen peroxide in the IMS since hydrogen peroxide might directly react with reduced cysteine residues in these proteins. We therefore determined the redox state of Cmc1 and Coa4 in wild-type mitochondria using the alkylating reagent MM(PEG)24. When mitochondrial proteins were denatured and reduced with the thiol-free reductant triscarboxyethyl phosphine (TCEP), treatment with MM(PEG)24 caused a large size shift of Cmc1 and Coa4 (Fig. 7G). However, in the absence of TCEP, both proteins were not shifted, indicating that their thiol residues were oxidized. In a “reverse shift” setup, that is, when samples were initially treated with N-ethylmaleimide (NEM) to block all accessible thiols, then reduced and treated with MM(PEG)24, Cmc1 and Coa4 shifted to a larger size. Thus, Cmc1 and Coa4, like Mia40 or Tim10, are present in an oxidized state in the IMS, making a direct function as quenchers of hydrogen peroxide unlikely.
The Δcoa4 mutant shows increased hydrogen peroxide levels in the cytosol
Next we tested whether the increased hydrogen peroxide levels produced in Δcmc1 and Δcoa4 mitochondria influence the redox milieu outside of mitochondria in these cells. To this end, we made use of a roGFP2-Orp1-sensor protein that was shown to serve as a tool to measure dynamic changes in cytosolic hydrogen peroxide levels (20, 40). We expressed this sensor protein in the cytosol of wild-type and mutant cells, which were then grown on galactose-based medium (i.e., fermentation conditions). Then, the cells were challenged by the addition of 5 mM hydrogen peroxide to the medium and the changes in the redox state of the sensor were followed by monitoring fluorescence in a plate-reader. In all strains, hydrogen peroxide treatment efficiently oxidized the sensor. However, there were considerable differences in the rate of sensor recovery between the strains. The sensor in the Δcoa4 cells recovered significantly slower compared to wild type, while in the Δcmc1 and Δcoa4Δcmc1 cells, probe recovery was slightly slower compared to wild type although not significantly so (Fig. 8).

While exact interpretation of the probe recovery data is complicated due to the oxidation state of the sensor being a product of both hydrogen peroxide-mediated oxidation and thioredoxin-mediated reduction, the data correlate well with both the differences in hydrogen peroxide production measured with the Amplex Red assay in Figure 7A and the growth phenotypes in Figure 9. Therefore, probably due to pre-existing increased endogeous hydrogen peroxide production, the capacity of the cells, particulary Δcoa4, to deal with additional exogenously added hydrogen peroxide is likely decreased.

It should also be noted that these measurements were carried out under fermentative conditions, thus when the respiratory chain was not fully active. The inability of the mutants to grow on glycerol prevented us from measuring the hydrogen peroxide levels under respirative conditions since nonproliferating cultures show strongly increased hydrogen peroxide levels at stationary phase (not shown).
Overexpression of cytosolic catalase suppresses growth defect of Δcoa4 mutants
Next we asked whether the increased hydrogen peroxide levels in the cytosol contribute to the growth defect observed in the Δcmc1 and Δcoa4 mutants. To reduce the levels of hydrogen peroxide specifically in the cytosol, we overexpressed the cytosolic catalase Ctt1 in these strains from a multi copy plasmid. This partially restored growth of the Δcoa4, but not of the Δcmc1 mutant (Fig. 9A). However, a slight improvement of respiratory growth of both mutants was observed when a fusion protein was expressed, which targeted Ctt1 to the IMS (Mia40-Ctt1, Fig. 9B). From this we conclude that the production of hydrogen peroxide in the absence of Cmc1 and Coa4 contributes to their inability to grow on nonfermentable carbon sources. At least in case of the Δcoa4 strain, the respiration-induced growth arrest is at least partially caused by cytosolic hydrogen peroxide, which was counteracted by overexpression of cytosolic catalase.
Discussion
The respiratory chain of mitochondria is a major source of ROS in eukaryotic cells (1, 13). Although there is still some debate about the degree of ROS production under physiological conditions, it is widely accepted that enzymes of the respiratory chain can transfer single electrons onto oxygen, giving rise to the production of superoxide, which is rapidly converted to hydrogen peroxide by superoxide dismutases. Hydrogen peroxide plays a physiological role as a spatially and temporally dynamic signaling molecule in the cell (2, 14). Under pathological conditions, the degree of mitochondrial ROS production can be significantly increased, as was reported, for example, for several neurodegenerative diseases, including Parkinson's disease (6), Alzheimer's disease (51), and amyotrophic lateral sclerosis (30).
To better understand the relevance of redox processes for mitochondrial activity, we performed a genome-wide mutant screen to identify yeast proteins that under respiring conditions are essential for cell viability unless reducing reagents like DTT or GSH are externally added. We thereby identified mutants that grow significantly better in the presence of reductants than in their absence. One group of mutants lacked nuclear components, in particular transcription factors and enzymes that modify or remodel histone complexes. For example, two components of the SAGA (Spt-Ada-Gcn5-Acyltransferase) complex were identified, which is critical for the metabolic adaptation to respiratory growth conditions (47). This suggests that an increased capacity of cellular redox buffer improves the ability of yeast cells to grow on nonfermentable conditions. The sensing of mitochondria-derived oxidative stress by a number of nuclear transcription factors or histone-modifying enzymes is well known; for example, histone deacetylases of the sirtuin family are strongly stimulated by low NADH levels to couple the redox state of cells to their metabolic activity (12, 15, 18).
The largest fraction of reductant-suppressed mutants lacked mitochondrial proteins (40%), two of which were specifically analyzed in this study, since the growth of respective mutants was strongly stimulated by DTT and GSH. Cmc1 and Coa4 are “twin Cx9C” proteins, which do not share any similarity at the level of their primary sequence except for the four cysteine residues of the Cx9C motifs. The data presented in this study confirm a—direct or indirect—role in the biogenesis of cytochrome c oxidase that had been proposed before (7, 22). Nevertheless, both mutants exhibit at steady-state levels about 20% to 50% of the cytochrome c oxidase activity found in wild-type cells. These reduced levels of complex IV are still high enough to enable cell growth under respiring conditions, at least in the presence of DTT, GSH, or ascorbic acid. Obviously, it is not the reduced activity of the respiratory chain, which prevents cell growth on nonfermentative carbon sources but rather the increased ROS production in these mutants (Fig. 9C). Oxidative stress was found to be toxic only under respiring conditions, and cell growth remained unaffected on glucose. It appears likely that assembly intermediates that accumulate in Δcmc1 and Δcoa4 mitochondria contribute to the growth defect (Fig. 9C). How this growth arrest is conveyed is unclear, but it is conceivable that the increased hydrogen peroxide levels are sensed and block cellular propagation via an intracellular signaling pathway, potentially including the nuclear components identified in this study. The observation that overexpression of cytosolic catalase partially rescued the growth of Δcoa4 cells on glycerol strongly suggests that the increased hydrogen peroxide levels in the cytosol are crucial for the observed growth arrest. It is conceivable that such an intracellular redox-dependent signaling pathway dampens the propagation of mutants that produce increased levels of mitochondria-derived oxidative stress in order to prevent the distribution of malfunctional mitochondria. A recent study showed that oxidative stress in mitochondria leads to cell death in yeast by inducing caspase-mediated apoptosis (19). The yeast metacaspase Yca1 is known to be activated by hydrogen peroxide although the details of this apoptotic signaling pathway in yeast are not entirely clear (33). It will have to be tested whether Yca1 is activated by mitochondria-derived oxidative stress to prevent cell growth of mutants in which the respiratory chain is not properly assembled.
A recent study systematically analyzed collections of yeast mutants to identify strains that are unable to grow on glycerol (35); it was found that in particular the growth of cytochrome c oxidase assembly mutants strongly depended on the strain background, which was interpreted by additional (epigenetic) cues that contribute to the growth phenotypes in these strains. This surprising observation might be explained by differences in the redox state or the redox sensitivity of the compared strains.
A cell-killing potential of assembly intermediates of cytochrome c oxidase was shown before, although only when cells were exposed to externally applied hydrogen peroxide (26, 53). In particular, assembly intermediates of Cox1 containing heme but no copper killed hydrogen peroxide-treated cytochrome c oxidase mutants on glucose medium. This cytotoxic potential might explain why mitochondrial Cox1 synthesis is under tight feedback control, which prevents the accumulation of assembly intermediates (5, 21, 37, 44, 45). At this stage it is not entirely clear where the ROS are produced. The strong genetic control of Cox1 and the large number of ROS-producing cytochrome c oxidase mutants suggest that the assembly intermediates of complex IV directly contribute to the generation of oxygen radicals. This cytotoxic effect is presumably not restricted to eukaryotes since it was shown before that bacteria lacking the cytochrome bd oxidase can be suppressed by addition of externally added glutathione (17). Nevertheless, it is also possible that the reduced levels of cytochrome c oxidase complexes lead to an accumulation of electrons at the level of complex III, which is known to have the potential to produce large amounts of superoxide (13). In vivo, ROS levels are presumably controlled by a number of mitochondrial reducing enzymes and by GSH, which diffuses across the outer membrane through porins (28, 34).
In summary, our study suggests that partially assembled cytochrome c oxidase complexes produce increased ROS levels under respirative conditions which trigger growth arrest of yeast cells. Presumably, these intermediates also form in wild-type cells but are rapidly converted into fully functional complexes and therefore produce ROS at much lower levels. Potentially, the significant levels of ROS that were reported to be produced by the respiratory chain are not exclusively byproducts of normal respiration but, at least in part, are generated by assembly intermediates of respiratory chain complexes. It will be interesting to monitor hydrogen peroxide production in mammalian cells that have cytochrome c oxidase assembly defects, for example, in cells from patients suffering from Leigh syndrome (43, 54). This might help to unravel the basic mechanisms of the ROS production by the respiratory chain under physiological and pathological conditions.
Materials and Methods
Yeast strains and media
All yeast strains were derived from the wild-type strain BY4742 (MATα; his3Δ1; leu2Δ0, lys2Δ0; ura3Δ0). The Δcmc1 Δcoa4 mutant was generated by replacement of the COA4 gene by a cassette containing the HIS5 gene of Schizosaccharomyces pombe in a Δcmc1 background.
The sequences of CMC1, COA4, and CTT1 from 300 bp upstream of the start codons to 150 bps downstream of the stop codons were amplified by PCR and cloned into the multi copy plasmid pRS426 (48). For the expression of Ctt1 in the IMS, the DNA sequence encoding for a fusion protein consisting of the N-terminal 75 amino acid residues of Mia40 and full-length Ctt1 was cloned into the expression vector pYX223 (Novagen) for expression under control of a GAL promoter. All strains were grown in YP (1% yeast extract, and 2% peptone) or synthetic medium with 2% glucose, galactose, or glycerol as carbon sources at 30°C. Mitochondria were prepared from galactose-grown cells as described (4).
Measurement of hydrogen peroxide production
Mitochondria (100 μg) were resolved in 600 mM sorbitol, 1 mM ethylenediaminetetraacetic acid (EDTA), 20 mM potassium phosphate (pH 7.4), 50 μM Amplex red, and 1 unit/ml horse radish peroxidase. By addition of 10 mM succinate respiration was induced. Hydrogen peroxide-induced formation of resorufin was measured in a fluorometer by excitation of 544 nm and emission of 590 nm for 10 min.
Measurements with the roGFP2-Orp1 probe
Yeast strains were grown for 24 h at 25°C in Hartwell's complete (HC) media containing 2% (w/v) glucose, followed by 1:50 dilution into HC + 2% (w/v) galactose and growth for a further 48 h. Following growth 4.5 OD600 units of cells were harvested by centrifugation and resuspended at a concentration of 7.5 OD600 units/ml in 100 mM Mes/Tris pH 6.0. The sample was divided into three equal aliquots which were either treated with 20 mM N,N,N′,N′-tetramethylazodicarboxamide (diamide) or 100 mM DTT for the fully oxidized and fully reduced controls respectively, or left untreated. Samples were loaded to a flat-bottomed 96-well plate (BD-Falcon; Product No. 353219) and centrifuged for 5 min at 20 g to form a loose cell pellet. Probe response in the untreated cells was induced by the addition of 5 mM hydrogen peroxide. Probe oxidation was followed by measuring fluorescence emission using a plate-reader system (FLUOstar Omega) with an emission filter of 510/20 nm and excitation filters of 390/10 nm and 480/10 nm. The degree of probe oxidation (OxDroGFP2) was determined according to Equation 1, where I is the fluorescence emission intensity at 510 nm, following excitation at either 390 or 480 nm, for the fully reduced (red) and fully oxidized (ox) controls, and the sample.
Equation 1:
Protein carbonylation assay
Mitochondria (150 μg) were lysed by addition of 6% sodium dodecyl sulfate (SDS). The samples were incubated with 3.96 mg/ml 2,4-dinitrophenylhydrazine, 0.154 g/ml trifluoroacetic acid for 10 min at 25°C. After precipitation with trichloroacetic acid (TCA) the pellets were resuspended in SDS-loading buffer (60 mM Tris/HCl pH 6.8, 10% glycerine, 2% SDS, 0.01% bromophenol blue). The proteins were separated on SDS–polyacrylamide gels, transferred to nitrocellulose membranes, and analyzed by Western blotting with a 2,4-dinitrophenylhydrazone-specific antibody. For the analysis of carbonylated translation products, proteins were synthesized in isolated mitochondria as described (16) in the presence of [35S]-methionine. Then mitochondria were washed, lysed with SDS, treated as described above, and used for immunoprecipitation with the dinitrophenylhydrazone-specific antibody or preimmune serum for control.
Analysis of redox state of proteins
To determine the redox state of Cmc1 and Coa4, cells were grown to an optical density (OD) of 0.6 and harvested by centrifugation. Cell pellets were resuspended in 12% TCA. For the “inverse shift” sample, the cell pellet was incubated in 50 mM NEM for 15 min at 30°C before addition of TCA. Then, cells were broken by sonication and glass bead homogenization. TCA pellets were resuspended in 80 mM Tris pH 7.0, 10% glycerol, 2% SDS, and 0.05% bromocresol purple. For reduction of disulfides, samples were incubated with 10 mM TCEP for 20 min at 96°C followed by addition of 15 mM MM(PEG)24 (Thermo Scientific). After incubation for 1 h at 25°C in the dark and 1 min at 96°C samples were analyzed by Western blotting.
Oxygen consumption
Mitochondrial oxygen consumption was measured basically as described previously (26, 53) using a Clark electrode (Hansatech Instruments). Mitochondria (100 μg) were incubated in 0.6 M sorbitol, 1 mM MgCl2, and 20 mM HEPES, pH 7.4. Oxygen consumption was induced by addition of 5 mM NADH and measured for 2 min, and then 1 mM ascorbate or 1 mM GSH was added and the measurement was continued for 2 min. Each measurement was repeated twice.
Statistical analysis
T tests were performed to calculate p-values. Confidence levels are shown in figures as follows; *p<0.05, **p<0.01, and ***p<0.001.
Miscellaneous
In vivo labeling of translation products (16), enzyme activity assays (49), oxygen consumption, and generation of antibodies against Cmc1 and Coa4 (32) were performed as described.
Footnotes
Acknowledgments
We thank Sabine Knaus for experimental assistance, and Jan Riemer and Benedikt Westermann for discussion and advice. This work was supported by grants from the Landesschwerpunkt für Membrantransport and the Deutsche Forschungsgemeinschaft (IRTG1830 and He2803/4-1) to J.M.H. and by a Kekulé fellowship of the Fonds der Chemischen Industrie to K.B.
Author Disclosure Statement
No competing financial interests exist.