
Abstract
Introduction
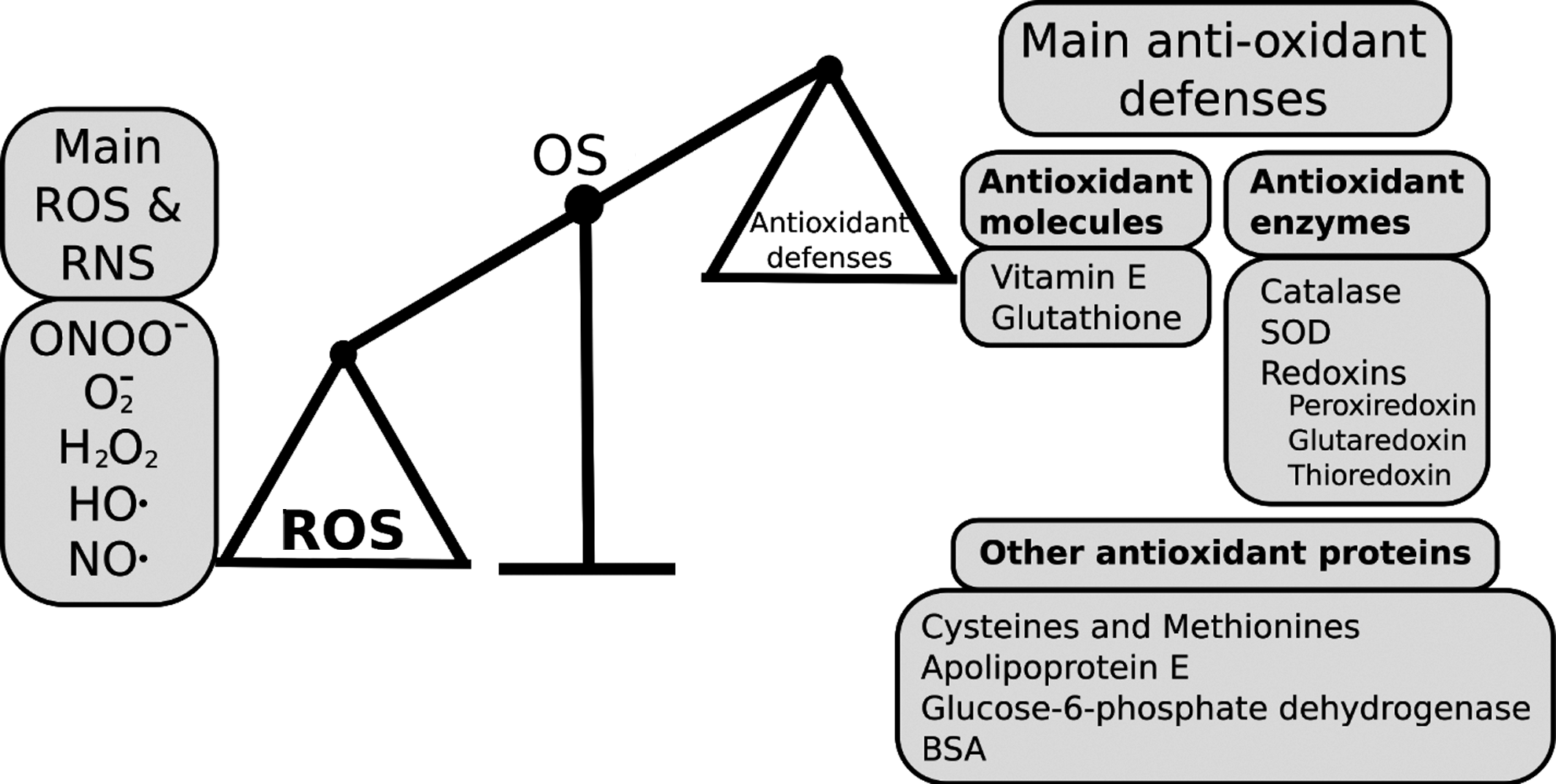
A key physiological consequence of kidney pathology is elevated blood pressure (hypertension) that can result in stroke and cardiac disease (20). Oxidative stress (Fig. 1) arises when levels of reactive oxygen or nitrogen species (ROS or NOS) exceed the cell/tissue's antioxidant capacity and has consistently been associated with hypertension (51, 60) and stage of renal malfunction (15). ROS/RNS can interact directly with important cell components such as lipids, nucleic acids, and proteins (82), and lead in particular to glomerular filtration barrier injury (52).
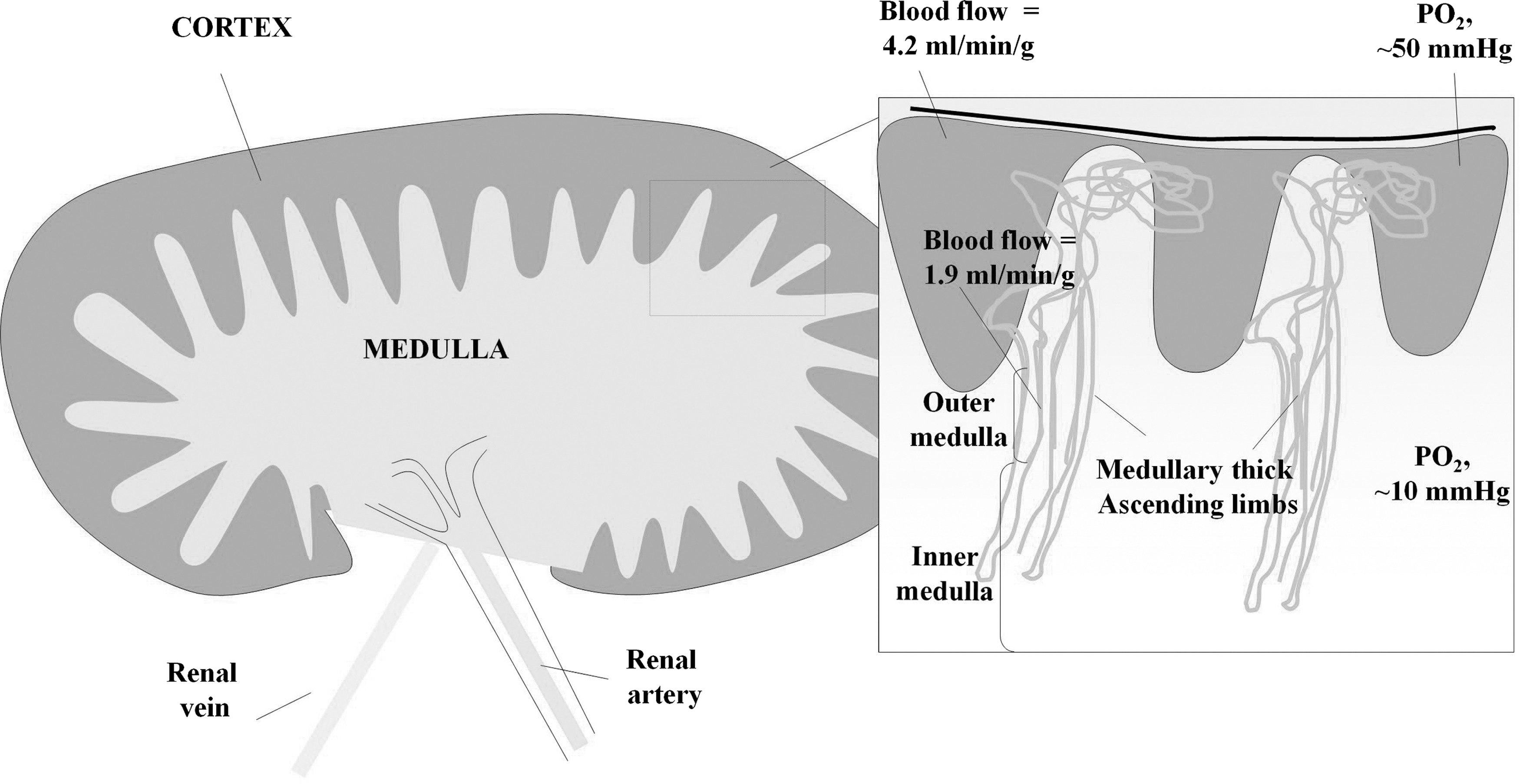
In comparison with other organs, the kidney has an unusually wide range of oxidative status, ranging from the well-perfused cortex to near-anoxic medulla (Fig. 2). The medulla and cortex also fulfill distinct functions. In the cortex, fluids and electrolytes are filtered from proteins at the glomerulus, and glucose, electrolytes, and water are reabsorbed along the nephron. The medulla, on the other hand, is almost anoxic and has the principal function of concentrating urine. Oxidative/nitrosative stress arising from ROS/RNS is thought to be differentially regulated in the cortex and medulla (55). NO-mediated signaling is more important in medulla, but cortex may be more liable to ROS-induced damage (60). Onset of kidney dysfunction seems to arise from progressive loss of redox homeostasis associated with overproduction of ROS/RNS, with consequent effects on renal proinflammatory/proapoptotic/profibrotic pathways. These lead to changes in vascularization, fibrosis, and diminished kidney function (51, 52, 60). Proteins are key quantitative targets for ROS/RNS; many proteins are known biomarkers of kidney pathology (17). This review focuses on redox-related proteomic studies of chronic kidney disease and related conditions. We will describe general approaches taken in human samples and associated model systems and assess the potential of redox proteomics to contribute new insights to chronic kidney disease.
Triggers for Oxidative/Nitrosative Stress in Kidney Disease
Physiological factors external to kidney are capable of triggering events within the organ, resulting in excess production of ROS/RNS and causing oxidative/nitrosative stress (Fig. 3). This can affect proteins, and oxidative stress increases with the stage of chronic kidney disease (15). Regardless of the initial cause, fibrosis is a common proximate cause for end-stage kidney disease, and proteomic profiling may provide novel early biomarkers with sufficient predictive power for clinical outcomes (32, 59). Hyperglycemia due to diabetes mellitus is commonly associated with onset of hypertension; this is the most common clinical cause of chronic kidney disease (12). Chronic kidney disease represents a persistently inflamed state often associated with pro-inflammatory stimuli such as interleukins-1, −6, −18, tumor necrosis factor alpha (TNF-alpha), and other cytokines (49). This inflammation contributes to progression of the disease by increased activity and production of adhesion molecules that contribute to adhesion of T cells and their migration into the interstitium, leading to attraction of profibrotic factors (78). Growth factors external to kidney contribute to the process of scarring or fibrosis which features excessive deposition of extracellular matrix causing loss of renal function (38). Fibrosis has been suggested to be a process of phenotypic transition from epithelium to mesenchyme (1), although the definitiveness of this has recently been questioned (34). The role of the endocrine or circulating renin–angiotensin–aldosterone system (RAAS) in stimulating fibrosis during chronic kidney disease is well established (52, 83). Decreased renal perfusion causes secretion of renin from the juxtaglomerular apparatus of the kidney. This cleaves circulating angiotensinogen from liver to form the ten-residue peptide hormone angiotensin I. This, in turn, is cleaved by removal of two C-terminal residues catalyzed by angiotensin-converting enzyme (ACE) from lung, to form angiotensin II. This binds to angiotensin receptors, triggering effects within the kidney, including reabsorption of sodium and protein. Angiotensin II also triggers release of aldosterone from adrenal glands and has increasingly been associated with numerous other proinflammatory functions (5). Overexpression of angiotensinogen attenuates apoptosis in model animals (21). Overstimulation of the RAAS cascade leads to kidney damage, resulting in diminished function (52, 83). All RAAS components are also produced locally in kidney, and it is thought that a local renal RAAS contributes to hypertension (52). RAAS inhibitors have protective effects on kidney in diabetics, most probably through reduction of blood pressure (85).

These triggers activate proinflammatory, profibrotic, and proapoptotic pathways, with consequent changes at the level of the proteome, the total protein complement of kidney cells and related tissues. Thus, proteomics has become a popular approach to explore these processes further (reviewed in Refs. 10, 32, 59, 64). Since proteins absorb the bulk of ROS/RNS, redox proteomics in particular offer opportunities to follow redox-mediated post-translational modification of proteins (73 –75). A fundamental difference between the traditional proteomics and redox proteomic approaches is that reversible redox modifications can alter protein activity even in cases where the relative protein abundance may not necessarily change. Given the importance of oxidative stress in kidney pathology, it is intriguing that redox proteomics have not been applied more extensively to its investigation.
Kidney Proteomics
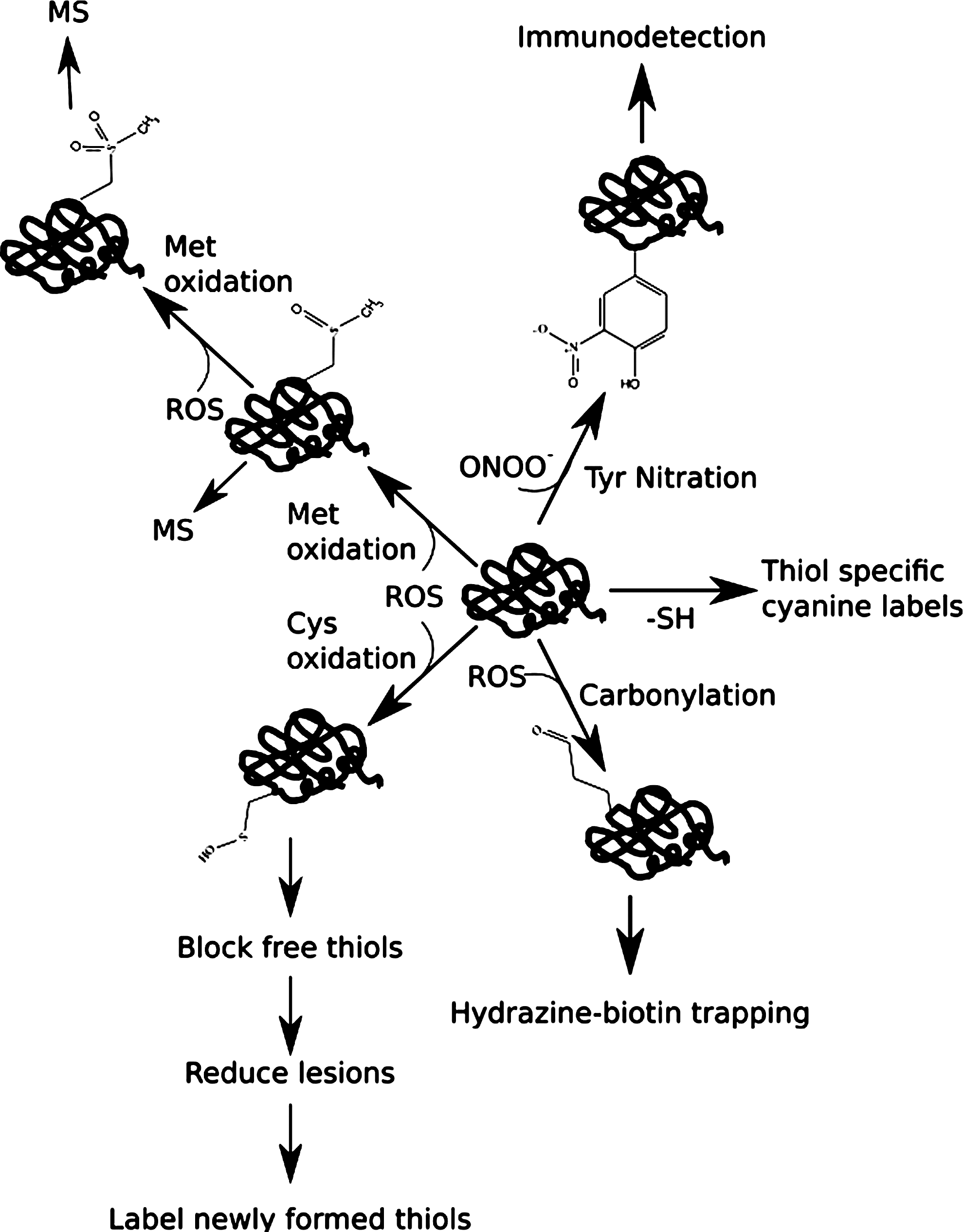
A range of experimental approaches are used in proteomic studies. These include two-dimensional electrophoresis (2DE) that combines isoelectric focusing with orthogonal sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE). Individual proteins are separated from complex mixtures based on differing isolectric point (pI) and Mr (32, 73 –75). When combined with immunodetection, it is often possible to find redox modifications to proteins in 2DE separations such as carbonylation and nitration (73, 75). Quantitative data can be obtained from 2DE using difference gel electrophoresis (DIGE) with DeCyder software (77). This has allowed differential identification of several proteins in animal models (6) and in human urine (56). Mass spectrometry (MS) approaches have also proved useful in research on kidney diseases (53) (Fig. 4). As with DIGE, quantitative MS is increasingly being used (80). This may exploit labeling (either chemical or metabolic) or label-free methods. Labeling approaches include isotope-coded affinity tags (ICAT), stable-isotope labeling of amino acids in cell culture (SILAC), and isobaric tags for relative and absolute quantification (iTRAQ). Thiol-specific ICAT reagents have been exploited to measure oxidation of cysteines in the total proteome (18). SILAC offers a potentially powerful tool in the quantification of redox proteomic targets and their modification in kidney samples (35). iTRAQ has only yet been used in a few studies such as alteration of the phosphoproteome in the epithelial-to-mesenchymal transition induced in HK-2 cells by transcription growth factor-beta-1 (11). As of yet, ICAT is a relatively under-exploited approach in kidney proteomics.
Once potential redox-modified proteins have been detected within samples by a discovery approach, they can be validated and quantified by selective reaction monitoring (SRM) (13). This selects proteotypic peptides of the protein of interest and specific fragment ions (or transitions) upon fragmentation of the parent ion. This is the most sensitive proteomic quantification method available and detects low-abundance proteins with a dynamic range of up to 4–5 fold (13). SRM has potential in the quantification of redox protein biomarkers but is not ideal for the quantification of a specific redox modification within proteins. Peptides containing redox modifications are not suitable as proteotypic peptides as they can decrease or eliminate the signal of the corresponding precursor ion. Nevertheless, this approach has potential for quantification of redox-responsive proteins.
In addition to the two “workhorse techniques” of classical proteomics (2DE and MS), novel selection/screening methods are being used, sometimes in conjunction with MS. Surface-enhanced laser desorption ionization (SELDI) uses functionalized chips (carrying affinity or ion exchange groups) to select subproteomes. Large numbers of samples can be analyzed quickly, which is ideal for screening of clinical samples. For example, this has been used in studies of interstitial fluid of kidney biopsy material (41). A disadvantage of SELDI is that it only allows Mr determination rather than specific protein identifications. A novel recent selection approach has involved the use of aptamers, random libraries of single-stranded oligonucleotides which can adopt an enormous number of structural conformations with protein-binding properties. This selected 60 proteins in the serum of patients with chronic kidney disease, many of which were low Mr proteins often observed in this pathology (22). Fractionation of serum samples on magnetic beads allows MALDI-MS analysis (67). Another approach is to use protein microarrays; a recent study of serum revealed angiotensinogen and PRKRIP1 as novel autoantibody targets in kidney (9).
Sample preparation is a key aspect of proteomics experiments and it is increasingly evident that this step needs to be standardized for discovery of reproducible biomarkers (80). Traditional diagnosis of kidney disease often depends on performance of a biopsy. However, this may not always be possible for clinical (e.g., obesity, severe hypertension, or bleeding disorders) or ethical reasons and could result in a poorly defined sample. Proteins are well represented amongst extant biomarkers of chronic kidney disease (17). Therefore, many proteomic studies relevant to kidney disease focus on plasma or serum, tissues (e.g., from animal models), and urine (66). In particular, there have been major developments in studies of urine because of the sample quantity, possibility of repeat sampling, and its noninvasive nature (10, 48). A public domain proteomic database for kidney-associated proteomes is curated by an international consortium (Human Kidney and Urine Proteome Project, HKUPP;
Studies on Human Kidney Tissue
Difficulties inherent in human kidney tissue proteomics have been eloquently described (59). Samples must be obtained invasively (e.g., by percutaneous biopsy) which yields small amounts of material, making comparison with healthy tissue ethically and logistically problematic. Such biopsies may not faithfully reflect the differing compartments/cell types in the composition of kidney. It has recently been pointed out that proteomic study of renal structures and compartments offer many opportunities for insights to kidney pathologies (10). A key technique is microdissection of defined kidney compartments such as glomeruli (87). This approach successfully identified autoimmune antigens in patients suffering from membranous glomerulonephritis, and suggested that oxidative stress in this pathology may drive superoxide dismutase 2 expression (58). An alternative is to prepare glomeruli by sieving. Using tissue from healthy cortex of four patients undergoing nephrectomy for renal tumors (renal cell carcinoma), this approach allowed proteome profiling by PDQuest™ image analysis and protein identification by 2DE and MS (88). Including novel pre-fractionation by one dimensional electrophoresis and solution-phase isoelectric focusing coupled with SDS PAGE, these workers identified more than 3500 proteins with at least two tryptic peptides (28). Using “microsieving,” it has even proved possible to isolate whole glomeruli from single needle biopsy samples suitable for protein profiling (33).
Podocytes are kidney epithelial cells that wrap around the capillaries of the glomerulus which have been implicated in the early stages of kidney disease. MS analyses on five separate MS instruments identified a profile of 332 distinct proteins in a 1 μg biopsy sample of human glomerulus, including podocyte-essential proteins such as podocin and nephrin (89). Expression profiling of podocytes exposed to high glucose as a model for diabetic nephropathy identified up-/downregulated genes including biomarkers of chronic kidney disease such as neutrophil gelatinase-associated lipocalin (26). A recent proteomic study of cultured podocytes revealed that RAAS activation caused downregulation of the antioxidant protein peroxiredoxin 2. This led to production of ROS, protein oxidation, inhibition of the Akt pathway, and onset of apoptosis (25). This is consistent with the observation in biopsy samples that SOD 2 is induced by oxidative stress in membranous nephropathy (58).
Studies on Blood Plasma/Serum
Plasma and serum samples derived from patient blood provide a less invasive sample source for proteomics that is also amenable to re-sampling. A particular problem with blood-derived samples is the broad dynamic range of blood protein concentration, with some proteins being very abundant whilst others are rare. The most abundant proteins can be depleted by immunoaffinity fractionation. After immunodepletion for the six most abundant proteins (albumin, transferrin, IgG, IgA, haptoglobulin, and antitrypsin), proteomic comparison of serum from diabetic nephropathy identified two biomarkers (selenium-containing extracellular glutathione peroxidase and apolipoprotein E) which were downregulated in this pathology (31). Both of these proteins are implicated in antioxidant defense (31). An alternative to immunodepletion involves use of a library of hexameric peptides that allows selection of the total proteome, including very low abundance proteins. This is based on creating at random an enormous repertoire of protein-binding sites which makes possible identification of a greater number of urinary proteins. Chronic kidney disease patients have a higher risk of premature atherosclerosis. An MS-based approach combining capillary electrophoresis (CE) coupled to MS analysis of serum of chronic kidney disease patients identified protein biomarkers for cardiovascular disease (62). 2DE of non-immunodepleted serum also identified four proteins (alpha-1-microglobulin, apolipoprotein A-IV, gamma-fibrinogen, and haptoglobin) associated with atherosclerosis in patients with chronic kidney disease (39). Chronic kidney disease is a common feature in patients after liver transplant, and proteomic analysis of serum has been reported using a commercial ELISA-based technology [multi-analyte panels (MAPs)] of 188 serum proteins and a kidney MAP of 14 kidney-associated proteins, which was predictive of later onset of disease, even in patients with only mildly diminished glomerular filtration rate at the time of transplant (36). A protein microarray study of serum recently discovered angiotensinogen and PRKRIP1 as novel autoantibody targets (9).
Advanced oxidation protein products are oxidized variants mainly of albumin and fibrinogen that have been identified in plasma/serum and are elevated in chronic kidney disease (63). More than 70% of the free radical-trapping capacity of serum is attributed to albumin. The oxidation status of serum proteins correlates with systemic inflammation during chronic kidney disease (43). Redox-induced post-translational modifications (PTMs) of protein residues include thiol oxidation and carbonylation (formation of aldehyde or ketone groups in side-chains) (43, 82). Redox proteomics approaches can identify oxidized proteins in serum/plasma. An elegant approach to profiling of carbonylated proteins in human plasma involved trapping this subproteome on hydrazine-biotin with avidin selection (40). A combination of MALDI-TOF-TOF and electrospray-tandem MS identified 65 carbonylated proteins, many of which were from kidney. Interestingly, several of these proteins carried multiple redox lesions and oxidation of methionine was the most commonly found modification. In studies of this type, kidney is consistently one of the largest contributors of plasma proteins (40).
Another category of redox modification found in proteins (Table 1) is nitration, in particular, modification of tyrosine to 3-nitrotyrosine (73, 76). Increased 3-nitrotyrosine in both chronic and end-stage kidney disease patients was found in serum proteins by both immunoblotting and LC-tandem MS (57). Cysteine, the second least abundant residue in proteins, is also subject to extensive redox modification and (with methionine) contributes the bulk of antioxidant activity of human serum albumin. Cysteine can exist in a variety of disulfide-bridged (e.g., homocysteinylated, glutathionylated), nitrosylated, and other oxidized variants (e.g., sulfenic, sulfinic, and sulfonic acids) (82). Serum proteins such as low density lipoproteins have been reported to have elevated homocysteinylation in chronic kidney disease patients (90), whilst irreversible oxidation of Cys-34 of serum albumin and increase in albumin carbonylation correlates with increasing stage of chronic kidney disease (43). Given the availability of ready methods for detecting redox lesions at methionine (40) and thiols (75), it is surprising how few studies exploit such approaches in serum proteins. Iodoacetamido-substituted cyanine labels are a possible route to profiling of cysteine-containing proteins in serum (8).
Denotes proteins only identified in one study presented in this table.
As only healthy subjects were used in this study, no protein can be associated to kidney disease. Carbonylated proteins, as well as many other OS-related PTMs, were present in the subjects, many of kidney origin.
Studies on Urine
Kidney disease is commonly diagnosed by albuminuria, reflecting diminished retention of protein by the kidney. Urine therefore offers potential as “readout” for diminished kidney function, which is also available noninvasively and may be repetitively sampled. Recent proteomic research in kidney disease has accordingly focused on urine (reviewed in Refs. 2, 46). In particular, small peptides, the peptidome, consisting of peptides largely of extracellular matrix origin are informative about the onset of chronic kidney disease (2). Considerable progress has been made in standardizing sample preparation and data analysis protocols for the kidney peptidome and proteome (44). Comparison of the plasma and urine proteomes revealed that the average Mr of the urine proteome is indeed smaller than that of plasma (27). This arises from the filtration and secretion activities of kidney plus proteins secreted or shed from downstream glands and the urinary tract. The analysis identified 2280 plasma proteins that were blocked by kidney, 394 proteins were common to plasma and urine (suggesting kidney allows them to pass through), while 1128 proteins were unique to urine, including proteins secreted/shed by kidney, downstream glands, and the urinary tract (27). Abundant plasma proteins can enter the urine once the glomerular filtration barrier is altered in chronic kidney disease, making fibrosis-associated proteins difficult to detect (32).
A significant body of literature has arisen on exosomes in urine (79). These are small (40–100 nm) endocytic vesicles originating from a range of cell types and secreted into body fluids (including urine) or the extracellular space. Exosomes carry proteomes reflecting their cells of origin and provide a ready means of profiling urine in a range of kidney pathologies (79). A compendium of exosome proteins is now available online (
As with serum/plasma, virtually no systematic investigation of redox protein variants has been performed on the proteomes of either urine or exosomes. Given that these sources frequently contain proteolyzed and otherwise modified proteins, it would seem that redox PTMs would offer a fruitful comparator across samples from differing kidney pathologies.
Studies in Animal Models
An alternative to human renal tissue is offered by animal models that are widely used in kidney disease research (reviewed in Ref. 32). These often allow detection of events on a relatively short time scale that might take years to develop in humans. In general, animal models are genetic, may have arisen spontaneously, or else are caused surgically or by drug treatment (Table 2). The spontaneously hypertensive rat (SHR), originally developed from Wistar rats by selective breeding, spontaneously displays hypertension emerging in weeks 12–14 and persisting for more than a month (54). The SHR lifespan (1.5–2.5 years) is significantly less than that of normotensive rats (2.5–3 years), but this difference makes life-long studies feasible in a practical time-frame. SHRs do not absolutely mirror hypertension in humans (e.g., onset is in the equivalent of young adulthood in SHRs but in middle age in humans). Nevertheless, SHRs are a sufficiently good animal model for the testing of novel antihypertensives (37). Redox proteomics studies have been performed in SHR kidney tissues, including identification of kidney proteins containing 3-nitrotyrosine (73), sulfenic acid (75), and carbonylation (74).
Other animal models for kidney disease research in which proteomics have featured include diabetic rats (6, 61), acidotic rats (14), passive Heymann nephritis rats (50), nephrectomized rats (86), ischemic-reperfusion injury in rat kidneys (16), sepsis in rats (24), stenosis in rats (65), genetically-modified mice (7), mice with hypokalemic nephropathy (70), diabetic mice (4), mice with immune nephritis (84), and dogs with inherited nephropathy (47). These studies often point to oxidative stress as a key feature of chronic kidney disease and agree well with knowledge of disease progression in humans. For example, downregulation of superoxide dismutases (58) and upregulation of the NAD(P)H oxidase (NOX) family (19), a ubiquitous source of superoxide, are found both in animal models and humans. An elegant illustration of the power of animal models in the study of kidney pathology is the overexpression of catalase along with angiotensinogen in a double-transgenic mouse (21). Catalase overexpression in renal proximal tubules resulted both in decreased oxidative stress and decreased fibrosis, albuminuria, and hypertension (21).
Future Directions
Proteomics is now an established route to study of important kidney pathologies (10, 32, 59, 64). Some proteins are oxidized due to oxidative/nitrosative stress associated with kidney disease (43, 57). However, this review documents that redox proteomics is a heretofore relatively under-utilized approach which nonetheless has enormous potential to offer insights to kidney pathology. Oxidative stress is a key factor in many kidney pathologies, especially chronic kidney disease (15, 60), and reversal of oxidative stress often alleviates clinical manifestations such as hypertension (21, 37). Interception of routes to oxidative stress has been suggested as a novel drug therapy approach for chronic kidney disease (68), and beneficial results have recently been found in a rat diabetic nephropathy model with the oral hypoglycemic drugs metformin (3) and pioglitazone (71). These drugs also have beneficial effects in human diabetics (29). As redox proteomics now allows routine fingerprinting of oxidative PTMs, there is potential here for early-stage detection of disease onset in complex samples such as plasma/serum and urine. Kidney proteins are particularly well represented in serum/plasma proteomes (40), and appearance of proteins in urine is an early indicator of chronic kidney disease, making urine a key sample source for kidney proteomics (2, 46). Amongst proteins found to be modulated by pathology within kidney tissues, antioxidant proteins are often prominent (25). These findings suggest that analyses of urine and exosome proteomes may be especially informative in the future.
A number of recent technical developments could underpin systematic redox proteomic study of clinical samples such as plasma/serum and urine. Standardized protocols for sample preparation, especially from urine, are now available (see:
Footnotes
Acknowledgments
The Sheehan laboratory is supported by the Programme for Research in Third Level Institutions of the Higher Education Authority of Ireland. We also acknowledge our long-term collaborator, Prof. Edward Johns, Department of Physiology, University College Cork, Cork, Ireland.
Author Disclosure Statement
No competing financial interests exist.