
Abstract
Introduction
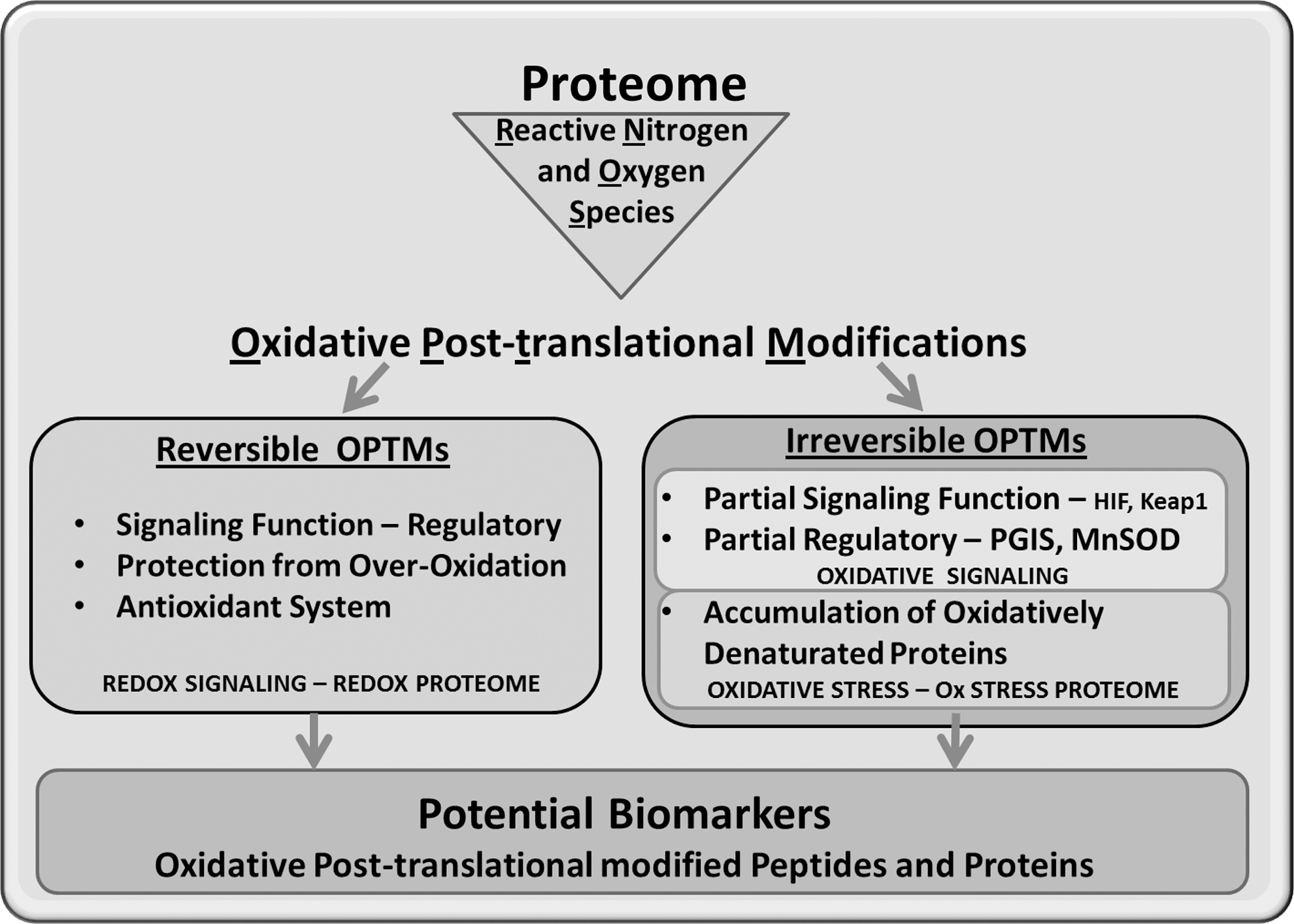
Redox Signaling Versus Oxidative Stress
Compared with typical cellular signals, RNOS were initially thought to be too unstable and short lived to have relevant biological effects or to function as signaling molecules. However, with the discovery of the antioxidant enzyme superoxide dismutase (SOD) by Fridovich and McCord (182, 183), the physiological relevance of RNOS was established. At the beginning of this new research era, it was believed that the oxidation of biological molecules was a side reaction derived from the necessity of utilizing oxygen to generate cellular energy in the form of adenosine-5′-triphosphate (ATP). During the generation of cellular ATP, ∼0.1% of oxygen consumed by mitochondria is converted into RNOS. This phenomenon is due to the misdirection of electrons from the respiratory chain to molecular oxygen, resulting in the one-electron reduction product superoxide (•O2 −). In the presence of transition metals ions such as iron (Fe), copper (Cu), organic radicals, or enzymes such as peroxidases, •O2 − can be quickly converted into more reactive RNOS, which readily oxidizes biological molecules. This group of RNOS includes the hydroxyl radical (•OH), carboxyl radical (CO2 •−), nitric oxide (•NO), and (•NO2) as well as the nonradical species hydrogen peroxide (H2O2), hypochlorous acid (HOCl), singlet oxygen (1O2), carbon monoxide (CO), and nitrogen dioxide (NO2) (94, 128, 265, 292).
In contrast to the assumption that RNOS were solely derived from oxidative metabolism, the immunology field recognized the antiseptic properties of RNOS for cellular host defense. Neutrophil granulocytes generate large amounts of superoxide through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (containing the cytochrome binding subunit called gp91phox or NADPH oxidase [NOX]-2). The spontaneous decomposition of •O2 − or catalysis by SODs results in H2O2 formation. Myeloperoxidase further converts this reactive oxygen species (ROS) into HOCl, a highly bactericidal agent that is believed to kill phagocytosed pathogens and activate the proteases required for phagocytosis (109). This process was termed “oxidative burst” and in macrophages, an alternate second system consisting of inducible NO-synthase (iNOS or NOS-2) was found to produce reactive nitrogen species. Discovery of the oxidative burst demonstrated that cellular RNOS formation was controlled, and the resulting OPTMs were identified as markers of inflammation. Various irreversible OPTMs were discovered and utilized as molecular footprints of RNOS generation, including protein-bound nitrotyrosine, protein carbonyls, lipofuscin (35, 180, 258), malondialdehyde (MDA) (93), ceroid bodies (101), and dityrosine crosslinks. These markers led researchers to hypothesize that cardiovascular diseases (CVDs) such as ischemia reperfusion injury and atherosclerosis, ionizing radiation-induced damage, and xenobiotic metabolism may involve the damaging effects of RNOS on biological molecules contributing to the pathophysiological process (94, 301).
Discovery of the various isoforms of NO synthase and NOX in the cardiovascular system led to the widespread recognition of RNOS as important signaling molecules (other cellular sources of radicals are listed in Table 1). Physiologically, RNOS levels range in the submicromolar concentration (12, 137, 178, 220, 224, 262) and can directly react with various amino-acid residues to alter protein activity and function (112, 120). RNOS can also affect carbohydrates (98, 189) and lipids to generate oxidative by-products that react with proteins and modify cellular signaling (71). In order to produce distinct signaling events, sufficient quantities of RNOS should act locally and modify a protein reversibly or irreversibly, resulting in a defined functional change. These RNOS-controlled signaling events can be subcategorized into oxidative signaling (Fig. 1), which includes irreversible OPTMs that lead to specific signaling events. The activation of cellular antioxidant defenses as modulated by the Kelch-like ECH-associated protein 1 (Keap1)/Nrf2 complex is a prime example. Various genes that are important for type II detoxification of RNOS and electrophiles are under control of the antioxidant- or electrophile-response element (ARE), including SODs and enzymes involved in glutathione metabolism. Reactive electrophiles, such as 4-hydroxynonenal (HNE), or RNOS can react irreversibly with critical Cys residues on Keap1, leading to its dissociation from Nrf2 and to ubiquitin-mediated degradation of Keap1 (151). Free Nrf2 then translocates to the nucleus, heterodimerizes with the Maf protein, and interacts with the AREs to promote the transcription of genes involved in oxidant detoxification.
Arg, arginine; eNOS, endothelial nitric oxide synthase OR NOSIII; H2O2, hydrogen peroxide; iNOS, inducible NO-synthase; MAP, mitogen activated protein; NOX, NADPH oxidase; PKA, cAMP dependent protein kinase; SDS PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; VEGF, vascular endothelial growth factor.
The second category of RNOS-controlled signaling is redox signaling (Fig. 1), which should be, by definition, enzymatically reversible. S-glutathionylation and S-nitrosylation (nitrosation) of HRas are excellent examples of this type of signaling. HRas is a key regulator of the propagation and amplification of receptor tyrosine (Tyr) kinase-mediated growth signals via the mitogen activated protein (MAP) kinase pathway (114). HRas tethered to the plasma membrane are reversibly modified at a critical Cys residue by the RNOS derived from NOX and NO synthases, and this modification leads to enhanced guanosine diphosphate dissociation and activation (53). Enzymatic systems such as glutaredoxin (Glrx) can then remove the S-glutathione adduct from the HRas to cease signaling (3, 165). The example of HRas is a further illustration by which RNOS may increase locally and target specific microdomains in the cell. In order to properly signal, HRas should be shuttled from the Golgi apparatus to the plasma membrane, and on termination of the signaling, the HRas are transported back to the Golgi. One may speculate that oxidases localized at the plasma membrane oxidatively modify and activate HRas and that translocation back to the Golgi may require reversal of the oxidative modification in HRas by the cellular reducing enzymes, thus terminating the signal. A recent publication from our group demonstrates that the shuttling of HRas, which is controlled by the palmitoylation of C-terminal Cys residues, is redox controlled as well (36).
From these examples, it is evident that the transition from redox to oxidative signaling in a cellular environment may not be easily distinguishable. Furthermore, higher and uncontrolled RNOS levels, or oxidative stress (Fig. 1), leading to multiple sites of irreversible OPTMs in many proteins may interfere with signaling cascades.
Regardless of this, the inhibition of the formation of these irreversible OPTMs may be therapeutic in delaying the aging process and associated pathologies (194). In terms of functional consequences, one can speculate that oxidative signaling, redox signaling, and oxidative stress allow for distinct proteomes to be affected, each of which warrants a separate analytical investigation.
Types of OPTM—Consideration of Reactivity, Topology, and Biological Effects
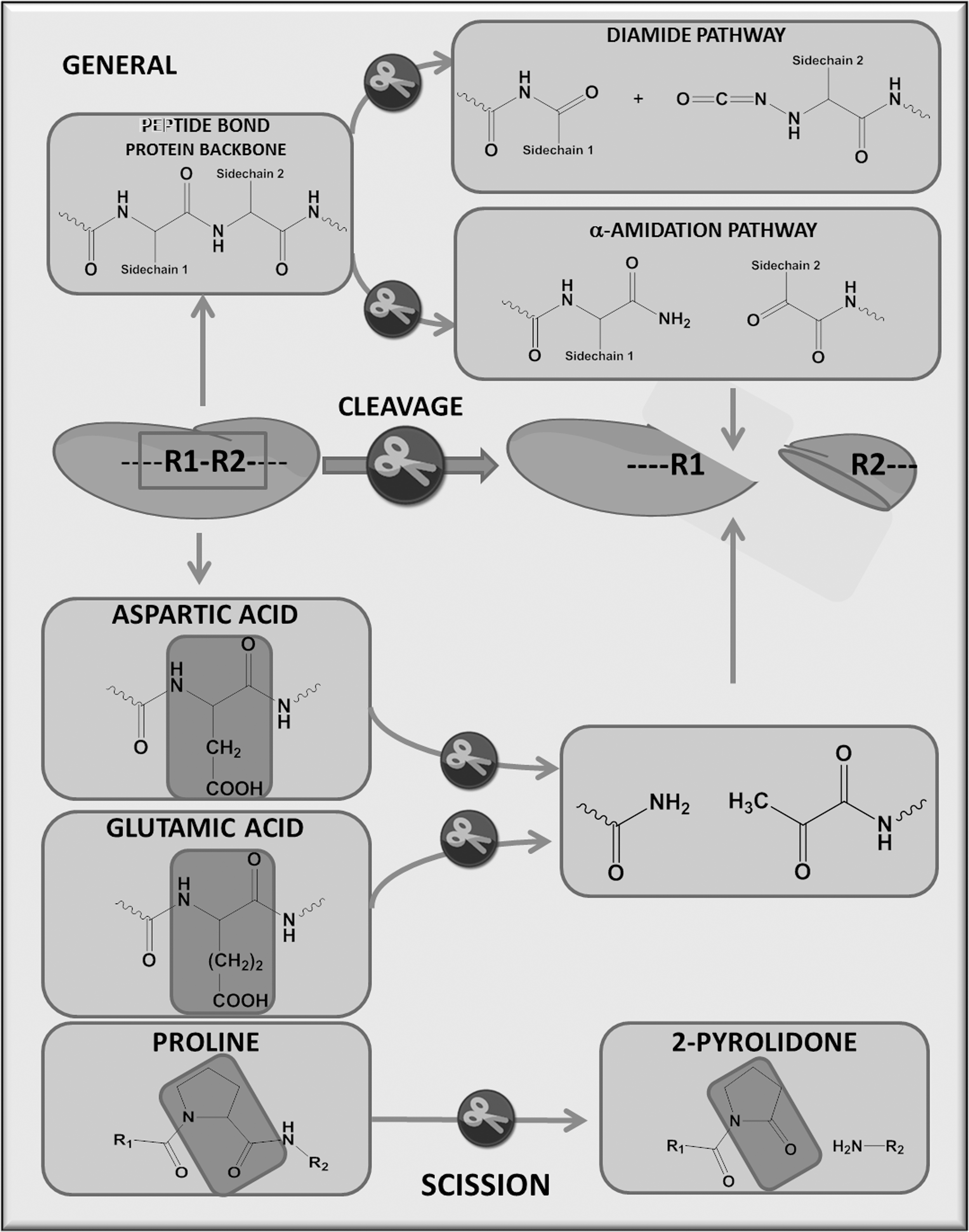
RNOS-induced protein changes can be classified as modifications that lead to cleavage of the peptide backbone (Fig. 2) or those that modify amino-acid side chains (27, 60, 78, 90, 263, 264). The former group includes the direct attack by a hydroxyl radical on glutamate residues, which converts the amino acid into pyruvic acid and ammonia or a reaction of the hydroxyl radical with a proline residue, forming aminobutyric acid and carbon dioxide (CO2). Also included in this category are the initial amino-acid side-chain modifications such as aldehyde production, which transfers the radical to the protein backbone via alkoxyl radicals (62, 77, 118, 119, 199, 202, 221, 312). Meanwhile, the latter includes direct oxidation of the amino-acid side chains as well as lipid and sugar oxidation by-products that form adducts with the side chains. All these oxidatively derived PTMs produce altered proteins that can be termed the redox proteome or redoxome (141). These modifications are most often described as functional changes, but, alternatively, some may act as antioxidant shields that prevent deleterious protein oxidation (162, 173, 231).
The proteome of OPTMs can be further subdivided into two groups as described in Figure 1. The reversible group, which will be referred to as the redox proteome, includes oxidative post-translationally modified proteins that mediate signaling events under normal physiological conditions and likely belong to the enzymatically reducible thiols of Met and Cys residues. The irreversible group would encompass OPTMs that occur under conditions of oxidative signaling or oxidative stress (Ox Stress proteome).
The oxidative modification of protein-bound amino acids depends on various biological and chemical aspects of the target amino-acid residues, including topology, steric accessibility, chemical reactivity, physiochemical properties, and subcellular localization of the protein (Table 2). RNOS preferentially react with amino acids that are in solvent or surface exposed and are less likely to reach residues that are buried inside the protein structure. Furthermore, proteins that exist in their constitutive form as a dimer or multimer will be less prone to modifications at the contact site of the interacting proteins.
Cu, copper; Cys, cysteine; Fe, iron; OPTM, oxidative post-translational modification; RNOS, reactive nitrogen and oxygen species
Nevertheless, OPTMs can be found on inaccessible residues, which suggests that the OPTMs may partially alter and unfold the protein to expose concealed residues, may target residues during protein synthesis and folding that are otherwise less accessible, or may transfer and tunnel radicals and electrons, respectively, from the amino-acid side chain to the protein backbone. This effect might be of particular interest for plasma proteomics, as a protein such as serum albumin could already be cotranslationally modified in the liver, revealing a local organ specific pathology; whereas the correctly folded circulating protein may be modified at another site as a “passerby” in the circulation. Thus, protein turnover may indirectly provide a footprint for the formation of RNOS and OPTMs at distinct cellular compartments.
A second aspect of factors that affect OPTM formation is that proteins may localize to different subcellular compartments that have a distinct redox state. For example, OPTMs of manganese SOD in the mitochondria would indicate mitochondrial dysfunction, which has been detected in various diseases (175) and the aging process (290, 291, 304). Depending on whether a protein is a transmembrane, membrane associated, or cytosolic protein, the subcellular location of the exposed amino acid can affect the protein residues that are targeted for modification by RNOS. RNOS-mediated lipid peroxidation products, including electrophilic aldehydes, would most likely target proteins that are in proximity to the site of peroxide formation, specifically at cellular membranes. Conversely, charged RNOS species are unlikely to affect amino-acid residues that are surrounded by membrane lipids. In addition, as demonstrated by Radi et al. (223), the membrane and its interface may promote a different chemistry for RNOS reactions, as certain species, including nitric oxide and various peroxides, are lipid soluble. These observations apply all the more to different cell types and tissues in which OPTMs in cell- or tissue-specific proteins are being investigated as meaningful and specific biomarkers.
A third aspect is the physiochemical properties and microenvironment of protein amino-acid residues, which may largely affect their reactivity. Cys residues, as explained in greater detail in a later section, are highly reactive as a thiolate and, in some proteins, can exert a peroxidase-like function. OxyR, a H2O2 sensing protein in E. coli, possesses a reactive thiolate within a lipophilic pocket. The geometry of the pocket, its lipophilicity, and the reactivity of the Cys determines its specificity for H2O2 as a substrate compared with other RNOS. Such structural arrangements of the protein are not only advantageous for cellular redox signaling but also make these residues prone to oxidative stress.
Lastly, the redox state of the cell plays a crucial role in the formation of OPTMs on proteins. For instance, the cellular redox state can dramatically change during the cell cycle, allowing for cell division, DNA synthesis, metabolic adaptation, and DNA dissemination to the daughter cell. During mitosis, the nuclear envelope dissolves, and the nuclear segregated content is adjusted to the cytoplasmic redox conditions. Moreover, the differential activation of cellular •NO and superoxide sources may change the type of reactive species formed. The cell can transition from primary •NO signaling, which is, for example, used by the vascular endothelium to control the vascular tone under homeostasis, to a more oxidative state during endothelial inflammation, which promotes the formation of more reactive RNOS through the activation of superoxide sources. In addition, leukocyte recruitment during the inflammatory process results in the concomitant release of high levels of superoxide and nitric oxide, leading to the formation of the very reactive peroxynitrous acid. This species nitrates Tyr residues, and in the case of the endothelium, inhibits the formation of the important homeostatic mediator prostacyclin.
Depending on these various aspects, signaling or pathologic events can be induced by OPTMs, with each separate protein amino acid serving as a sub-proteome for oxidative modifications. In addition, the irreversible oxidation of proteins can hypothetically occur before cellular dysfunction, and, thus, these OPTMs can be used as early biomarkers of disease. We will discuss the chemistry of each oxidatively altered protein amino acid and the corresponding sub-proteome, the tools used to measure these and the in vivo significance of the specific Redox and Ox stress proteomes as they pertain to the cardiovascular system.
Cysteine
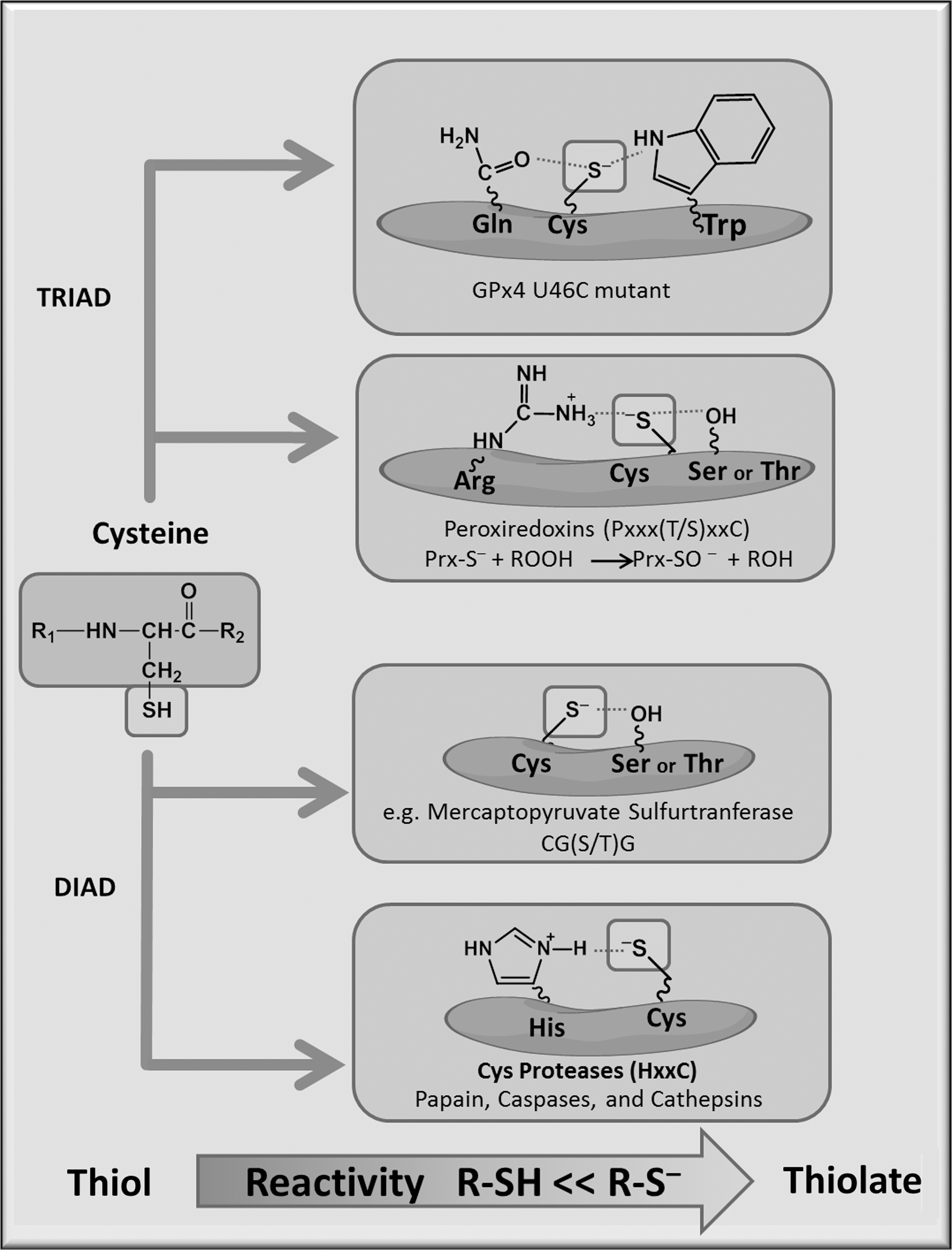
When considered from an evolutionary point of view, Cys is one of the most conserved amino-acid residues within protein sequences from species to species, which highlights its importance in overall protein structure and function (80). Cys modifications can lead to changes in catalytic activity by altering enzyme protein stability through changes in the protein structure. Redox reactivity of Cys is governed by the thiol (R-SH) group on its side chain (Fig. 3). Generally, Cys involved in redox reactions are located in the vicinity of arginine (Arg), lysine (Lys), or histidine (His) residues, resulting in a lower pKa, which is the pH at which 50% of the thiol is deprotonated and, thus, more reactive. Adjacent hydroxyl groups from serine or Tyr can also stabilize the thiolate by hydrogen bonding. The thiolate anion (R-S−) is the most reactive cellular nucleophile, and it can change enzyme activity, substrate recognition, and allosteric regulation.
Cys modifications have been identified by the use of antibodies and labeling techniques. The thiol-containing side chain reacts with numerous chemical reagents and allows for the labeling of oxidized Cys in a sample. This is generally done by alkylating with a thiosulfanate, reducing the sample to remove the reversible modification and then re-alkylating with a tagged thiosulfanate. These may then be used for immunoblotting, immunohistochemistry, or mass spectrometry. As a word of caution, one thiosulfanate, maleimide, at higher concentrations can undergo nonspecific side reactions with Lys or His, complicating the analysis of specifically tagged Cys (this is unlikely to occur while using nanomolar or low micromolar concentrations of the alkylating agent). A direct measure of peptide mass changes in a Cys can be used for OPTM detection by MS, and these include S-glutathionylation (+305 Da), S-sulfonylation (+80 Da), S-cysteinylation (+119 Da), di-sulfide (−2 Da), sulfinic acid (+32 Da), and sulfonic acid (+48 Da) (115).
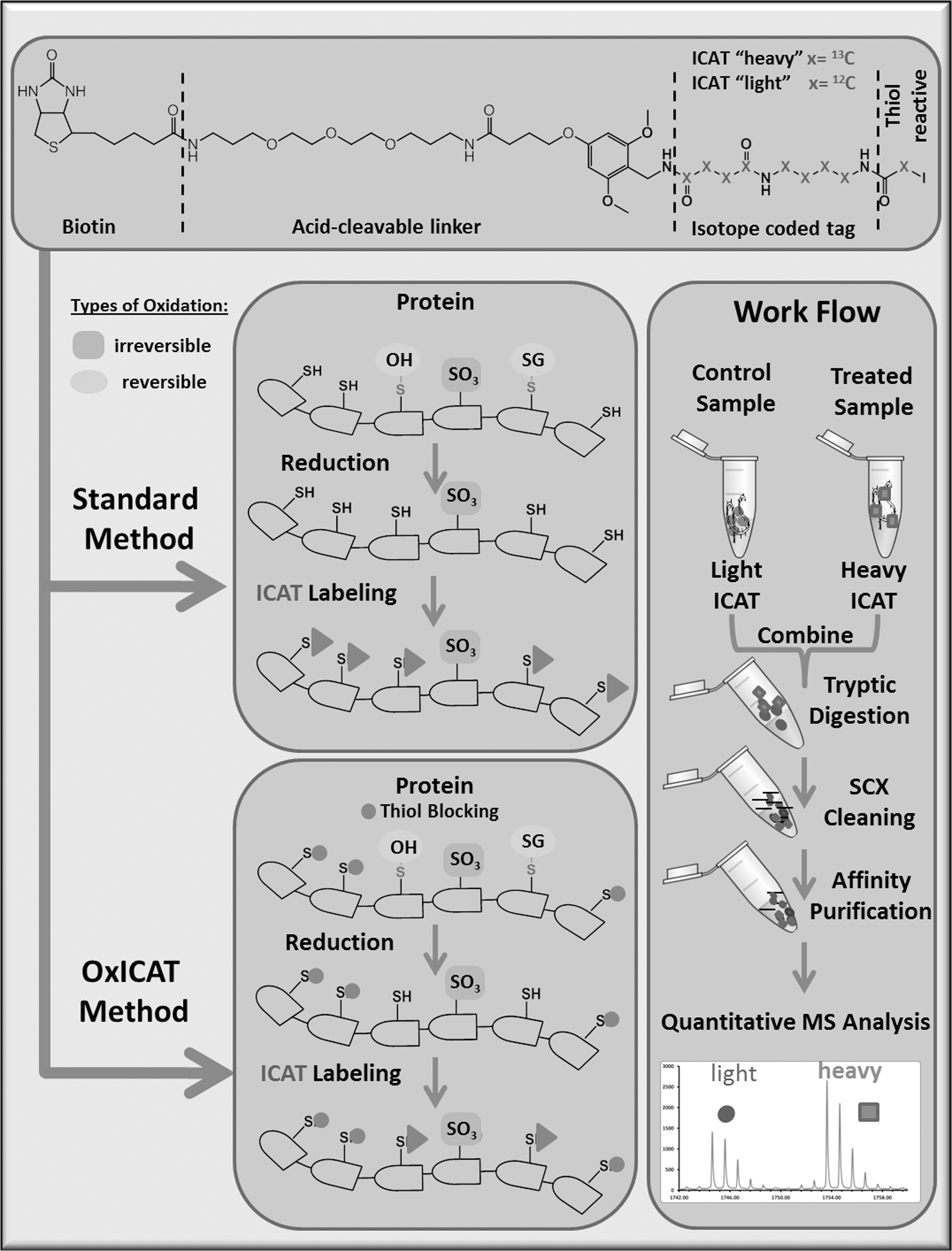
Most detection methods, including MS, incorporate the use of labeling techniques that selectively enrich the fraction of oxidized Cys residues. Using an iodoacetomide-based fluorescent probe that labels reversibly oxidized Cys (19), 50 reactive thiol-containing proteins were identified, separated by two-dimensional gel electrophoresis (2DE), and the extent of oxidation was evaluated with the light emitted from fluorescently probed proteins. Using a similar approach with indocarbocyanine, Cy3 and indocarbocyanine, and Cy5 labeled maleimide, each of which have a distinct excitation and emission wavelength, differentially oxidized free thiols were labeled, compared by differences in emission, and extracted from the gel for MS/MS detection (86). Quantitative MS approaches can utilize thiol-reactive isotopic or isobaric tag based, including isotope-coded affinity tags (ICAT), as shown in Figure 4 (105) and can eliminate the short comings of gel extraction methods (61). ICAT is available in two forms, heavy or light, depending on the carbon isotope present in the thiol-reactive iodoacetamide. With ICAT, one sample can be labeled with heavy ICAT and the other, with light ICAT, introducing a 9 Da difference between the Cys-labeled peptides. The mass difference is then detected by MS, and the relative quantities of the two peptides can be obtained. Using this technique, oxidized creatinine kinase was identified by Sethuraman et al. by liquid chromatography tandem mass spectrometry (LC-MS/MS) (249) and since then, has been utilized to identify modified myocardial proteins after exposure to H2O2 as well as myocardial proteins sensitive to thioredoxin (Trx) and nuclear proteins in mice (86, 97, 250). The advantage of using ICAT is that it allows for the simultaneous identification and quantification of redox-sensitive Cys residues (157). ICAT is also advantageous for (i) screening hydrophobic proteins, for example, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) (86), which are poorly resolved by 2DE; (ii) evaluating proteins with high pI values exceeding the limits of 2DE, for example, cytochrome c oxidase (pI 8.96); and (iii) identifying numerous peptide ratios from a single sample. ICAT is limited by its poor peptide sensitivity either due to retention on the reverse-phase LC or poor ionization and/or fragmentation of certain peptides in the mass spectrometer. The interpretation of results using ICAT can be confounded by inadequate peptide identification due to the low intensity of one of the ICAT pairs or by improperly accounting for expression changes that could be misinterpreted as oxidation. Changes in expression, though, could be corrected by using another method in parallel, such as isobaric tag for relative and absolute quantitation (iTRAQ) (87). Further refinement of the methodology may allow for ICAT to be the approach of choice when determining oxidative changes in vivo under physiological oxidative stress. ICAT, however, does not allow a comparison of more than two samples in parallel. To perform multiple comparisons, thiol-specific cysteine specific tandem mass tags (CysTMT), which are pyridyldithiol-based reversible tags with similar masses (isobaric) that result in the generation of a reporter ion with different masses, and that can vary by 1 Da and be detected by tandem MS, could be utilized (198).
Novel mapping of reactive Cys have been proposed using an IsoTOP-ABPP (isotopic tandem orthogonal proteolysis activity-based protein profiling) that uses an iodoacetamide reactive probe coupled to an azide functionalized TEV protease recognition site containing a biotin tag. The tag contains a site that can then accommodate either a light (standard) or (heavy) labeled valine which can allow the differential labeling and quantification of peptides (296, 297) by MS. In addition to this, computational or in silico methods have been developed to determine the most likely redox-active Cys residues present (82).
S-Nitrosylation
The thiolate anion can undergo a direct oxidative modification through the action of nitric oxide to form an S-nitrosylation modification. S-nitrosylation can occur chemically on thiols via NO-derived species such as N2O3 (NO+), transfer or transnitrosation reactions, or by interaction with metalloprotein-linked enzymes (248). The principle method used to detect S-nitrosylation is the biotin switch assay in which free thiols are initially blocked with an alkylating agent or reactive thiolsulfonate, such as MMTS; the S-nitrosylated thiols are reduced with ascorbate; and then, the free Cys thiols are labeled with a biotinylated alkylating agent, such as biotin N-(6-[biotinamido]hexyl)-3′-(2′-pyridyldithio) propionamide for subsequent pull-down and immunoblotting (131). This assay is fairly specific but should be performed in the absence of UV light, in the presence of metal chelators, and with fresh materials (83). Fluorescent labels have also been utilized to improve the detection of SNO peptides (146). Antibodies that detect S-nitrosylation also exist; however, these antibodies suffer from nonspecific binding to unmodified Cys residues (52). Since the SNO adduct is too labile to remain intact during purification schemes with reliability, the proteomic identification of S-nitrosylated proteins involves variations of the biotin switch assay in combination with mass spectrometry (131). Either this or novel labeling techniques using resin-assisted capture (RAC) or labeled tags in protein SNO can be used to evaluate RSNO by mass spectrometry (38, 51, 84). SNO-SID (SNO-site identification) was described to identify SNO peptides, rather than proteins, by introducing a proteolytic step before protein pull-down (116). Forrester et al. have developed an assay combining the labeling and pull-down steps with a thiol reactive resin, thus creating an RAC for S-nitrosylation (SNO-RAC) (84). The use of SNO-RAC, which forms a covalent disulfide between the SNO and the resin, allows for trypsinization and peptide labeling directly on the resin that is rapidly followed by LC-MS/MS identification methods. SNO-RAC appears to be more sensitive than the biotin switch assay for high mass proteins and at least comparably sensitive for proteins smaller than 100 kDa.
Lastly, two methods have been developed to detect the endogenous S-nitrosothiol proteome. One method utilizes an organomercury resin that reacts with S-nitroso protein adducts to form a stable thiol-mercury bond, and then, the modified proteins can be identified with MS/MS (66).
The second method ESNOQ (endogenous SNO quantification) combines stable-isotope labeling of amino acids in cell culture (SILAC) with detergent free lysis followed by the biotin switch assay and protein identification by LC-MS/MS (313) The latter two methodologies have the advantage of direct measurement of SNO-modified sites, as the other methods measure putative reaction sites (313).
The reversal of S-nitrosylation is accomplished by Trx (which also reduces intramolecular dithiols) in the cytosol or mitochondria (24, 25). In addition to the Trx system, S-NO adducts can form S-nitrosoglutathione (GSNO), which itself may undergo transnitrosation reactions with reactive thiols, or be reduced by GSNO reductase or alcohol dehydrogenase class III (168, 169). By using Trx reductase ablated mice, an association between S-nitrosylation and poor vascular relaxation was observed (50). This finding has been further buttressed by the GSNO reductase-deficient mouse that has diminished peripheral vascular tone and depressed β-adrenergic inotropic responses due to the denitrosylation of cardiac ryanodine receptor 2 (RyR2) (21). In addition, the deletion of GSNO reductase also increases tissue damage (222) and mortality after endotoxic exposure with more hypotension after anesthetic challenge, suggesting impaired responses of the vasculature (169). These mice also have attenuated myocardial infarct size due to increased angiogenesis that may be driven by HIF1α S-nitrosylation-induced vascular endothelial growth factor (VEGF) expression (166). Similarly, the overexpression of Trx, which can reduce RSNO, attenuates infarct size by the same mechanism, further suggesting that infarct-related dysfunction may be modulated by S-nitrosylation (5). Novel tools such as mitoSNO may allow for the directed modulation of the S-nitrosylation of proteins in specific compartments (51) and better identification, localization, and role of specific SNO proteins.
Sulfenylation
The thiolate can also undergo thiol shuffling reactions (203) or react with H2O2 to form a sulfenic (R-SOH) acid, also referred to as sulfenylation. Sulfenylation has been detected in the structural and mitochondrial proteins of cardiac myocytes treated with H2O2 (46, 241) and may be enzymatically reversed by Trx (277). At present, it is uncertain whether sulfenic acid can act as a signaling molecule, and speculation is that it acts as an intermediary for further oxidation only.
Sulfenylation can be analyzed and trapped by using the specific alkylating agent, 5,5-dimethyl-1,3-cyclohexanedione (dimedone) (9, 26, 46, 218). Typically, proteins forming a putative sulfenic acid are incubated with dimedone, and the protein containing the dimedone-modified Cys is then isolated and identified by tandem mass spectrometry, tagged purification, or antibody identification. The recent advent of newer dimedone analogs, based on the removal of two methyls from the ring of dimedone, DAz, which is 1,3-cyclohexadione coupled to azide, and the 3-(2,4-dioxocyclohexyl)propyl (DCP) analogs, which couple Cys reactive DCP fluorophore groups to 1,3 cyclohexadione, have been synthesized as well (9, 283). The labeling of sulfenylation using these compounds should be performed as described by Poole and colleagues (149). The major limitation of dimedone is its poor cell permeability; thus, high concentrations of sulfenic acid are needed or elevated concentrations of dimedone or analogue are required (149), and in complex mixtures, it competes with endogenous-free small-molecule thiols such as Cys, homocysteine, and glutathione, which react much faster with protein Cys-SOH (228). Other reagents used in the detection of sulfenic acid include 2-nitro-5-thiobenzoic acid (TNB), which may be a better agent for the identification of protein Cys-SOH, as it reacts faster than endogenous thiols or dimedone. For example, it can react with albumin Cys-OH at a rate of 105 M −1 s−1, which is more than 3000 times faster than unsubstituted dimedone and 5 times greater than free Cys (236, 286). Care should be taken when using this reagent, as exposure to air for prolonged periods of time can turn TNB into 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), which may react with free Cys (228).
Lastly, a chimeric tool that measures sulfenyl binding partners in cellular models has been developed using an 85 AA portion of the Cys rich C-terminal portion of YAP-1, which is a Saccharomyces cerviasiae transcription factor known to form a sulfenyl adduct. Its specificity was determined using an inhibitory assay with dimedone (276). The proteomic identification of sulfenylation is typically based on the specific reduction of sulfenic acid with arsenite or the specific labeling of sulfenic acid by dimedone (72). With these approaches, multiple cardiac proteins, including cytoskeletal components, undergo sulfenylation after exposure to H2O2, as detected by matrix-assisted lazer desorption ionization time of flight mass spectrometer (MALDI-TOF) analysis (241). Labeling with dimedone has allowed for both improved detection for protein enrichment (46) and evaluation of sulfenylated proteins using more highly cell permeable azide-linked analogs, such as DAz-2 by LC-MS/MS (160). In addition, novel isotope-coded dimedone and iododimedone have been applied in the quantitative analysis of sulfenic acid modifications with the use of LC-MS (247).
S-Glutathionylation and disulfides
In addition, Cys can lead to the formation of intra- or intermolecular disulfides and mixed disulfides [S-glutathionylation; see review for further info (215)]. Interestingly, RSNO has been postulated to form glutathione-mixed disulfides and may act as an intermediary for S-glutathionylation. S-glutathionylaton, inter- and intramolecular disulfides can occur under RSOH formation, enzymatic disulfide formation, thioltransferase activity from oxidized glutathione (GSSG) to a thiolate, sulfenylamide bond interactions with reduced glutathione (GSH), and condensation of GSSG to a glutathione disulfide S-oxide. Enzymatic disulfide formation has been reported for glutathione S-transferases (GST), ERV1, sulfhydryl oxidases, protein disulfide isomerase (PDI), and glutathione peroxidases (88). Glrx-1 specifically reduces glutathione-mixed disulfides in the presence of NADPH and glutathione reductase; while Trx and PDI reduce intra-intermolecular disulfides (163, 238). These modifications are detected with the use of labeling techniques as reviewed (215). The methods used to look at oxidative changes of thiols generally rely on creating derivatives with iodoacetamide, maleimide, and thiosulfates because of their tendency to alkylate thiol groups selectively (67). Labeling can be carried out in several ways: The first is using tagged alkylation of free thiols and determining the reduction of tagged labeling, which would be indicative of oxidation. The second involves the alkylation of free thiols with subsequent reduction of the initially oxidized thiols and re-labeling with a labeled alkylation agent, be it with fluorescent, biotin, isotopic, or radioactive tags (95, 303). An increase in tagged labeling is indicative of increased oxidation.
Some other approaches include utilizing (127) activated thiol sepharose to enrich thiol-containing proteins, eluting with a reducing agent, and separating by 2DE to visualize the difference between samples. The problems with these techniques are the specificity of the reducing agents, as they can reverse other Cys modifications such as S-palmitoylation and cannot distinguish between a mixed disulfide and an intramolecular disulfide. More novel glutathione analogs that specifically identify thiol-modified compounds are being used as well to detect S-glutathionylation (31, 67, 270, 308). An antibody is also available but was limited by its sensitivity with differing specificity for glutathione adducts on different proteins, and, therefore, large quantities of a particular S-glutathionylated protein may be required for detection (215). The proteomic detection of S-glutathionylation has been accomplished by either direct or indirect means. The former utilizes a reactive thiosulfanate to initially block free thiols and LC-MS/MS to determine a 305 Da shift that would be suggestive of protein-SG formation. This method has been used for individual proteins but not for a whole proteome analysis. Indirect measurements of S-glutathionylation uses an initial labeling step with a thiosulfanate, a subsequent reduction of the mixed disulfide by glutaredoxin (Glrx) and, finally, labeling with biotinylated N-ethylmaleimide or other thiol tags. Measurements of intermolecular disulfide formation have been performed with diagonal gel electrophoresis (32). The principle of this method is that proteins with intermolecular disulfides move away from the diagonal protein band as a result of a shift in the molecular weight, geometry, and charge under reducing conditions. Generally, samples are first separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis under nonreducing conditions, and then, the whole lane is excised and further separated under reducing conditions in the second dimension. Distinct protein spots are then analyzed by LC-MS/MS for an evaluation of protein modifications.
The in vivo effects of S-glutathionylation are probably best assessed in Glrx knockout mice, which have worsened ischemia reperfusion injury in the cardiovascular system (124). In contrast, it appears that the effects of angiotensin II (Ang II) infusion on cardiac and aortic medial hypertrophy are inhibited by Glrx deletion (15). This effect is attributed to attenuation of NOX activity by interference of upstream signaling components. Alternatively, the effects of overexpression of Trx, which reduces sulfenic acid, S-nitrosylation, and intramolecular disulfides but not mixed disulfides, inhibits Ang II infusion by regulating mir-98 (307). Similarly, the overexpression of Trx2 also inhibits Ang II-induced vascular and cardiac hypertrophy with decreased mitochondrial RNOS production (300). Whether these effects are due to a change in mitochondria to nuclear signaling needs further evaluation. Lastly, deletion of Trx leads to the potentiation of pressure overload hypertrophy and is thought to be secondary to changes in histone deacetylase 4 (HDAC4) disulfide formation at sites 667–669 (7, 305).
Sulfinylation and sulfonylation
Two other Cys thiol modifications include sulfinylation and sulfonylation. Sulfinylation is reversible with sulfiredoxin; but the only protein known to have a reversible sulfinyl group is peroxiredoxin (233). The last modification, sulfonylation, is a completely irreversible modification that is widely present on SERCA2 in cardiovascular pathologies, as identified with a site-specific antibody (281). Recent methods have been described as selectively enriching the quantity of sulfopeptides such as Cys sulfonic acid. Similar to other acidic peptides, a sulfopeptide shows inefficient ionization in MALDI-MS and electrospray ionization (ESI)-MS (230). Given these difficulties, several methods have employed ammonium salts (148) or basic polypeptides to improve the isolation of acidic peptides. Wu and colleagues (45) have used polyarginine, which is known to bind to sulfonate groups, as a complementary ligand. After affinity ionic capture had been performed using polyarginine-coated nanodiamonds, the peptides from a complex mixture of proteins were analyzed by MALDI-TOF-MS (45), and the particles demonstrated a propensity for peptides binding to multiple sulfonic acid residues. This technique would allow for further evaluation of the sulfonic acid sub-proteome.
Methionine
Met, similar to Cys, is another amino acid that contains a sulfur group at its side chain and is sensitive to oxidative changes by either a one-or two-electron oxidation. The former will produce a methionine sulfoxide by direct oxygen transfer from an RNOS, while the latter produces a disulfide cation that can then react with H2O to result in methionine sulfoxide (244, 294). The oxidation of Met alters its hydrophobicity, which can lead to conformational changes that affect protein-protein interactions or enzymatic activity (266). Two distinct stereoisomers are formed around the sulfur-S- and R-epimers, and these are reversed by methionine sulfoxide reductase A (MsrA) and B (MsrB), respectively (99, 195 –197). MsrA can also act independently of redox signaling as an S-epimer oxidase (164). The reducing equivalents needed for this reaction are provided by the Trx system. Protein carbonyl adducts had been used as a marker for methionine sulfoxide modifications, as the current assumption is that the structural changes associated with sulfoxidation increase the likelihood of further modification and promote protein carbonyl formation (195). Recently, antibodies to methionine sulfoxide have been developed and demonstrate increased Met oxidation in the cardiac tissue after treatment with an oxidant (206). The measurement of Met modifications has been difficult due to their chemical instability, detection methods that inadvertently reduce Met oxidation, and a sample preparation that can introduce additional artifacts, as quenching cations and heavy metals are necessary to prevent the formation of spurious RNOS in the isolation procedure (34). MS techniques have been developed for detecting methionine sulfoxides (34, 102, 103, 126). Methionine sulfoxide in acid-labile and protective carriers such as methane sulfonic acid or chemical derivatization such as by CNBr cleavage are necessary to preserve this modification. The latter technique is frequently used and involves CNBr cleavage of Met on the carboxyl side of the amino acid, but if a methionine sulfoxide is present, then this cleavage can no longer occur. These methionine sulfoxide residues can be detected by MS as a 16 Da shift (126, 299) and a missed peptide cleavage. However, the interassay variability of chemical modification by bromination can differ by approximately 20% (34). Another technique that has been used is collision-induced dissociation (CID) and tandem MS, which along with gas-phase fragmentation can determine the sequence and position of methionine sulfoxide. During this procedure, a specific 64 Da loss from the product precursor, methane sulfenate (CH3SOH) occurs that is unique to methionine sulfoxide and helps differentiate between phenylalanine (Phe), which has the same mass of 165 Da. The problem with this method is that only small peptides can be evaluated (132, 242). The use of this technique in combination with electron capture dissociation (ECD) allows for improved detection of slightly more complex peptide mixtures (103). One last method for methionine sulfoxide detection utilizes combined fractional diagonal chromatography (COFRADIC) with SILAC and a subsequent analysis by LC-MS-MS. Ghesquiere et al. have described more than 1600 modified proteins from H2O2-treated Jurkat cells using SILAC and 27 protein methionine sulfoxide modifications in serum from a mouse sepsis model using the COFRADIC technique alone. Interestingly, the sulfoxide modifications appear to have occurred more commonly near polar groups on proteins (91).
MsRA knockout mice were initially observed as having a prolonged life span, but subsequent studies were unable to confirm this result and found the mice to be more susceptible to oxidative stress (194, 240). In the cardiovascular system, these MsrA knockout mice were found as having lower sarcomeric shortening and peak calcium transients in cardiomyocytes along with larger mitochondria (201). In addition, MsrA is necessary for preventing further ventricular remodeling post-infarct, as it counteracts the oxidation and activation of calmodulin kinase II (CaMKII), which controls postinfarct apoptosis and aldosterone-mediated matrix remodeling (74, 311).
The complimentary enzyme MsrB is composed of three subtypes: B1, which is located in the cytosol and the nucleus; B2, which localizes to the mitochondria or endoplasmic reticulum; and B3, which is solely located in the endoplasmic reticulum (1, 143). Although there is an up-regulation of MsrB1 in cardiac hypertrophy (125), the MsrB1 deletion animal shows no cardiac pathology or increases in cardiovascular protein oxidation at baseline (81). With regard to human pathology, loss-of-function mutations of MsrB3 are associated with deafness (8), but no connection has been made to CVD. Further studies involving models of cardiovascular pathology and MsrA are required to delineate the role of Met oxidation. Interestingly, while reduction may be carried out directly by thionein for MsrB2 and B3, the effects of Msr proteins are highly dependent on Trx, and, thus, Trx deletion or overexpression may indirectly influence Met oxidation as well (113, 144, 239). Further oxidation of methionine sulfoxide may form sulfone (RSO2CH3), which is irreversible and damaging.
His and Lys
His and Lys are the two most reactive amino acids that undergo metal-catalyzed oxidations (Fe2+ or Cu2+/O2/ascorbate). His forms 2-oxo-histidine and can undergo another oxidation reaction to produce ring-opened products such as aspartate, aspartylurea, and formylasparagine. 2-oxo-histidine occurs in vivo on MnSOD (136), which alters its activity, and regulates PerR, a metal-dependent sensor of H2O2 in Bacillus subtilis (282). On the other hand, Lys is subject to substantial post-translational modifications, including acetylation, methylation, SUMOylation (small ubiquitin-like modifier), and ubiquitination. These post-translational modifications have been shown to control cellular signaling and protein degradation. Oxidative modifications have been proposed to affect these modifications, thus interfering with signaling functions (135). Lys can react to form allysine (adipic semialdehyde) in a metal-catalyzed oxidation (Cu2+/O2/ascorbate) that occurs in tissue culture after treatment with high glucose (232) and is increased in the human lens with both aging and diabetes (76). His and Lys side chains can also react to form protein carbonyls. One significant difference between the two in-forming protein carbonyls is that histidine, similar to Arg and proline, forms aldehydes, while the Lys oxidation product, adipic semialdehyde leads to direct carbonylation. The reader is referred to several recent excellent reviews for further information on this topic (18, 195, 302). Carbonylation has been detected by antibodies, enrichment-labeling techniques, and mass spectrometry (271). The antibody utilizes the reactivity of carbonyls to 2,4-dinitrophenylhydrazine (DNPH) and measures the conjugates of DNPH with proteins. In addition to DNPH, labeling can be performed with biotin hydrazide, Girard's P reagent, or oxidation-dependent, element-coded affinity tags and coupled with MS for identification or in the case of biotin, hydrazide immunoblotting. Protein carbonylation may lead to changes in signaling and function and could be used as an oxidative biomarker. In a rat model of diabetes-induced cardiovascular dysfunction, the carbonylation of SERCA2a at specific Lys residues inhibits enzymatic activity (253) and the cardiac contractile proteins myosin heavy chain α and β were carbonylated, which was associated with a decline in cardiac inotropy (254). A recent work demonstrates that endothelin-1 selectively increases carbonylation of the annexin A1 protein (257) in smooth muscle cells, leading to increased proteosomal degradation of the receptor and subsequent cellular growth. In addition, protein carbonylation has been used as a general marker of protein oxidative damage in Alzheimer's disease, cystic fibrosis, diabetes, and systemic amyloidosis (59). In the cardiovascular system, ischemic injury has been noted to increase carbonyls in both coronary bypass surgery patients and post-infarct patients (210) and (190). Interestingly, heart failure patients were found to have a 10-fold increase in protein carbonyls in their plasma (39). Lastly, these two amino acids can undergo a reaction with lipid aldehydes, which are covered in detail in the section entitled lipid peroxidation-derived electrophilic aldehydes.
Tyr and Phe
The aromatic phenol side chain of Tyr characterizes the hydrophobic and fairly unreactive nature of this amino acid. Tyr residues are well-studied components of receptor Tyr kinases that are important in cancer biology, and their oxidative modifications during CVD is a growing area of research. Tyr forms a tyrosyl radical at the active site of cyclooxygenase, initiating the controlled peroxidation of arachidonic acid to prostaglandin G2 (284). Various oxidative modifications have been reported for Tyr, including hydroxylation to form 3,4-dihydroxyphenylalanine, often referred to as protein-dihydroxyphenylalanine (DOPA), (92) as well as ortho-quinones, nitration (212, 272), dityrosine-forming intraprotein and interprotein crosslinks, and halogenation such as chlori- and iodination. Halogenation is catalyzed by peroxidases occurring under physiological conditions in the thyroid follicle during the synthesis of the thyroid hormones thyroxine-3 and −4 (T3, T4). Thyroglobulin, a Tyr-rich protein, is released into the luminal colloid and a specialized NOX (dual oxidase [DUOX]), which also contains a peroxidase domain, reduces I− to iodine and iodinates Tyr residues in thyroglobulin. It should be noted that Tyr iodination may be artificially produced while working up samples for proteomic analysis, and can be observed when blocking Cys with decomposed iodoacetamide (brownish color). Furthermore, chlorination and nitration of Tyr residues occurs during inflammation and is mainly catalyzed by myeloperoxidase, but other peroxidases can also mediate this reaction. Nitration, however, is also mediated by peroxynitrite (PN), an RNOS that is formed in the reaction of nitric oxide with superoxide. At low concentrations, this reaction requires enzymatic catalysis, which has also been observed with prostacyclin synthase or manganese SOD. Interestingly, reports have suggested the existence of a reducing denitrase enzyme, but the protein has yet to be identified (2, 129, 138, 257). Nevertheless, nitration should be analyzed with caution, as acid-catalyzed nitration from nitrate via a nitrosotyrosine intermediate has been reported as a freezing artifact, and nitration might occur postmortem when acidosis is apt to reduce nitrite in tissues. By virtue of the phenolic group, the Tyr residues can also form tyrosyl radicals by reacting with RNOS that in a radical-radical reaction leads to dityrosine formation and crosslinks (104). The hydroxylation of Phe is a well-known reaction that leads to Tyr formation, and subsequent hydroxylation of Tyr leads to DOPA formation. These reactions are either catalyzed with free amino acids by specialized mixed functional monooxygenases (Phe and Tyr hydroxylase) or can be caused by very reactive species, including the hydroxyl radical or PN.
The detection of Tyr and Phe oxidations has been challenging, as RNOS reactions produce a variety of modifications, including nitrotyrosine, dityrosine, DOPA, hydroxyl ortho-, and meta-Tyr (96, 156, 235). Nitrated proteins can be probed for with various commercially available antibodies, but detection is problematic and should always be confirmed by a reduction of nitrotyrosine to aminotyrosine with dithionite. Since the quantity of these modified amino acids can be quite low, nitrated proteins have been enriched using anti-nitrotyrosine antibodies (204) followed by a partial transfer of 2DE separated proteins with MALDI-TOF and ESI-MS/MS identification of the sites (2, 139, 140). Using this method, Kanski et al. have shown that nitration appears to increase in metabolic proteins during cardiac aging. Amoresano et al. (11) have developed an assay called reporter ion generation tag (RIGhT) for the detection of nitrotyrosine-containing proteins (10). This method involves the selective modification of nitrotyrosine to dansylaminotyrosine using dansyl chloride and then monitoring proteins by generating specific ions that are unique to dansyl derivatives by MS. Another quantitative proteomics approach to detect 3-nitrotyrosine developed by Sharov et al. (255) is based on the selective reduction of 3-nitrotyrosine to 3-aminotyrosine. The reduced modification is then labeled with a fluorescent tag in a reaction with 4-(amino-methyl) benzenesulfonic acid.
Dityrosine can be detected in a sensitive and quantitative way by the use of mass spectrometry (63, 207). Furthermore, most other modifications of Tyr and Phe are relatively stable and can be detected using high-performance (pressure) liquid chromatography (HPLC), GC-MS, and LC-MS-based methods.
In contrast to nitrotyrosine modifications, the other protein Tyr oxidation products have only been analyzed by MS due to the fact that no antibodies have been developed for their evaluation. 3-Chlorotyrosine is a stable oxidative Tyr product derived from myeloperoxidase, and has been identified as a modification of ApoAI from high-density lipoprotein cholesterol using LC-ESI/MS/MS (252).
Tryptophan
Tryptophan (Trp) can be oxidized to hydroxytryptophan, dihydroxytryptophan, N-formylkynurenine (NFK), and kynurenine (KYN). A hydroxyl radical can interact with the aromatic nucleus of Trp, and in the presence of Fe, forms hydroxylated Trp at the 4, 5, 6, and 7 positions in the ratio of 4:2:2:3. 5-hydroxytryptophan, which can be enzymatically formed in the presence of tetrahydrobiopterin and Trp hydroxylase, is a precursor to serotonin (181). Using 500 μM to 1 mM H2O2, hydroxytryptophan modifications can be induced in cytochrome C oxidase, subunit 1 (158). Other oxidation end-products of Trp include NFK and KYN, which are formed after a direct interaction with 1O2, or ozone (70). Two other radical reactions by which NFK and KYN can be formed are (i) the Trp radical interacting with superoxide to form Trp hydroperoxide and (ii) the Trp radical interacting with molecular oxygen, leading to a Trp peroxyl radical. The trytophan hydroperoxide and Trp peroxyl radical can be rearranged to form NFK and KYN (70). Similar to some amino acids such as Met, Trp may also theoretically act as a long-range electron transfer shuttle between other amino acids such as Tyr and Cys, which would allow for the formation of tyrosyl and thiyl radicals, respectively, on the peptide backbone (256). Antibodies have been designed for the detection of NFK on protein side chains (70, 267), but MS detection suggests that oxidized Trp products can sometimes be a result of artifacts, as they have been observed with proteins isolated by gel electrophoresis, but not with in-solution digests (214).
LC-MS has identified oxidation products of Trp in the serum in association with atherosclerosis and in cardiac and skeletal muscle tissue (78, 108). Several of these modified proteins include actin, troponin, H-FABP, and complex V. However, due to the multiple isomeric oxidative degradation products (e.g., serotonin or 5-hydroxytryptamine [5-HT] and oxyindolylalanine) that occur, it has been difficult to distinguish these in a complex protein mixture. Hoffman and colleagues (280) have shown that by using high-energy ESI-CID with isolated peptides, the differences in masses within a given ion series and specific marker ions could differentiate the isomeric forms of 5-HT and oxindolylalanine. This suggests that in complex protein mixtures, the possibility of identifying these isomeric forms is feasible.
KYN has been proposed as a direct endothelium-derived relaxing factor that is produced during inflammation (295). The oxidation of this amino acid has also been suggested as playing a role in atherosclerosis, as KYN has been identified in oxidized low-density lipoprotein and apolipoprotein B (apoB)-100 (205, 306). Patients with coronary heart disease may have an elevated ratio of KYN: Trp in the serum, which could function as a potential oxidative stress biomarker of disease (142, 211). KYN also seems to lower the mitochondrial ATP synthesis by significantly increasing state IV respiration, and reducing the respiratory control index and adenosine-5′-diphosphate (ADP)/oxygen ratio of glutamate/malate-consuming heart mitochondria (17). Human heart mitochondrial proteins have these oxidative modifications of Trp without pathology (278), and whether this can act as a means of controlling the ATP/ADP ratio is unclear and requires further investigation. Although oxidatively modified Trp residues are generated in some CVDs, no direct evidence exists to prove the role of Trp oxidation in causing cardiac pathology.
Lipid peroxidation-derived electrophilic aldehydes
This diverse series of α,β-unsaturated aldehydes (Fig. 5) is derived from endogenous lipid peroxidation, xenobiotic metabolism, food, or environmental pollution (209). Members of this group include acrolein (217), which is the most reactive molecule, and prominent lipid peroxidation products such as HNE (37) and MDA (75). RNOS can interact with polyunsaturated fatty acids and initiate lipid peroxyl radicals that lead to a radical chain reaction. Usually, the cellular antioxidant system, which includes the lipid soluble vitamin E, quenches these radical chain reactions. However, lipid peroxidation can occur either under inflammatory conditions or during the reperfusion phase after ischemia/anoxia. This is often accompanied by an increase of free Fe in the cell, catalyzing Fenton-like reactions, further potentiating oxidative stress. Enzymatically catalyzed reactions, such as myeloperoxidase or cyclooxygenase in the presence of RNOS, can also efficiently peroxidize polyunsaturated fatty acids. The decomposition of lipid peroxyl radical species generates various α,β-unsaturated aldehydes by β-cleavage (cleavage at the second carbon atom). The toxicity of these compounds is mainly based on their reactivity with proteins and peptides to form a stable adduct, but DNA modifications (mutagen) and the depletion of cellular reducing equivalents, including glutathione, may also contribute. The cellular detoxification of reactive aldehyde occurs either by glutathione S-transferase-catalyzed phase II conjugation reactions or aldehyde dehydrogenases.
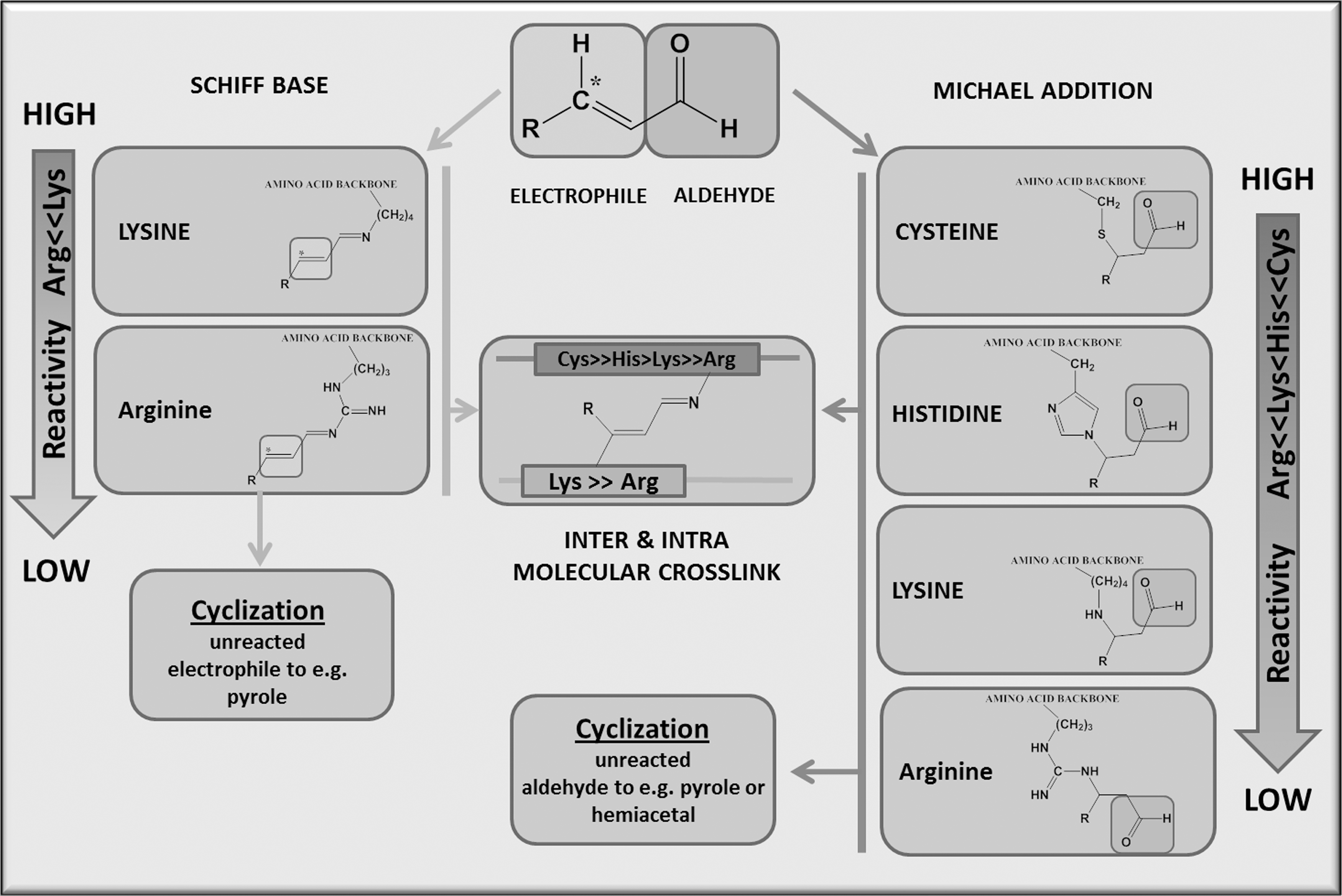
The α,β-unsaturated aldehydes (Fig. 5) are comprised of an aldehyde group that forms a conjugated system with the β-double bond, thus withdrawing electrons from the β-double bond (π-electrons) which forms a soft electrophile (electron-deficient species) on the C-atom (asterisk). This effect can be further enhanced by neighboring electron withdrawing groups such as the hydroxyl group in HNE. Chemistry suggests that soft electrophiles preferentially react with soft nucleophiles (electron-rich species), including the thiolate of Cys. The soft electophiles conjugate the reactive aldehyde at its electrophilic center to the nucleophilc moiety of the amino acid; this reaction, termed the Michael adduct, is named after Arthur Michael. In contrast, strong nucleophiles, including the imidazole moiety of His or the ɛ-amino group of Lys residues, will react much slower. Therefore, the following general sequence of reactivity for amino-acid side chains is generally observed: Cys>> His>Lys>Arg. However, vicinal amino acids, as mentioned earlier, can greatly affect this reaction by steric interference or by modulating the nucleophilicity of the side chain moiety. The Michael addition leaves the aldehyde group intact, which can either further undergo cyclization or react in a Schiff base reaction with amino moieties of neighboring amino acids, leading to intra- and intermolecular crosslinks. The complex chemistry of α,β-unsaturated aldehydes illustrates the multitude of protein modifications and the intricacy that arises for MS analytics. Currently available search algorithms can at best map simple adducts but are unable to identify inter- and intramolecular crosslinks and cyclization reactions.
HNE, which is the most studied α,β-unsaturated aldehyde, was first observed in peroxidized liver microsomal lipids as the major cytotoxic lipid peroxidation byproduct (23) and has since been observed in the serum (145), brain (193), kidney (134), and the heart (69). An early method used to identify the targets of HNE modification was the spectrophotometric detection of a 2,4-dinitrophenol (DNP) derivative that reacts with carbonylated proteins (159, 161). Other carbonyl reactive probes, such as biotin-linked hydrazide (121), allows for identification by mass spectroscopy and electrochemical detection of proteins that are oxidatively modified in vitro (188) or in vivo (100, 259). The major limitation of these methods is the nonspecificity of hydrazide and 2,4-DNP (54), which can react with other aldehydes (i.e., MDA) and the possibility that native biotin-containing proteins can be extracted with the biotin pulldown (20). To increase the specificity of using hydrazide labeling, carbonylated proteins isolated using biotin hydrazide can then be enriched using stable isotope labeling (185). Immunochemical detection of HNE-proteins has been used with antibodies specific to the His and Lys dihydropyrrol HNE adducts (285, 288). The antibody specific for HNE-bound His has identified cytochrome c oxidase modifications in the rat heart subjected to ischemia/reperfusion (49), and the HNE-Lys antibody has detected modifications of mitochondrial subunits in the diabetic rat heart (153). Antibody detection is sufficient to demonstrate the presence of HNE, but these antibodies lack specificity and may detect other Michael adducts such as 4-hydroxy-2-decenal and 4-hydroxy-2-octenal (288, 289). To obviate these shortcomings, experiments with azido or alknyl-derivatized HNE coupled with biotin click chemistry could be utilized to investigate proteins that have a propensity for modification (293). To determine endogenous HNE adducts, enrichment techniques should be used with either antibody-bound sepharose (79) or biotin hydroxylamine or hydrazide to selectively isolate Michael adducts (47, 225). The problem with each of the latter techniques is that both of these compounds react with all carbonyl adducts. Other lipid-derived aldehyde species, including carbonyls and acrolein adducts, have been identified during CVD states and may even mediate cellular signaling (274).
GC-MS has been used to detect HNE-adducted products when applied to human plasma (179, 184, 260) as well as urine (246), although more accurate methods have been performed with other MS methodologies to obtain peptide sequence information. By incorporating labeling probes, reverse-phase chromatography MS/MS has been used to identify modified-adducted proteins in adipocytes (100). Using an ICAT reagent that is linked to a hydrazide functional group, differential labeling with light and heavy carbon identified HNE-modified cardiac mitochondrial proteins by reverse-phase chromatography coupled with MALDI-MS/MS (110). A similar means of identifying carbonylated skeletal muscle mitochondrial proteins used biotin-hydrazide enrichment and iTRAQ labeling with LC MS/MS (185). Furthermore, an MALDI-TOF/MS analysis of mouse myocardium treated directly with the unsaturated aldehyde acrolein was found to have several carbonyl-modified proteins (172). Recently, methods that required no enrichment strategy were described for ex vivo HNE-treated mitochondrial proteins using a neutral loss- MS3 on a hybrid linear ion trap-Fourier transform ion cyclotron resonance mass spectrometer (LTQ-FT) that identified histidine-modified HNE peptides (269). In this protocol, neutral loss of HNE is observed on the MS/MS of HNE-modified peptides, thus triggering MS3 analysis of the neutral-loss product ion to reveal the sequence of the peptide. By coupling this with ECD-fragmentation, retention of the HNE adduct on the product ions is feasible for identification, and, thus, may be a more meaningful peptide fragmentation than CID fragmentation-based MS/MS analysis (170, 176, 192, 227). Lastly, the use of electron transfer dissociation fragmentation may improve the detection of HNE modifications and may be superior to CID in the characterization of modification sites due to the production of c and z ions (85, 226).
Pathophysiological concentrations of HNE have been reported to range from 0.3 to as high as 10 μM in tissues (246), but the cumulative oxidative effects of HNE may be underappreciated, as it can induce peroxide formation (266) and mitochondrial-derived ROS (151) at doses as low as 10 μM. In patch-clamp studies, isolated cardiac myocytes perfused with supra-normal concentrations HNE were shown to undergo membrane depolarization and loss of excitability (28). Other works using high concentrations concluded that HNE can induce intracellular calcium overload in cardiomyocytes (200). At 40 μM, HNE induces adducts that inhibit the tumor suppressor protein LKB1 to promote signaling changes which are indicative of myocyte hypertrophy (64). At high physiologic levels of HNE (10 μM), transient treatment (1–3 h) increases both oxygen consumption and glycolysis, while sustained HNE treatment (8–16 h) greatly reduces cell viability (123). At 5 μM, HNE activates the oxidant-sensitive Nrf2 transcription factor in cardiomyocytes (310) and can protect against further oxidative injury by increasing GSH content through its modifications of glutamate- Cys-ligase, the rate-limiting enzyme in de novo GSH biosynthesis (16).
Detoxification of free HNE involves either oxidation by aldehyde dehydrogenase (primarily the mitochondrial isoform, aldehyde dehydrogenase 2 [ALDH2]) (229) or reduction by aldose reductase (AR) (261). A single nucleotide polymorphism of ALDH2 (E504K) first identified in Asian populations inactivates ALDH2. Since ALDH2 functions cooperatively as a homo- or heterotetramer, all tetramers that contain 1 or more mutated subunits are functionally inactive. In a transgenic loss-of-function mouse model, overexpression of E504K-ALDH2 significantly increased HNE adduction of mitochondrial proteins (73). Similarly, AR-deficient mice fed a high fat diet exhibit increased plasma HNE adducts, advanced glycation endproducts, and acrolein formation (13). Interestingly, the loss of AR had no effect on HNE adduct formation in mice fed a chow diet, suggesting that lipid peroxide accumulation may be minimal or readily detoxified by other mechanisms under physiological conditions.
From these studies, the quantity of HNE used and the treatment duration are critical parameters to be considered when studying the signaling changes induced by HNE. When assessing the effects of HNE on cellular signaling, it is suggested that investigators use pathophysiologically relevant doses of HNE (≤10 μM) for short-term treatments (≤2 h). Furthermore, since low doses of HNE can initiate RNOS production, the possibility of RNOS-mediated effects needs to be addressed. All these studies have used exogenous sources of HNE to study its effect on signaling, but a system by which HNE is endogenously produced would be more pathophysiologically relevant in determining its biological role. With such a system, the localized concentrations of HNE produced would be more likely to reflect native cellular signaling events induced by this species.
Analytics of OPTMs
Several methods exist for the detection of OPTMS. These can range from antibody detection, UV-vis evaluation, labeling techniques for enrichment, and mass spectrometry. Each of these methods have their strengths as well as weaknesses for analyzing complex mixtures of proteins. OPTMs have been detected using chemical labeling or antibodies raised against specific oxidative amino-acid modifications (see Table 3 for specific OPTMs). Antibodies allow for a rapid evaluation of a large number of proteins and samples using either protein electrophoresis, enzyme-linked immunosorbent assays (ELISA), immunoblot, or immunohistochemistry. However, several of the available antibodies that study oxidative modifications suffer from the lack of specificity (52, 214, 215). To overcome this, several OPTMs are evaluated via selective enrichment techniques using compounds or peptides with a high specificity to the oxidized amino acids. Examples of these include anti-DNPH antibodies used in labeling carbonyls and reactive thiolsulfanates used in labeling Cys. These allow for the detection of lower quantities of modified peptides but do not directly measure the modification (e.g., S-nitrosylation, S-gluthionylation, or sulfenylation will all change thiolsulfanate binding). In addition, if an affinity tag is used, then protein complexes may be bound to the affinity column that is not necessarily oxidized (234). Another method used to detect oxidative modifications in proteins includes changes in UV-Vis spectra. Although the utilization of a spectroscopic analysis of protein modification has been used for isolated proteins in solution, it would work poorly for complex mixtures. The final technique is mass spectrometry which allows for a direct detection of the peptide residue affected by oxidant modification. Several limitations of mass spectrometry exist, however, and include inadequate peptide digestion, multiple PTMS that make database mining difficult to achieve, incomplete and variable databases, low-abundance proteins, and environmental contamination. The reader is referred to an excellent review (171) on this topic. Similarly, our experience while identifying OPTMs has shown that (i) the proportion of oxidatively modified protein is small in relation to the unmodified form and both should be compared and reported; (ii) OPTMs occur on multiple sites delivering complex signals that result from the modified digestion of the protein due to an adduct that precludes fragmentation; and (iii) there can be multiple modifications on a single tryptic peptide. These factors are not taken into consideration by many search algorithms, and as a result, these modifications are missed due to low confidence. Moreover, some oxidative modifications on, for example, Lys, may result in a missed cleavage with trypsin, which might make identification of the tryptic peptide difficult due to the increased peptide length. We have found that a combination of manual and computer-aided searches are necessary for the adequate identification of peptides. This may help explain a lack of reproducibility and, at times, a general inability to identify standardized samples among MS laboratories, even though peptide identification for the correct proteins is present (22). Despite these limitations, this technique has very sensitive detection capabilities and is a potent platform for the analysis of multiple types of OPTMs simultaneously. Readers are referred to the following reviews for a more in-depth description of mass spectrometry (6, 56, 57, 268, 309).
ADP, adenosine-5′-diphosphate; ATP, adenosine-5′-triphosphate; GDP, guanosine diphosphate; Glrx, glutaredoxin; HNE, 4-hydroxynonenal; KYN, kynurenine; NFK, N-formylkynurenine; SOD, superoxide dismutase.
The overall workflow of MS-based proteomics for biomarker discovery is depicted in Figure 6, and first involves protein extraction from cells or tissue. The conditions of extraction and choice of buffer are crucial, as a sample preparation might produce artificial oxidation (150). In fact, appropriate storage is also a concern for the evaluation of oxidative modifications. For example, carbonyls are noted to undergo Schiff base reactions with Lys even at −80°C; thus, the quantities of the modification may lessen over time. It is unclear whether other OPTMs are changed after storage, and ideally, samples should be used immediately. After extraction, the proteins are digested using specific proteases (e.g., trypsin or chymotrypsin) in solution or after gel isolation. Although trypsin is the most commonly used enzyme for digestion, some proteins do not fragment well with this enzyme and result in a reduced protein sequence coverage. In addition, the oxidation of Lys and Arg residues may interfere with tryptic digestion and cause missed cleavages, resulting in larger MS fragments. Using different enzymes such as Glu C or chymotrypsin in conjunction with trypsin may allow for a better coverage. Enrichment and fractionation using different approaches such as HPLC with reversed phase, strong cation exchange, or hydrophilic interaction LC produces a purer fraction for MS analysis. The list of mass peaks can then be evaluated on search engines such as Mascot or Sequest, where they are compared with theoretical masses generated from protein databases. By comparing this information, the analysis can provide information on the amino-acid sequence and modifications on that peptide (65, 268).
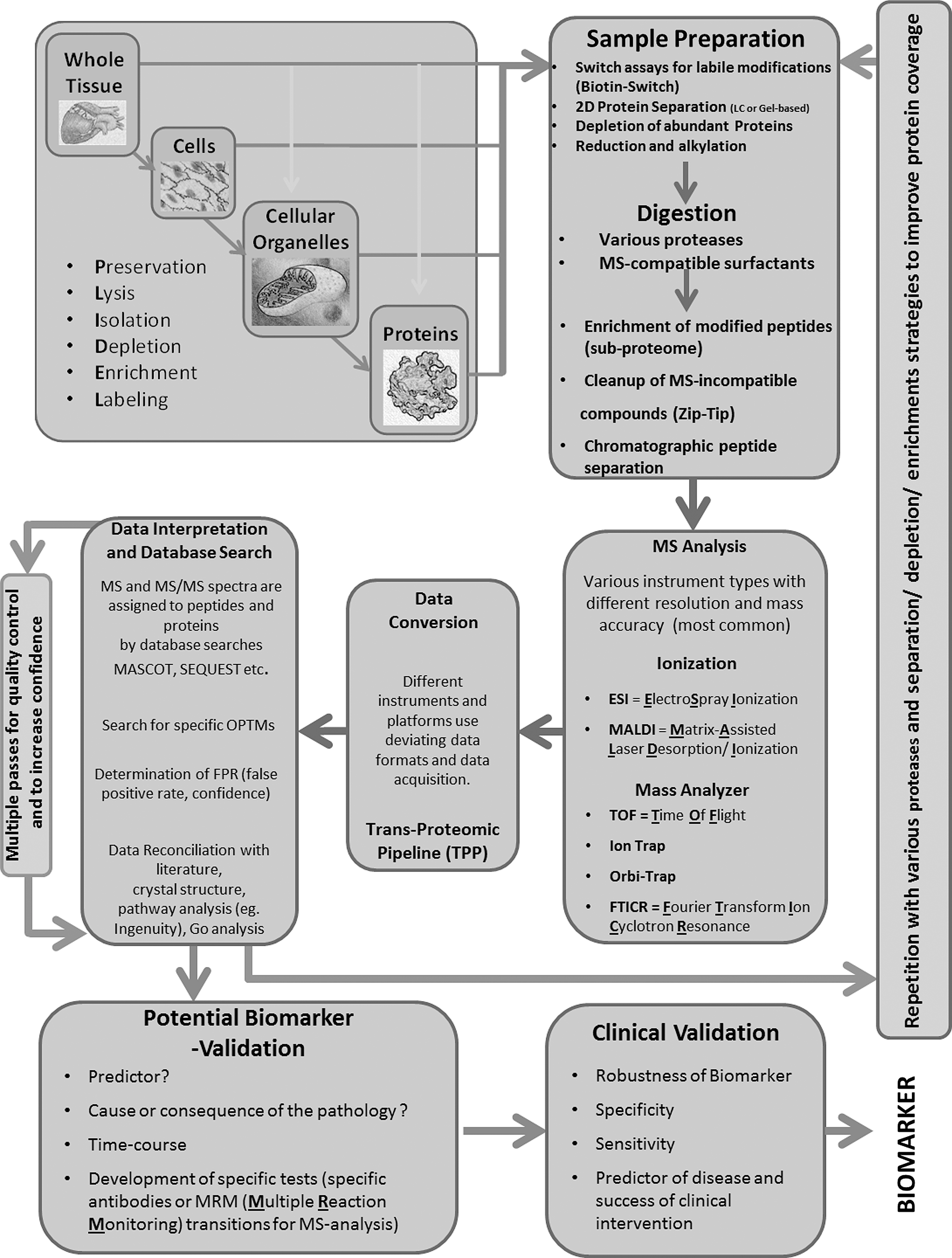
OPTMS As Biomarkers in Humans
Due to their sheer quantity and proximity to oxidant sources, proteins are a principle target of RNOS and may be a part of the antioxidant cellular defense system (117, 162, 173, 231). This direct effect of RNOS allows for the accumulation of oxidative modifications over the protein's lifetime. Theoretically, these can be used as specific biomarkers in human disease. Although oxidation affects both the protein backbone and amino-acid side chains, an in vivo analysis of back bone fragmentation is limited due to the presence of proteases and peptidases, which could lead to protein or peptide cleavage and produce variable peptides, thereby further increasing the complexity of in vivo mixtures (219). Therefore, a potential oxidative biomarker should be based on changes to the amino-acid side chain groups. In searching for an oxidative marker, a stable, nonenzymatic, and irreversible modification would be preferred, as adequate absolute quantification of protein oxidation could be performed. Thus, oxidative side-chain modifications that could undergo further modification or can be enzymatically produced, such as carbonyls and lipid peroxidation products, would be difficult to quantify during progressive disease states and may not be the ideal biomarker. For example, protein carbonyls are initially formed under metal-catalyzed oxidations, can undergo further modification, act as a signal for protein degradation, and have also been suggested as being a reversible enzymatic modification. Thus, the levels of carbonyls measured may vary depending on their further reactivity, cellular antioxidant status and detoxification capacity, and presence of degradative enzymes (177). These factors would indicate that the carbonyl content may be in constant flux, and, therefore, would not reflect the total oxidative state of the cell. By extension of this logic, other reversible OPTMs may also undergo fluctuations in their levels and could not reliably be used to quantify the total oxidative state of the organ. We will review the most likely R-group oxidative modifications that may reasonably comprise an oxidative biomarker.
The R-groups include the aliphatic, aromatic, amidic, acidic and basic amino acids. Aliphatic amino acids, which include Gly, Ala, Val, Pro Leu, and Ile, form alcohols, peroxides, or carbonyls. Amidic, Acidic, Basic, and hydroxylic amino acids can lead to carbonyl groups. In the case of Lys, hydroxylation or adipic aldehyde can be generated, but both these can be generated enzymatically as well. The sulfur-containing amino acids, Cys and Met, form reversible modifications that may rely on factors other than radical flux in determining its quantification, and this is partially due to the presence of Cys and reductive enzymes. However, the irreversible Cys, sulfinic and sulfonic, and methionine sulfone oxidation may be reasonable modifications for studying biomarker identification.
Aromatic side chains of Phe, Tyr, and Trp can be easily oxidized and result in ring oxygenation; Trp can also undergo ring cleavage. Another ring structure, Oxo-histidine, is formed from metal-catalyzed oxidation of histidine (243), but it is unclear whether this product is specific for radical chemistry. Trp forms oxidative by-product species that can also be enzymatically derived. Thus, His and Trp would be poor candidates for biomarkers. Phe can be modified by hydroxyl radicals to ortho- and meta-Tyr, which are stable and can be readily detectable by HPLC; these modifications have been used to demonstrate the presence of oxidative stress in the plasma and urine (89, 208, 213). However, no human studies have been performed to evaluate specific proteins that undergo either changes in ortho-Tyr or meta-Tyr. Similarly, Tyr modification to chlorotyrosine, dityrosine, and 3-nitrotyrosine are specific for radical-induced modifications and would function well as oxidative biomarkers. The nitrotyrosine proteome has been evaluated in uremic patients and has identified fibrinogen chains, alpha-1 antitrypsin, transferrin, ceruloplasmin, and haptoglobin (216). For chlorotyrosine, atherosclerotic patients have been analyzed by Thomson, but no differences in the modifications were noted (279). Thus, particular focus on methionine sulfone, Cys sulfonic acid, and oxidative Tyr modifications for oxidative biomarker discovery should be considered, as these are stable, irreversible, nonenzymatic changes.
Although these studies were performed with plasma, the complex mixture of proteins in serum and the action of proteolytic enzymes activated during clotting may make it difficult to identify oxidatively modified serum proteins. Conversely, urine samples have far fewer components and can contain oxidatively modified peptides, indicating that they may also be used for analyzing excreted oxidative peptides (154, 155).
Traditional biomarkers of cardiac injury, troponin I and T can be detected in the plasma from patients with myocardial injury using sensitive immunoassays (287). Increased levels of three novel markers of subclinical myocardial injury (mid-regional pro-atrial natriuretic peptide, mid-regional pro-adrenomedullin, and C-terminal pro-endothelin-1) were independently associated with the risk of cardiovascular death or heart failure (237). While these biomarkers are quite useful in determining whether a patient is in the midst of myocardial injury and an increased amount of protein has been released in response to cellular injury, the development of cardiovascular pathologies resulting from persistent elevations in oxidative damage may only be detected early by evaluating OPTMs.
A number of oxidant-sensitive proteins have been reported either during a pathological disease state or with direct oxidant treatment (Table 3), and among this group, several have the potential to be used as biomarkers. Creatine kinase and troponin I are already known cardiac biomarkers (29), but only the nitration of creatinine kinase would make it an ideal oxidative marker. The heart-specific myofibrilllar proteins tropomyosin and actin could be candidates, as they have been shown to undergo disulfide cross-bridge formation during ischemia/reperfusion (42) and are carbonylated in human failing hearts (41). However, the modifications that these proteins undergo are enzymatically reversible, and, thus, the quantities can vary. The modulation of acyl-CoA dehydrogenase activity could indicate changes in the fatty acid oxidation status of failing hearts (167), and enzymes such as aspartate aminotransferase were observed to be indicative of diabetes and associated as risk factors for CVD (107), but both these undergo reversible modifications as well. On the other hand, aconitase 2, a Krebs cycle enzyme localized to the mitochondria, has been recognized as a potential biomarker (245), and its sulfonated modification should remain stable and measurable.
Conclusions
For more than a hundred years, the role of OPTMs has been studied. Antibody and MS analytics have been extensively used in the identification of OPTMs, and the functional significance of these modifications has been defined by the genetic modulation of reductive enzymes in animal models. A further discovery of the RNOS sources of several OPTMS will be needed to validate the importance of these modifications in vivo. In order to make further advances in the field, refinements in detection that allow for improved recognition of OPTMs, such as antibody recognition or better labeling techniques, will need to be developed.
The field of MS-based proteomics is constantly evolving and taking more sophisticated approaches to answer biological questions, yet it is still unable to resolve multiple modifications found in a complex proteome. In a typical MS analysis from a tissue extract or a cell lysate, there are many peptides that fail to be identified either due to their poor quality or because search algorithms are incapable of matching them to a known pattern. Recent developments in the field hold promise for improving on peptide identification. For instance, the new Q Executive instrument is a combination of a quadrupole mass filter with an Orbitrap analyzer and with its advanced configuration, has the capability to identify more than 2500 proteins in a single 90 min gradient from a mammalian cell lysate having undergone trypsin digestion (186). High-throughput instruments with both ESI and MALDI ionization capabilities have been advantageous because of their complementary nature (30). To better match mass peaks, error-tolerant searches with Mascot version 2.2 or later can perform analyses with semi-specific proteases, and can consider all the possible modifications and substitutions that can arise from a single base substitution in the nucleic acid sequence. Although very helpful, these search parameters are limited foremost by our knowledge of variable modifications and technically by the number of variations that can be specified per search.
Currently, the cardiovascular evaluation of both the Redox and Ox stress proteomes are rarely delineated in the literature, and the terms are interchangeably used. Most fundamental evaluations have been done on the Redox sub-proteome of Cys (S-glutathionylation and S-nitrosylation) and the Ox stress sub-proteome of Tyr (nitration) and Lys/histidine (HNE). Each of these demonstrates importance as markers of CVD and appears to be pertinent for disease modification in myocardial infarction, hypertension, and inflammation. Further work, however, needs to be performed to determine the functional significance on other members of the redox proteome, such as methionine sulfoxide and carbonyls, both of which would be more likely to form under conditions of increased H2O2 production, a purported major mediator of cardiovascular phenotypes such as hypertension, cardiac hypertrophy, and heart failure (14). The role of the Ox stress proteome has mostly been evaluated on an associative basis (199). Animal models such as the aldehyde dehydrogenase, AR, and glutathione S-transferase transgenics and deletion mice may provide a better insight regarding the role of HNE in cardiovascular pathology. Lastly, screening for several of these Ox stress proteomes should be done in transgenic mice expressing catalase, mito-catalase, SOD, etc., to further delineate the roles of these OPTMs in disease states. Since analyses from MS can be incomplete, the creation of a specific OPTM database that collects and annotates published MS data would allow for future analysis when novel search methods are developed to aid in further discovery (147).
Footnotes
Acknowledgments
This work was supported by NIH grants PO1 HL 068758, R37 HL104017, R01 HL31607, and T32 HL007969 (TDC) as well as by NHLBI, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN268201000031C, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the awarding offices.