
Abstract
Chronic Inflammatory Diseases
Positional Identification of a Polymorphism in Ncf1—A Major Gene Associated with Arthritis
One of the first genes to be positioned from a quantitative trait locus was the Ncf1 gene. This was done in crosses between DA rats, susceptible to both rheumatoid arthritis (RA) and multiple sclerosis (MS) models, and E3 rats resistant to these diseases. The strongest locus, Pia4, was found to control inflammation severity (82). Through a straightforward recombinant congenic approach, the underlying gene was identified to be Ncf1 (54) (Fig. 1). This came as a complete surprise; the gene was in fact the least suggested candidate, as it led to a higher production of reactive oxygen species in the congenic DA.Ncf1E3 rat that was protected from developing disease. The Ncf1 gene encodes the p47phox/NCF1 protein, an essential component of the phagocytic NADPH oxidase complex (NOX2). Upon phosphorylation in the cytosol, NCF1 complexes with NCF2 (p67phox) and NCF4 (p40phox), which then adhere to the membrane and form the NOX2 complex in cooperation with NOX2 (CYBB)/CYBA. Subsequently, the NOX2 complex transports electrons from NADPH to oxygen, resulting in the generation of a variety of ROS, the so-called oxidative burst. The allelic differences between DA and E3 rats were pinpointed to three point mutations (SNPs). Today, we know that the responsible amino acid position leading to impaired ROS production is T153M (31).

The finding that impaired ROS production is associated with more severe disease is, however, in contrast to the longstanding dogma that the release of ROS is pro-inflammatory. In compliance with this dogma, antioxidative agents are commonly believed to have therapeutic potential. So, how could it be explained that a reduced ROS production leads to dramatically more severe inflammation?
Interestingly, rats with genetically determined high ROS response or rats with a low response treated with NOX2 activating substances inducing oxidative burst (e.g., phytol), not only were less susceptible to arthritis but also had a lower T and B cell autoimmune response (18, 29). T cells from DA rats (with a lower ROS response) had a higher density of −SH (thiol) groups (i.e., more reduced) on their cellular membranes, and this change in T cell phenotype led to a higher activity and induction of arthritis, whereas T cells from DA.Ncf1E3 rats (with a high ROS response) could not induce arthritis even if transferred to DA rats (18). Importantly, no expression of Ncf1 could be detected in the T cells, nor could a NOX2-dependent ROS response be detected (18). Increase of thiols by glutathione treatment turned T cells from DA.Ncf1E3 rats into arthritogenic T cells and, vice versa, T cells from DA rats treated with oxidized glutathione (or H2O2) became less arthritogenic (18).
A spontaneous mutation in the mouse Ncf1 gene resulting in a truncated protein and nearly absent ROS production led to similar observations as in the rats, that is, a reduced disease activity in models for rheumatoid arthritis (collagen-induced arthritis, CIA) and multiple sclerosis (experimental autoimmune encephalomyelitis, EAE) (30). The mutation in Ncf1 also caused a reduced immunological tolerance to type II collagen (CII) in mice (24, 28, 30). These effects were at least partly dependent on Ncf1 expression in antigen presenting macrophages (19, 58). Interestingly, it was observed that Ncf1 mutated mice can also spontaneously develop autoimmune arthritis and autoimmunity to CII, but only during the postpartum period (30), a period when the mice are more susceptible to arthritis (50). Spontaneous development of arthritis has also been reported recently in NOX2 KO mice due to an altered Th17/Treg cell development (45). However, it is important to exclude the possibility that arthritis is caused by staphylococcal infections, which occur more frequently in Ncf1- and NOX2-deficient mice (59).
Taken together, these data show that Ncf1 alleles leading to a reduced oxidative burst enhance autoreactive T cell activation and allow the development of severe chronic inflammation. Importantly, these observations stem from hypothesis-free genetic findings in rat models, were confirmed in mouse models, and challenge some of the current dogmas concerning the role of ROS during inflammation.
Physiological Role of NOX2-Dependent ROS Production by Phagocytes
ROS are derived both from the cellular metabolism and through specific induction by oxidases such as the NOX2 complex. The classical ROS-producing oxidase, the NOX2 complex, is however dominating quantitatively as an induced ROS producer in the body. The NOX2 expressing cells are mainly phagocytes with polymorphonuclear neutrophils as the cell type with highest NOX2 expression and ROS-inducing capacity, but also phagocytes with antigen-presenting functions, such as macrophages/monocytes/dendritic cells and B cells, can be induced to give a NOX2-dependent oxidative burst.
Using the luminescent probe L-012 (40), we could show in vivo that the mutation in Ncf1 significantly impaired the induced ROS production. This new method (40) revealed that Ncf1 mutant mice fail to produce a signal, whereas a signal in both naïve mice or those expressing Ncf1 in macrophages only, can be detected (Fig. 2). As expected, the ROS response increases dramatically by injecting agents such as LPS that trigger inflammation.
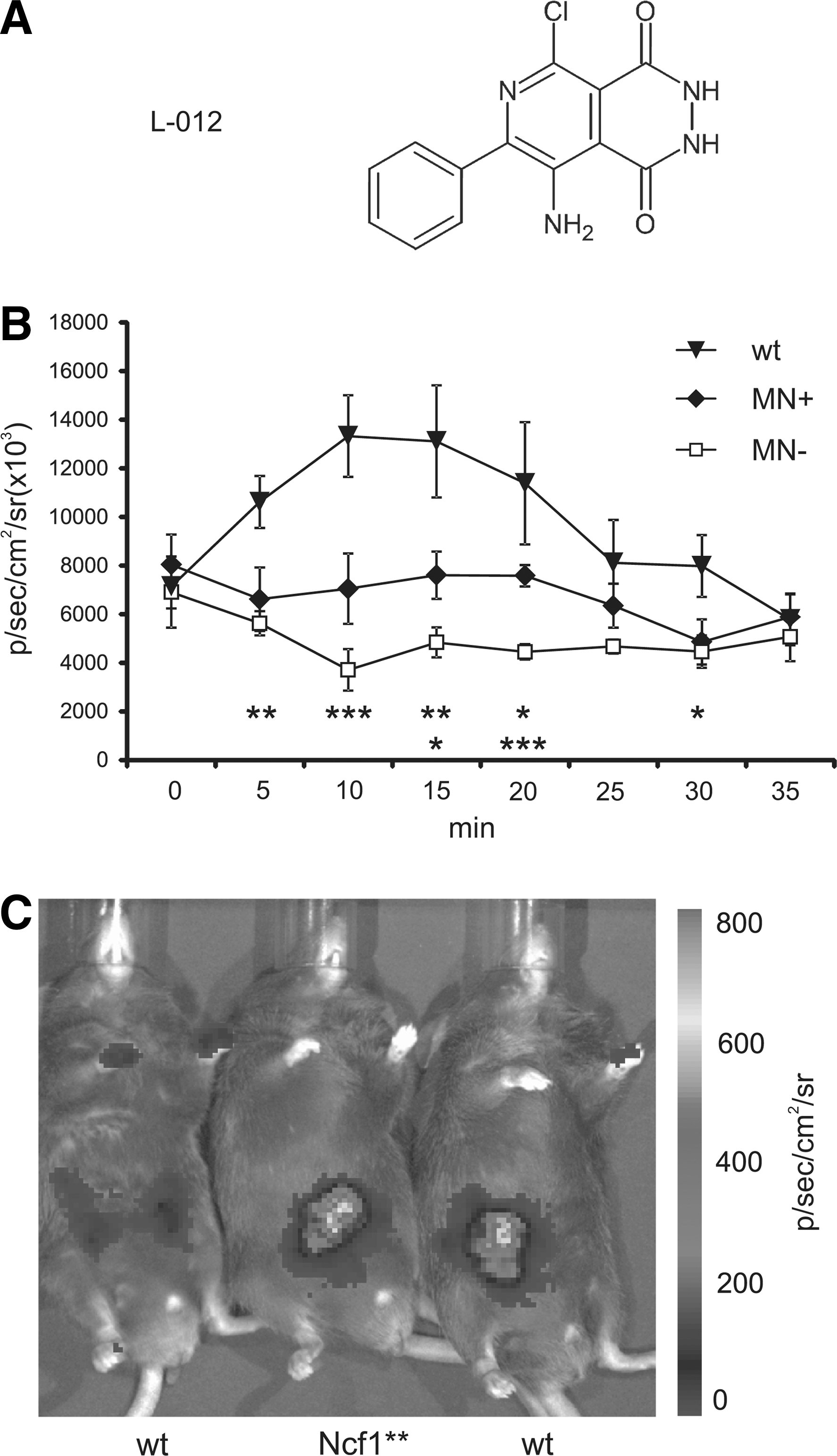
Clearly the NOX2-induced ROS have a fundamental role in both physiological intracellular regulation, as well as in the defense against pathogens and tumors. During the last 10 years, more data have been accumulated supporting a role of ROS as anti-inflammatory: ROS produced by phagocytes and monocytes also regulate protection against autoimmunity, cancer, and viral infections. In cancer, in the absence of NOX2 activity, myeloid-derived suppressor cells lose their ability to suppress T cell responses (12). During infections, viral or bacterial peptides may stimulate ROS production by monocytes, which in turn reduces T cell activation (7, 25, 78). In accordance with these data, NOX2-deficient mice develop a less severe influenza with decreased viral titers (74) and with increased inflammatory side reactions (33). In contrast, ROS also protect against bacterial and fungal infections (36, 59). Therefore, ROS produced by phagocytes seem to have a dual role: killing certain bacterial and fungal pathogens but also to limit activation of inflammatory macrophages and T cells. Evolutionary pressure to survive bacterial/fungal and viral infections may have acted on the ROS production by phagocytes from two opposite directions. Challenge by bacterial and fungal infections or autoimmune manifestations would have favored survival of individuals with high ROS production. Interestingly, wild rats are highly polymorphic within Ncf1, approximately 50% of wild rats have a reduced oxidative burst (55). This observation is difficult to explain, as these rats are likely to have an increased risk of developing autoimmune chronic inflammation, as well as a higher susceptibility to bacterial and fungal infections. Possibly other factors, such as some virus infections, can have a balancing effect (33). An alternative explanation was recently provided, as it was found that the Ncf1 polymorphism profoundly changed the risk behavior of rats, something that is highly likely to be of importance during natural selection (Schiavone et al. Transl Psych, in press) (Fig 3).

To conclude, NOX2-dependent ROS production has several immunological functions, which have been fine-tuned to maintain a healthy balance between an effective immune response against pathogens and an excessive adaptive inflammation that would result in damage for the host.
The Genetic Control of NOX2-Dependent Oxidative Burst and Autoimmune Disease Associations
Deficiency of any of the NOX2 complex proteins not only leads to increased infection susceptibility but also to a higher frequency of inflammatory autoimmune diseases (9). Interestingly, increased autoimmune diseases are also seen in family members of CGD patients (39). A role for the NOX2 complex in human autoimmune disorders has recently been reported (37, 56, 57, 62, 75) (Table 1). An association to NCF2 polymorphism has been reported for systemic lupus erythematosus (SLE) (17). The causal allele was shown to disrupt the binding to Vav1, thereby reducing the Vav1/3-dependent NOX2 activation initiated by cross-linking of the Fcγ receptor (37). A rare variant in NCF2, which reduces the binding capacity of p67phox to RAC2, is associated with very-early onset inflammatory bowel disease (53). SNPs in NCF4 are associated with Crohn's disease (CD) (62), at least in the subgroup of ileal CD (63), and with multiple sclerosis (MS) (34). An association analysis of RA with matched controls identified an association in NCF4 with rheumatoid factor-negative male RA patients (56).
The data of natural genetic variations is collected from UniProt (
Spontaneous mutation m1J in Jackson Laboratory mouse colony,
From unpublished sequence data.
CD, Crohn's disease; CGD, chronic granulomatous disease; MS, multiple sclerosis; RA, rheumatoid arthritis.
The region surrounding the NCF1 gene is structurally complex, which has hindered large-scale association analysis of NCF1 using genome wide platforms. The NCF1 gene is located in a chromosomal segment duplicated three times, resulting in three copies of NCF1 (4). Two of these gene copies have a 2 bp deletion in exon 2, resulting in a shifted reading frame and a truncated protein (23). In addition, also the functional NCF1 gene is copy number variable, and an estimated 10% have at least one extra copy of NCF1 (57). Combining both genotyping and copy number variation analysis of NCF1 revealed an association of NCF1 with RA (57). This suggests that having an extra copy of NCF1 is protective, possibly as a back-up mechanism to ensure sufficient ROS production.
Genetic control of ROS in autoimmune diseases is likely to be not limited to NOX2 complex genes, but possibly includes genes associated with autoimmunity that could either regulate or be regulated by oxidation, for example, the second strongest associated gene in RA, the PTPN22 gene (20).
Role for NOX2 in Antigen-Presenting Cells
Many NOX2-expressing phagocytes have antigen presentation capacity. Dendritic cells (DCs) are professional antigen-presenting cells (APCs), involved in selecting T cells in the thymus and priming naive T cells in lymph nodes. B cells present antigen to T cells and thereby regulate T–B cell interaction and entry into germinal centers. Tissue macrophages can be induced to express MHC class II molecules, and these cells play a dual role in both activating entering T cells and dampening T cell responses in the resolution phase of the inflammatory response (Fig. 4).
NOX2 in the APCs are activated through several different stimuli mediated through various receptors stimulated by inflammatory (PAMP receptors), danger (DAMP receptors), and immune stimuli (Fc receptors) to produce superoxide, which is rapidly converted into other ROS (reviewed in Ref. 69). In general, the cellular activation of the NOX2-dependent induced ROS is similar in all phagocytes, but in antigen-presenting cells there are several issues that could be of relevance. The NCF4 protein adheres to phospholipids typical of cellular organelles such as endosomes rather than the cellular membranes (16). Thus, it will steer the oxidative burst into endosomes in which antigen processing and loading of MHC class II molecules occur. The endosomes also communicate with the immunological synapse—the compartment formed when antigen-presenting cells and T cells interact for antigen presentation. Both antigen peptide-loaded MHC class II molecules and NOX2 complexes are likely to be transported and integrated into lipid rafts, forming the APC membrane in the immunological synapse. The release of ROS will also lead to generation of hydrogen peroxide (H2O2). Among different ROS, only H2O2 can diffuse across membranes, while others have low membrane permeability (reviewed in Ref. 85). Hydrogen peroxide is a two-electron oxidant that acts as an electrophile and can react with protein thiol moieties to produce a variety of sulfur oxidation states, including disulfides, sulfenic (−SOH), sulfinic (−SO2H), or sulfonic (−SO3H) acid products. This reactivity affords (mostly) reversible post-translational modifications of proteins involved in cell signaling (reviewed in Ref. 11). Hydrogen peroxide is relatively stable with an estimated half-life of 0.1 msec and a diffusion distance as far as 1.5 mm (85). H2O2 will thus have a localized effect revolved around the activated NOX2 complex. As it passes lipid membranes, it will be regulated by diffusion to form a gradient from its production center. It will be rapidly neutralized by peroxide-inhibiting enzyme systems, whereas the more long-term redox equilibrium is regulated by thiols. Hydrogen peroxide itself has a relatively low reactivity compared to other oxidants, but the reaction rate is remarkably accelerated by peroxiredoxins. Two models of H2O2 action on target proteins have been proposed. H2O2 may directly oxidize cysteines to promote formation of disulfide bridges. The alternative model proposes that H2O2 oxidizes peroxidases (such as peroxiredoxins). Oxidized peroxidases then subsequently act as catalysts for oxidation of regulatory thiols on cellular proteins (reviewed recently in Ref. 32). This will lead to a nonuniform spreading of H2O2, concentrating it into organelles such as endosomes and lysosomes as well as into the immunological synapse. The release of ROS into DC phagosomes has been shown to affect the peptide processing and loading onto MHC molecules during cross-presentation to CD8+ T cells, through increase of the phagosomal pH that affects the activity of processing enzymes (70). This effect has, however, not been observed in macrophages or during MHC class II-dependent presentation to classical CD4+ T cells (47). Recently, it has also been reported that antigen processing in DCs is dependent on ROS through oxidation of cystein cathepsins rather than increase of endosomal pH (65). ROS production into endosomes is also likely to regulate antigenic proteins and peptides that contain free thiols that may affect antigen presentation and T cell stimulation (83). However, an alternative major effect could be that NOX2-derived H2O2 affects pathways within the antigen-presenting cells in localized compartments. It has been shown by Rhee and co-workers that peroxiredoxin (Prx) enzymes are regulated through both phosphorylation and oxidation and create compartments within the cell that allow enzyme regulation by peroxide (87). Thus, it is likely that APC are affected by their own NOX2-derived oxidative burst, both by modulating their intrinsic inflammatory activity as well as their antigen presenting capacity. Both effects lead to a dampening of chronic inflammation.
Logically, the induced ROS will affect not only the APC but also the interacting T cell. If ROS is released into the immunological synapse and since peroxide is long-lived and passes membranes, they will affect both the T cell and the APC itself. In fact, it is known that peroxiredoxin I gets inactivated by phosphorylation around the TCR signalosome (87). This inactivation may allow ROS to oxidize proteins downstream of the TCR, which is suggested to regulate the TCR signaling, as discussed in following paragraphs (Fig. 5).

However, all the studies conducted so far are based on experimental evidence from biochemical assays or cell culture studies in vitro. In vivo studies are warranted and potentially very promising, as suggested by the preliminary data reviewed below.
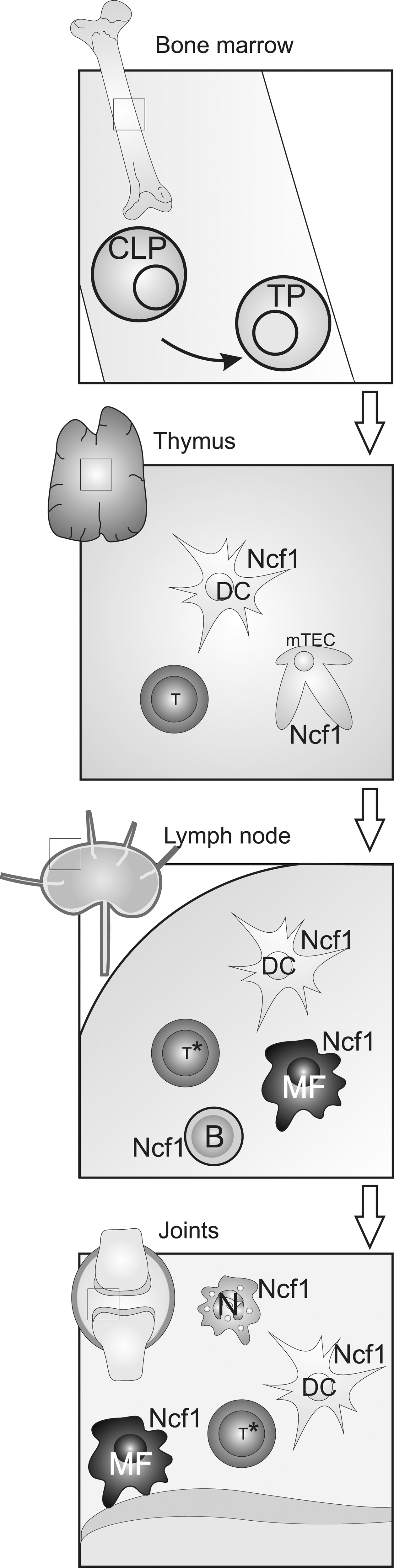
Selection in Thymus
Bone marrow-derived T cell precursors are selected in the thymus by interactions with APCs. First they will be positively selected (i.e., only T cells able to recognize the self MHC molecules on thymic epithelial cells will survive). In the second step, T cells are negatively selected; T cells with high affinity to self peptides bound to the same MHC molecules on other APC will be deleted and only those with intermediate or low affinity survive. In the negative selection, the development of T cells critically depends on the interactions with both thymic epithelial cells and bone marrow-derived dendritic cells (DCs). The available repertoire of self-peptides displayed by thymic APCs critically determines the efficiency of this quality control step. Routes of antigen supply include intrathymic self-proteins, blood-borne antigens, extrathymic organ-specific antigens imported via a steady influx of peripheral DCs, and promiscuously expressed genes coding for extrathymic antigen either presented by medullary thymic epithelial cells (mTECS) or cross-presented by DCs. To which extent thymic DCs complement promiscuous gene expression is still not known. During tolerance induction, potentially autoreactive thymocytes are either deleted, anergized, or converted into ‘natural’ regulatory T cells (Tregs) (27). Importantly, a suppressed TCR signaling may in fact lead to rescue of autoreactive T cells instead of deleting them or in fact also lead to less Treg activity. For example, a mutation in the TCR signal regulator ZAP70 leads to weaker TCR signaling but an expansion of autoreactive T cells and the development of autoimmune arthritis (66, 67). Deletion of the peroxiredoxin 2 (PrxII) gene, affecting ROS levels, leads to a disturbed thymic selection (52). Interestingly, a reduced ROS response has been shown to lead to reduced Treg function in both rats, susceptible to autoimmune arthritis, and humans with CGD (41). It will be challenging to understand the role for ROS in thymic selection since ROS effect on developing T cells during negative selection is likely to affect both T effector cells and Tregs.
Priming in Peripheral Lymphoid Organs
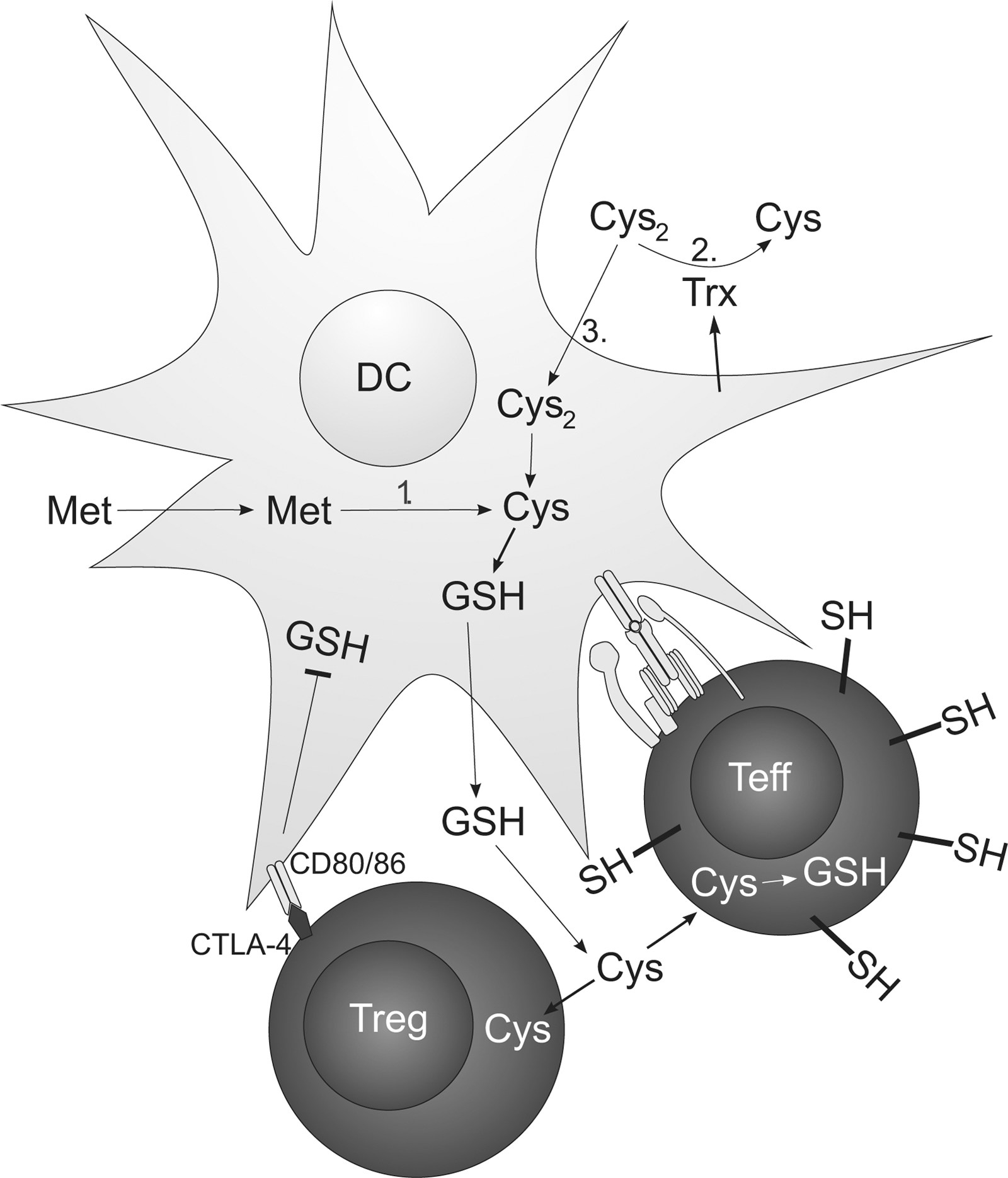
Naïve T cells exported from the thymus circulate and will eventually reach peripheral lymphoid organs where they might encounter DCs that prime them with specific antigen. Priming of naïve T cells is a critical and well-controlled step in the adaptive immune response. The DCs are known to operate in a reduced milieu during priming (1) and to produce cysteine needed for T cell activation (Fig. 6). This will also lead to a reduced synapse formed between a DC and a T cell. Extracellularly, the cysteine/cystine system represents the major thiol/disulfide redox buffer. Naïve T cells lack a cystine transporter but require cysteine in order to synthesize GSH, which serves as an important proliferative signal (76). DCs, however, can import cystine from the extracellular compartment via a cystine/glutamate antiporter and convert it to GSH, which is then exported and subsequently degraded to yield cysteine. Enhanced synthesis of cysteine from methionine via the trans-sulfuration pathway in DCs and reduction of extracellular cystine by thioredoxin released from DCs after contact with Ag-specific T cells could also contribute to the extracellular accumulation of cysteine (1, 90). Subsequently, extracellular cysteine accumulation produces a reduced extracellular redox potential conducive to T cell proliferation and provides T cells with cysteine required for the synthesis of GSH. Interestingly, the redox status of secondary lymphoid organs such as lymph nodes is more reduced than that of nonlymphoid organs and an increase in free (nonprotein) thiols inside cells and in the extracellular space has been observed in lymph nodes following immunization (10). The specific membrane targets of redox remodeling still remain to be elucidated. Recent data suggest that ICAM-1-dependent cell–cell contacts between DCs and T cells contribute to a DC-induced increase of cell surface and intracellular thiols in Ag-specific CD4+ and CD8+ T cells and that T cells activated by DCs, but not by B cells, are protected from H2O2-induced apoptosis (49). This is in line with a previous study that had shown that DCs protect T cells and NK cells from H2O2-driven apoptosis by a catalase-dependent mechanism (77). NOX2-dependent ROS also regulate signaling pathways that control DC cytokine expression (e.g., IL-12p70), which in turn influences cell fate decisions during CD4 T helper cell commitment (38). Interestingly, DCs in Listeria-infected Ncf1-deficient mice enhance immune activation, leading to an increased specific antibody response (80). In summary, DCs seem to neutralize their own ROS to create a reduced environment favorable for activation of naïve T cells.
Macrophages in Inflamed Tissue
Besides neutrophils, macrophages are the main ROS producers. Tissue macrophages are derived from monocytes and can have diverse roles in the tissue, depending on the inflammatory context. During the initiation of an immune response, some macrophages will be inflammatory, mainly by activating other cells to perform effector functions, such as fibroblasts, neutrophils, and T cells. At subsequent steps, macrophages play a role to limit and downregulate the inflammatory response and to clean up dead cells and tissue debris (Fig. 7). This also leads to activation of other inflammatory and regulatory cells but now with different and sometimes counteracting roles as compared to the initiation phase. Macrophage nomenclature is based on cellular phenotypes in vitro, thought to mirror functions in vivo. The most common classification distinguishes between macrophages type 1 (M1) and type 2 (M2) (3). M1 macrophages are the “classical” inflammatory macrophages that can be differentiated in vitro from monocytes in the presence of IFN-γ, LPS, or GM-CSF. They are characterized by a high IL-12 production and the promotion of T helper cell type 1 (Th1) responses. M2 macrophages are the alternatively activated macrophages, which can be differentiated in culture with M-CSF or IL-4/IL-13. M2 macrophages produce generally low pro-inflammatory cytokine levels and promote T helper cell type 2 (Th2) responses (48). M2 macrophages have also been described as anti-inflammatory macrophages that downregulate co-stimulatory molecules upon activation and suppress T cells (81). It is mainly the M2 macrophage that expresses NCF1/NOX2 complexes and produces ROS, but it is still unclear whether ROS activate or differentiate these cells in vivo (42).

From a mouse strain expressing NCF1 in macrophages only, we know that macrophages are critical cells for regulating both autoimmune-dependent chronic inflammation and typical CGD infections such as with Staphylococcus and Aspergillus (59). In this model, NCF1 expression is targeted to CD68+ cells, which represent predominantly classical macrophages and may also contain dendritic cells but not granulocytes or B cells. Interestingly, restricted expression of the MHC class II molecule Aq on CD68+ cells in this model led to priming of a collagen II-specific T cell response and development of arthritis but only if the mouse lacked a NOX2-dependent ROS response (58).
ROS production from macrophages is a double-edged sword, as it seems to be important for the acute defense against pathogens but also important to limit chronic inflammation. It is not clear whether these outcomes are dependent on the same or similar mechanisms or even executed by the same subtype of macrophages.
Expression of NCF1/NOX2 and ROS Production in T Cells
Not only phagocytes respond by ROS production when activated. Generation of ROS has also been detected in T cells when activated through the TCR (35). T cells are capable of producing various types of ROS, as detected by sensitive methods in vitro after stimulation with anti-CD3 using both murine and human T cell lines and purified T cells (15, 35, 86). A first rapid response generates ROS from DUOX1 oxidases, whereas a later sustained response involves NOX2 oxidases. Specific inhibition of DUOX1 showed that this NADPH oxidase generates ROS during activation of human CD4+ T cells that enhance TCR signaling (44). On the other hand, TCR-induced ROS stimulation dampens ERK activation (43). From these findings, it can be concluded that ROS act as second messengers regulating TCR signaling. There are, however, several remaining questions. It is difficult to exclude the occurrence of small numbers of phagocytes among purified T cells; these will have strong bursting capacity in cell cultures and it is likely that hydrogen peroxide can diffuse to neighboring cells in an in vitro culture. Another problem is that there are T cell subpopulations, such as gamma delta T cells, which phenotypically are related to phagocytes (26). The expression of the NOX2 complex in T cells is very weak and difficult to detect and not at all comparable to the expression in interacting APCs. It is therefore likely that a more important ROS influence is mediated by hydrogen peroxides intercellularly rather than intracellularly. This is supported by the finding that mice expressing NCF1 in macrophages only and not in T cells, displayed suppressed autoreactive T cell responses and were protected from development of arthritis (19). Recent data suggest that APCs, mainly macrophages, suppress T cell responses and functionally activate Tregs in a ROS-dependent manner also in the human system (41).
Oxidation Pathways Regulating T Cell Activity
Regardless of whether the most important ROS-mediated regulation in T cells is derived from the interacting APC or generated intracellularly, ROS likely have a profound regulatory effect on T cell function. The outcome of this regulation, and the precise mechanisms are, however, very complex and we are only in the beginning of our understanding.
It was originally suggested that ROS (i.e., hydrogen peroxide), are generated to enhance TCR and B cell receptor (BCR) signaling (60). This is supported by findings showing a regulatory effect of ROS on protein tyrosine phosphatases (PTPs) (68). The PTPs are regulated by a cysteine in the active site with a low pKa value and therefore accessible for oxidation by hydrogen peroxide and oxidative enzymes. Oxidation of PTPs inhibit their activities, and it is known that PTPs negatively regulate TCR and BCR signaling (2, 61, 84). This is of particular importance since the protein tyrosine phosphatase, nonreceptor type 22 (PTPN22) in both T and B cells, has been identified to genetically regulate several autoimmune diseases, including rheumatoid arthritis (6). The disease-promoting allele enhances TCR signaling due to reduced function of this negative regulator (91). The oxidative regulation of PTPN22 is, however, not clear in vivo, and there are also cysteines outside the catalytic site that could modify the effect (79). Furthermore, the role of PTPN22 on TCR signaling might have different effects in different types of lymphocytes, for example, oxidation of PTPN22 might functionally activate regulatory T cells rather than helper T cells and thereby suppress the T cell-dependent immune response. In addition, there are numerous cysteines in T cells and a few of them are likely to be regulatory targets for ROS or oxidative enzymes like glutathione peroxidases and peroxiredoxins. It has, for example, been shown that oxidation of LAT (linker for activated T cells), an important regulator of TCR signaling interactions (46), is inactivated by oxidation (22). Exposure of T cells to hydrogen peroxide led to inactivated LAT and suppression of T cell activity. This phenomenon occurs in T cells from rheumatoid arthritis synovium, normally exposed to high levels of ROS, which showed an inactivated LAT and are hyporesponsive (21). Thus, if LAT is affected also in vivo, this could contribute to a reduced T cell activation. There are numerous other targets of oxidation in T cells, such as calcium channels, potassium channels, integrins, CD2, the TCR/CD3 complex, ZAP70, or lipids in the membranes that could be regulated and they will all contribute to the downstream effect of hydrogen peroxide secreted from APCs and ROS from inside the T cell.
The effects of ROS on T cells are likely to be complex and highly context-dependent. Thus, in vitro results with selected cells and a high oxygen pressure will be different from in vivo where also the tissue context and timing of operations play an important role.
Footnotes
Acknowledgments
This work was supported by the Swedish Research Council, the Swedish Strategic Science Foundation, the Sigrid Juselius foundation, the European Union grants BeTheCure, Masterswitch, and Neurinox.
Author Disclosure Statement
None of the authors have conflicting financial interests.