
Abstract
Macrophages are present throughout the human body, constitute important immune effector cells, and have variable roles in a great number of pathological, but also physiological, settings. It is apparent that macrophages need to adjust their activation profile toward a steadily changing environment that requires altering their phenotype, a process known as macrophage polarization. Formation of reactive oxygen species (ROS), derived from NADPH-oxidases, mitochondria, or NO-producing enzymes, are not necessarily toxic, but rather compose a network signaling system, known as redox regulation. Formation of redox signals in classically versus alternatively activated macrophages, their action and interaction at the level of key targets, and the resulting physiology still are insufficiently understood. We review the identity, source, and biological activities of ROS produced during macrophage activation, and discuss how they shape the key transcriptional responses evoked by hypoxia-inducible transcription factors, nuclear-erythroid 2-p45-related factor 2 (Nrf2), and peroxisome proliferator-activated receptor-γ. We summarize the mechanisms how redox signals add to the process of macrophage polarization and reprogramming, how this is controlled by the interaction of macrophages with their environment, and addresses the outcome of the polarization process in health and disease. Future studies need to tackle the option whether we can use the knowledge of redox biology in macrophages to shape their mediator profile in pathophysiology, to accelerate healing in injured tissue, to fight the invading pathogens, or to eliminate settings of altered self in tumors. Antioxid. Redox Signal. 19, 595–637.
III. The Distinguished Redox Species: Nitric Oxide and Superoxide Radical Anion
V. Redox Systems Modulate and Determine the Macrophage Phagocyte Function
VII. Targets of the NO/O2
− Redox Biology in Macrophage Function
VIII. Redox Signals and Hypoxia-Inducible Macrophage Reponses
IX. Nrf2 at the Transition from Classical to Alternative Macrophage Activation
XI. ROS Support Classical Activation in Response to Modified Lipoproteins
XII. ROS: Linking Classical Macrophage and Inflammasome Activation
XIII. Autophagy Dampens ROS-Dependent Inflammasome Activation
XIV. The Redox Sensor AMPK Favors Regulatory Macrophage Activation
I. Introduction
The term oxidative stress was defined as a disturbance in the prooxidant–antioxidant balance in favor of the former (264). This term describes the conditions when the redox state of main cellular redox systems, for example, ascorbate, glutathione, vitamin E, lipoic acid, NADPH, or NADH, is shifted to the oxidized state. Cells cannot tolerate this condition for extended periods and may respond with pathophysiological events such as protein oxidation, DNA damage, and lipid oxidation. Consequently, cells either repair the damage or die by necrosis or by more subtle ways subsumed by the apoptotic pathways. Prooxidants encompass a wide range of molecules, including superoxide radical anion, hydrogen peroxide, alkoxy- and peroxyradicals, and peroxynitrite, often combined under the term reactive oxygen species (ROS) (79). A redox balance is achieved by the activity of generating enzymes such as the mitochondrial electron transport chain NADPH oxidases, peroxisomes, xanthine oxidase, cytochrome P450, lipoxygenases, and cyclooxygenases, as well as scavenger enzymes such as superoxide dismutase, catalase, glutathione peroxidase, thioredoxin, glutaredoxin, and others (78). Oxidative stress, being associated with an irreversible cell/tissue damage and linked to a variety of diseases, has long been acknowledged as a detrimental consequence of (chronic) inflammation. However, the mediators once thought to cause oxidative stress, often followed by an increase of the intracellular concentration of oxidized glutathione (GSSG) or NAD+/NADP+ and thus causing toxicity, are now recognized as critical players in physiological signal transmission systems. Changes elicited by ROS are often fully reversible and more subtle, and often not associated with a generally disturbed cellular redox balance. These signaling events are known as redox signaling or redox regulation. Redox regulation is integral to cellular communication and, like other universal networking systems, controls the physiological cellular responses. It is presumed that most, if not all, of the classical transcriptional events within cells are modulated or critically depended on redox signaling [for references, see (73)]. Here we will summarize the essential components of the redox control system that operates in macrophages to guarantee diverse functions in evoking chemical toxicity and immune-regulatory functions. Formation of ROS, expression and function of the natural antioxidant antidote system, targets of redox signaling, operating macrophage responses, and potential therapeutic opportunities will be addressed. Specifically, we discuss the formation of redox species, their primary targets and actions, redox-controlling mechanisms during macrophage development and/or polarization, transcriptional/translational networks that contribute to inflammation, and its antagonization as well as the impact of lipid mediators in shaping the role of macrophages during the onset, propagation, and resolution of metabolic diseases.
II. Macrophage Development
Macrophages are long-lived innate immune cells of the mononuclear phagocyte system that ubiquitously populate distinct organs and tissues and can be considered as a prototypic immune cell (176). Monocytes have the capacity to differentiate into elicited macrophages within tissues. They develop from hematopoietic stem cells in the bone marrow and migrate to the circulation, from where they are recruited to the periphery (86). However, in vertebrates, macrophages are generated through at least two distinct mechanisms of hematopoiesis. For instance, embryonal macrophages develop in the yolk sac of mice from mesenchymal progenitors from embryonic day 8, without passing through a monocytic stage (12, 281). Due to this developmental difference and/or dependent on environmental cues in local environments, macrophages in adult animals exhibit a varying morphology and function.
A reflection of this diversity can be found in the different names, that is, Kupffer cells, microglia, marginal-zone macrophages, red-pulp macrophages, subcapsular sinus and medullary macrophages, metallophilic macrophages, Langerhans cells, alveolar macrophages, or osteoclasts that have been given to specific tissue macrophage populations. It is important to stress that some investigators question the classification of Langerhans cells (skin-resident phagocytes) and microglia (brain-resident phagocytes) as macrophages. These cells differ from some other macrophage populations with regard to their ontogeny and steady-state renewal conditions as outlined below. However, their major outstanding feature is their dependency on interleukin 34 (IL-34) as opposed to colony-stimulating factor 1 (CSF1), which are both ligands for the CSF1 receptor (316). While CSF1 receptor-deficient mice lack virtually all macrophage populations, CSF1-deficient mice are depleted of various macrophage populations, but not Langerhans cells or microglia. Unchallenged CD34-deficient mice lack Langerhans cells and microglia, but do not show alterations in, for example, alveolar macrophages or Kupffer cells (316). Langerhans cells are often associated with a functionally distinct group of cells within the mononuclear phagocyte system, the dendritic cells (DCs). DCs themselves are divided into conventional DCs (cDCs) and plasmacytoid DCs, of which only cDCs will be discussed here. The defining functional property distinguishing cDCs from macrophages is their superior capacity to initiate adaptive immune responses. Therefore, cDCs in an immature stage take up the extracellular antigen, migrate to the draining lymph nodes, while differentiating to a mature state, where they cross-present the acquired antigens to T cells and finally succumb to apoptosis (109, 337). Whether cDCs can be easily distinguished from macrophages is a matter of debate. Macrophages cross-present antigens to T cells and show migratory potential and the initially proposed cell surface markers for discriminating macrophages from cDCs, such as expression of CD11c or major histocompatibility complex II on cDCs and CSF1 receptor or F4/80 on the macrophage side, thus failed to fulfill their promise (109, 123). However, recent progress in the field allows the discrimination of at least certain cDC subsets from macrophages, although being currently limited to the murine system. These cDCs develop from terminally committed cDC precursors (Fig. 1) in an fms-related tyrosine kinase 3-ligand (Flt3-L)-dependent manner as opposed to the CSF1 receptor ligands and show a higher migratory potential, a higher capacity to cross-present antigens, and a higher susceptibility to undergo cell death (109, 337). Apart from this, a core cDC signature was established, which revealed cDC-specific cell surface markers such as Flt3 and CD26. Interestingly, these markers were not sufficiently coexpressed by certain phagocyte subsets (including Langerhans cells) that were previously regarded as cDCs, thus enlarging the macrophage subset family (193). It will be important to investigate whether these findings can be translated to the human system.

The term macrophage was first introduced by Metchnikoff in 1887 (141). Since then, the origin of tissue macrophages in the adult organism has been an issue of debate. After the introduction of the mononuclear phagocyte system by van Furth, Cohn, and others (303), the notion that macrophages in each tissue develop from monocytes also under steady-state conditions became prevalent, although early conflicting data existed (309). Evidence supporting the mononuclear phagocyte system arose from adoptive transfer studies or administration of clodronate liposomes, showing that monocytes contribute to tissue macrophage pools in the spleen and the intestines (74, 305, 328). Further, mice deficient for factors regulating the development of precursor cells into monocytes and their differentiation into macrophages, such as the transcription factor PU.1 (PU.1−/−) (187, 252), lack tissue macrophage populations. However, recent studies indicate that the origin of macrophages in adult tissues and during steady state versus inflammation is heterogeneous. Langerhans cells and microglia, as well as Kupffer cells, alveolar macrophages, and peritoneal macrophages, proliferate and sustain their presence in vivo independent of the bone marrow or monocytes (252). Further, humans with a combined deficiency in monocytes, DCs, and lymphocytes do not show defects in Langerhans cells and other macrophage populations (14). Bone marrow transplantation studies and in vivo tracing of yolk sac-derived primitive macrophages strongly suggested that the microglia are generated during early hematopoiesis in the yolk sac (90). Finally, two ontogenetically distinct populations of macrophages were identified, based on differential expression of the specific mouse macrophage marker F4/80. Yolk sac-derived macrophages expressed high levels of F4/80, whereas monocyte-derived macrophages expressed low levels of F4/80 (252). Bone marrow transplantation and fate-mapping strategies demonstrated that in adult mice, yolk sac-derived macrophages contribute significantly to the pools of Kupffer cells, Langerhans cells, alveolar macrophages, and macrophages of the pancreas, the spleen, and the kidney, as well as microglia (252). Thus, at least in the mouse, two systems of macrophage development overlap and two ontogenetically distinct macrophage populations persist in adult animals. It will be exciting to determine whether distinct functional properties can be assigned to yolk sac-derived versus bone marrow-derived macrophages and whether targeting one population over the other might be of benefit for therapeutic approaches. There is evidence that monocyte-derived macrophages in the peritoneal cavity are more potent regarding antigen presentation and in generating NO upon activation (89), whereas yolk sac-derived macrophages may show an altered response to hypoxia, evident by high basal expression of the transcription factor hypoxia-inducible factor (HIF)-3α (252). Unfortunately, we might not be yet at the end of the road of macrophage developmental diversity, as even lymphoid progenitor cells retain potential to differentiate into macrophages (142). However, the contribution of lymphocyte progenitors to macrophage pools in vivo remains to be determined. The complexity of macrophage differentiation is summarized in Figure 1.
III. The Distinguished Redox Species: Nitric Oxide and Superoxide Radical Anion
A. Formation of NO and associated species
In the following paragraph, we recapitulate the essentials on the formation of the classical radical products that are produced upon macrophage activation, that is, nitric oxide and superoxide radical anion that account not only for toxicity but also for redox signaling.
The notion that a structurally simple free radical is produced in cells and functions as a messenger molecule initiated a major paradigm shift in discriminating oxidative stress versus redox signaling. Table 1 lists the nitrogen oxides that arise during redox biology of NO by causing its either oxidation or reduction and provides some of the targets that interact with those species.
For more information of potential target interactions, please see the text.
sGC, soluble guanylyl cyclase; COX, cytochrome-c oxidase.
It was Hibbs et al. (115), who first observed NO production by immune cells, that is, macrophages, and reported that arginine was required for the antipathogen and antitumor activity of these cells. The same group connected cytokine-inducible NO production from
The major source of NO is its enzymatic production in a reaction using
The chemical biology of NO can be divided into direct and indirect effects as shown in Figure 2. Direct effects are the reactions that allow NO to directly react with biological target molecules and are reported at concentrations of NO at a low nM range [for references see, (290)].

At low NO, with very limited O2 − being around, nitrosylation and thus activation of soluble guanylyl cyclase (sGC) occur (Fig. 3). sGC is considered the natural NO receptor and the most sensitive direct target, responding to roughly 1–30 nM NO. Next in the sensitivity are other heme and nonheme iron targets such as COX or prolyl hydroxylases (PHD), which, when inhibited, account for the interference with the respiratory chain or stabilization of HIF-1α or HIF-2α. This is achieved at concentrations of roughly 30–100 nM.
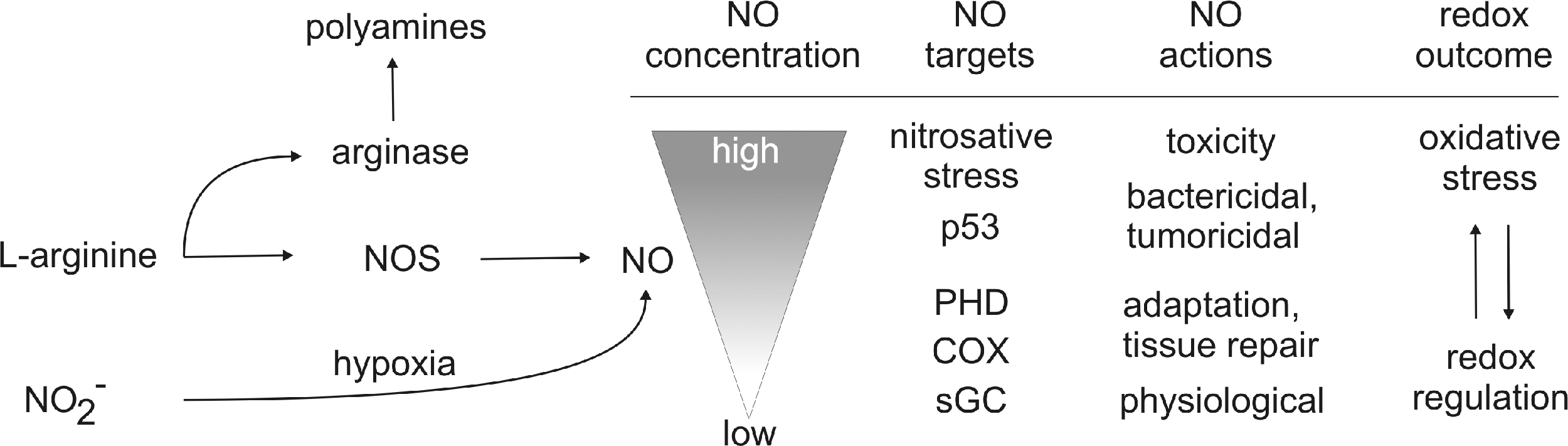
Indirect reactions occur at NO concentrations above roughly 400 nM and require that NO reacts with oxygen or a superoxide radical anion to generate reactive species, which subsequently target biological molecules. Indirect effects are further divided into oxidative and nitrosative stress. During oxidative stress, the oxidation state of a target molecule increases, whereas nitrosative stress implies the formal addition of NO+ (nitrosonium) to a thiol, secondary amine, or hydroxyl group. The formation of N2O3 in the reaction of NO with O2 is a mild oxidant and prefers to nitrosate nucleophiles such as thiols and amines. Peroxynitrite and nitrogen dioxide, which can be formed in the reaction of NO with O2 −, are potent oxidants and may provoke oxidative stress associated with the oxidation and nitration of proteins, lipids, or DNA. The balance between oxidative and nitrosative reactions was found to largely depend on the flux rate of NO formation, as autoxidation of NO first generates NO2, which in the presence of increasing NO concentrations is converted to N2O3 (290). Thus, when NO levels are above those of O2 −, the CO2-OONO− (nitrosoperoxocarbonate) intermediate is converted to NO2 and N2O3, which is accompanied by changes in the redox profile from oxidative to nitrosative reactions (134). Formation of relatively large amounts of NO and O2 − would favor nitration via peroxynitrite. Thus, O2 − generation during NO signaling can regulate the concentration of NO, thereby ablating many of the NO-target interactions. During the interaction of ROS with NO, the dominant outcome of the reaction is scavenging NO by its simple conversion to nitrite/nitrate. When more NO than O2 − is produced, NO2 and N2O3 are formed (Fig. 2). Conversely, when O2 − is in excess, oxidative and nitrosative stress is quenched, suggesting that the direct ONOO− chemistry has a limited role, although N2O3 formed under these conditions can oxidize, nitrate, and nitrosate proteins. Apparently, the reaction of NO with O2 − consumes most of the NO and produces the intermediates associated with indirect signaling effects. This scenario lowers NO concentrations, while creating potentially new pathways of signal transmission. During the course of macrophage activation, the flux of these radicals constantly changes, which makes prediction on a redox-regulated reaction extremely difficult and indeed limits more generalized conclusions. During in vitro macrophage activation with lipopolysaccharide (LPS)/cytokine, the early O2 − formation normally is separated from the later NO formation (226, 248). The temporal changes in radical formation in microcompartments where these radical meet will then affect the resulting signal quality, which at present is not fully accessible due to methodological limitations. It should also be mentioned that NO can ablate the oxidation chemistry mediated by H2O2 or ROS (330). The attenuation of metal/peroxide oxidative chemistry, as well as lipid peroxidation, appears as a rational explanation by which NO may limit oxidative injury to cells.
With NO increasing to ∼400 nM or higher, the tumor suppressor p53 is stabilized, and concentrations are reached, where NO can induce apoptosis, become cytostatic as well as cytotoxic (Fig. 3).
Clearly, this marks the transition to indirect effects, but can be causatively linked to iNOS activation. NO concentrations at which S-nitrosation occur are not well defined, but overlap with the iNOS activity as well. S-nitrosation has been shown to regulate a broad spectrum of cell functions during the transport, storage, and delivery of NO in cell signaling and inflammation (265).
Another important regulatory feature of NO formation is the availability of oxygen. The concentration of oxygen at which the activity of the NOS is half-maximal, that is, the KM, is considerably different for the three isoforms. With oxygen around the KM or lower, NO formation is a direct result of the oxygen concentration. The KM for iNOS is 135 μM, making NO production from iNOS under hypoxia extremely inefficient (274). Importantly, iNOS can become a source of superoxide radical anion/H2O2 in the absence of arginine, thus contributing to the NO/O2
− balance. Moreover,
B. Formation of superoxide radical anion and associated species
Besides NO, macrophages are known for their capacity to generate ROS, which comprise superoxide radical anion, H2O2, and other species (161). Activation of the NADPH oxidase (NOX) and Dual-oxidase (DUOX) family of enzymes are the primary source of superoxide radical anion/H2O2. The ROS-generating family members comprise NOX1 to NOX5 as well as DUOX1 and DUOX2. The respiratory burst observed in macrophages is predominantly attributable to NOX2, which generates large amounts of ROS in the phagosome as an innate immune process to kill the invading pathogens. ROS formation in macrophages requires the assembly of a large aggregate, composed of a membrane-bound cytochrome b558 complex, which contains gp91phox (now known as NOX2) and p22phox catalytic subunits and four cytosolic proteins, that is, p47phox, p67phox, p40phox, as well as the GTPase Rac1 (166). Activation of NOX2 is achieved by immunogens interacting with the macrophage Fc-receptors, with the notion that priming agents such as IFNγ or IL-1β significantly increase the amount of superoxide radical anion formation. Bacterial killing by NOX2 is compartmentalized to the phagosome, which may limit collateral damage by superoxide radical anion production inside this organelle. Constitutive and low-level superoxide radical anion and/or H2O2 production is primarily linked to NOX4 and DUOX activation (169). As iNOS-generated NO is freely diffusible, the level of NO inside the phagosome depends on the NOS activity. Likely, NO and O2 − will combine to increase peroxynitrite, which under acidic conditions provides nitrosative stress. Thus, in the phagosome of macrophages, redox reactions, initiated by the formation of NO and O2 −, work in concert to kill pathogens.
IV. Macrophage Plasticity in the Light of Redox Biology
Metchnikoff described phagocytes as cells that eat (phagocytose), digest, and feed other cells, coupled by a remarkable migratory capacity. He designated these functional properties as a prerequisite to cope with physiological inflammation, supporting development and harmonizing the integrity of the organism. The term pathological inflammation comprised the recognition and phagocytosis of nonself and unwanted self to protect against injurious agents (288). The defense function has been the focus in macrophage research for a long time, while the metabolic function (e.g., secretion of trophic factors), which is closely coupled to regulating tissue development and repair, became a focus of intense research recently. Details concerning the roles macrophages play under physiological and pathological inflammation have been reviewed elsewhere (204, 228, 267). Briefly, macrophages sense cellular debris and pathogens, phagocytose them, and subsequently may present the acquired antigens to adaptive immunity, although with a limited efficiency as compared to professional antigen-presenting cells such as DCs. After contact with pathogens, macrophages produce a wide array of inflammatory mediators that determine immune responses.
The local tissue environment dictates specific tasks performed by macrophages. However, although macrophages are well adapted to the needs of their present locality, they retain a remarkable functional plasticity, enabling them to respond to the disturbances in tissue homeostasis. Macrophage activation patterns in a skin wound may serve as an initial illustrating example. Macrophages are rapidly recruited to the sites of tissue damage, as was recognized by Metchnikoff in one of his key experiments, when he introduced a splinter into a starfish larva. Interestingly, a key molecule for rapid macrophage recruitment to wounds might be H2O2, which is produced by the wound epithelium in zebrafish larvae through DUOX (212). In a similar model system, H2O2 was required to recruit phagocytes to early-transformed cells (68). The connection between an evolutionary-conserved function (phagocyte recruitment) and an evolutionary stable simple molecule is certainly charming, but the relative impact of H2O2 compared to other phagocyte chemoattractants as well as its function in higher vertebrates [where recruitment of monocytes as compared to resident macrophages prevails (204)] remains to be proven. Once macrophages reach the site of injury, they face pathogen-associated molecular patters (PAMPs) or intracellular constituents released by dead cells known as alarmins or damage-associated molecular patterns (DAMPs) (13), to which they respond utilizing similar sensing mechanisms by inducing an inflammatory reaction. This inflammatory reaction, which again involves the generation of NO and/or ROS, serves to clear the site of tissue injury from harmful agents. In a next step, macrophages switch their phenotype to promote repair and resolve the inflammatory response (5, 167). Accordingly, macrophage depletion shortly after introducing skin wounds in mice resulted in delayed re-epithelization and angiogenesis, as well as prolonged inflammation (177). Resolution of inflammation likely requires macrophage interaction with apoptotic resident cells (5, 128), leading to secretion of anti-inflammatory mediators such as TGFβ or IL-10 and modulation of ROS and NO production, as outlined in more detail below. Finally, to restore the tissue to its original state, newly recruited macrophages leave the wounded site mainly by emigrating to the lymphatic system, again underscoring the high motility of these cells (256).
It has to be noted that there is still uncertainty in the literature whether macrophages readily switch phenotypes in vivo or whether inflammatory versus healing or regulatory macrophages are derived from different monocytic precursors (86). At least two monocyte subpopulations are known in mouse and human blood, which are distinguished based on the expression of different cell surface markers and migratory patterns (85, 342). In the murine system, inflammatory monocytes are Ly6C+ CCR2+ and CX3CR1−. These cells are short-lived and upon infection are rapidly recruited from the bone marrow to the site of inflammation in response to the CCR2 ligand CCL2, where they differentiate to inflammatory macrophages or monocyte-derived DCs (85, 86, 94). Resident Ly6C− CCR2− and CX3CR1+ monocytes are long-lived cells that initially may not migrate to the sites of infection due to the absence of CCR2 expression. However, they may be recruited at later stages [e.g., when the CX3CR1 ligand fractalkine is produced from resident macrophages due to interaction with dying cells (99)], when tissue regeneration is required, and when they differentiate preferentially to macrophages with an anti-inflammatory phenotype. Under steady-state conditions, these monocytes are thought to replenish the tissue-resident macrophage populations (86, 94). The notion that Ly6C+ and Ly6C− monocytes develop preferentially to inflammatory or anti-inflammatory macrophages, respectively, may simply reflect the impact of the current microenvironment at the time of their recruitment to the inflammatory site, as both subsets can be activated to support inflammation as well as its resolution and repair. Nevertheless, selective depletion of Ly6C− monocytes due to genetic ablation of NR4A1 enhanced the development of atherosclerosis, which was attributed to enhanced generation of inflammatory macrophages (104). Thus, the intrinsic potential of the monocyte subpopulations may influence the resulting macrophage phenotype, although further studies are required to support this conclusion and to evaluate the model in the human system.
Despite the evidence summarized above, macrophages retain plasticity after differentiation from distinct monocyte subsets and switch the phenotypes readily at least in vitro, which provoked a considerable effort to classify macrophage activation states systematically, as summarized in Figure 4. Acquisition of different phenotypes is referred to as macrophage polarization (182).
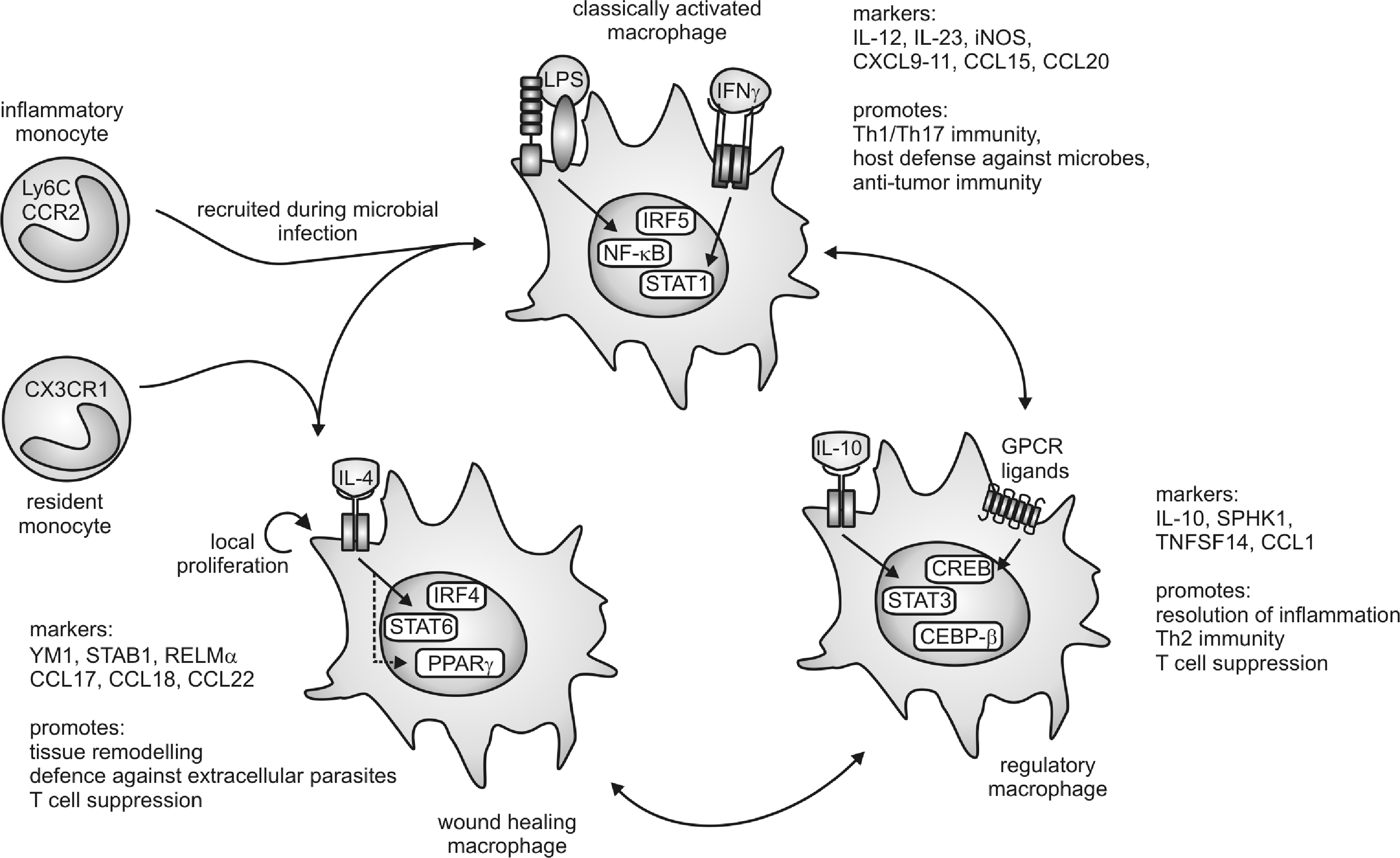
Initially, proinflammatory macrophages (now termed classically activated macrophages) were described as the main responders to infection (178). Classical macrophage activation requires priming by IFNγ (50), followed by an appropriate stimulus indicating infection or danger, such as the microbial cell wall component LPS, certain IL-1 family proteins, or TNFα. Downstream of these effectors, transcription factors such as nuclear factor-kappa light-chain-enhancer of activated-B cells (NF-κB), signal transducer and activator of transcription 1 (STAT1), as well as interferon regulatory factor 5 (IRF 5) are activated to induce inflammatory gene transcription (163). Classically activated macrophages produce a wide range of proinflammatory cytokines such as TNFα, IL-1β, IL-12, IL-23, and IL-6, as well as inflammatory chemokines, and are believed to show a higher capacity to present antigens (199). The latter might indeed reflect their origin from inflammatory monocytes, as discussed above. Overall, classical activation endows macrophages with the potential to recruit inflammatory cells and to support Th1/Th17 immunity (83). Thus, their critical function is defense against invading microbes, for which they are further endowed with a high bactericidal capacity through their production of high levels of NO and ROS (302). Similar mechanisms might be responsible for the antitumor potential of classically activated macrophages.
Macrophages exhibiting tissue repair properties, also termed alternatively activated macrophages, were first described in the 1990s (268). These cells arise from IL-4 receptor activation by IL-4 or IL-13 and show attenuated production of proinflammatory cytokines, ROS, and NO, the latter due to upregulation of arginase. Further, alternatively activated macrophages secrete immunosuppressive cytokines such as IL-10 or IL-1ra (IL-1 receptor antagonist) and chemokines such as CCL17, CCL18, and CCL22 to attract anti-inflammatory leukocytes, upregulate phagocytic receptors, and produce extracellular matrix (ECM) components and growth factors such as fibroblast growth factors, vascular endothelial growth factor (VEGF), and others (83, 93, 199). These mediator profiles enable alternatively activated macrophages to limit inflammation, to produce the ECM, to induce angiogenesis, but also to combat extracellular parasite infection (83, 93, 199). Transcription factors involved in shaping the wound-healing macrophage phenotype are STAT6, IRF4, and peroxisome proliferator-activated receptor (PPAR)-γ (163). An interesting feature of the wound-healing macrophages is their potential to proliferate in vivo upon induction of Th2 inflammation (129). This might be required to enhance their numbers for effective killing of extracellular parasites in the absence of inflammatory monocyte recruitment. Mechanistic explanations how IL-4 triggers macrophage proliferation await their identification.
In an analogy to the T-helper-cell nomenclature, where Th1 cells are associated with the response against bacteria or viruses and Th2 cells are associated with the response to parasitic infection and with limiting Th1-associated immunity, classically activated opposed to alternatively activated macrophages were denoted as M1 and M2 macrophages, respectively (182, 195). However, after the identification of alternatively activated macrophages, several other functional macrophage phenotypes were described, mostly generated in vitro by stimulation with immunomodulatory agents such as glucocorticoids, immune complexes, immunosuppressive cytokines such as IL-10, as well as various G protein-coupled receptor ligands or dying cells (93, 199). These stimuli generate macrophages that regulate immunity and tissue homeostasis in discrete, but also sometimes overlapping, ways. Common markers and transcription factors involved in shaping the phenotype of regulatory macrophage populations are therefore naturally diverse. However, CCAAT/enhancer-binding protein (CEBP)-β, cAMP-responsive element-binding protein (CREB), and STAT3 emerge as critical transcriptional regulators of these cells (81, 163), and the production of high levels of IL-10 as well as upregulation of sphingosine kinase 1 and TNF superfamily member 14 are associated with regulatory macrophage polarization (199).
To account for the resulting complexity, especially with regard to the notion that different stimuli provoke similar macrophage activation patterns, a classification focusing on the physiological outcome, that is, the functional properties of activated macrophages, has been proposed (199). Macrophages were separated into three functional states analogous to the three primary colors in a color wheel, allowing the occurrence of hybrid-type macrophages, analogous to secondary colors in the color spectrum, and thus an infinite number of functional macrophage subtypes. The defined functional states comprise classically activated (M1), wound-healing (alternative-activated or M2), and regulatory macrophages (Fig. 4) (199). We will follow this model in the present review, as macrophage subtypes will be described that do not fit into the M1/M2 nomenclature. A prominent example of hybrid-type macrophages are tumor-associated macrophages (TAMs) that exhibit healing and tissue remodeling properties that produce inflammatory mediators (IL-6 and IL-23), but also negatively regulate other tumor-associated immune cell populations through IL-10 (230), thus combining the functional properties of all three primary macrophage populations. It is therefore not surprising that transcription factors such as NF-κB and STAT1 (classically activated macrophages), PPARγ (wound-healing macrophages), as well as STAT3 and CREB (regulatory macrophages) are connected to the TAM phenotype (319).
There are still important open tasks/questions with regard to macrophage plasticity. A major drawback is the lack of definite markers to identify functional macrophage subpopulations (markers, which are unambiguously associated with a specific function) in tissues. This would allow predicting individual macrophage function, for example, in tissue sections. Recent transcriptome studies resulted in the identification of gene signatures for murine macrophage subpopulations (204). However, mRNA expression at a certain timepoint (mostly in vitro) does not necessarily correspond to stable expression of the gene product in a tissue. Further, gene profiles derived from mouse macrophages are difficult to transfer to human cells, as, for instance, classically activated human macrophages may not express iNOS, whereas alternatively activated human macrophages do not express arginase, which are put forward as prototypic markers for the respective macrophage populations in mice. Techniques such as membrane proteomics of macrophages sorted from tissues in a discrete pathological state might result in the identification of the elusive phenotype markers.
Importantly, the impact of redox modification of signaling or structural proteins on macrophage function is largely unexplored, although data with regard to classical activation are available. The role of ROS (H2O2, HOCl, O2 −, and reactive nitrogen species) in NF-κB activation in different cell types is controversially discussed, owing primarily to the unspecific effects of antioxidants (butylated hydroxyanisole, glutathione (GSH), and N-acetyl-cysteine) used in respective studies (92). However, it seems clear that ROS are involved in the activation process of classically activated macrophages. Downstream of signaling through the LPS receptor complex consisting of Toll-like receptor (TLR)4, CD14, and NOX4-dependent ROS production was involved in the activation of NF-κB as well as p38 mitogen-activated protein kinase (MAPK), which is required for LPS-induced activator protein 1 (AP-1)-dependent transcription (92). Further, mitochondrial ROS seem to foster inflammasome activation as outlined below. Various redox-sensitive proteins are involved in the signaling cascades triggered by inflammatory mediators (75). It will therefore be a challenging task to elucidate in detail how redox reactions regulate polarization of macrophages into different functional subpopulations. Of specific interest will be to determine whether ROS generated upon classical activation interfere with priming macrophages to become tolerant or activated toward an anti-inflammatory or regulatory macrophage phenotype. For instance, the induction of LPS tolerance in macrophages requires chromatin remodeling (76), which is a redox-sensitive process (232). Further, human individuals with chronic granulomatous disease (CGD), having a genetic defect in ROS production, show hyperinflammatory responses (26). At this point, it is interesting to note that classically activated versus wound-healing macrophages were also denoted as reductive or oxidative, respectively, due to differences in glutathione levels (203).
V. Redox Systems Modulate and Determine the Macrophage Phagocyte Function
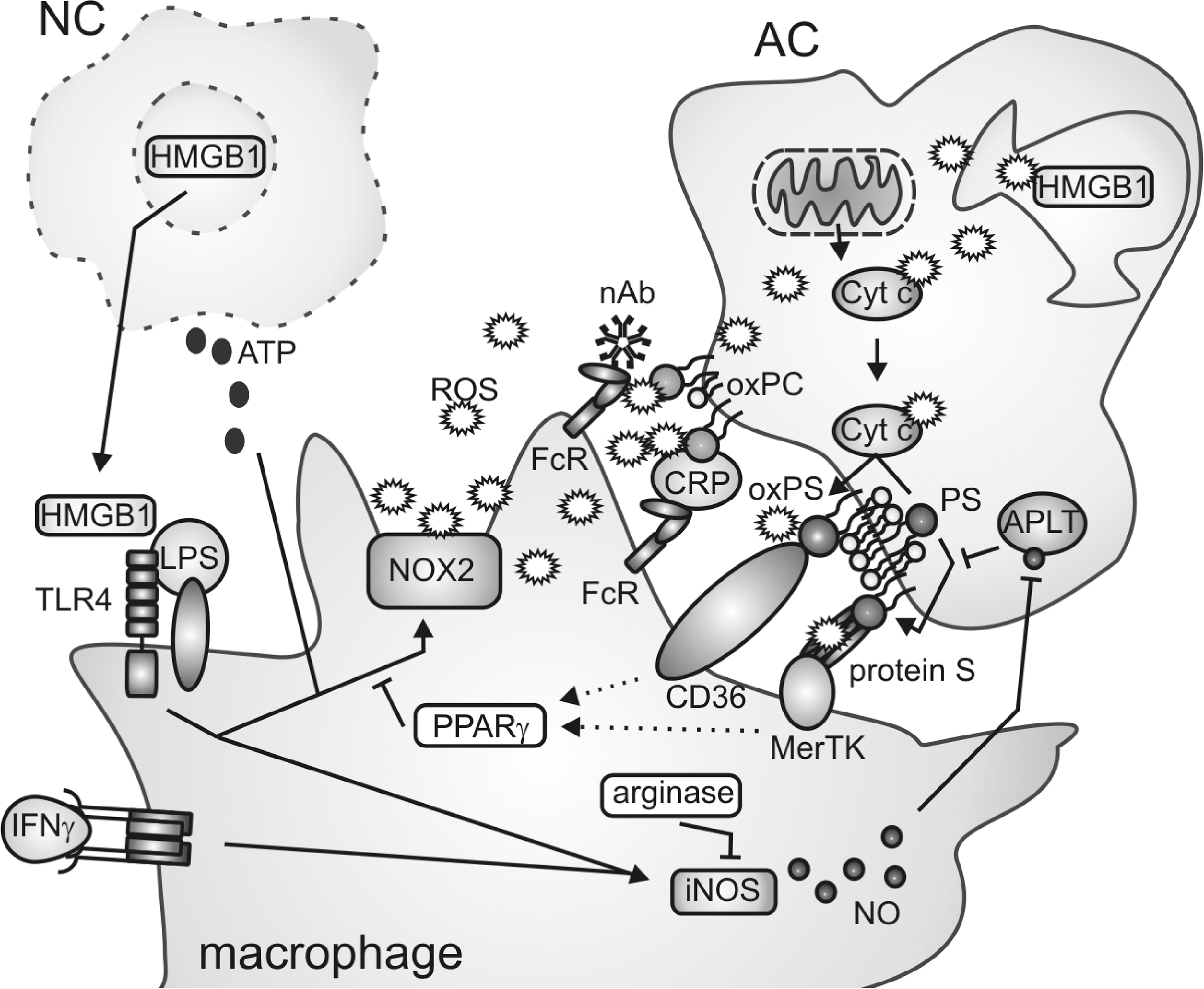
In the following paragraph, we will concentrate on the interaction of macrophages with pathogens and dying cells and summarize, as schematically outlined in Figure 5, the mechanisms how this interaction is controlled by and how it controls redox signals.
A. Redox signaling in macrophages upon phagocytosis of pathogens
As outlined, upon pathogen recognition, macrophages assemble NOX2 to produce O2
−, its dismutated product H2O2 as well as OH
Macrophages rely primarily on the NOX2 complex to produce ROS (26), as opposed to neutrophils, which possess myeloperoxidase to generate highly bactericidal HOCl from H2O2 (64). Thus, the oxidative burst mounted by macrophages can be viewed as less intense compared to neutrophils. This may be one reason why classically activated macrophages, as opposed to monocytes or neutrophils (10), do not themselves succumb to oxidative stress. However, two additional mechanisms are discussed to account for this phenomenon. First, monocyte-to-macrophage differentiation results in increased expression of DNA repair proteins (10) as well as molecules involved in free radical scavenging (158). Second, classical activation upregulates antioxidant enzymes downstream of NO generation (65) or NF-κB activation (92), or directly activates DNA damage-activated kinases (321). Therefore, macrophages seem to be uniquely equipped to survive oxidative stress resulting from endogenous production of free radicals. This might be a biological necessity enabling them to survive classical activation and to persist at the sites of infection to re-establish tissue homeostasis upon switching their functional program.
NO also participates in pathogen killing by macrophages through direct generation of ROS (318). However, recently, another function of NO in conveying the immunological outcome of pathogen phagocytosis was suggested. NO was shown to delay fusion of phagosomes with lysosomes to form a functional phagolysosome, which might serve to enhance the time for processing and presentation of antigens (40). This might be of considerable importance for macrophage function, since macrophages present antigens less effectively as compared to DCs, which was attributed to rapid generation of phagolysosomes and thus inefficient processing of potential antigens (109).
B. Redox reactions in dying cells modify their uptake by macrophages
Besides extracellular pathogens, macrophages are professional scavengers of endogenous dying cells. Although a variety of different cell death modalities are known, we will discuss only the interaction of phagocytes with apoptotic and necrotic cells. Phagocytosis of dying cells is a three-step process, requiring the secretion of find-me signals or alarmins by dying cells to attract and preactivate phagocytes, the exposure of eat-me or the degradation/masking of do-not-eat-me signals by dying cells to ensure specific recognition, and finally the uptake process itself (229, 344). Redox processes at each level (Fig. 5) intricately regulate the interaction of the phagocyte with the dying cell, which will be discussed next.
Oxidation of lipids and proteins regulates specific recognition of apoptotic cells by macrophages. After the finding that oxidized low-density lipoprotein (oxLDL) competes with apoptotic cells for binding to phagocytes, it was demonstrated that monoclonal antibodies recognizing oxidation-specific epitopes on oxLDL (primarily oxidized phospholipids such as the phosphatidylcholine [PC] species) bind to apoptotic cells and inhibit their uptake by peritoneal mouse macrophages (31). Additionally, oxidized PC (oxPC) and related species were identified on the surface of early- or late-apoptotic cells, being specifically recognized by C-reactive protein, which is bound by macrophage Fcγ receptors and facilitates apoptotic cell clearance (32). Strikingly, 30% of the population of natural antibodies, which are conserved during natural selection, also bind specifically to oxidation-specific epitopes and facilitate the uptake of apoptotic cells. Thus, a primary function of natural antibodies may be ensuring apoptotic cell phagocytosis through recognition of oxidized lipid epitopes (35). Besides oxPC, oxidation of phosphatidylserine (PS) occurred during apoptosis (139). PS is confined to the inner leaflet of the plasma membrane in live cells due to the action of aminophospholipid translocase, which is inhibited during apoptosis, resulting in PS exposure. PS is then recognized by a multitude of phagocytic receptors (229). Cytochrome c, whose release from the mitochondria is a critical step for the execution of apoptosis, was suggested to directly facilitate PS oxidation (139). Oxidized PS (oxPS) on the surface of apoptotic cells as well as living cells facilitated their uptake by macrophages. Further, apoptotic stimuli such as etoposide that prevent PS oxidization attenuated PS externalization as well as apoptotic cell uptake by macrophages (298). The receptors recognizing oxidized epitopes such as oxPS on the surface of apoptotic cells may be diverse, in analogy to the multitude of receptors that recognize oxidized epitopes of oxLDL or PS in general. However, the major importance was attributed to the scavenger receptor CD36. CD36 was essential for apoptotic cell clearance by macrophages in vivo, recognizing different oxPS species and, to a lesser extent, oxPC (97). Redox reactions were further connected to the recognition of nonoxidized PS. Binding of the blood anticoagulant factor protein S to PS on apoptotic cells triggered auto-oxidation of cysteine residues in protein S and the formation of oligomers, which are ligands for the phagocytic receptor Mer tyrosine kinase on macrophages (300).
C. Redox signals in macrophages modify apoptotic cell uptake
Redox reactions in phagocytosing macrophages also influence apoptotic cell clearance. Classically activated macrophage-derived NO contributed to PS externalization in dying cells by S-nitrosation-dependent inhibition of aminophospholipid translocase (297). Further, primary macrophages from patients with CGD, lacking functional NOX2, were defective for apoptotic cell engulfment (247). These results were confirmed in NOX2-deficient murine macrophages, which interestingly showed a higher capacity for phagocytosis of bacteria (22). NOX2-dependent generation of extracellular free radicals was required for apoptotic cell uptake, suggesting a role for oxidative modifications on the phagocyte or apoptotic cell plasma membrane structures (22). Thus, prominent functions of classically activated macrophages are required for apoptotic cell recognition and clearance. This was surprisingly also true for macrophages polarized to a wound-healing phenotype (22). Clearance of apoptotic cells by macrophages however is a noninflammatory event also with respect to avoiding presentation of self-antigens. In a mouse peritonitis model, uptake of apoptotic cells was confined to a population of resident macrophages expressing 12/15 lipoxygenase (299), which do not express a highly functional antigen presentation machinery as opposed to monocyte-derived macrophages (252). Interestingly, resident macrophages blocked the uptake of apoptotic cells by newly recruited monocytes and thus, likely their progeny, through 12/15 lipoxygenase-dependent oxidation of phosphatidylethanolamine (PE) species on apoptotic cells. Oxidized PE (oxPE) species were suggested as a sink for apoptotic cell recognition by inflammatory monocytes/macrophages that rely, other than resident macrophages, on certain PS-binding bridging molecules such as milk fat globule-EGF factor that also bind to oxPE. Depletion of 12/15 lipoxygenase resulted in apoptotic cell uptake by inflammatory nonresident cells, subsequent antigen presentation, and lupus-like autoimmunity (299). Together, there is strong evidence that redox reactions during apoptosis or upon apoptotic cell–phagocyte interactions secure specific apoptotic cell recognition by macrophages and avoid inflammatory reactions resulting from secondary necrosis, which, as outlined below, would induce autoimmunity. It will be interesting to determine if redox modifications of intracellular epitopes in apoptotic cells modify the phagocytosis program or the metabolism of cell corpses on the macrophage level and how this may interfere with antigen presentation to induce tolerance or immunity. As outlined in this paragraph, protein nitrosation versus lipid oxidation may result in either enhanced or diminished antigen presentation.
D. Redox reactions determine the outcome of cell death
It is the general notion that the cells undergoing necrosis enhance immune responses, whereas apoptotic cells inhibit immunity. Necrosis can be regarded as a signal for overshooting tissue damage, which requires an inflammatory reaction for its clearance. Thus, as described above, necrotic cell-derived DAMPs serve a similar purpose as PAMPs.
DAMPs comprise molecules that are normally confined to intracellular structures, such as high-mobility group box 1 (HMGB1). HMGB1 is a nuclear protein that is released to the extracellular space after membrane rupture (344). HMGB1 forms complexes with various inflammatory mediators, whereupon it interacts with, for example, TLRs to trigger potent inflammatory signaling in macrophages (13). Interestingly, HMGB1 can be oxidized by mitochondrion-dependent ROS during apoptosis resulting in its inactivation, which possibly enables the discrimination of primary versus secondary necrosis (143). The importance of HMGB1 in determining the outcome of cell death is illustrated by the observations that triggers of immunogenic apoptosis such as certain chemotherapeutic agents induce release of HMGB1 in its active form (4). This supports the notion that differences in redox reactions shape the immunological outcome of cell death. Other redox-sensitive proteins released from necrotic cells (predominantly neutrophils) are the calgranulins S100A8, S100A9, and S100A12. Calgranulins initially function as potent antioxidants, thus protecting the tissue from oxidative stress, whereupon they undergo multiple oxidative modifications. These modifications contribute to generating multimeric calgranulin complexes that are recognized among others by TLR4 to induce activation of NOX2 in macrophages (175). Moreover, necrotic cell-derived adenosine triphosphate (ATP) serves as a potent trigger/enhancer of NOX2 activity in macrophages, leading to rapid cellular oxidation (196). However, the consequences of cellular oxidation with regard to macrophage polarization are unclear.
As opposed to necrotic cells, which mainly activate defensive redox systems in macrophages, the interaction with apoptotic cells rather modifies the property of phagocytes to trigger the production of ROS and/or reactive nitrogen species. Formation of ROS in macrophages was inhibited by apoptotic cells as well as oxPS-enriched viable cells (257). We established a mechanistic link showing that macrophage PPARγ was activated upon the interaction with apoptotic cells and interfered with NOX2 assembly (132, 311). The impact of apoptotic cell recognition on macrophage NO production is more diverse. While iNOS was induced by the interaction with apoptotic cells in unprimed macrophages, which was accompanied by NO production, apoptotic cells suppressed iNOS expression in classically activated macrophages or did not alter iNOS expression in IFNγ-stimulated macrophages (152). On the other hand, apoptotic cells provoked arginase I upregulation or arginase II expression in macrophages, thus limiting, but probably not completely abrogating, NO production (9). It is important to stress that different concentrations of NO regulate inflammatory signaling in a contradictory manner. While a high local NO production during inflammation is nonselectively toxic to bystander cells, comparatively lower levels of NO may specifically target lymphocytes, and low intracellular amounts of NO enhance inflammatory NF-κB signaling (152).
In conclusion, the impact of the redox biology on the macrophage phagocyte function is determined by the level of oxidation and localization of the redox signal, which agrees with the observations that molecules such as H2O2 or NO may account for antithetic functional outcomes. Therefore, it will be critical to develop reliable localization (organelle)-specific probes, allowing measuring quantitative changes in free radical generation in live cells.
VI. Redox Systems and Regulatory Macrophage Function
As discussed in the previous chapter, apoptotic cell interactions limit the production of ROS and NO by macrophages. Apoptotic cell-stimulated macrophages can be regarded as regulatory macrophages, and there is more information in the literature, indicating that the functional properties of regulatory macrophages are determined by their limited, but not absent, potential to produce ROS and NO. The clearance of apoptotic neutrophils as well as the stimulation with LPS and immune complexes generated regulatory macrophages that produced NO and primed Th2 responses (63, 69). The glucocorticoid dexamethasone increased ROS production in alternatively activated macrophages, thereby inducing immunosuppressive regulatory T-cells (157). Along this line, macrophages from patients with CGD, lacking functional NOX2, showed hampered regulatory T-cell induction and thus reduced T-cell suppression (156). Interestingly, tumors contain a population of immunoregulatory myeloid cells, myeloid-derived suppressor cells (MDSCs), which express iNOS and arginase simultaneously, thereby producing low NO levels, and also generate ROS. These macrophage-related cells, which can differentiate into TAMs, inhibit antitumor T-cells and/or generate regulatory T-cells (80). It is tempting to speculate that apoptotic cells may induce a similar response in macrophages at sites of tissue injury and inflammation, such as in tumors, to limit adaptive immune responses. Besides NOS and NOX, another enzyme completes the redox arsenal of regulatory macrophages that aims to limit T-cell responses. Indoleamine 2,3-dioxygenase, an intracellular heme enzyme that is involved in tryptophan catabolism, is expressed by apoptotic cell-stimulated (233, 322) and other regulatory macrophages (21). Depletion of tryptophan, the least-abundant essential amino acid, blocks effector T-cell proliferation and enhances the suppressive activity of regulatory T-cells (7). These data indicate that an essential feature of regulatory macrophages, the modulation of T cell responses, is shaped by redox processes. As studying the regulatory macrophage biology progresses, new redox-dependent functions of this therapeutically highly interesting cell population are likely to emerge (21, 72).
VII. Targets of the NO/O2 − Redox Biology in Macrophage Function
The NO/ROS redox biology not only helps to eliminate pathogens or to kill tumor cells but also shapes other aspects of macrophage biology. Here we describe some of the established NO targets in macrophages and discuss potential functional consequences.
A. sGC and cGMP formation
The highest affinity interactions of NO with biological targets in macrophages are those with metalloproteins such as sGC or COX (see Fig. 3). sGC forms a 5-coordinated Fe2+-NO complex to activate the enzyme with the concomitant production of cyclic guanosine monophosphate (cGMP). Generally, it appears that cGMP in macrophages is linked to cell-protective mechanisms. A cGMP increase in macrophages protects against NO-, ONOO−-, or oxLDL-induced toxicity (112), attenuates proinflammatory cytokine production in LPS-stimulated macrophages (147), reduces high-fat-evoked proinflammatory cytokine production as well as the recruitment of macrophages into adipose tissue (103), and limits Kupffer cell activation during high-fat feeding (287). In addition, cGMP formation activates latent TGF-β1 to enhance wound healing (192). Generally, a cGMP increase in macrophages favors survival, limits cell destruction, and attenuates inflammatory responses. Unfortunately, these studies remain descriptive and fail to provide molecular insights of how cGMP evokes these responses. Even when assuming that phosphorylation by cGMP-dependent kinases is involved, we lack identification of distinct targets. Moreover, we have only limited knowledge on how NOS activation in macrophages signals via cGMP in an autocrine or paracrine manner to affect inflammatory conditions associated with iNOS activation. In response to LPS/cytokine stimulation, one might assume activation of NOX enzymes with the concomitant formation of O2 −, which would trap low NO levels and thus restrict cGMP formation. With time progressing, the NOX activity declines, whereas iNOS expression increases. Now, with an excess of NO, over O2 − activation of sGC may occur. Based on the scattered information summarized above, one would speculate that any cGMP increase in macrophages should dampen their proinflammatory, that is, classical activation, eventually contributing to tolerance development. Easier to understand at the molecular level are those protective/anti-inflammatory mechanisms attributed to NO that do not necessarily require cGMP formation. As discussed later, these mechanisms may comprise activation of PPARγ, which antagonizes NOX2 assembly/activation (310) and attenuates the prototype inflammatory transcription factor NF-κB, or activation of the nuclear erythroid-2-p45-related factor 2 (Nrf2) antioxidant-response system.
B. Redox signals and transcriptional/translational regulation
As innate immune cells, macrophages are designed to respond to incoming signals to coordinate inflammatory as well as resolving mediators. It has long been appreciated that NO regulates gene expression (17) with the notion that potential targets comprise transcription factors, upstream signaling cascades, mRNA stability, translation, as well as processing of the primary gene products. Studies using human macrophages exposed to NO donors for times from 3 to 24 h identified specific categories of NO-sensitive genes (295, 296). Candidate genes comprise transcription factors, cell cycle regulators, components of mitochondrial oxidative phosphorylation, and many more, with a number of proinflammatory genes among them. To circumvent the problems arising from mRNA stability, gene arrays have also been performed by looking at newly transcribed (nascent) mRNAs by nuclear run on, followed by a microarray analysis (124). Experimental conditions favoring an increase in nitrosating species (N2O3) showed that nitrosation chemistry is a primary driving force for the expression of many genes, including proinflammatory ones, that is, TNFα (295). Despite the useful information on gene regulatory features, uncertainties remain how gene regulation is facilitated at the molecular level, that is, which targets are affected, and which of the NO redox-related species are involved.
It is proposed that the post-translational protein modification by S-nitrosation conveys an ubiquitous mechanism of how NO affects signal transduction in eukaryotic cells (114). Although the list of potential targets that become S-nitrosated still increases and the feature of a dynamic regulation by nitrosation/denitrosation emerges, we still lack the proofs for a general relevance of this regulatory concept in macrophage biology. This is evident for two classical targets known to be S-nitrosated, that is, NF-κB and caspases. NF-κB serves as a critical element in immune and inflammatory responses with central roles in acute and chronic immune dysfunctions. Its activation contributes to iNOS expression, while the impact of NO on NF-κB is not uniform. A burst of NO augments NF-κB in inflammatory macrophages, whereas long-lasting NO exposure rather attenuates its activity [references in (329)]. NF-κB subunits p50 and p65 are subjected to S-nitrosation, with iNOS-derived NO contributing to this post-translational modification, while at the same time, the amount of S-nitrosated p65 is depleted in inflammatory-stimulated cells (145). If NO formation in macrophages indeed contributes to deactivate NF-κB via S-nitrosation to counteract classical macrophage activation, it remains to be determined to which extent this limits the classical macrophage responses. With an inflammatory response progressing, rather than limiting NF-κB by S-nitrosation, other mechanisms such as Nrf2 activation or NF-κB p50/p50 dimer formation upon a macrophage phenotype shift may become equally or even more important (341). NFκB is also modulated by ROS, with the notion that low-level H2O2 enhances, while high H2O2 suppresses its activity. Moreover, oxidation of inhibitor of NF-κB (IκB) attenuates its degradation to limit NF-κB activation [references in (329)]. For macrophages, a unifying concept may predict NF-κB activation at low-level NO/ROS and its inhibition at higher radical production rates. The precise role of S-nitrosation of NF-κB subunits or redox-regulated upstream targets in achieving significant shifts in macrophage polarization upon endogenous iNOS or NOX isoform activation awaits further clarification.
The limitations to generalized predictions on the physiological significance of S-nitrosation in macrophages are reflected at the level of caspases. S-nitrosation of caspases is well documented and reported to attenuate apoptotic responses under the impact of NO. However, in macrophages exposed to various NO donors or after activation of iNOS, apoptotic cell death occurs, a process accompanied by activation of the tumor suppressor p53 and caspase-3 [for references, see (23)]. Thus, in macrophages the endogenous formation of NO per se elicits a proinflammatory response and may initiate the pathways of cell demise. However, NO also activates protective mechanisms aimed at limiting cell destruction. Interestingly enough, for hypoxic mitochondria, it has been proposed that electrons from the respiratory chain reduce nitrite to NO to compete with oxygen at the COX, which allows O2 − formation and subsequent S-nitrosation as well as inhibition of mitochondrial Krebs-cycle enzymes (301). Upon reoxygenation, electrons from glycerol-3-phosphate selectively reduce the ubiquinone pool to generate O2 −, which at the mitochondrial matrix could provoke one-electron reduction and reactivation of S-nitrosated proteins. The chemistry of S-denitrosation has been investigated when nitrosoglutathione was treated with O2 − to generate a considerable amount of GSH, instead of the expected GSSG (133, 301). If this sequence of events holds its promise and can be defined under cellular conditions, a burst in O2 − in the transition from hypoxia to normoxia would trigger mitochondrial reactivation. A general role of O2 − in causing S-denitrosation needs to be proven as well as a potential impact of S-denitrosation of transcriptional responses that are initiated by NO.
VIII. Redox Signals and Hypoxia-Inducible Macrophage Responses
Redox-sensitive transcription factors with a relevance to classical and alternative macrophage activation are the HIF-1 and HIF-2. Macrophages accumulate at sites of inflammation such as wounded areas or tumors where hypoxia prevails, activating HIF-1 and HIF-2. Macrophages and in particular the HIF-system within these cells play decision-making roles of how tissues respond to environmental and cell autonomous signals to deliver cell-adaptive responses. Here we will briefly characterize how redox signals impinge on the HIF system, and how this system in turn affects the macrophage biology.
A. NO and the hypoxia-responsive system in macrophages
HIF proteins are composed of HIFα subunits that are often kept at low levels due to continuous 26S proteasomal degradation. Mechanistically, both HIF-1α and HIF-2α are continuously hydroxylated at proline residues by PHDs, whose activity is regulated by oxygen availability, thus serving as oxygen sensors [reviewed in (138)]. Hypoxia attenuates PHD activity, stabilizes the HIFα subunits, and allows their dimerization with HIF-1β and gene regulation. In some analogy to PHDs, factor inhibiting HIF (FIH) hydroxylates a C-terminal asparagyl residue in the HIFα subunits, which impairs the transcriptional activity of HIF. Readers are referred to excellent reviews for further details of HIF regulation [for review, see (98, 138)].
It emerges that the PHD activity is affected not only by oxygen but also by NO and/or ROS (Fig. 6).
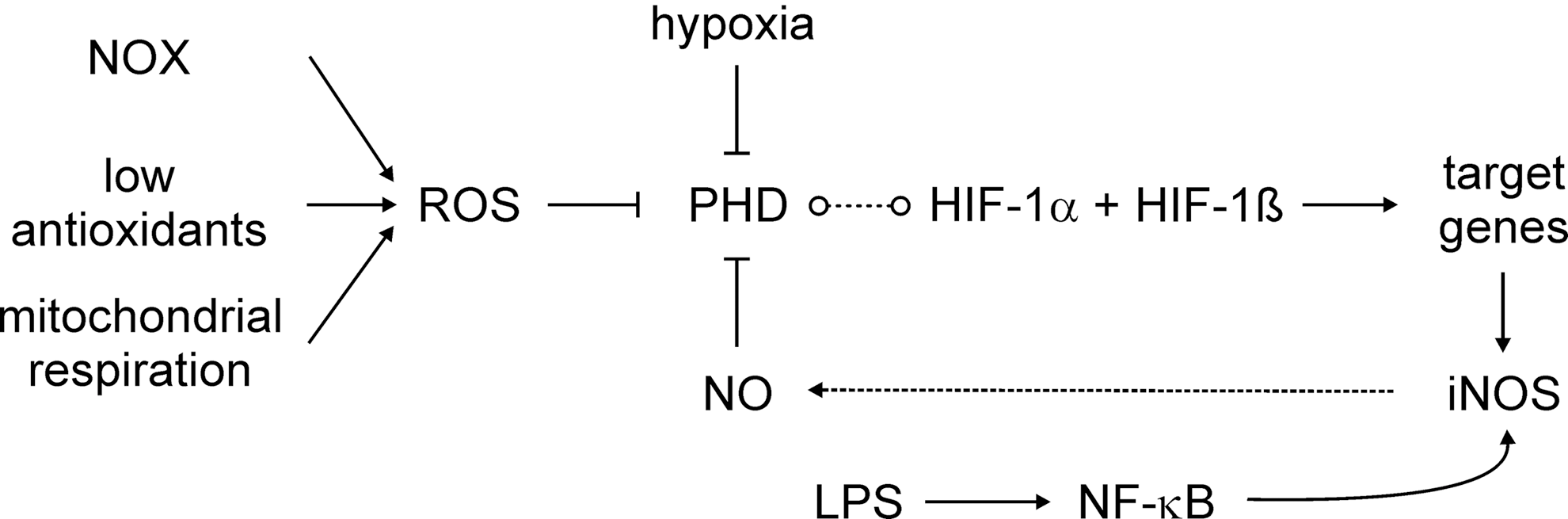
NO-releasing drugs or NO derived from iNOS stabilizes HIF-1α and activates HIF-1 [for review, see (25)]. Recently, it has been shown that NO reacts with a truncated PHD2 catalytic domain (PHD2181–426), both by coordinating the active Fe(II), as shown by electron paramagnetic resonance analysis, and/or via S-nitrosation of cysteine residues, as shown by mass spectrometry studies (36). Although the biological significance of S-nitrosation is not fully elaborated, it is possible that liberation of NO from S-nitrosated PHD2 can result in inhibition via reaction at the active site Fe(II). Stabilization of HIF-1α by NO might be relevant in certain forms of human cancer, where inhibition of iNOS interferes with the accumulation of HIF-1α (231), with the notion that lowering the HIF-1 activity reduces tumor progression (317). Considering the similarity in reactions catalyzed by PHDs and FIH, it is not surprising that the FIH activity is also blocked by NO [reviewed in (24)]. In addition, stabilization of HIF-1α through S-nitrosation at Cys533 within the oxygen-dependent degradation domain, with NO being generated from neighboring macrophages, was shown in tumor tissue after ionizing radiation (170).
The NO-stabilizing effect changes to a destabilizing one, when NO is supplied under hypoxia (25, 217). This paradox can be resolved, considering that NO competes with O2 for the binding to mitochondrial COX, which consumes most of the oxygen within a cell (Fig. 7). At a low pO2, when NO competes with oxygen at COX, oxygen will be spared by mitochondria and instead can support the PHD activity [reviewed in (24)]. In analogy, also mitochondrial respiratory chain inhibitors or eNOS-generated NO lowers accumulation of HIF-1α under hypoxia (59, 102). This intriguing argument in sparing oxygen is not without limitations, as NO binding to COX may trigger O2 − formation, which leads to not only the O2 consumption but also the trapping of NO. However, experiments looking for cellular hypoxia by pimonidazole staining showed that at intermediate oxygen concentrations (3% O2), hypoxia staining and HIF-1α expression were detectable, but strongly reduced after inhibition of the electron transfer chain (59), clearly indicating that more oxygen is available for the PHD reactivation, thus allowing the cells to return to normoxic conditions. Formally, pimonidazole staining experiments have not been carried out with NO under hypoxia, but as the PHD activity is reactivated and hypoxia staining correlated with PHD activity in other experiments, the potential inference by O2 − is of no or minor importance.
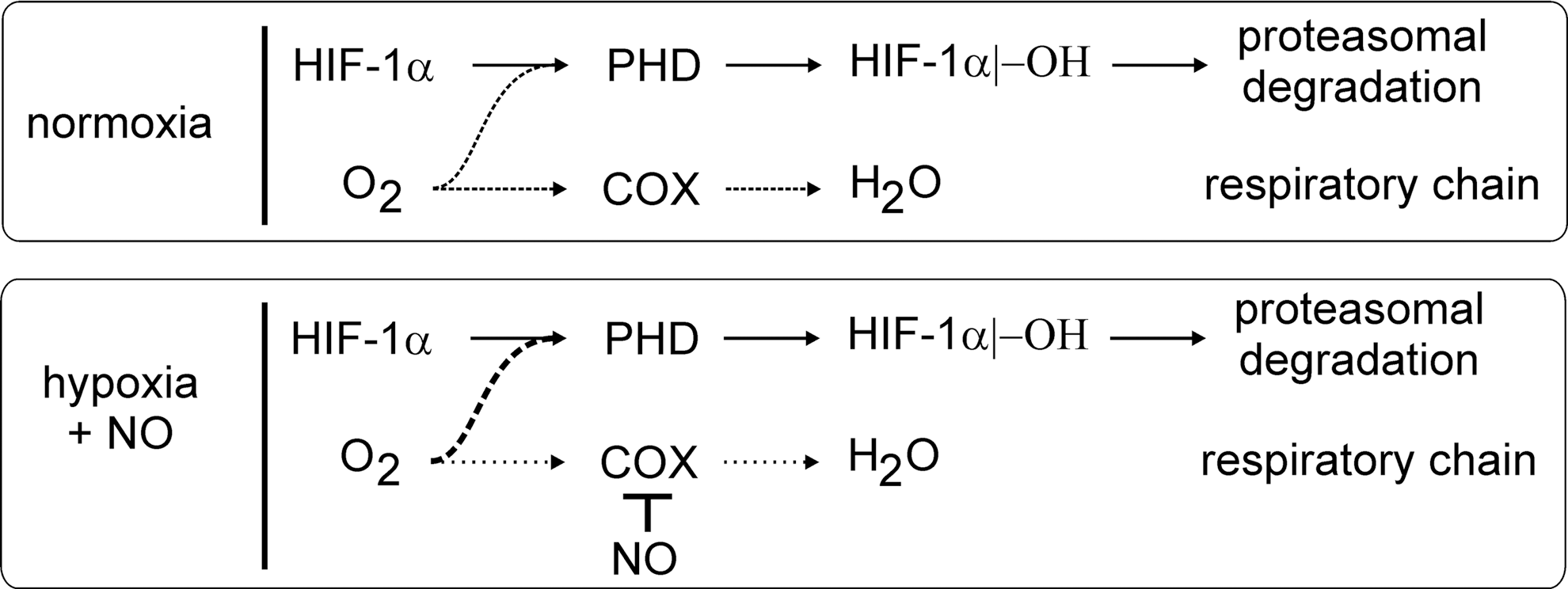
A more direct connection between hypoxia and the NO/iNOS system exists, as the iNOS promoter contains classical hypoxia-responsive element sites. However, despite the potential HIF-1-binding site, hypoxia alone only poorly (if at all) induces iNOS expression (58). Considering that the KM value of iNOS for oxygen is 135 μM, it is not surprising that NO formation under hypoxia (1% O2) is also attenuated (189), irrespective to iNOS expression. This becomes relevant during bacterial infections (225). Under normoxia, the bacterial cell wall components contribute to the expression of HIF-1α, likely via NF-κB activation, as discussed below. HIF-1, presumably with the help of supportive transcriptional regulators such as NF-κB, causes iNOS expression and NO formation, whereas interfering with NO formation lowers HIF-1α expression. Apparently, HIF-1 is central to an NO-dependent amplification loop during the response of murine macrophages to bacterial infection. The relevance of the HIF-1/NO-signaling axis might be restricted to the conditions that allow iNOS to be active rather than being dampened by hypoxia. The fact that iNOS expression in the human system is much more restricted compared to the murine system may further limit the relevance of the HIF-1/NO axis for human diseases.
B. ROS and the hypoxia-responsive system in macrophages
The catalytic activity of PHD requires iron in a reduced state. During redox stress, antioxidants such as ascorbate may become limiting, and, indeed, addition of antioxidants activates PHDs (150, 279). However, in contrast to collagen PHD, ascorbate is not limiting for PHD activity, as glutathione can sustain the catalytic iron in its ferrous state (214). Treatment of cells with H2O2 decreases the PHD activity, but interestingly, PHD sensitivity toward oxidants is time-dependent. H2O2 decreases PHD activity and enhances HIF-1α levels early after oxidant delivery, while later on, PHD activity increases to reduce HIF-1α expression (211). Early, the Fenton chemistry accounts for a reduced PHD activity, whereas later, the cellular iron redox state promotes PHD activity possibly by ferrireductases. Along those lines, the transcriptional regulator and proto-oncogene jun D increases antioxidant levels, whereas its deletion increases ROS and stabilizes both HIFα subunits (87). Mitochondria are a proposed source of ROS in activating HIF-1α during hypoxia (30). Initial data were met with skepticism, and the relationship between ROS production and HIF-1α activity has become increasingly complex, as FIH was recently demonstrated to be more sensitive to redox stress than PHDs (184). Genetic or pharmacologic approaches to block mitochondrial respiration and to prove a link between mitochondrial ROS formation and HIF-1α accumulation are challenged, because these manipulations not only interfere with O2 − production but also disturb mitochondrial oxygen consumption. Thus, it cannot be distinguished between ROS production and sparing mitochondrial oxygen consumption to promote PHD activity (37). More recently, it has been reported that the mitochondrial deacetylase sirtuin-3 (SIRT3) destabilizes HIF-1α by inhibiting ROS production, thereby promoting a maximal PHD activity (70). A knockout or knockdown of SIRT3 caused an intracellular ROS increase and stabilized HIF-1α, although direct inhibition of PHDs by ROS was not proven. SIRT3 may suppress ROS by deacetylation and activation of manganese superoxide dismutase (MnSOD), as well as activation of the trichloroacetic acid (TCA) cycle enzymes, particularly isocitrate dehydrogenase 2, which delivers NADPH for ROS-detoxifying enzymes [for references, see (98)]. However, modulation of SIRT3 under pathophysiological conditions and its impact on HIF activity in macrophages await further clarification.
C. The NO/ROS interplay in affecting the hypoxia-responsive system in macrophages
The near-diffusion-controlled interaction between NO and O2 − adds another perception in regulation of HIF-1α expression. The simultaneous generation of O2 − and NO increased dihydrorhodamine oxidative intermediates, with a decrease in steady-state NO concentrations and a proportional reduction of NO-evoked HIF-1α stabilization [reviewed in (25)]. To lower the HIF-1α protein amount under conditions of NO/O2 − coformation required proteasomal degradation and thus PHD activity. It is assumed that the primary outcome of producing O2 − and NO at the same time is a change in the cellular phenotype due to an altered bioavailability of either NO or O2 − [reviewed in (329)], rather than the resultant chemistry of newly formed higher nitrogen oxides. This notion may help to explain the discrepancies among various studies on the ability of either NO or O2 − to affect the amount of HIF-1α. Differences in the relative flux rates of NO or O2 − may drastically change under normoxia versus hypoxia and in macrophages under resting or stimulated conditions.
D. HIF in classically activated macrophages
Classical macrophage activation at the sites of inflammation increases the demand of ATP and O2 in an area facing a shortage in O2, thus being or becoming hypoxic. Under resting physiological conditions, oxygen delivery and consumption are balanced, whereas during inflammation, the oxygen demand exceeds oxygen delivery, and consequently, HIFα subunits are stabilized (119, 326). Besides oxygen, additional cofactors such as iron, 2-oxoglutarate, and ascorbate (138) regulate PHD activity. Iron chelation, but also oxidation of the iron, either directly or indirectly by reducing cellular antioxidants attenuates PHD activity (Fig. 8). Bacterial- or LPS-activated macrophages produce oxidants via activation of NOX or iNOS and promote HIF-1α protein accumulation even under an ambient oxygen level [for references, see (53)]. Macrophage responses to eukaryotic microorganisms also affect HIF, as Leishmania-infected macrophages accumulate HIF-1α and HIF-2α (52). Oxidants such as oxLDL provoke HIF-1 activation as well (227, 260), and proinflammatory cytokines such as TNFα or IL-1β induce HIF-1α protein accumulation by increasing either ROS production or phosphoinositide-3-kinase (PI3K)-dependent translation [Fig. 8, (53)].
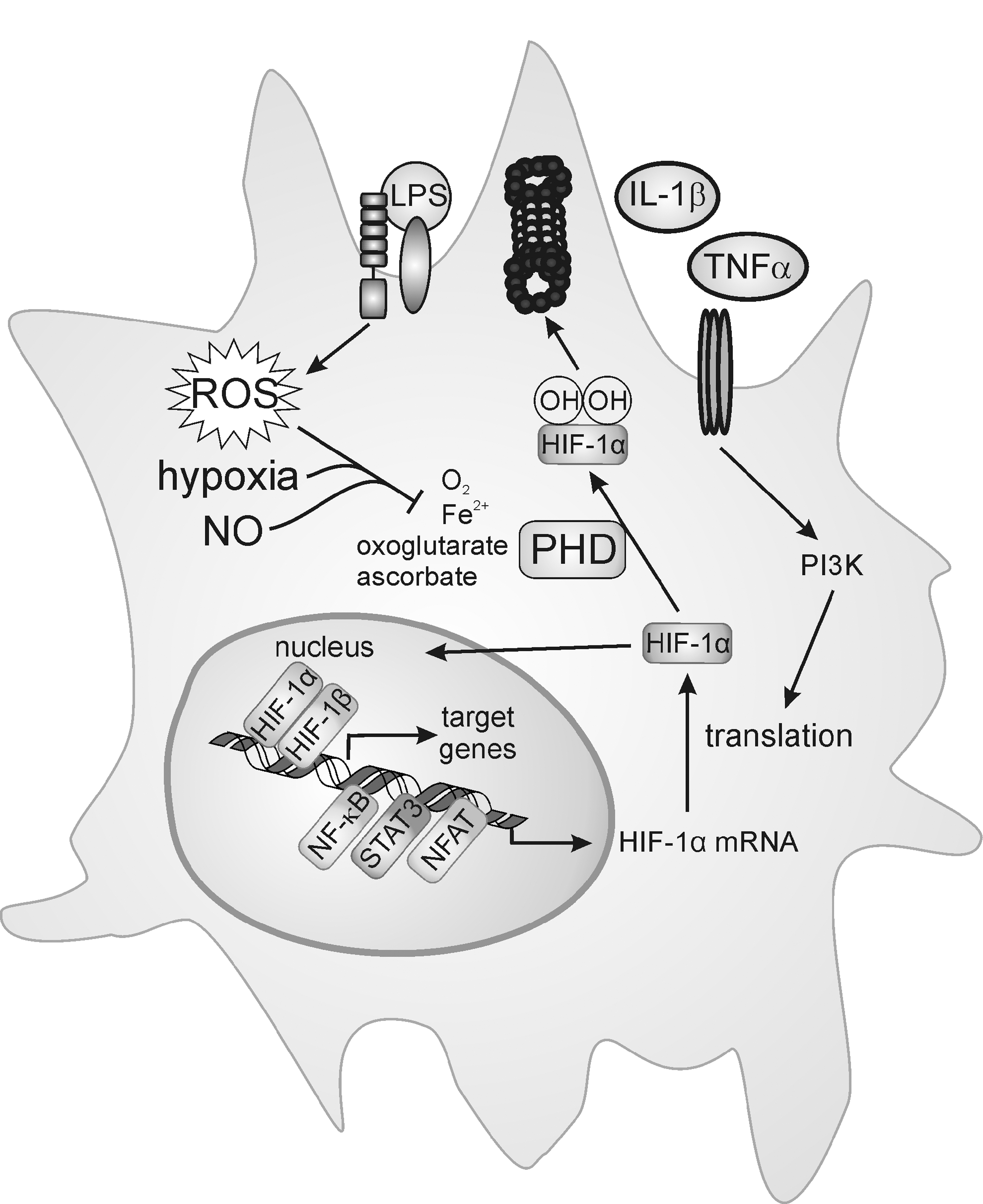
Other small-molecule mediators of the inflammatory microenvironment, such as adenosine, may also induce PI3K-dependent HIF-1α translation. Although the steady state of HIF-1α protein expression is predominantly determined by PHD-mediated degradation, increased protein translation may shift the balance and overwhelm the degradation system, adding another level of HIF-1 regulation.
At the transcriptional level, classically activated macrophages show, among others, NF-κB activation, which is facilitated via the classical/canonical or alternative/noncanonical pathway. Canonical NF-κB activation requires the assembly of an I-kappa-B kinase (IKK) complex consisting of the catalytic kinases IKKα and IKKβ, and the regulatory component IKKγ (NEMO) (Fig. 9). This complex phosphorylates IκB and targets it for proteasomal degradation, releasing the p50/relA (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1/v-rel reticuloendotheliosis viral oncogene homolog A) heterodimer for nuclear translocation (221). IKK complex formation is usually induced after ligand binding to TLR, TNFα, or cytokine receptors. Noncanonical NF-κB activation is stimulated by specific TNF receptor family members, which releases p52 (nuclear factor of kappa light polypeptide gene enhancer in B-cells 2) (221). Like HIF, NF-κB is activated by ROS [for overview, see (197)] and hypoxia. Hypoxic activation of NF-κB is weaker compared to proinflammatory activation, but still is detectable in vivo (71). Mechanistically, several options have been proposed, for example, tyrosine phosphorylation of IκB, hypoxic-induced transactivation via p42/44 (MAPK 1/3), and PI3K (155). Recently, NF-κB activation under hypoxia was linked to the HIF-system. FIH hydroxylates p105 and IκBα at conserved ankyrin repeat domains, and IKKβ is hydroxylated by PHDs, which causes their inactivation (39, 48) (Fig. 9).

Analyzing HIF-1α expression in classically activated macrophages shows an increase in HIF-1α mRNA [see (53)]. An NF-κB binding site was identified within the HIF-1α promoter, and binding of several NF-κB members was confirmed (19, 304). Studies in mice lacking IKKβ pointed to NF-κB as a mandatory transcriptional activator of HIF-1α. Apparently, a basal NF-κB activity is required for HIF-1α protein accumulation under hypoxia (239). Transcription factors such as Krüppel-like factor 2 that are able to modulate NF-κB activity also alter HIF-1α accumulation in macrophages (179). Therefore, HIF-1α accumulation/HIF-1 activation in classically activated macrophages integrates the signals of PHD inactivation, increased PI3K-mediated HIF-1α translation, and/or an NF-κB-induced HIF-1α mRNA increase (Fig. 8). Hypoxia-induced NF-κB activation may cooperate with activated HIF-1, which can support/enhance macrophage function in the hypoxic inflammatory microenvironment.
The interaction of NF-κB and HIF-1 continues at the level of transcriptional activation. Copper metabolism (Murr1) domain-containing 1 is transported out of the nucleus during hypoxia, and this transport is required for activation of NF-κB and HIF-1 (200). After DNA binding, HIF-1 recruits cofactors such as E1A-binding protein p300 (p300/CBP) to initiate target gene transcription. Binding of cofactors is inhibited by FIH-dependent asparagyl hydroxylation. In macrophages, FIH activity is suppressed by Mint3, which sequesters FIH to the perinuclear region and consequently enhances target gene expression (246). The interaction of HIF-1 with p300 is also disturbed by SUMOylation of p300. Micromolar concentrations of H2O2 enhance stability and abundance of SENP3 (SUMO1/sentrin/SMT3-specific peptidase 3) in the nucleoplasm to initiate the removal of the SUMOylation sites in p300, thereby activating the HIF-1 transcriptional activity, which might be of particular relevance in the pro-oxidant milieu created by classically activated macrophages (121). On the other side, in classically activated macrophages, NF-κB and HIF-1 share the coactivator p300, and NF-κB competes for coactivator binding, to repress HIF-1 target gene expression (190). In contrast, genes such as iNOS are induced by both HIF-1 and NF-κB and are synergistically induced under hypoxic and proinflammatory conditions (189). Therefore, it seems to be a gene-specific effect whether the interaction of HIF-1 with NF-κB acts synergistically or inhibitory. To generalize, in classically activated macrophages, NF-κB and HIF-1 are activated in parallel and share transactivation mechanisms to induce common target genes, which implies a concerted biological action in these cells (for summary see Fig. 9).
This is reflected in an in vivo colitis model, where administration of dimethyloxallyl glycine (DMOG) activated HIF-1 as well as NF-κB and conferred protection (49). Although HIF is considered to mediate prosurvival signals under hypoxia, Shah et al. demonstrated that activation of HIF-1 increases damage in this dextran–sodium sulfate-induced colitis model (258), implying that NF-κB accounts for the protective effect of DMOG. Protection by DMOG was also confirmed in TNFα-induced loss of an ileal barrier function by reducing apoptosis in enterocytes (117). In vitro experiments suggest an HIF-dependent inhibition of Fas-associated death domain protein expression that is required for TNFα-induced apoptosis, showing that HIF is at least partly involved in the protective effects of DMOG in the intestine. On the other hand, HIF is involved in the induction of several cytokines in response to LPS, and lethality of LPS-induced endotoxic shock is largely abolished in myeloid-specific HIF-1α and HIF-2α knockout mice (46, 125, 224). The phenotype is reversed by knockout of PHD3 with enhanced lethality in endotoxic shock or sepsis, accompanied by the enhanced production of cytokines, migration of macrophages into organs, and enhanced organ damage (149). The increased production of cytokines resulted from an enhanced activation of HIF-1 and NF-κB, but not of HIF-2, in PHD3 knockout cells, again showing a close relationship of HIF-1 and NF-κB in classically activated macrophages. Surprisingly, the knockout of PHD1 did not alter the LPS-induced lethality at all, while a haplodeficiency of PHD2 slightly decreased the survival of animals, leaving the possibility that a complete knockout of PHD2 might also enhance lethality in septic mouse models. In most cells, PHD2 was established as the most important PHD to regulate HIFα protein expression as reflected by the embryonic lethality of a complete PHD2 knockout in mice (282). Nevertheless, in classically activated immune cells, PHD3 seems to have a prominent role.
Parallel to bacterial infection, eukaryotes such as Leishmania induce classical activation of macrophages (302). The resulting production of oxidants such as O2 −, H2O2, or NO is designated to destroy the phagocytosed parasite. Although some parasite stages are sensitive toward oxidants, parasites may survive within the macrophage by eliminating ROS via their own antioxidant defense system. Hypoxia allows macrophages to control Leishmania, an effect likely involving ROS, because treatment with antioxidants such as N-acetylcysteine or ebselen inhibits these activities (52). Although accumulation of both HIFα subunits in infected macrophages was demonstrated, the details of this process are unclear.
E. HIF in alternatively activated macrophages
Besides fighting invaded pathogens, wound healing is a major function of macrophages. Wound-healing macrophages are characterized by a lower ability to present antigens and to produce proinflammatory cytokines, but they secret components of the ECM and polyamines. Administration IL-4 to a wound in vivo decreased the wound size and increased collagen expression (29). HIF-1α is expressed in wounds (336), and a knockout of HIF-1α in myeloid cells enhanced early wound closure, generating an increased number of activated keratinocytes (218).
Macrophages are able to accumulate HIF-1α and HIF-2α. While HIF-1α accumulates in classically activated macrophages, HIF-2α is barely detectable (283). In contrast, macrophages polarized by IL-4 toward a wound-healing phenotype show increased HIF-2α, but reduced HIF-1α, accumulation, implying opposing functions of macrophage HIF-1 and HIF-2. This is in contrast to the studies indicating impaired acute inflammatory responses in myeloid-specific HIF-2α knockout mice that parallel the findings in myeloid-specific HIF-1α knockout mice (46, 125, 224). An explanation might be that during inflammation, macrophages change their phenotype resulting in a mixed macrophage population in vivo, allowing the simultaneous accumulation of HIF-1α and HIF-2α. This is particularly well established for tumor tissue, where TAMs express both HIF isoforms and acquire a mixed phenotype with features of all three macrophage phenotypes (199, 285). In tumor cells and TAMs, STAT3 is required for HIF-1α expression and necessary for the proangiogenic function of these cells (96, 213). In addition, STAT3 directly interacts and stabilizes HIF-1α and increases its hypoxic accumulation (136). Apoptotic tumor cells, which are abundant in tumors, provoked transcriptional nuclear factor of activated T-cell (NFAT)-dependent upregulation of HIF-1α mRNA, subsequent protein expression, and HIF-1 activity in macrophages (113). Mechanistically, sphingosine-1-phosphate and transforming growth factor-β were identified as the activators of NFAT to cause transcription of HIF-1α mRNA. In contrast to HIF-1α, no information is available whether HIF-2α can also be transcriptionally regulated.
Growing evidence suggests that we urgently need to understand the role of tumor-infiltrating cells such as macrophages to entirely explain tumor formation and progression. The tumor microenvironment impairs the antitumor activity of macrophages and other immune cells, but enhances their tumor-supporting properties (146). This is reflected by the phenotype of associated macrophages, characterized by low arginase1 and high iNOS expression, observed in tissues surrounding various tumors as early as 1 week after carcinogen treatment (236). Similar activation is also observed in bone marrow-derived macrophages of these mice, but not in tissue-resident macrophages of intact organs, limiting the influence of the tumor microenvironment to the tumor site and the bone marrow. Hypoxia is characteristic for tumors, and exploring tumor models using either myeloid-specific HIF-1α or HIF-2α knockout mice revealed that each knockout reduced tumor mass and/or tumor grade by different mechanisms (58, 125). HIF-1α knockout macrophages show a limited ability to reduce T-cell proliferation, which was likewise reduced in wild-type macrophages by inhibiting iNOS. Using an in vitro spheroid model, Werno et al. demonstrated that HIF-1α knockout macrophages invading tumor spheroid showed enhanced signs of alternative activation (325). In contrast, HIF-2α-deficient macrophages are impaired in their ability to invade the tumor sites (125). Apparently, in TAMs, both HIF isoforms are important for a macrophage tumor-promoting function.
An important, but unresolved, question is whether hypoxia or HIF contributes to macrophage polarization. Their inflammatory products characterize classically activated macrophages. As NOX2 and iNOS use O2 to produce ROS or NO, their activities decrease in a hypoxic atmosphere, limiting macrophage proinflammatory functions (186, 240). On the other side, HIF-1 increases the expression of markers for classical activation such as iNOS. Haplodeficiency of PHD2 skews macrophages toward an alternative and/or proangiogenic phenotype (284). Analysis of HIF-1/-2 and NF-κB activation showed that HIF activity was not significantly enhanced in haplodeficient cells, compared to complete knockouts, while NF-κB was activated and seemed to convey the phenotype shift. Macrophages bearing a genetic alteration toward enhanced ROS formation and HIF-1 activation showed increased classical, but decreased alternative, activation markers (314). An HIF-1α knockdown indicated that indeed HIF-1 may enhance classical activation, but still autonomous cell activation was necessary to unleash the HIF-1-promoting effect. In line, the expression of alternative activation markers such as scavenger receptor 1/CD204 was suppressed by HIF-1 (263). As summarized in Figure 10, one may hypothesize that HIF-1 promotes classical activation, while NF-κB more likely causes alternative polarization under hypoxia.
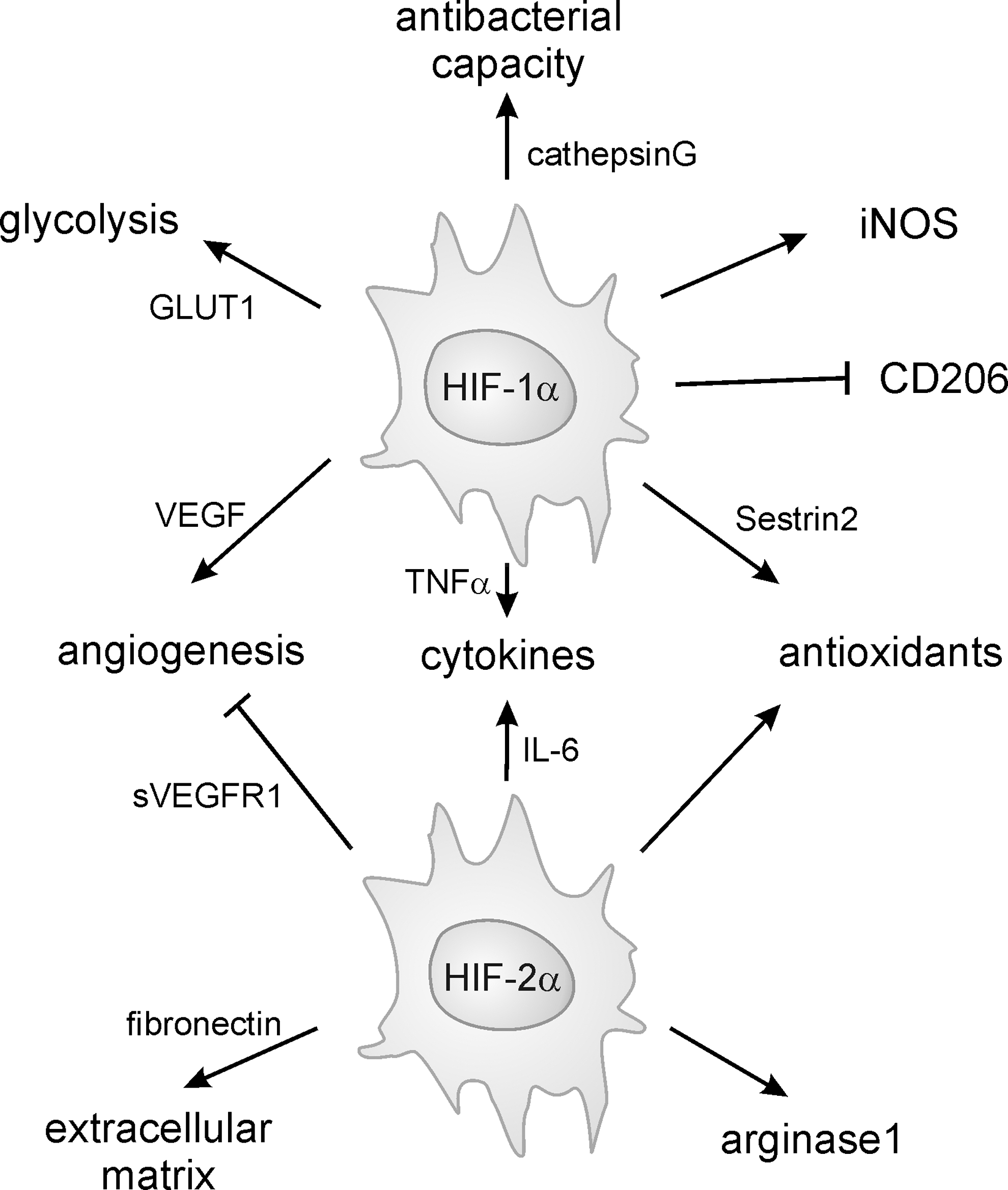
F. HIF target genes in macrophage biology
Comparing gene expression in hypoxic or NO-treated macrophages displayed several identical highly regulated genes, including VEGF, BCL2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3), but also antioxidant genes such as sestrin2 (67, 124, 295). Sestrin2 is induced in macrophages treated with NO or hypoxia via HIF-1 and protects cellular peroxiredoxins against H2O2-induced sulfinylation (65). A global knockout of HIF-2α pointed to a prominent role for HIF-2 in regulating antioxidant-related proteins (249, 253). Further studies are needed to establish whether the selective knockout of HIF-1α or HIF-2α particularly affects the antioxidant status in macrophages.
To sustain the periods of limited oxygen availability, cells adapt their metabolism from oxidative toward glycolytic energy production. Similar to most cells, macrophages use HIF-1 to upregulate glycolytic enzymes to increase lactate and ATP production (46). Macrophages stimulated with the TLR4 or TLR7/8 ligands increase lactate production, but demonstrate reduced ATP levels, which is further impaired by a knockout or knockdown of HIF-1α (46, 209, 242). A considerable part of the glycolytically produced ATP is used to maintain the mitochondrial membrane potential to prevent apoptotic cell death (84). Genes such as pyruvate dehydrogenase kinase, isoenzyme 1 (PDK1), and BNIP3 are also a part of the hypoxic adaption and are regulated in macrophages (own unpublished observation). PDK1 is important to block the entry of pyruvate into the mitochondrial TCA cycle, while BNIP3 induces autophagy of mitochondria [compare (24)]. Both steps successfully attenuate an increase in mitochondrial ROS under hypoxia to promote cell survival and/or proliferation. The profound metabolic adaption by HIF-1 toward glycolytic energy production is paralleled by decreasing mitochondrial metabolism, in part by simply reducing the mitochondrial mass.
HIF activation in various tissues is linked to proangiogenic responses. Studies using myeloid-specific HIF knockouts established this principle also for macrophages (313, 325). Interestingly, although VEGF is considered an HIF-2 target in most cells, in macrophages, VEGF expression is HIF-1 dependent, while HIF-2 regulates soluble VEGF-receptor1, a negative regulator of VEGF signaling (66, 144) (for overview, see Fig. 10). This can be translated to tumor growth in vivo by using an HIF-2α-specific PHD inhibitor reducing tumor growth and vessel density (241).
Two enzymes closely linked to the macrophage biology and HIF are arginase1 and iNOS. Both are induced by HIF, and both require
IX. Nrf2 at the Transition from Classical to Alternative Macrophage Activation
Macrophages recognize and fight pathogens in part by creating a toxic environment. Therefore, self-protective mechanisms are required to guarantee their survival. These should be turned on tightly coupled to redox changes occurring during macrophage activation. The most prominent redox-sensitive transcription factor accounting for protective mechanisms is Nrf2. Nrf2 is a CNC-bZIP (Cap‘n'collar-basic region leucine zipper) transcription factor (127). As shown in Figure 11, under resting conditions, Nrf2 binds to the cytosolic inhibitor Kelch-like ECH-associated protein 1 (Keap1)/INrf2, an adaptor protein to the cullin-3 E3 ubiquitin ligase complex (308), which targets Nrf2 for proteasomal degradation to keep the cytosolic level of Nrf2 low. Keap1 is a sensor of electrophiles and oxidants (333), containing 27, in part highly redox-active cysteine residues (Fig. 12).


Apart from ROS-dependent modifications of Keap1 cysteine residues, electrophilic-dependent stress responses acting via inhibition of phosphatases can provoke Nrf2 phosphorylation and subsequent stabilization. Moreover, the protein human DnaJ homolog 1 inhibits Nrf2 proteasomal degradation by blocking binding of Keap1 to Nrf2 (38). Stable Nrf2 translocates to the nucleus and binds to the antioxidant-responsive element sites in the promoters of target genes with other bZIP transcription factors such as small Maf proteins (173), activating transcription factor 4 (ATF4) (111), c-Fos/Fra (148, 306), or JunD (294). Moreover, binding of the coactivator p300/CBP (275) enhances transcription of target genes such as glutathione S-transferase, NADP(H) quinone oxidoreductase 1, heme oxygenase-1 (HO-1), uridine 5′-diphosphate-glucoronyl transferase 1a6, epoxide hydrolase, glutathione peroxidase-2, peroxiredoxins, glutathione reductase, or ATF3 (34, 118). These are members of the phase II antioxidant and detoxifying systems, which are important for macrophage survival. Moreover, ATF3 lowers cytokine expression to protect against endotoxic shock (118). Considering the protective role of Nrf2, it is not surprising that a conditional LysM-Cre-driven Nrf2 knockout in the myeloid lineage increases inflammatory response after LPS challenge (153). LPS-treated Nrf2-deficient macrophages enhance ROS formation (154), which augments the TLR4 transport from the trans-Golgi network to the cell surface, consequently amplifying TLR4 downstream signaling (153). This effect was absent in Nrf2−/− NOX2−/− double-knockout mice (154). Likely, Brutons tyrosine kinase mediates TLR4-dependent activation of Nrf2 (307). The importance of Nrf2 in macrophages is evident for several diseases, often associated with macrophage malfunction and frequently characterized by chronic inflammation. For example, chronic obstructive pulmonary disease (COPD) improves when Nrf2 is activated or constitutively overexpressed in mice (108, 278). Enhancing Nrf2 activity stimulates the phagocytic capacity of alveolar macrophages, which allows the efficient removal of pathogens and probably apoptotic cells. Pulmonary Nrf2 dominates in the epithelium and in alveolar macrophages, but the mechanisms why Nrf2 is lost during COPD progression remain unclear (100, 181). However, when Nrf2 is exogenously activated in a COPD mouse model by sulforaphane or an imidazole derivative of 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid, lung emphysema is clearly reduced (181, 277).
Another chronic inflammatory disease affected by Nrf2 is atherosclerosis. Disruption of Nrf2 attenuates apoplipoprotein E (ApoE)-mediated atherosclerosis in mice (276). During atherosclerosis, a specific macrophage phenotype develops in response to atherogenic lipids via Nrf2-dependent gene expression (1). These Mox macrophages are, compared to classically activated or proresolving wound-healing macrophages, characterized by a unique expression pattern of antioxidative and detoxifying genes. Among others, decreased phagocytosis and reduced migratory properties are two features, although as of yet, it has not been clarified whether these macrophages are pro- or antiatherogenic. Understanding the role of Nrf2 in deactivating oxLDL-generated foam cells, we noticed attenuated ROS as well as proinflammatory cytokine production (159). In foam macrophages from Nrf2−/− mice, LPS-induced proinflammatory responses were restored. Mechanistically, oxLDL induced Nrf2-dependent antioxidative responses, which in turn attenuated the ROS-dependent activation of CCAAT/enhancer-binding protein (C/EBP) family members to reduce proinflammatory cytokine production. In experimental animal models of atherosclerosis, deletion of Nrf2 is protective. Therefore, Nrf2 seems to have a proinflammatory function as well. In line, cholesterol crystals activate Nrf2 and the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome and thus IL-1β production (77).
During acute inflammation, like sepsis, macrophage Nrf2 appears in the focus as well. Sepsis is characterized by a hyperinflammatory-phase with a cytokine storm of classically activated macrophages (266), contributing to a systemic inflammatory response. During this phase, NOX2 generates ROS (154), which stabilize Nrf2 to initiate self-protection of macrophages (148, 154). Normally, a second, hypoinflammatory phase, also known as immune paralysis, follows, because macrophages acquire a proresolving, tolerant phenotype (151). In these macrophages, NOX2 is inhibited (132, 311), making Nrf2 stabilization by ROS unlikely.
Often, oxidative signals activate more than one redox-sensitive transcription factor (335) with the option that they may substitute each other (95). One example is HIF-1α, which contributes to HO-1 expression during alcohol-induced inflammation and liver injury in Kupffer cells. The knockdown of HIF-1α and Nrf2 completely blocked HO-1 expression, suggesting a mutual effect of both transcriptional regulators (335). Interestingly, in smokers with a primary spontaneous pneumothorax, the Nrf2 target HO-1 is exclusively induced by HIF-1 (95). Moreover, there is also a link between Nrf2 and the second transcription factor family described here, the PPARs.
X. Alternative Macrophage Activation and PPARs
Three members of the PPAR nuclear receptor family have been described. PPARα is predominantly expressed in the liver, controlling fatty acid oxidation (56). PPARβ/δ is ubiquitously expressed, regulating fatty acid metabolism, mitochondrial respiration, thermogenesis, muscle lipid metabolism, and macrophage phenotype switching (56). PPARγ is also expressed in adipocytes, macrophages, and DCs (205, 293). All three members are activated by nonesterified fatty acids, polyunsaturated fatty acids, eicosanoids, or prostanoids, provoking binding to an identical PPAR-responsive element (PPRE) sequence motif. Predominantly, PPARγ has been established as an anti-inflammatory macrophage modulator. Therefore, we focus on this family member. In macrophages, PPARγ causes a transrepression of proinflammatory genes. As shown in Figure 13, ligand-activated PPARγ binds as a heterodimer with retinoid x receptor to the PPRE of target genes (ABCA1, CD206, and CD36) (20, 210), which mainly affects lipid uptake and export.

However, as identified in thioglycolate-elicited macrophages, PPARγ initiates a potent anti-inflammatory response (238), with several potential explanations. PPARγ not only transactivates the genes by binding to their PPRE, it also acts in a non-DNA-bound manner. This involves protein–protein interactions between PPARγ and proinflammatory transcription factors such as AP-1, NF-AT, NF-κB, and STAT-1 (237), as shown in Figure 14A. By scavenging these transcription factors, expression of proinflammatory genes is blocked. Binding to transcriptional coactivators, for example, steroid receptor co-activator 1 or p300/CBP, PPARγ inhibits AP-1, and reduces NF-κB-dependent gene induction (Fig. 14B).
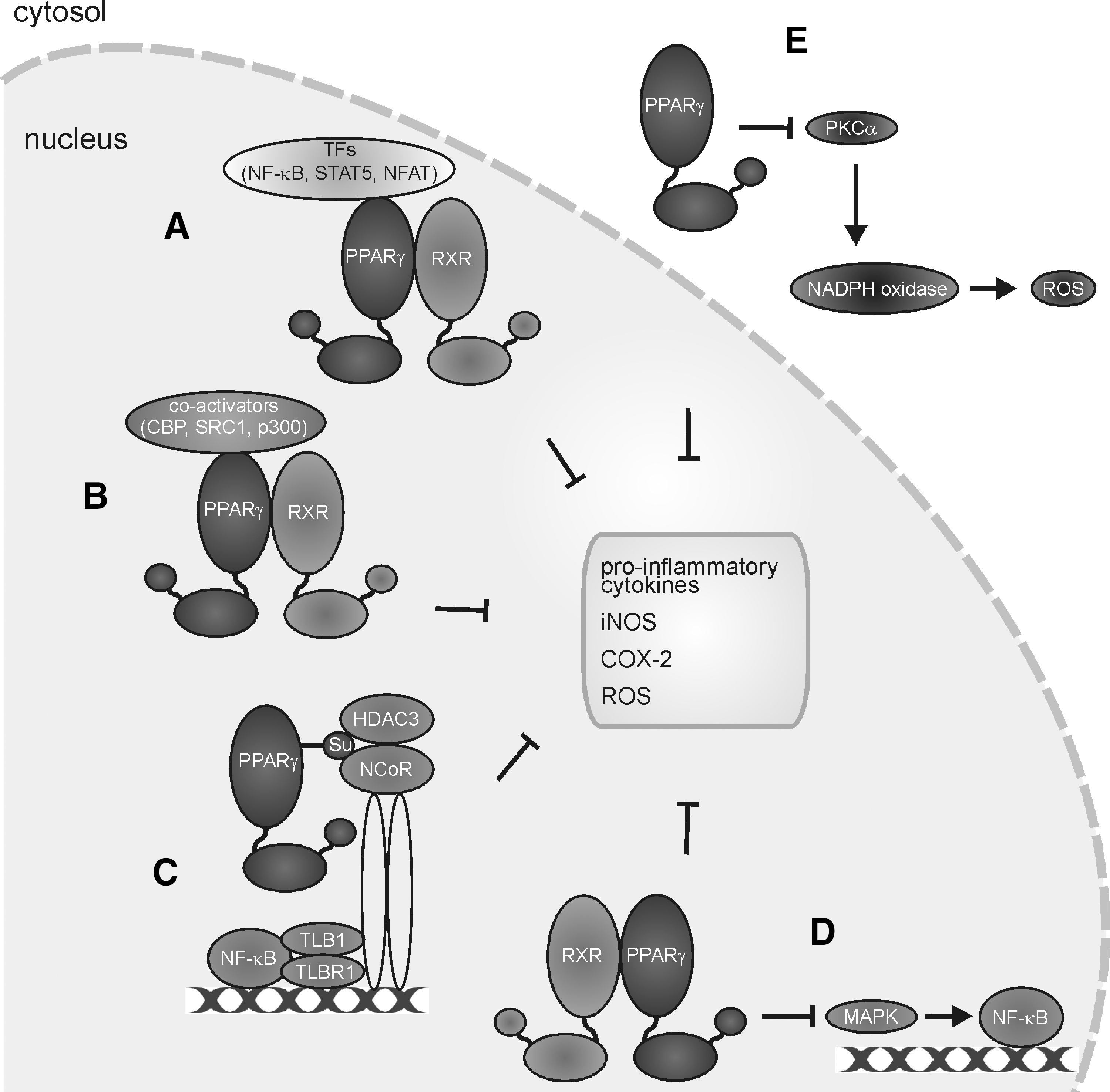
These coactivator complexes possess histone acetyltransferase activity, allowing the transcription machinery to gain access to the promoter region (51). As shown in Figure 14C, SUMOylated PPARγ transrepresses promoter activation of the transcription factor NF-κB by inhibiting the removal of the corepressor complex nuclear receptor corepressor/silencing mediator of retinoid and thyroid hormone receptors (130). In addition, PPARγ can block MAPK by a so-far unknown mechanism (Figure 14D). MAPK inhibition prevents phosphorylation and thus activation of downstream transcription factors that are necessary for MAPK-dependent proinflammatory gene expression (55). Casein kinase-II phosphorylation of PPARγ makes PPARγ accessible for chromosomal region maintenance 1/exportin1 nuclear export machinery, to localize PPARγ to the cytosol (312). There, PPARγ can directly bind to protein kinase C (PKC)α to interfere with the PKCα translocation to the cell membrane and thus its activation (Fig. 14E). Blocking the PKCα activity attenuates NOX-dependent ROS formation, which contributes to monocyte/macrophage desensitization because of apoptotic cell phagocytosis (132, 311). It is assumed that this inhibitory function of PPARγ reduces redox stress of these desensitized, compared to classically activated, macrophages. However, a detailed description of the macrophage redox status related to different macrophage phenotypes remains elusive.
The mechanism how apoptotic cells activate PPARγ in macrophages is unknown. One explanation might be redox regulation of PPARγ. PPARγ contains nine cysteine residues, eight located in the two zinc-finger motifs of the DNA-binding domain and one is present at the 3′-terminus of the hinge domain. Hypothetically, redox-modified cysteine residues in the zinc finger can block DNA binding and favor protein–protein interactions. A redox-modified cysteine residue located in the hinge domain might affect ligand binding, consequently altering downstream signaling. Interestingly, two studies, although not performed in macrophages, observed activation or deactivation of PPARγ-dependent gene expression in response to lower (291) or higher (16) ROS, thus supporting the redox hypothesis. Future work is necessary to analyze to which extent PPARγ redox modification alters its function in distinct macrophage subtypes.
As outlined for Nrf2, PPARγ is linked to atherosclerosis as well. During the initiation phase of atherosclerosis PPARγ activation may reduce disease progression by attenuating the expression of proinflammatory genes (131). At later stages of the disease, PPARγ activation induces the expression of CD36, a scavenger receptor that facilitates the uptake of oxLDL into macrophages already located in the intima, consequently supporting their differentiation into foam cells. Recent data support the concept that CD36 is an Nrf2 target under proinflammatory conditions, whereas PPARγ agonists seem to be ineffective (216), perhaps because PPARγ DNA-binding is blocked due to redox modified cysteine residues. However, PPARγ indirectly stimulates, via liver x receptor, the expression of ABCA1, a reverse cholesterol transporter (33). Moreover, caveolin-1, another player in reverse cholesterol trafficking, is also a PPARγ target gene in macrophages (120). In vivo studies support the assumption of a protective role of PPARγ (205) in various mouse models of atherosclerosis (6, 33, 41, 168). PPARγ deletion in macrophages provokes disease progression, whereas PPARγ activation reduces lesion formation. From these results, an anti-inflammatory role of PPARγ is obvious. In line, experiments using a PPARγ knockout model identified PPARγ to control alternative macrophage polarization. Mice with a functional deletion of PPARγ in the myeloid lineage failed to generate a wound-healing macrophage phenotype (215). However, in a different mouse strain, PPARγ seems to be dispensable for alternative macrophage polarization (183). These apparent contradictions may rather suggest a modulator function of PPARγ in alternative macrophage polarization. A summary of PPARγ and Nrf2 in macrophage polarization is shown in Figure 15. Interfering with the PPARγ activity in macrophages will be a challenging task for the future.

Although ligands such as the thiazolidinediones are pharmacologically available and already in use, the development and characterization of drugs belonging to the new class of selective PPARγ modulators, which should have a cell-type-specific effect, will allow to bias PPARγ dependency of macrophage polarization.
XI. ROS Support Classical Activation in Response to Modified Lipoproteins
A. NOX and mitochondrial-derived ROS in response to modified lipoproteins
The concept of oxidative modification of lipoproteins as a driving force of atherosclerosis emerged more than 30 years ago [reviewed in (269)]. Cells oxidize LDL by 12/15-lipoxygenase to generate minimally modified LDL (mmLDL), which contains cholesteryl ester hydroperoxides as biologically active components (106). mmLDL binds and activates TLR4 to elicit inflammatory responses in macrophages (194), including NOX2-dependent ROS formation (8). A second mechanism of LDL oxidation involves myeloperoxidase and yields a variety of oxLDL products, which are ligands for the macrophage scavenger receptor CD36 (110). Recent data show that CD36 may form a signaling complex with TLR4 and TLR6 in endosomes (270). While NOX4 was suggested to transmit oxLDL signals in endothelial cells (165) and CD36-mediated signaling may involve ROS-dependent oxidative inactivation of the tyrosine phosphatase SHP2 (219), the distinct NOX isoform responsive to oxLDL in macrophages remains unclear, although NOX4 can be induced in human macrophages by oxLDL (164). Oxidative modifications of LDL may also give rise to reactive aldehyde products, exemplified by 4-hydroxynonenal (HNE). 4-HNE may cooperate with other phospholipid oxidation products in macrophages to increase the expression of CD36 (140). Redox signaling by 4-HNE may involve Nrf2 (126) or modulation of the mitochondrial redox status (62).
The role of mitochondrial ROS in regulating cell responses to oxLDL is also evident (101). An interesting link involves c-Jun N-terminal kinase(s) (JNK)-mediated ubiquitination and degradation of MnSOD by oxLDL in endothelial cells (280), which may increase mitochondrial ROS generation and apoptosis. However, in macrophages, low-level oxLDL may also induce MnSOD expression as a part of a cytoprotective circuit (259). A further link between mitochondria and atherosclerosis was suggested from studies using mice deficient for mitochondrial uncoupling protein 2 (UCP2). Bone marrow transplants from UCP2 knockout mice reduced atherosclerosis in LDL receptor (LDLR) knockout mice (15). Protective actions of UCP2 may be linked to decreased mitochondrial ROS (207), although the exact mechanism of UCP2 function remains to be clarified (180). The role of macrophage ROS in atherosclerosis development was analyzed in several animal models of atherosclerotic disease with genetic modification of ROS-generating or ROS-scavenging systems. Currently, no conclusive evidence regarding a pro- or antiatherosclerotic role of specific NOX isoforms exists. Total NOX2 knockout mice crossed with atherosclerotic ApoE knockout mice revealed either no effect on atherogenesis or reduction of early lesions in the descending aorta (135). Future studies employing macrophage-specific knockout of NOX2 and other isoforms would help to unravel the contribution of macrophage ROS-generating systems to atherogenesis.
B. Modified lipoproteins and endoplasmic reticulum stress
Endoplasmic reticulum (ER) stress is a major mechanism underlying macrophage cell death during atherosclerosis development (254). ER stress may be triggered by disturbed ER functions, for example, an increase of misfolded proteins, inhibited glycosylation, altered ER calcium homeostasis, or perturbed ER membrane structures (Fig. 16). Three signaling proteins sense ER stress: inositol-requiring protein-1α, protein kinase RNA–like ER kinase (PERK), and ATF-6. Activation of these proteins triggers a homeostatic unfolded protein response (UPR), aimed at relieving ER stress by reducing general protein synthesis and increasing folding chaperones.
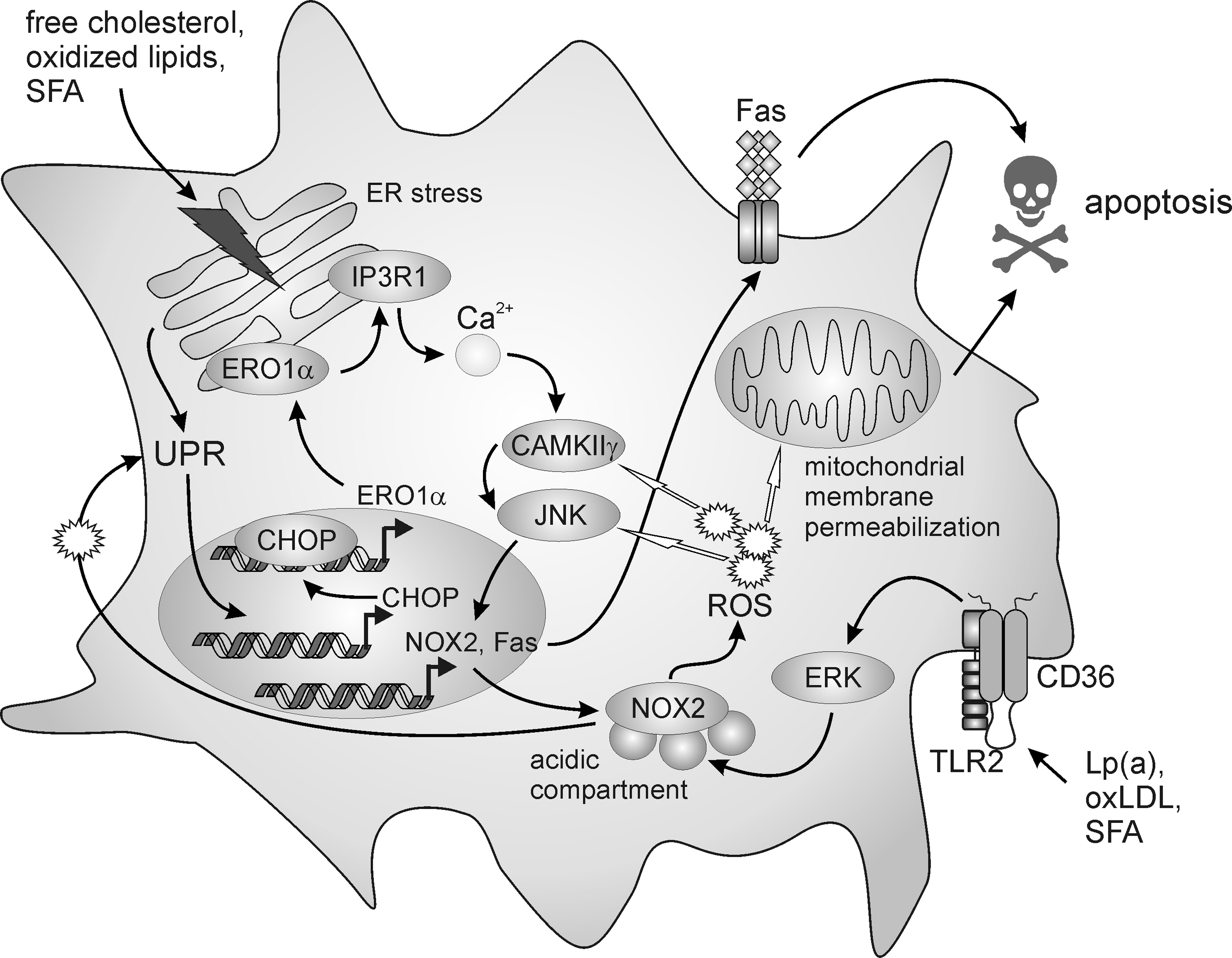
A prolonged and excessive UPR can stimulate apoptosis, where C/EBP-homologous protein (CHOP), a downstream target of PERK, plays a major role. Macrophage ER stress by atherogenic stimuli linked to cell death has extensively been characterized. This integrates ER signals and CD36/Toll-like receptor 2 signaling in response to oxidized phospholipids or saturated fatty acids (255). A signal emanating from the ER due to accumulation of unesterified cholesterol upregulates intracellular calcium, via the CHOP-ER oxidation 1α (ERO1α) pathway (171). This signal in turn activates calcium-/calmodulin-dependent protein kinase IIγ (CAMKIIγ), which triggers apoptosis (292). A recent study highlighted the key role of ROS, generated by NOX2, in transmitting detrimental ER signals (172). ER stress activators, such as free cholesterol or 7-ketocholesterol, induced the expression of NOX2 via a CHOP-ERO1α-CAMKIIγ-JNK-pathway, and NOX2-deficient macrophages showed reduced apoptosis upon ER stress induction. Further, NOX2 was necessary for ER stress-induced IP3 receptor activation and mitochondrial calcium loading as well as depolarization, preceding mitochondrial cell death induction. NOX2 was also involved in scavenger receptor/TLR-activated proapoptotic signaling, provoking the sustained clustering and activation of NOX2 in acidic cellular compartments (255). Although the role of NOX4 in macrophage ER responses is unknown, it induces ER stress and apoptosis in vascular smooth muscle cells (220). In contrast, recent data suggest a cytoprotective role of NOX4 in endothelial cell via activation of Nrf2 (250). Collectively, current evidence supports an important role of NOX2-derived ROS in ER stress-induced macrophage apoptosis. This likely contributes to the formation of the necrotic core and plaque destabilization in advanced atherosclerotic lesions.
XII. ROS: Linking Classical Macrophage and Inflammasome Activation
Inflammasome activation is a part of the innate immune response to pathogens, but also accompanies the development of autoimmune and chronic inflammatory diseases (271, 324). As presented in Figure 17, inflammasomes are multicomponent platforms that contain caspase-1 to process IL-1β and IL-18. Inflammasomes sense a variety of danger signals, including bacteria, viruses, pathogenic crystals, and aggregates, through a family of Nod-like receptors (NLR). Among these, NLRP3 is the most studied NLR, responding to pathogens, exogenous and endogenous danger signals such as extracellular ATP, uric acid, and cholesterol crystals, alum, or silica. Current concepts propose a two-step NLRP3 inflammasome activation. A priming signal, such as LPS, induces transcription of IL-1β and NLRP3 itself, whereas an activating signal causes caspase-1 and IL-1β proteolytic processing.

The nature of the activating signal is subjected to intensive investigations, with three potential events being implicated: K+ efflux, lysosomal rupture, and ROS production. The involvement of ROS in NLRP3 inflammasome activation was first suggested by ROS scavenger interventions (47, 223). A later study proposed mitochondrial ROS to activate the NLRP3 inflammasome (340). In unstimulated cells, NLRP3 colocalizes with ER structures, while another inflammasome component, apoptosis-associated speck-like protein containing a CARD, is cytosolic. Inflammasome-activating stimuli relocalized both proteins to the perinuclear ER and mitochondrion-containing structures, where they may sense ROS generation. This sensing may involve thioredoxin-interacting protein (TXNIP) (339), which dissociates from its binding partner thioredoxin in an ROS-dependent manner and translocates to mitochondria/ER structures, where it directly binds NLRP3. However, this pathway was questioned by experiments in TXNIP-deficient macrophages (185).
Redox regulation of inflammasome activation may be even more complex as outlined recently (244). Several studies suggested a role of the antioxidant defense system in regulating the inflammasome activity. A negative effect of ROS on IL-1β processing was reported for peritoneal macrophages in a study of SOD1-deficient mice (188). This was attributed to glutathionylation and inhibition of caspase-1. Another study suggested a biphasic, redox control mechanism of IL-1β processing in human monocytes (286), whereby initial ROS generation triggers thioredoxin upregulation and cystine/cysteine import/export, which then enhances IL-1β secretion after TLR activation. Differences in the basal redox state among primary monocytes, macrophages, and myeloid cell lines were suggested to explain the discrepancies in IL-1β secretion in response to TLR activation in these cells (28). Further evidence that antioxidant responses contribute to inflammasome activation came from Nrf2 knockout macrophages, which display reduced IL-1β secretion in response to cholesterol crystals, which was proposed to underlie an antiatherosclerotic phenotype of ApoE Nrf2 double knockouts (77). It is worth mentioning that also an excessive reductive environment causes mitochondrial stress and ROS release (338).
It has recently been suggested that ROS may participate in inflammasome priming rather than activation, based on diphenylene iodonium (DPI)- and N-acetylcysteine-sensitive induction of IL1β and Nlrp3 mRNA by LPS, whereas DPI addition after priming failed to modulate inflammasome activation (11). Although the use of the unspecific ROS-scavenging compound N-acetylcysteine points to ROS as an inflammasome activator, NOX-dependent ROS generation can be excluded, taking into consideration that DPI as a competitive inhibitor of flavin-containing cofactors was without effect. A crosstalk between ER stress and the inflammasome activation pathways may exist, as ER stress activators also stimulated IL-1β processing in human macrophages (191). Molecular details of this crosstalk remain elusive, but may involve induction of apoptotic mechanisms as discussed above.
In conclusion, most of the experimental evidence thus far supports a critical role of mitochondrial ROS for NLRP3 inflammasome activation. However, further research is needed to provide mechanistic details on how NLRP3 senses ROS signals and whether pathways involving lysosomal rupture crosstalk to mitochondria. Spatiotemporal changes in ROS generation or the activity of antioxidant defense systems may also modulate the components of the inflammasome-signaling cascade, such as redox regulation of caspase-1.
XIII. Autophagy Dampens ROS-Dependent Inflammasome Activation
Autophagy is a process whereby cellular components, including whole organelles, are degraded in specialized phagolysosomal compartments known as autophagosomes. Autophagy emerges as a fundamental process in many aspects of immune cell physiology (54). Autophagosome formation requires the autophagy-related gene (Atg) protein family members, is followed by autophagosome maturation, and fusion with the endosomal and lysosomal organelles for cargo degradation. Autophagy contributes to cell homeostasis, providing nutrients during starvation, and is critical for cell quality control and disposal of dysfunctional or damaged organelles. In immune cells, autophagy participates in pathogen elimination, either directly or downstream of TLR activation (54). Recent data also showed a critical role of autophagy in regulating the inflammasome activity. Thus, a study of Zhou et al. revealed a role of autophagy-mediated disposal of damaged ROS-producing mitochondria to suppress IL-1β secretion (340). When autophagy was inhibited by knockdown of beclin 1 or Atg5, inflammasome activation by monosodium urate crystals or nigericin was enhanced. Mitochondrial ROS were also critical to explain the effects of autophagy inhibition on inflammasome activation (206). It was shown that in the absence of autophagic proteins LC3B or beclin 1, defective mitochondria accumulated upon treatment with LPS and ATP, which was associated with increased mitochondrial ROS generation. The involvement of mitochondrial ROS in inflammasome activation was further confirmed by the lower IL-1β secretion in cells devoid of mitochondrial DNA or in macrophages treated with the mitochondrion-specific ROS scavenger Mito-TEMPO. Defective mitophagy may also contribute to inflammasome activation by saturated fatty acids (Fig. 17). Mitophagy decreased during combined LPS/palmitate treatment of macrophages, which was associated with increased ROS formation and inflammasome activation (323). In contrast, increasing autophagy by adenosine monophosphate (AMP)-dependent protein kinase (AMPK) activation may rescue cells and prevent ROS formation and inflammasome activation. Another role for autophagy may be the sequestration and degradation of pro-IL-1β (107). Thus, inhibition of autophagy would sustain pro-IL1β for processing by the inflammasome. It has been shown recently that several NLRP3-activating stimuli trigger autophagy of whole inflammasomes, thereby limiting IL-1β production (262).
In the context of atherosclerosis, macrophages deficient in autophagy promoted atherosclerosis in ApoE knockout mice by activating the inflammasome and increasing IL-1β production (234). Inflammasome activation during atherosclerosis proceeds through accumulation of cholesterol crystals and subsequent lysosomal destabilization (61). In another atherosclerotic mouse model, autophagy deficiency increased macrophage apoptosis, while decreasing efferocytosis of dead cells (174). In both cases, the proatherosclerotic effects of autophagy deficiency were linked to increased ROS generation, although the source of ROS may differ (mtROS in the first model and NOX in the second).
XIV. The Redox Sensor AMPK Favors Regulatory Macrophage Activation
A critical component of homeostatic cell responses toward a variety of stress signals is AMPK (105). AMPK senses energy depletion by means of regulation through the AMP/ATP and adenosine diphosphate/ATP ratios. AMPK activation switches off anabolic processes, such as protein and fatty acid synthesis, while promoting catabolic processes, for example, glycolysis, fatty acid oxidation, and autophagy. In macrophages, AMPK supports regulatory macrophage polarization induced by IL-10 and TGFβ (245), probably by activating Akt. Further, AMPK counteracts fatty acid-induced inflammatory responses of macrophages either by increasing β-oxidation (82) or by activating NAD+-dependent deacetylase SIRT1 (334), thus promoting deacetylation and inactivation of NFκB. These two mechanisms may actually be linked, since AMPK-driven changes of the NAD+/NADH ratio and SIRT1 activation were shown to demand β-oxidation (27). In addition, an AMPK-dependent mitophagy may play an important role in ROS-mediated inflammasome activation by saturated fatty acids as discussed above (323). Studies from other cellular systems have shown several levels of interplay between ROS and AMPK. AMPK can be activated by ROS directly through the redox modification of cysteines (345) as well as by redox regulation of its upstream activator liver kinase B1 homolog (2, 331) and redox-induced changes of intracellular calcium, activating the AMPK upstream kinase CAMKKβ (202). In turn, AMPK activity may affect ROS formation by upregulating MnSOD, thioredoxin (42, 160), or UCP2 (332). AMPK may also interfere with NOX activation by attenuating NOX2 expression in response to angiotensin (251) or attenuating NFκB-dependent expression of several NOX components by inhibiting IκB proteasomal degradation (315). Whether AMPK interferes with macrophage NOX activation is still unknown.
XV. Iron Availability Dictates Macrophage Polarization
Iron not only plays a central role in different metabolic pathways as an essential cofactor of proteins/enzymes, but also is of crucial importance for the regulation of immune cell function. Recently, a role of iron in immunity, specifically in macrophage polarization, emerged (Fig. 18). Macrophages have evolved multiple pathways to acquire, recycle, process, store, and transport iron, including expression of the receptor for the main iron carrier protein transferrin, the iron storage protein ferritin, and lactoferrin receptors to take up the antimicrobial iron transport protein lactoferrin.

Iron can also be ingested by transmembrane acquisition, but the major iron uptake route in macrophages is phagocytosis of aging erythrocytes. In addition, macrophages express the surface receptor CD91, the hemopexin receptor, which takes up heme bound to the heme-sequestering protein hemopexin. The uptake of heme iron induces HO-1 in macrophages. Iron is needed for cytotoxic immune defense mechanisms as a central catalytic compound for the production of toxic hydroxyl radicals (243). Cytokines and radicals produced and released by immune cells are in turn crucial to intracellular iron homeostasis. Estimated 10%–20% of ingested iron remains in macrophages as a labile iron pool, which is necessary to maintain cellular iron homeostasis and to fulfill their effector functions. These are often regulated at the post-transcriptional level by iron-binding proteins (IRP), which bind to the RNA stem–loop structures, the iron-responsive elements (IREs) in the untranslated region (UTRs) of their target mRNAs. IREs have been identified in the 5′-UTR of mRNAs coding for proteins related to iron storage (ferritin), iron incorporation, for example, erythroid aminolevulinic acid synthase (e-ALAS), which catalyzes the synthesis of
In response to inflammation, iron is sequestered specifically in macrophages, which leads to a state of systemic inflammation-associated anemia. Proinflammatory cytokines and acute-phase proteins provoke iron sequestration in macrophages. LPS and proinflammatory cytokines such as IL-6 are described to induce hepcidin, the main iron regulator for whole-body iron homeostasis (289). Hepcidin is an acute-phase protein, mainly produced in the liver, which regulates iron transport across the gut mucosa. Hepcidin is also implicated in iron retention in macrophages, as it directly binds to FPN, the only known cellular iron exporter, to provoke its internalization and degradation. It also seems that iron retention in macrophages plays a crucial role in combating bacterial infections (208). The availability of iron influences the production of cytokines and thus significantly modulates macrophage polarization (see Fig. 18). In this sense, iron chelation results in marked suppression of mononuclear cell activation by inhibiting TNFα production (327). Polarization of macrophages plays a crucial role for their function in iron metabolism. Wound-healing macrophages are characterized by a rapid mobilization of their intracellular iron pools releasing it to the iron-lacking environment (235). During inflammation, proinflammatory cytokines downregulate the expression of the iron exporter FPN (43), whereby iron remains sequestered in macrophages. This seems to be a defense mechanism to withdraw iron from invading pathogens. Polarization of macrophages dictates the expression of genes involved in iron metabolism. Classically activated macrophages express high ferritin, natural resistance-associated protein 1 (Nramp-1), HIF-1α, SOD1, and low TfR. In contrast, wound-healing macrophages upregulate TfRs, divalent metal transporter-1, IRP1, IRP2, FPN, and HO-1. Thereby, the macrophage iron status influences its immune effector functions by regulating cytokine formation and the induction of the antimicrobial machinery (Fig. 18). Since iron is an essential compound for bacterial growth, the control over iron homeostasis is central. Phagolysosomal proteins such as Nramp-1 that are associated to resistance toward infection can act as iron transporters. Iron-laden macrophages lose their ability to kill intracellular pathogens by IFNγ-mediated pathways due to reduced formation of NO. In the presence of iron, iNOS transcription is blocked because iron inhibits binding of C/EBPβ and HIF-1 to the iNOS promoter (3). Consequently, macrophages show a lower inducibility in response to cytokines. The induction of lipocalin-2 (Lcn-2) by LPS could represent an alternative pathway of sequestering iron. Lcn-2 acts as a potent bacteriostatic agent due to its high affinity to iron, thereby sequestering iron from bacterial siderophores (198).
Macrophages also deliver iron to their neighborhood (81, 235). It was demonstrated that these macrophages express higher levels of HO-1 and FPN to increase the iron pool within the local microenvironment. Iron release from wound-healing macrophages contributes to their roles in tissue repair and tumor growth (Fig. 18). Higher iron availability could influence the growth of adjacent fibroblasts, since iron represents an essential factor for cellular proliferation and DNA synthesis. Iron overload also enhances ECM deposition due to the induction of PHD, which is important for collagen biosynthesis, thereby promoting repair, but possibly also fibrosis. Additionally, we recently described a role of Lcn-2, produced by wound-healing macrophages, during regeneration of an ischemic kidney (137). High FPN iron release may also occur in TAMs. This causes higher iron availability in the tumor microenvironment, which might represent a novel mechanism of protumorigenic macrophage characteristics. Previous data from our group showed that TAMs express and release Lcn-2 to induce proliferation and growth of tumor cells. Lcn-2 is currently discussed as an alternative iron shuttle protein by binding to its specific receptor (NGALR) on the cell surface. In addition, it may function as a possible iron export system because Lcn-2 binds iron with a high affinity inside macrophages and exports iron via exocytosis (Fig. 19). Iron-laden Lcn-2 shuttles to the tumor cell, where the uptake of iron–Lcn-2 complexes ensures their survival and growth. Conversely, the internalization of iron-free Lcn-2 would induce apoptosis, which is driven by iron deficiency and increased expression of the proapoptotic protein Bim (57). It is therefore tempting to speculate that tumor cells specifically take up iron-laden Lcn-2 to ensure their own survival. However, a defined mechanism of the tumorigenic function of Lcn-2 is still missing.

It can be speculated that alternative iron transport mechanisms mainly occur under pathophysiological conditions, while transferrin-bound iron transport is coupled to physiology. A possible nontransferrin-dependent mechanism may be linked to Lcn-2, since it binds iron with a high affinity and can act independently of transferrin. Additionally, high Lcn-2 concentrations are found in chronic inflammatory diseases such as cancer. Therefore, it may be speculated that Lcn-2 can provide iron to cells under pathophysiological conditions, associated with bacterial infections, inflammation (45), ER stress (261), or anemia (18). Biological functions of Lcn-2 are determined by binding to iron. In contrast to iron-deficient Lcn-2, iron-laden Lcn-2 increases intracellular iron, thereby inhibiting apoptosis by inducing the protective, antiapoptotic protein Bcl-2. It can further be postulated that Lcn-2 may serve as a new iron-handling protein in macrophages, adopting the macrophage to different stimuli.
XVI. Conclusions
The unique feature of macrophages is their high functional diversity, which positions them at the center of pathways that promote inflammation and control its resolution. Sensing environmental and internal signals allows their polarization, which coins macrophage functional properties. Redox signaling contributes to shape the role of macrophages during the onset, propagation, and termination of inflammatory responses. A large number of redox-active components operate simultaneously, being produced by a class of redox-generating systems that comprise NOX enzymes, mitochondria, or NO synthases and are balanced by the antioxidative capacity of a cell. Redox biology adds to macrophage polarization and thus their role in health and, disease. Multiple redox targets, among them several transcription factors such as Nrf2, PPARγ, HIF-1, or HIF-2, transform redox signals into the gene and protein signatures and the associated characteristic functional consequences. Table 2 summarizes information on how distinct macrophage phenotypes are related to health and disease in man or mice (and zebrafish) and how distinct macrophage functional characteristics are linked to defined redox signals.
mmLDL, minimally modified LDL; NOX, NADPH-oxidase; ER, endoplasmic reticulum; ROS, reactive oxygen species; CGD, chronic granulomatous disease; COPD, chronic obstructive pulmonary disease; Nrf2, nuclear erythroid-2-p45-related factor 2; FPN, ferroportin; C/EBP, CCAAT/enhancer-binding protein; HIF, hypoxia-inducible factor; iNOS, inducible NO synthase; TLR, Toll-like receptor; MDSCs, myeloid-derived suppressor cells.
It is evident that macrophages not only add to control the onset of disease conditions but also determine the course of disease as well as the process of disease resolution. Formation of ROS and/or NO during classical macrophage activation may favor phagocytosis, while little is known whether these distinguished redox signals determine alternative macrophage activation, although NOX4 might contribute to desensitize cells. In the context of lipid overload, macrophages respond with the characteristics of classical activation in conjunction with ER stress and oxidation of mitochondrial DNA. Signatures of classical macrophage activation are connected with activation of HIF-1 and NF-κB, which may operate in concert to boost inflammatory reactions, with the notion that HIF-1 is under redox control. How HIF-2 affects macrophage polarization is less understood. Although a few alternative activation markers are regulated by HIF-2, a generalized concept did not emerge so far. Despite HIF-1 and HIF-2 are associated with macrophage polarization, their transcriptional capacity apparently does not directly drive the polarization program. The redox-sensitive Nrf2 may add to a macrophage safeguard mechanism. When stabilized in classically activated macrophages, it signals back to further limit inflammation and cell self-destruction. Opposed is the role of PPARγ. Initially, this transcriptional machinery is operating at a low level, while it gets active during alternative activation to directly interfere with proinflammatory signaling. Iron sequestration and its release may further add to the macrophage phenotype. Sequestering iron in macrophage depletes the microenvironment, to limit pathogen or tumor cell replication and fostering the production of inflammatory components. In contrast, iron release may be beneficial for cell replication, to promote wound healing with the same mechanism hijacked by tumor cells to stimulate their own survival and growth. In the future, it will be challenging to use this knowledge of redox-regulated processes to shift macrophage polarization and thereby use distinct phenotypes either to limit inflammation or to boost tissue repair and regeneration. In contrast to oxidative stress, redox regulation emerges as a unique navigating mechanism to shape the multiple facets of the continuum of the macrophage subtypes in our body.
Footnotes
Acknowledgments
We apologize to researchers whose primary observations that form the basis of current knowledge in the field could not be cited due to space limitations, or have been acknowledged indirectly, by referring to current reviews. Our work was supported by the grants from Deutsche Forschungsgemeinschaft (SFB 815, BR999, and Excellence Cluster Cardiopulmonary System), Deutsche Krebshilfe (109599), TRIP (Translational Research Innovation Pharma), Hans Kröner-Graduate School, LOEWE-Schwerpunkt: Anwendungsorientierte Arzneimittelforschung, and a young investigator start-up funds by the Goethe-University Frankfurt, Faculty of Medicine (to MJ).