
Abstract
The Anthracyclines—History, Chemical Structure, and Pharmacology
The introduction of ANTs into clinical practice has been one of the major successes of modern oncology. This is particularly evident in pediatric oncology, where the 5-year survival rate for childhood cancer has increased from ≈30% in the 1960s to ≈80% seen today (236), whereas it is estimated that over 50% of the childhood cancer survivors have received ANTs (144). Even though now half-a-century old, ANTs remain among the mainstays of the modern combination cancer therapy. Rather than being supplanted by various novel targeted agents, there is a tendency to combine them in various schedules to maximize the therapeutic response (206).
As seen in Figure 1, ANTs are composed of a rigid planar tetracyclic structure with adjacent quinone and hydroquinone moieties, a short side chain with a carbonyl group at C-13, and an aminosugar daunosamine attached by a glycosidic bond to the C-7 of the tetracyclic ring. DOX differs from DAU only by the presence of a C-14 hydroxyl group. EPI is an epimer of DOX differing only in the orientation of hydroxyl group on daunosamine. Idarubicin is a DAU analog that lacks a C-4 methoxy group (292).

For a long time since their introduction, the precise mode of ANT anticancer action has not been well defined. The first proposed mechanism was DNA intercalation (185). The planar aromatic aglycone part of the molecule intercalates between two base pairs of the DNA, while the six-membered daunosamine sugar is positioned in the minor groove, and interacts with flanking base pairs immediately adjacent to the intercalation site (207). ANTs may also be toxic to cancer cells by reactive oxygen species (ROS) formation and site-specific DNA damage (190). Today, topoisomerase IIα (TOP2α) is generally recognized as the main target of ANT antitumor action (34). ANTs belong to the TOP2 poisons, a group of cytotoxic agents stabilizing a reaction intermediate in which DNA strands are cut and covalently linked to tyrosine residues of TOP2, eventually impeding DNA resealing. Failure of relaxing the supercoiled DNA results in block of DNA replication and transcription. Double-strand DNA breaks could ultimately then trigger apoptosis of cancer cells, apparently via the p53-dependent pathway (220).
ANTs are administered strictly by intravenous infusions. They are known for their large volumes of distribution (∼800 L/m2 for DOX) due to the wide distribution of the drug into the tissues and their binding within the intracellular compartment. Marked intracellular accumulation can result in concentrations 10–500-fold higher than extracellular drug levels, and this corresponds with considerably long terminal half-life of elimination (24–50 h). DOX is ∼75%–80% plasma protein-bound, and plasma clearance is mediated mostly through hepatic metabolism and biliary excretion (75).
With respect to adverse/toxic effects, myelosuppression predominantly leading to neutropenia is the major dose-limiting complication of ANT-containing chemotherapy (75). Nausea, mucositis, diarrhea, and alopecia are common, but reversible. Accidental extravasation of the drug during administration can cause severe necrosis of affected tissue (75). The main complication associated with ANT treatment is however the risk of irreversible life-threatening cardiomyopathy and congestive heart failure (CHF) (75).
ANT-Induced Cardiotoxicity
Classification, incidence, and risk factors
ANTs are clearly the most dangerous anticancer drugs with respect to cardiovascular toxicity and are now perceived as the prototypic drugs responsible for Type I of chemotherapy-induced cardiac dysfunction (60). This type of cardiotoxicity is characterized by mostly irreversible morphological changes and cell death, which differ considerably from largely reversible Type II cardiac dysfunction induced by trastuzumab or tyrosine kinase inhibitors (60).
While undetected during preclinical animal studies (101), cardiotoxicity of ANTs has been first documented during early clinical evaluations of DAU (261) or DOX (22, 156). In 1979, the seminal study by Von Hoff et al. demonstrated cumulative ANT dose as the key risk factor for cardiotoxicity development (284). This study recommended a cutoff for DOX at a cumulative dose of 550 mg/m2, based on a perceived relatively low incidence of cardiotoxicity (≤7%) in doses up to that level (284). However, in 2003, a meta-analysis by Swain et al. found that an estimated 26% of patients would experience DOX-related manifest CHF at a cumulative dose of 550 mg/m2 (258). Furthermore, data from the Childhood Cancer Survivor Study showed that as little as 100 mg/m2 of DOX might cause a trend toward an increased risk of detectable asymptomatic abnormalities, whereas 270 mg/m2 of DOX induced a measurable 4.5-fold excess risk of such abnormalities (119). Today, ANT cardiotoxicity is viewed as a multifactorial process that primes the heart to a life-time risk of systolic and/or diastolic dysfunction. The ANTs may interact and eventually synergize with comorbidities, unfavorable lifestyle, and cardiovascular toxicity of other anticancer drugs (182). Therefore, additional risk factors such as hypertension, early or advanced age, obesity, diabetes mellitus, coronary artery disease, prior irradiation, exposure to other cardiotoxic drugs, or prior heart failure have been defined (59, 294).
Several types of ANT cardiotoxicity have been recognized with a rather variable classification (62, 116, 131, 296): Acute cardiotoxicity occurs during or soon after the ANT administration (116, 131). It is often subclinical, but vasodilatation with hypotension and transient cardiac rhythm disturbances may be observed (116, 131). These changes are usually not dose dependent, resolve spontaneously, and do not preclude further ANT use (62). In addition, no connection to chronic ANT cardiotoxicity has been established (72, 185). Pericarditis–myocarditis syndrome may occur within 1–3 days; however, it was particularly seen in early trials employing high individual doses of ANTs and is uncommon today (27, 83). Clinically, the most significant are the chronic forms of ANT cardiotoxicity with the early- and late-onset manifestations. The early-onset form develops later in the course of the chemotherapy or within the first year after its completion. It is typically characterized by development of left ventricular (LV) systolic dysfunction, which progresses toward dilated cardiomyopathy and CHF (116, 131). Histopathological hallmarks of the ANT cardiotoxicity are found predominantly in the left ventricle and interventricular septum, and consist of distension of the sarcoplasmic reticulum, cytoplasmic vacuolization within the myocytes, mitochondrial swelling, myofibrillar disarray, and eventual loss (62). These latter ultrastructural changes were quantitated by Billingham et al. and later by Mackay (21, 174). The second category of the chronic ANT cardiotoxicity is the late-onset (or delayed) form. It was described at the beginning of the 1990s among survivors of childhood cancers (76, 167, 253), and it is now well established that ANT cardiotoxicity may manifest even decades after the anticancer treatment (235a).
Chronic forms of ANT-induced cardiotoxicity are associated with poor prognosis, and patient survival seems to be worse than in other types of CHF (61). At present, there is no specific evidence-based treatment of ANT-induced cardiotoxicity (159), and the treatment follows the guidelines for chronic HF. Angiotensin-converting enzyme inhibitors (ACEI) and β-adrenergic receptor-blocking drugs (β-blockers) may be used already upon asymptomatic drop of the LV systolic function (159). For patients with end-stage CHF, heart transplantation remains the last option (268).
In addition, there is a considerable interindividual variability in the susceptibility to chronic ANT cardiotoxicity. Some patients develop cardiotoxicity at a total cumulative ANT dose of 300 mg/m2 or even lower, whereas some early studies suggested that some patients had no significant cardiac complications despite achievement of the doses as high as 1000 mg/m2 (284). These findings strongly suggest the involvement of a genetic component and pharmacogenomic approach pointed to a potential role for polymorphisms in several candidate genes that include carbonyl reductase 3, glutathione S-transferase, multidrug resistance proteins 1 and 2, or NAD(P)H quinone oxidoreductase (48).
Cardiac assessment before, during, and after the ANT-containing treatment is essential. Among the first approaches employed for sensitive and reliable detection of the chronic ANT cardiotoxicity was the endomyocardial biopsy (21). However, its invasive nature hinders and in fact nearly prevents its routine use in modern clinical practice. Current recommendations for cardiac monitoring of ANT-treated patients are thus mostly based on the noninvasive examination of the LV systolic function, since its decline is a well-known hallmark of ANT cardiotoxicity (129). Echocardiographic assessment of diastolic function has been reported as a more sensitive tool for the detection of asymptomatic early myocardial damage (70). Furthermore, selective biochemical markers of myocardial damage (particularly cardiac troponins T and I and natriuretic peptides such as NT-pro-BNP) are also valuable for early detection of cardiotoxicity (180, 225).
ANT-induced cardiotoxicity can be influenced by concomitant application of other anticancer drugs. Trastuzumab, a humanized monoclonal antibody against the HER2 (human epidermal growth factor receptor 2, also known as c-erbB-2 or neu), is an important component of current chemotherapy protocols for women suffering from HER2-positive breast cancer. However, its use has been associated with a higher incidence of symptomatic or asymptomatic cardiac dysfunction that is apparently related to interference with HER2-dependent prosurvival signaling in the cardiomyocytes (73). Even though the trastuzumab-related cardiotoxicity is far less dangerous than that of ANTs, their concomitant use is associated with CHF development at significantly lower cumulative ANT doses (73). Furthermore, ANT cardiotoxicity can be exacerbated also by chemotherapeutics that possess limited cardiotoxicity per se, such as in the case of concomitant administration of paclitaxel (73).
Strategies for cardiotoxicity prevention
In the light of difficult treatment, there has been a continuous effort to prevent or reduce the risk of cardiotoxicity development (296). The most effective, simple, and widely employed tool to prevent the ANT-induced cardiotoxicity is the limitation of cumulative ANT dose (258, 296). For each ANT, there are now recommended total cumulative doses that may be exceeded only with very careful cardiac monitoring (2) and/or pharmacological cardioprotection. However, lower ANT doses may imply the risk of lower cancer response rate, and considerable interindividual variability hinders reliable risk prediction in individual patients. Furthermore, it is increasingly recognized that there are no entirely safe ANT cumulative doses (258).
Over the past five decades, more than 2000 modified ANT chemical structures have been synthesized and tested. The main effort was to reduce cardiotoxicity while retaining their unique cytostatic effectiveness. While several ANT derivatives have been evaluated in clinical trials (e.g., amrubicin or pirarubicin), only EPI and idarubicin received approval for clinical use. Unfortunately, clinical trials have not confirmed lower cardiotoxicity of idarubicin as compared to DAU (16, 198). In some chemotherapy regimens, EPI is favored over DOX due to the perceived lower risk of cardiac injury. However, as EPI is administered in higher individual and cumulative doses, there is a question that a distinct difference between these two drugs exists at the equipotent doses (67). Despite the limited success, the search for less-cardiotoxic ANTs still continues.
The next important strategy to ameliorate the risk of ANT cardiotoxicity is the modification of ANT administration. Several reports have suggested that continuous infusion with reduced peak ANT concentrations is associated with a lower risk of cardiotoxicity than administration as a bolus (157, 235d). However, literature is not conclusive, as several studies failed to find a relationship between the infusion duration and cardiotoxicity risk (161, 168, 251). Another popular and widely tested strategy is to alter the pharmacokinetics (but also pharmacodynamics) of ANTs by their passive targeting into the tumors that may reduce myocardial exposure. This can been achieved by liposomal encapsulations of the drugs, and such delivery systems are available for DAU (DaunoXome™) as well as DOX (Myocet™, Caelyx™, and Doxil™). These formulations have distinct properties, such as stability, drug-release rate, plasma half-life, sites of deposition of the liposome, or toxicity profiles (160). Several controlled trials have been published that demonstrated a lower overall risk of cardiotoxicity of liposomal ANTs in adults (14, 197). In children with cancer, several observational studies described the use of liposomal ANTs, but no randomized controlled trials have been reported. Hence, definite data on cardiac safety of these formulations need longer follow-up of both cardiotoxic effects as well as the anticancer response (237). Of note, liposomal ANTs also have specific side-effects, such as allergic reactions and hand-foot syndrome, a dermatologic toxic reaction that can lead to considerable discomfort (82). Finally, the method for ANT cardiotoxicity prevention with the strongest evidence from clinical trials is the pharmacological cardioprotection with dexrazoxane (DEX) (257).
Experimental models of ANT-induced cardiotoxicity
Clearly, the best and the most relevant data should be obtained from randomized controlled clinical trials. However, regarding cardiotoxicity assessment, one may usually evaluate only noninvasive parameters, as human myocardial biopsy or necropsy samples are hard to obtain. Furthermore, the majority of cancer patients are treated with multidrug regimens, which together with overall heterogeneity of their clinical picture make the interpretation of clinical data troublesome. Some approaches are also either ethically or practically unfeasible in clinical settings. Therefore, the use of experimental model systems is unavoidable.
In all nonclinical studies, the selection of an appropriate experimental model is an essential point determining translatability of research outcomes into clinical medicine. The simplest are cell-free in vitro experiments, such as with combinations of chemicals in buffered solutions, purified proteins, or subcellular fractions (e.g., microsomes or mitochondria). Very common is then the use of cellular models using isolated cardiac myocytes—mostly, primary neonatal rat cardiomyocytes (10, 89, 112, 146), and less often, adult cardiomyocytes (136). During the last decade, studies with immortalized cardiomyocyte-derived cell lines (in particular the H9c2 rat embryonic cardiomyoblasts) have emerged (138, 186, 279). Ex vivo studies often use isolated atria (although these are not a major target for ANT cardiotoxicity), papillary muscles, or whole-heart preparations perfused according to Langendorff (217). It should be noted that the majority of these biological models enable only limited time of exposure, which hinders simulation of the chronic ANT cardiotoxicity. In addition, increasing ANT concentrations to supratherapeutic levels help to achieve distinct toxicity, but again, it may hinder translatability of such data to in vivo settings. For in vivo studies, mouse, rat, rabbit, pig, and dog have been the commonly used animals (101). Importantly, these whole-animal experiments enable repeated administration of ANTs and chronic cardiotoxicity development, relevant to what is seen in clinical practice (71, 101, 139, 239). Nevertheless, numerous studies have unfortunately used regimens where animals were treated only with single or few very high ANT doses. The outcomes from these studies have to be treated cautiously. Not only there is a risk of oversimplification, but also importantly, the induced myocardial impairment need not share many common denominators with the chronic ANT cardiotoxicity feared in clinical practice (242).
Molecular mechanisms of ANT-induced cardiotoxicity
Despite the four decades of vigorous research, the pathophysiology of ANT-induced cardiotoxicity is still not fully understood, and it is a subject of debate and considerable controversy (11, 35d, 72, 131, 184, 185, 235a, 312). The prevailing and traditional hypothesis is linked to an iron-catalyzed induction of cardiac oxidative stress (242) and will be discussed in more detail in the following sections. In addition, multiple other possible mechanisms have been proposed such as impairment of calcium homeostasis (250) that can be related to direct effect of either ANTs or their more polar hydroxyl metabolites (183). The calcium overload may lead to sarcomere disruption due to the calpain-dependent degradation of titin, a giant myofilament protein, crucial for force modulation in the myocytes and a scaffold for assembly of myofilamentous proteins (164). Some other suggested cardiotoxicity mechanisms include, for example, impaired gene expression of various important cardiac proteins (25), dysregulation of protein degradation by the ubiquitin–proteasome system (247), induction of mitochondrial DNA lesions (154), or interference with TOP2 (173) (Fig. 2).
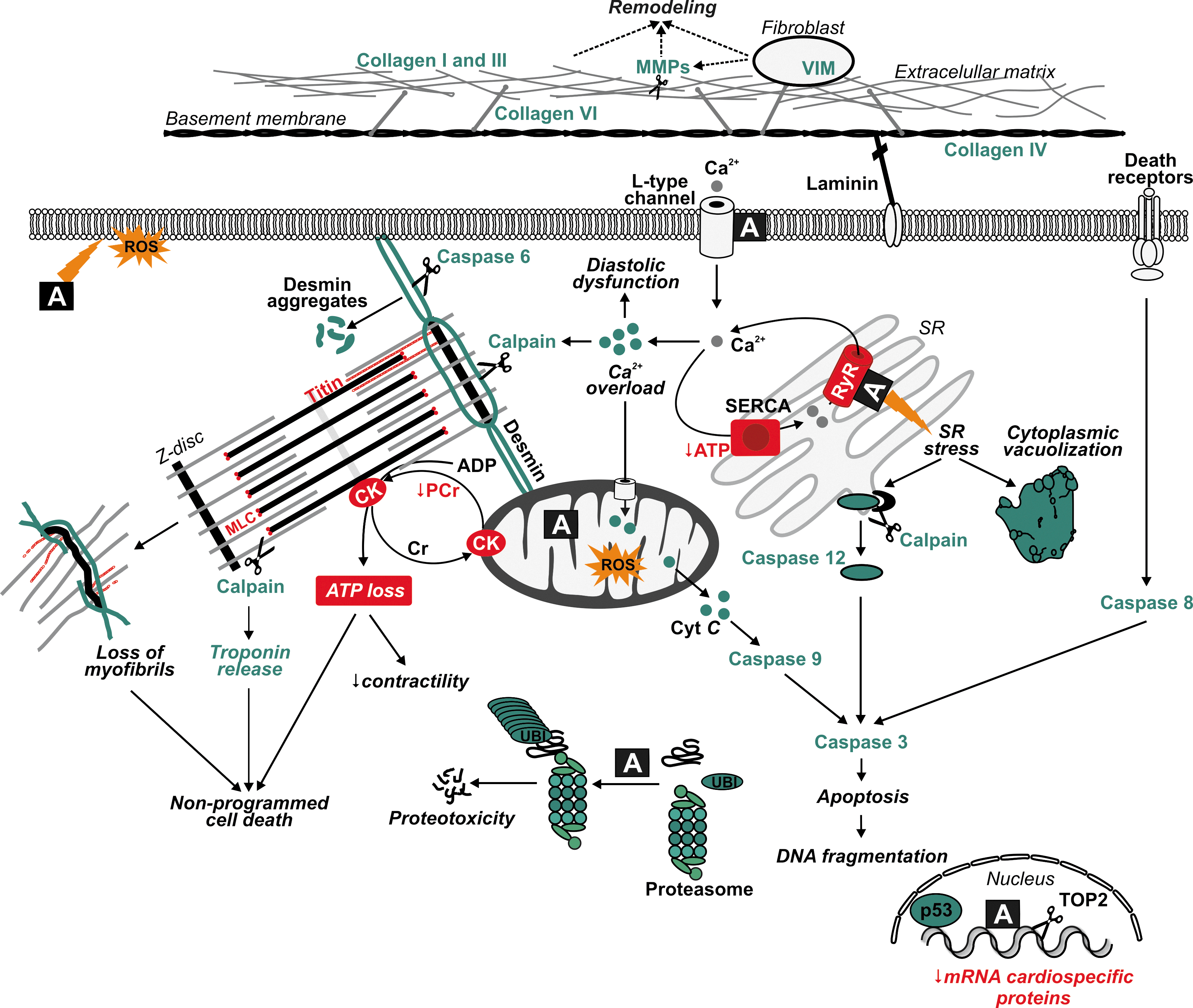
ANT-induced ROS production
ANTs are well known for their ability to produce ROS through multiple pathways (116, 137, 242) (Fig. 3). First, one-electron reduction of ring C of the ANT tetracycle leads to the formation of a semiquinone free radical. Its unpaired electron can be donated to oxygen forming superoxide radicals (•O2 −). Suitable flavoproteins catalyze the formation of reduced semiquinone radicals by accepting electrons from NADH or NADPH and passing them to ANT in a sequence of reactions known as the redox cycling. The dismutation of •O2 − to hydrogen peroxide (H2O2) is catalyzed by superoxide dismutase (SOD) or may occur spontaneously. H2O2 is a relatively stable and low-toxicity molecule, and under physiological conditions is eliminated by catalase or glutathione peroxidase. However, H2O2 and •O2 − may generate highly reactive and toxic hydroxyl radicals (•OH). This takes place during the Haber–Weiss reaction, which is very slow unless catalyzed by transition metals—especially iron (84).
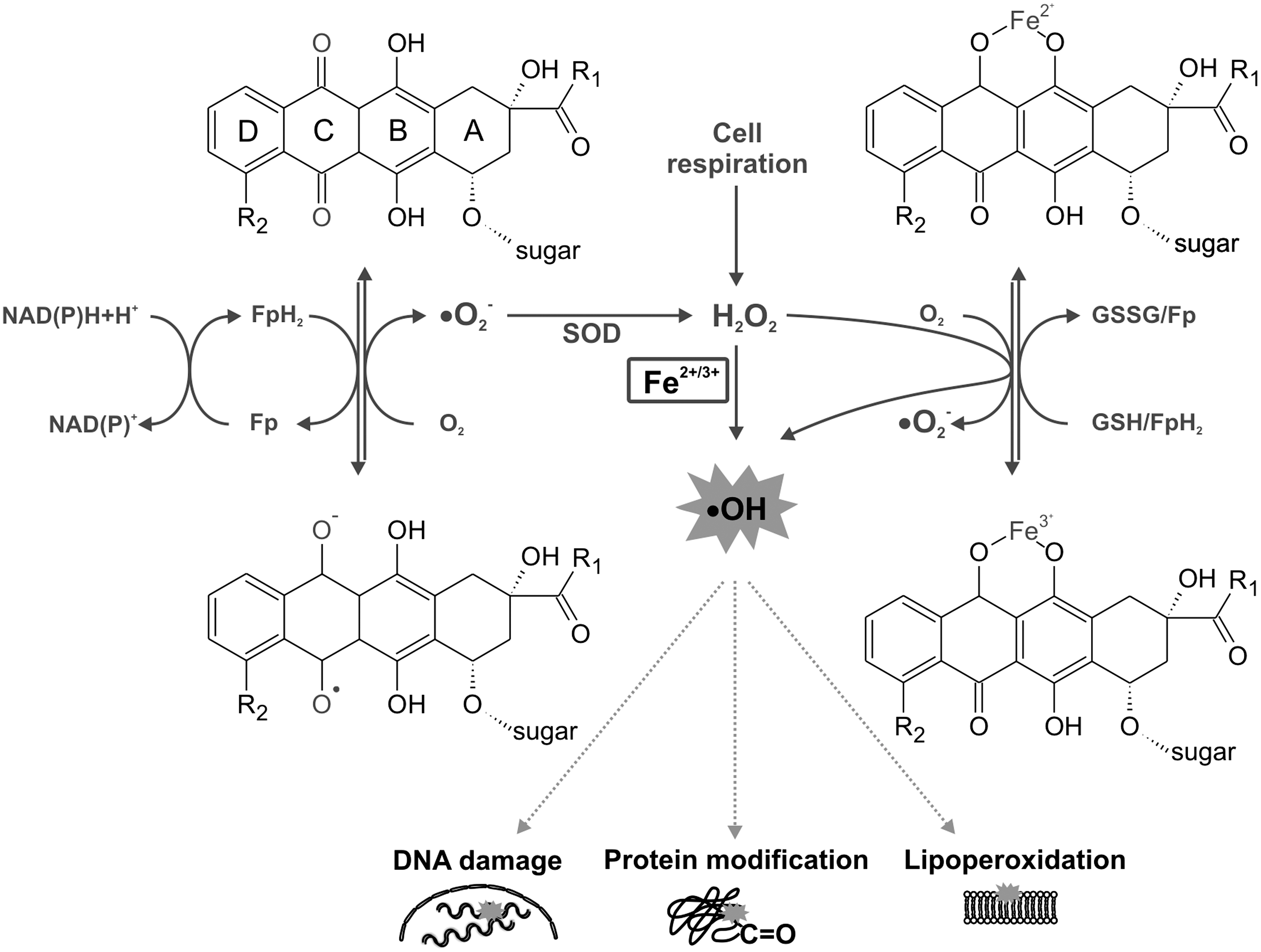
Thomas and Aust have shown that ANTs may increase the amount of free redox-active iron by generating •O2 −, which mediates an iron release from ferritin (267). The formed •OH has a very short half-life and extreme high reactivity and toxicity. It reacts with any oxidizable compound in its vicinity and thus can induce damage to all types of macromolecules, including lipids, nucleic acids, and proteins (84).
The second basic mechanism by which iron may promote the oxidative stress induced by ANTs is via the formation of ANT-Fe complexes (137). In the presence of a reducing system (NADH cytochrome P450 reductase or thiols, such as cysteine or glutathione), the ANT-Fe3+ complex is reduced to ANT-Fe2+, which can react with O2 to form •O2 −, which in turn dismutates to H2O2 and/or enters the Haber–Weiss reaction, resulting in •OH. Alternatively, ANT-Fe2+ can react with H2O2 yielding directly •OH. In the absence of reducing systems, ANT-Fe3+ can reduce its chelated iron by an intramolecular redox reaction, by oxidation of either the side chain on C-9 or the hydroquinone moiety at ring B, forming a free radical •ANT-Fe2+. In the presence of O2, this complex is oxidized, yielding •O2 − and •ANT-Fe2+ (116, 137, 242). Furthermore, apart from the ROS, cardiac exposure to ANTs may be also connected with the deregulation of the nitric oxide (NO) network. Overproduction of ROS and NO yields reactive nitrogen species (RNS), particularly the powerful oxidant—peroxynitrite (ONOO−) (64).
Involvement of iron in the ANT-induced oxidative stress and cardiotoxicity has been supported by rich experimental evidence as reviewed in detail elsewhere (242). Already in the early 1980s, Myers et al. have shown formation of the ANT-Fe complexes and resulting potentiation of oxidative damage of biomembranes in vitro (194). The ANT-Fe complex-mediated damage could not be blocked by classical ROS scavengers. Later on, augmentation of ANT cardiotoxicity by iron has been demonstrated in isolated rat cardiomyocytes, where lactate dehydrogenase (LDH) release and changes in cell contractility have shown a marked augmentation of DOX toxicity by prior iron loading (111). In vivo, using DOX-treated rats, iron loading resulted in severe weight loss and a twofold increase in the rate of mortality (166). In both studies, the unfavorable effects of iron on DOX cardiotoxicity could be eliminated by the iron chelator deferoxamine (DFO). Substantially increased DOX cardiotoxicity by dietary iron loading in rats was also reported by the study of Panjrath et al., concluding that body iron stores as well as iron bioavailability in tissue may be important independent predictors of susceptibility to DOX cardiotoxicity in man (204). Miranda et al. have shown that Hfe-deficient mice (a model of human hereditary hemochromatosis, a disorder resulting in body iron overload) exhibited significantly greater sensitivity to DOX-induced cardiotoxicity. Increased mortality rate after chronic DOX treatment was observed in both Hfe −/− and Hfe+/ − mice compared with wild-type animals. DOX-treated Hfe −/− mice had a higher degree of mitochondrial damage and iron deposits in the heart than did the wild-type mice (187).
ANT-induced formation of ROS/RNS within the cell context
The investigation on intracellular sites of ANT redox cycling and production of ROS/RNS pointed to a potential involvement of the mitochondria, sarcoplasmic reticulum, perisarcolemmal space, and cytoplasmic compartments (Fig. 4). Although the flavin-containing reductases are located in all these compartments, mitochondria have attracted the main interest. Nevertheless, the involvement of NADP oxidases (Nox) located in the cytosol and attached to the sarcolemma cannot be excluded (309).
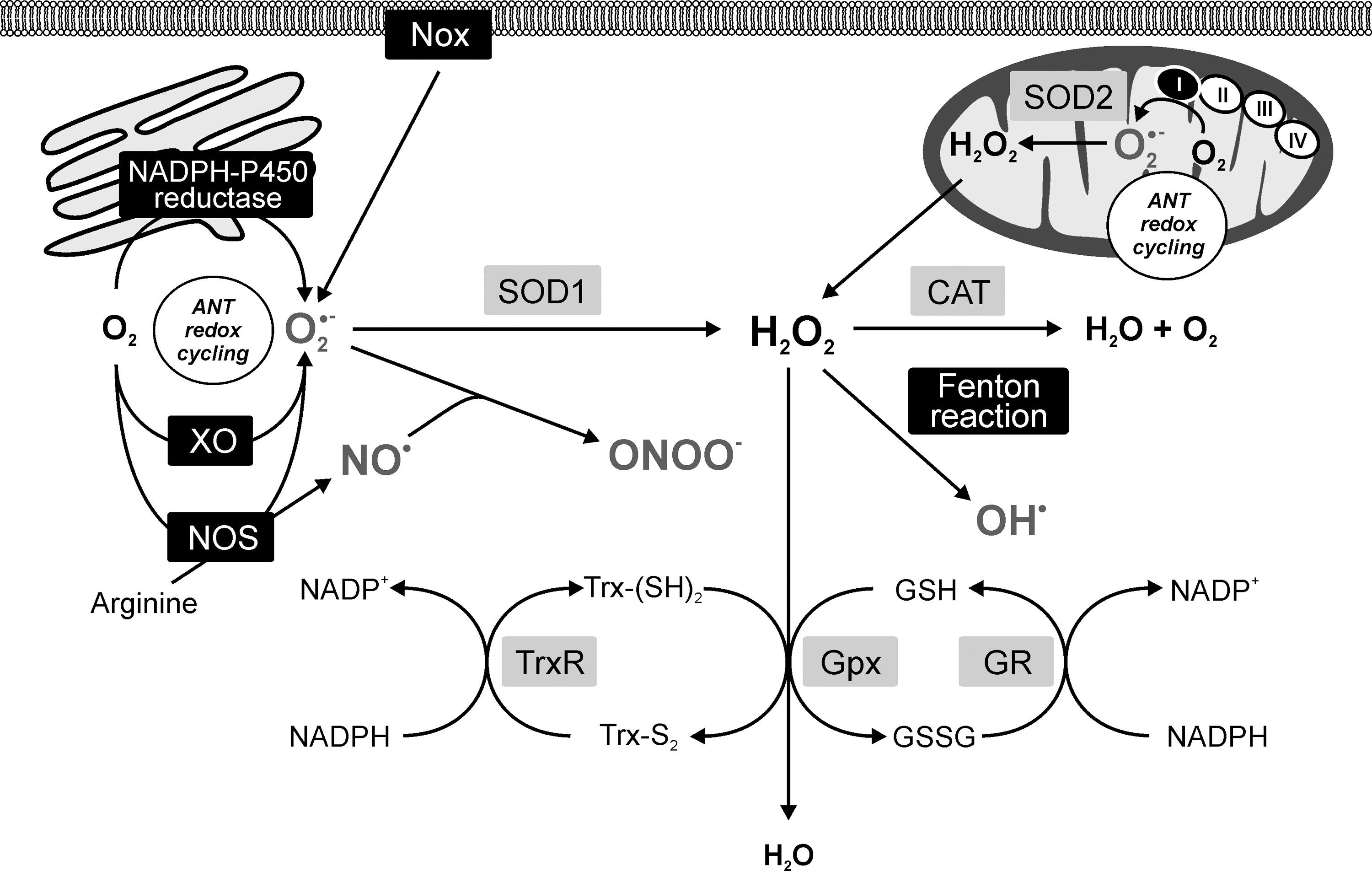
ANT cardiotoxicity, ROS, and mitochondria
Mitochondria are among the most important sources of ROS under both physiological and pathological conditions (192). Hence, ANT-induced ROS production was quickly attributed to the mitochondria. In addition, unlike other organelles, mitochondria contain their own genome, which is quite prone to oxidative damage, and this has been also linked to ANT toxicity development (19, 155). The connection between ANTs and mitochondria is not only mechanistic. ANTs possess a high affinity for cardiolipin, a negatively charged phospholipid of the inner mitochondrial membrane (77). Thereby, ANTs are retained at high concentrations in the mitochondrial compartment, particularly in the proximity of cardiolipin-associated proteins, such as the NADH dehydrogenase—complex I. Mitochondria have been repeatedly suggested as the most important target for ANT cardiotoxicity (18, 43, 175, 271, 285), and similar results have been also suggested by recent unbiased proteomic and phosphoproteomic investigations (Fig. 5) (79, 254).

ANTs have been reported to directly and indirectly interact with mitochondrial oxidative phosphorylation. In vitro, ANTs increase basal (state 4) respiration (78), which is explained by electron-accepting properties of the ANT molecule, causing partial diversion of electrons from the respiratory chain (18). This process results into one-electron reduction of the ANT molecule to the semiquinone radical with subsequent production of •O2 − as described above. Further experiments identified that this process takes place at complex I of the respiratory chain, and this site has been proposed as the most important site for ANT redox cycling within the cardiomyocytes (43, 44, 52). The ANTs interact with the electron flow through complex I proximally to the rotenone-binding site (285), but molecular details of this interaction remain obscure.
Complex I is not only the major source of ROS but also an important target of their toxic action. This has been found not only in acute cardiotoxicity models (302), but more importantly, also in several well-established chronic ANT cardiotoxicity models (199, 201, 254). Furthermore, there are some indices that complex I impairment could be an early event preceding CHF development (199). ANTs may also impair the function of other components of the respiratory chain as reviewed in detail by others (271). However, it is unclear whether these components are primary targets for ANT cardiotoxicity or mere consequence of previous perturbation in the electron transport chain or induced by elevated mitochondrial ROS due to the ANT redox cycling at complex I (18). Furthermore, some of these effects have been found only in rather high (supratherapeutic) doses or concentrations of ANTs, and thus their clinical relevance is unclear.
Importantly, using a chronic ANT cardiotoxicity model, Serrano et al. have described that ANTs induce a cumulative oxidative damage to mtDNA, and the change was detectable several weeks after the treatment (235b). Furthermore, Adachi et al. have shown that chronic ANT cardiotoxicity is associated with deletion mutations in mtDNA, which appeared to be at least partially oxidative stress dependent (1). Using low individual and total cumulative doses (DOX 1 mg/kg, weekly for 7 weeks) and a long follow-up period (30 weeks), Lebrecht et al. have reported a decrease in mtDNA/nDNA content and presence of common deletion mutations in mtDNA in rat hearts (153). These authors also found a relative decrease in activity of complex IV along with impaired expression of mtDNA-encoded complex IV subunits at this timepoint. Interestingly, these changes were not significant a week after the last drug administration, which highlights the importance of the time factor.
ANT-induced qualitative and quantitative mtDNA lesions with subsequent impairment of expression or compromised functionality of mtDNA gene products may explain the perpetuation of tissue damage for weeks, months, or even years after the exposure to drug (19, 155). Considering that mtDNA encodes 13 subunits of the respiratory chain, long-lasting defects in mitochondrial respiration, energy deficit, and sustained oxidative stress are expectable. If the ANT-induced oxidative stress plays a pivotal role in this process, the above-described process may roundup its complex and vicious role in ANT cardiotoxicity. On the other hand, using human autopsy myocardial samples, it has been found that the above-described mtDNA lesions and increase in thiobarbituric acid-reactive substances (TBARS) were typical only for DOX-treated patients, whereas patients treated with EPI or idarubicin did not show even a trend toward such changes (152). These data may question the key importance of mtDNA damage and mutations for cardiotoxicity development, as it is well established that chronic ANT cardiotoxicity is not unique to DOX; instead, it is a class effect with very similar morphological and functional features for all derivates (185). However, small groups of patients, heterogeneity in the administrated cumulative doses, coadministrations of other anticancer drugs, as well as autopsy character of the samples may complicate interpretation of these outcomes.
ANT-induced oxidative stress has been also implicated in the alterations in the mitochondrial creatine kinase (mtCK). The sarcomeric isoform of this enzyme (smtCK) is crucial for mitochondrial energy channeling and creation of energy reservoir. In vitro, DOX has been shown to induce mtCK dissociation from octamers to dimers together with impaired anchoring of the enzyme to the mitochondrial membrane, and smtCK has been shown particularly sensitive to this action of ANTs (269). The enzyme may undergo ANT-induced oxidative modification of cysteine at the active site (269). An isolated heart experiment (2 μM DOX, perfusion for 1 h) suggested that such changes might take place even under clinically relevant conditions (270). Unfortunately, rather limited data on smtCK are available from chronic models of ANT cardiotoxicity. Robison et al. have shown decreased total myocardial CK activity along with cardiomyopathy development in rats after repeated administration of DOX (2 mg/kg, weekly for 13 weeks, 1–6-week follow-up). However, the authors have found no increase in myocardial TBARS content, although they detected increased TBARS in the kidneys along with the development of nephrotoxicity (218). Marked decrease in smtCK abundance has been found in the myocardium of DAU-treated rabbits (3 mg/kg, weekly for 10 weeks), and this was at least in part connected to downregulated gene expression (254). In addition, using 31P NMR, Maslov et al. have detected 32% decline in the myocardial creatine phosphate-to-ATP ratio (PCr/ATP) after repeated DOX dosing (5 mg/kg, weekly for 5 weeks, and 5-week follow-up) (178). Of note, these changes preceded a significant decline of the LV systolic function.
Both decreased mtCK expression and activity may result in seriously impaired energy channeling, especially when it has been demonstrated that ANT treatment may disrupt expression and function of adenine nucleotide translocase-1 (ANT1) (254, 286), which is in a functional complex with smtCK at the inner mitochondrial membrane. In addition, impaired energy channeling may have a significant downstream negative effect, due to the inadequate stimulation of the mitochondrial respiration, and further exacerbate the failure to meet the energy requirement of the cardiomyocytes.
ANTs have been shown to have a profound impact on mitochondrial superoxide dismutase (MnSOD, SOD2), a manganese-containing enzyme located within the mitochondrial matrix. This enzyme is responsible for prompt conversion of mitochondrial •O2 − to H2O2, which is less reactive, and better crosses cell membranes. Interestingly, Li and Singal have shown that repeated ANT treatment (DOX 2.5 mg/kg, six doses over 2 weeks) markedly downregulates expression of MnSOD both at mRNA and protein levels and decreases its enzymatic activity 1 to 24 h after the last ANT dose with persistence for up to 3 weeks (162, 163). Similar outcomes have been reported from a proteomic analysis of chronic DAU-induced cardiotoxicity (3 mg/kg, weekly for 10 weeks) (254). This finding may also imply that oxidative stress can be substantially compartmentalized in ANT cardiotoxicity, as ANT-induced •O2 − produced within mitochondrial matrix cannot cross the inner mitochondrial membrane as easily as H2O2. Furthermore, increased local concentrations of •O2 −might also favor its interaction with NO to form extremely reactive peroxynitrite, which may in turn cause local nitrosative stress. Interestingly, ANT-induced nitrosative stress has been documented to modify MnSOD to diminish its enzymatic activity (263), and this could round up the inability of the mitochondria to face the increased stress. Further studies should demonstrate whether this takes place in the chronic ANT cardiotoxicity settings.
ANT-induced oxidative and nitrosative stress in the mitochondria may have numerous consequences. Not only they may deregulate mitochondrial respiration and bioenergetics, but it may also result in mitochondrial calcium overload, which has been described by several groups as reviewed elsewhere (151, 286). Mitochondrial matrix calcium overload has been well connected with mitochondrial permeability transition pore (mPTP) opening and depolarization of the inner mitochondrial membrane, which lead to ATP depletion and further oxidative stress aggravation (128). In addition, mPTP opening is associated with mitochondrial swelling, which has also been observed in electron microscopic studies of hearts suffering from ANT cardiotoxicity (21). Unfortunately, the in-depth study of mPTP opening in chronic ANT cardiotoxicity is complicated by the inability to simulate appropriately these conditions in vitro on one hand and potential selection bias toward ANT-damaged cells upon isolation of the cells from chronically treated animals on the other hand.
Mitochondrial swelling can result in rupture of the inner mitochondrial membrane and release of apoptosis-inducing proteins. The latter event may be also triggered by the proapoptotic Bcl-2 family proteins creating nonselective channels in the mitochondrial membrane. Indeed, clinically relevant doses and concentrations of ANTs have been repeatedly shown to activate apoptotic pathways (8, 196, 208, 226, 301), and successful cardioprotection with DEX has reduced this process (208). Hence, mitochondrial oxidative stress as well as other above-described events may trigger apoptotic cell death, which may be coresponsible for CHF development. Although, mitochondrial autophagy is likely to take place in ANT cardiotoxicity, and one cannot exclude its impaired regulation, as very little is known so far about involvement of this process in chronic ANT coardiotoxicity (246). Quite obscure is also the role of mitochondrial fission and fusion as well as mitochondrial biogenesis. Hence, all these processes deserve further studies preferably using chronic models, as this investigation may provide more complex and significant insights into the global picture of the chronic ANT cardiotoxicity.
ANT-induced ROS production outside the mitochondria
Besides mitochondrial complex I, other flavin-containing enzymes located outside the mitochondria have been implicated in ANT redox cycling, including NADPH oxidase, CYP450 reductase, or nitric oxide synthase (NOS) (50) (Fig. 4). Interestingly, the location of some of these enzymes matches with the location of ultrastructural damage to cardiomyocytes induced by chronic ANT cardiotoxicity.
Over the last decade, NADPH oxidase (Nox) has been suggested as one of the most important sources of ROS in the cardiovascular system. While five Nox isoforms have been distinguished, Nox2 and Nox4 are believed to play a major role in the heart, where they are bound to the sarcolemma of the cardiomyocytes (245). Wojnowski et al. have revealed that genetic polymorphisms in three cytoplasmic subunits of Nox2 make patients more prone to develop cardiotoxicity after the ANT treatment (295). However, only polymorphisms in p40phox have been found associated with chronic ANT cardiotoxicity. In addition, using the Nox2 −/− mice, the authors have shown that genetic disruption of Nox2 protects the heart from LV dilation and systolic dysfunction induced by DOX (4 mg/kg, weekly for 3 weeks, and 5-week follow-up). Furthermore, using a similar DOX treatment, no significant changes in LV systolic and diastolic function, LV diameter, cardiac fibrosis, and Nox4 expression were found in Nox2 −/− animals (309). Mukhopadhyay et al. have detected no change in expression of Nox1, Nox2, and its regulatory subunits after single high-dose DOX treatment (20 mg/kg) (191). These authors also did not observe any effect of Nox inhibitors on DOX-induced toxicity in H9c2 cells (DOX 1 μM, 16 h exposure). However, Zhao et al. have found a marked increase in Nox2 and Nox4 expression at mRNA levels after repeated in vivo treatment (309). Although further studies are definitely needed, available data suggest that Nox might be implicated in chronic ANT cardiotoxicity development.
NADPH-P450 reductase is another flavin-containing enzyme implicated in ANT redox cycling (50). The intracellular location of this enzyme within the endoplasmic reticulum suggests its connection to pathological distension of the sarcotubular system, which is believed to be behind cytosolic vacuolization observed in chronic ANT cardiotoxicity. Liver microsomes rich in NADPH-P450 reductase have been shown effective in catalysis of ANT redox cycling and ROS production (9). However, Doroshow et al. have demonstrated 14-fold lower relative effectivity of redox cycling induced by cardiac sarcosomes as compared to cardiac submitochondrial particles, which may diminish the importance of this pathway (50). Furthermore, genetic disruption of the NADPH-P450 reductase expression within the heart has been reported to show no protection against acute ANT cardiotoxicity (DOX 10–20 mg/kg) (50). As there is a lack of data from the chronic ANT cardiotoxicity models, one cannot draw a definite conclusion on involvement of this pathway in clinically important forms of cardiotoxicity. Nevertheless, available data do not argue for the key importance of this ROS generation pathway.
Besides the mitochondria and endoplasmic reticulum, important ANT redox cycling activity has been found also in the myocardial cytosolic fraction, and this has been mainly attributed to xanthine oxidase (50). Pan and Bachur have demonstrated that bovine milk xanthine oxidase can catalyze ANT redox cycling and ROS production (203). Using H9c2 cells, Mukhopadhyay et al. have found a significant increase in xanthine oxidase expression at the mRNA level after the DOX (1 μM) treatment, whereas only an insignificant trend was detected at the protein level. However, allopurinol failed to provide significant protection from DOX-induced cell death under the same conditions (191). Quite surprisingly, there is a lack of in vivo data about its potential against chronic ANT-induced cardiotoxicity.
ANT redox cycling can be also promoted by NOS (278), and all three NOS isoforms are able to catalyze these reactions. Conflicting data have been published on the impact of genetic and pharmacological manipulation of activity of NOS isoforms on acute ANT cardiotoxicity models (35a, 37, 202, 289). Nevertheless, repeated dosing with DOX (3 mg/kg, weekly for 4 weeks, and 4-week follow-up) strongly suggested distinct roles of different NOS isoforms in ANT cardiotoxicity. While nNOS has been suggested to play rather a protective role, eNOS expression did not correlate with the cardiotoxicity severity. In wild-type animals, the authors have not found induction of NOS isoforms at the mRNA level due to the ANT treatment, whereas a gradual decrease of abundance has been shown in eNOS at the protein level along with decrease in NO production. Interestingly, despite a significant decline in systolic function and increase in the LV diameters after the chronic ANT treatment, the authors have not found an increase in lipoperoxidation. Although complex phenotype of animals with genetic manipulation of individual NOS isoforms may complicate the interpretations of data, it seems that NOS are not among the main catalysts of ROS production following the clinically relevant ANT doses (47).
ANT cardiotoxicity and myocardial antioxidant defense
Oxidative stress takes place when production of ROS/RNS oversaturates the capacity of cellular antioxidant defense (84). Of note, lower levels of physiological antioxidants such as catalase have been speculated as responsible for particular vulnerability of the heart to toxicity of ANTs (53). Although many studies have examined the effects of acute and chronic ANT treatment on physiological antioxidants, it is very difficult to draw a definitive conclusion due to the enormous heterogeneity of models, tissue sampling times, and outcomes of these interventions. Unfortunately, genetically modified animals, which might shed a light on mechanistic importance of antioxidant defense and signaling cascades, have been employed solely in acute ANT cardiotoxicity settings so far (69, 135, 298, 302, 303). Therefore, the translatability of these findings to clinically important chronic forms of ANT cardiotoxicity remains unsure.
As described above, chronic ANT cardiotoxicity has been associated with a marked drop in expression of the mitochondrial SOD isoform (163, 254). Interestingly, the CuZnSOD (SOD1) isoform has not been found regulated in the similar manner in these studies, and therefore signaling pathways responsible for selective ANT-induced downregulation of MnSOD deserve further study. Several studies have reported no significant change in total myocardial SOD activity due to chronic ANT treatment (5, 81, 218, 249). A role of MnSOD in ANT cardiotoxicity has been probed with transgenic mice overexpressing MnSOD. In these animals, a short-term treatment with supratherapeutic doses of DOX (10–25 mg/kg) has been found to induce less morphological ultrastructural changes (particularly in the mitochondria), and it preserved mitochondrial respiration (302, 303).
Several studies have found no change in expression and/or activity of myocardial catalase in chronic ANT cardiotoxicity models (5, 42, 81, 249), whereas others have found an increased activity of the enzyme due to the treatment (163, 218). The mechanistic role of catalase has been probed with the transgenic mice overexpressing the enzyme. Unlike the wild-type controls, transgenic mice with 60–100-fold higher myocardial catalase activity were found free of myocardial lipoperoxidation, elevation of total CK activity in plasma, and functional deterioration of the atria after the supratherapeutic dose of DOX (20 mg/kg). In contrast, transgenic animals with the highest (200-fold) activity in the heart showed similar results as the wild-type controls (135).
Different regulation of glutathione antioxidant system enzymes has been observed on chronic ANT cardiotoxicity models. Robison et al. (218) and Alderton et al. (5) have found no change of glutathione peroxidase activity a week or more after the chronic weekly ANT treatments, while others have reported a slight, but significant, increase at this time point (279). A persistent decrease of expression and activity of this enzyme has been reported from compressed chronic ANT cardiotoxicity models where a total cumulative dose of DOX (13.5–15 mg/kg) was administered over 2–3 weeks only (163, 215, 249). The involvement of glutathione peroxidase in acute ANT cardiotoxicity has been probed using genetically modified organisms (69, 298). While knockout of the enzyme has not affected sensitivity of the mice to acute in vivo treatment with high DOX dose (22.5 mg/kg), significant enhancement of the toxicity has been found in the hearts isolated from these animals and perfused with DOX (5 μM) (69). Conversely, the overexpression of the enzyme protected the isolated hearts from toxicity induced by perfusion with DOX (5 μM), and it showed cardioprotection against single high dose of DOX (15 mg/kg) (298). While some studies reported no change in glutathione reductase activity in the heart a week after repeated ANT treatment (279), others have shown either a slight decrease (81) or even a significant elevation of the activity (218).
Several authors have focused on examination of oxidized and/or reduced forms of glutathione in the myocardium after chronic ANT treatment. Some studies have reported no change in availability of reduced glutathione or unaltered GSSG/GSH ratio within the myocardium a week after chronic ANT treatment (5, 279) despite the cardiomyopathy development. One of these studies examined also a functional role of glutathione in ANT toxicity in vitro using H9c2 cells exposed to DAU (0.1–10 μM). While a decrease of cellular glutathione content induced by preincubation of the cells with buthionine sulphoximine (BSO) sensitized the cells toward H2O2 (60–1000 μM) toxicity, it had no impact on the toxicity of DAU (279). Others have found either increase (218) or slight decrease of glutathione content after repeated in vivo ANT treatments (81).
Antioxidants As Cardioprotectants in ANT-Induced Cardiotoxicity
As ANT-induced cardiotoxicity has been hypothesized to be linked with oxidative stress, many classic as well as novel antioxidants have been tested in vitro or using acute cardiotoxicity models in vivo with reported positive outcomes in most of cases. Such investigation has been performed with vitamin A (265), vitamin C (66), vitamin E (288), N-acetylcysteine (195), amifostin (126), a metalloporphyrine peroxynitrite decomposition catalyst (202), different flavonoids (223), and many others. However, direct translatability of such data into clinical medicine might be an issue, as the acute ANT cardiotoxicity is rarely clinically significant, and extrapolation to chronic ANT cardiotoxicity may be skewed by employment of supratherapeutic doses/concentrations, lacking the factor of time and absence of reactions associated with repeated exposure (72).
Numerous antioxidants have been also studied using different chronic ANT cardiotoxicity models with considerably less positive outcomes, especially when long-term DEX-validated animal models were employed. Furthermore, many findings were contradictory to the outcomes described from acute ANT cardiotoxicity models. This includes negative findings on cardioprotection with vitamin E (17, 26, 277), N-acetylcysteine (102, 272), and amifostin (107, 216). Interestingly, although Berthiaume et al. have found suppression of DOX-induced oxidative stress due to the dietary supplementation of vitamin E, the treatment has little-to-no effect on the cardiotoxicity parameters (17). In addition, while significant protection has been found in two flavonoid derivatives (mono-HER and frederine) (273, 274), further experiments showed that cardioprotective effects lose their power after longer follow-up (29). Although vitamin C has been suggested to decrease the mortality rate induced by both acute and subchronic DOX treatment, cardioprotective effects have been evaluated against repeated ANT treatment only in a very low DOX cumulative dose (2 mg/kg) (66). Moderate experimental evidence has been found on cardioprotective effects of ebselen,
Interestingly, a significant cardioprotection has been described on the compressed chronic ANT cardiotoxicity model (DOX 2.5 mg/kg, six doses over 2 weeks, and 3-week follow-up) with carvedilol, a beta-blocker with vasodilatatory and antioxidant properties (179). The protective effects were not found in atenolol, which suggests that cardioprotection can be β1-receptor independent. These findings have been confirmed further by others on the models utilizing weekly DOX dosing (2 mg/kg for 7 weeks) (200, 224), and these investigations showed that the cardioprotective potential of this agent may be connected with the preservation of mitochondrial function and prevention of mitochondrial calcium overload.
Data from clinical trials have also yielded negative results regarding the cardioprotective potential of several antioxidants, namely vitamin E (158), N-acetylcysteine (54, 193), or mono-HER (28). Although these agents have been so far tested in rather small clinical trials with numerous imperfections, accumulating body of evidence argues against the value of these agents as clinically effective cardioprotectants in ANT cardiotoxicity settings.
Of note, relatively encouraging results have been reported from a clinical trial with carvedilol (133). Here, the potential cardioprotective effects of carvedilol (12.5 mg orally on the daily basis) were investigated in 25 patients undergoing ANT-based chemotherapy. Interestingly, while control patients showed significant worsening of both systolic and diastolic cardiac performance after DOX or EPI (cumulative dose 525 and 788 mg/m2, respectively), the carvedilol-treated patients were free of significant changes. These optimistic findings merit further study in large randomized controlled trials with longer follow-up.
DEX-Afforded Cardioprotection Against ANT Cardiotoxicity
DEX (ICRF-187, ADR-529) and the corresponding racemic mixture—razoxane (ICRF-159, ADR-159)—belong to bisdioxopiperazine agents originally developed by Creighton et al. as potential anticancer agents (39). The authors aimed to find cell-permeable EDTA prodrugs that may target biometals of vital importance within the cancer cells. However, anticancer effects of DEX and razoxane have been later attributed to TOP2 inhibition (36, 262). Razoxane has passed through extensive preclinical development as an anticancer drug. However, it has never advanced beyond clinical trials.
Herman et al. has initiated research on potential cardioprotective effects of DEX and razoxane (105), but most of later studies preferred DEX due to the better solubility. Despite the intensive research, a mechanism of cardioprotective effects of DEX against ANT-induced cardiotoxicity is not completely understood. However, the most accepted hypothesis proposes the key role to iron chelation properties of a hydrolytic product of the drug with subsequent prevention of myocardial oxidative stress (41, 87). DEX had passed all the stages of preclinical and clinical research and has been finally approved in Europe and the United States for cardioprotection in patients treated with ANTs (Cardioxane™ and Zinecard™) with several generic preparations recently available (Procard™ and Cardynax™). More recently, DEX has been also approved for treatment of accidental extravasation of ANTs (Savene™).
Chemistry
DEX is a (+)-(S)-enantiomer of racemic 1,2-bis-(3,5-dioxopiperazine-1-yl) propane (razoxane). Racemic razoxane is poorly soluble in water, which precluded preparation of intravenous formulation, and therefore this drug was available only for oral administration. Unfortunately, dose-dependent fluctuations in razoxane bioavailability complicated its therapeutic use (38). Nevertheless, Repta et al. have observed nearly five-times higher solubility of DEX, which enabled intravenous formulation of the drug and resolved the pharmacokinetic issues (214).
In aqueous solutions of physiological pH and temperature, DEX undergoes chemical hydrolysis of dioxopiperazine rings with a half-life of 9.3 h (85) (Fig. 6a). This reaction yields intermediates with only one ring opened (B and C), which then hydrolyze further to a final product—a diamide of EDTA (ADR-925) (Fig. 6a). Interestingly, multivalent ions may facilitate the hydrolysis of DEX and its intermediate degradation products. Fe3+ has been shown to increase hydrolysis of DEX, whereas Fe2+ is particularly effective in promoting hydrolysis of one-ring-opened hydrolytic intermediates to the ADR-925. Significant enhancement of the latter reaction has been also demonstrated for physiological concentrations of Mg2+, Ca2+, Mn2+, Cu2+, or Zn2+ (87).

Chelating properties of DEX hydrolysis products
ADR-925, a product of total hydrolysis of DEX, has a significant affinity to both Fe2+ and Fe3+ with formation constants 1010 M −1 and 1018 M −1, respectively. Indeed, EDTA, which is lacking a diamide moiety, is a several-order-of-magnitude stronger iron chelator with the Fe2+ and Fe3+ formation constants 1014 M −1 and 1024 M −1, respectively. Nevertheless, ADR-925 is still relatively a strong hexadentate iron chelator (Fig. 6b) that chelates iron in a similar manner as EDTA, and the same applies on hydrolytic products of other bisdioxopiperazines (49). The resulting complexes [Fe(ADR-925)H2O]+ and [Fe(EDTA)H2O]− exhibit a distorted pentagonal bipyramidal geometry with the 7th coordination site on the Fe3+ center occupied by a water molecule (49). Of note, the labile water molecule bound to iron is thought to explain the redox cycling capabilities of EDTA complexes and associated production of damaging hydroxyl radicals. The same properties have been described to be analogically applicable on the complex of iron with ADR-925 (49, 266).
ADR-925 is a stronger iron chelator than ANTs, and thus it can effectively displace Fe3+ from the complex with these drugs. Also, the intermediate hydrolysis products (B and C) have been identified to act as iron chelators, although only as tetradentate ones (32). They have been also shown to displace the iron from complexes with ANTs, and the resulting complexes then hydrolyze further to form the ADR-925–iron complex. However, Thomas et al. have demonstrated that ANT semiquinone radicals drive redox cycling of racemic ADR-925 (ICRF-198) nearly as effectively as EDTA, and this process leads to production of •OH, which argues against simple displacement of iron from ANT–iron complexes as the main mechanism of cardioprotective action. It should be also noted that, similarly as EDTA, ADR-925 is far from being a selective iron chelator, as it has been found to bind also Cu2+, Mn2+, Zn2+, and Ca2+ with formation constants 1015, 1010, 1010, and 107 M −1, respectively, and one cannot exclude that it may be related to some of therapeutic effects of DEX (266).
General pharmacokinetics
In clinical practice, DEX is administered in a short (≈15 min) infusion, and its pharmacokinetics is best described by a two-compartment open model with first-order elimination kinetics (41). Clinically used doses of DEX (typically 500–1000 mg/m2) yield cmax of 136–280 μM (41). Plasma protein binding is negligible, and distribution into the tissues is relatively fast with a volume of distribution matching total body water, which implies significant penetration into the intracellular compartment. The plasma concentrations of DEX decline in two phases with a half-life of 15 and 140 min. The total body clearance is 0.29 L/h/kg, with renal excretion as a predominant route of elimination (42%–48% of a dose of DEX is recovered unchanged in the urine) (41, 55, 293). In experimental animals, the highest concentrations of the drug have been found in the kidney and liver 2 h after the DEX (12.5 and 100 mg/kg) administration, whereas the order-of-magnitude lower concentrations have been reported in the myocardium (94).
Metabolism and bioactivation
DEX is believed to be a prodrug that acts as a cardioprotectant through its chelating-active metabolite (41, 87). Hence, DEX metabolism and particularly its bioactivation within the heart are the points of key importance. Previously, a little attention has been paid to DEX metabolites in clinical studies, which may be due to the analytical difficulties with their selective and sensitive determination in biological matrices. Schoeder et al. (234) have shown detectable ADR-925 concentrations already at the end of DEX infusion (1000–1500 mg/m2 over 15 min). The cmax (≈30 μM) was reached soon, and after 4 h plateau, a slow decline of ADR-925 concentrations occured. The concentrations of the intermediates B and C reached their maximal concentrations very early and declined rapidly thereafter with half-lives 2.5 and 0.6 h, respectively. The maximal concentrations of the parent compound (211 μM) were much higher than those of metabolites (234).
Schroeder and Hasinoff have described the metabolism of DEX in rats. The authors have found a rapid metabolism of DEX with significant concentrations of all three metabolites in plasma, already 5 min after DEX administration (40 mg/kg, intravenously). The plasma concentrations of ADR-925 rose significantly to become comparable to DEX 60 min after the administration (232). The same investigators have determined the tissue concentrations of ADR-925 2.8 h after the administration of the same dose of DEX to rats (233). They found 51 μmol/kg of wet tissue of ADR-925 in the rat heart, whereas almost double values were found in the liver. A complementary study on ADR-925 (20 mg/kg) pharmacokinetics has shown a short elimination half-life (8.3 min) and relatively low myocardial concentrations (7.4 μmol/kg of wet tissue, 1.5 h after drug administration), which could imply limited distribution of the metabolite from the circulation (233). The latter finding rather suggested in situ bioactivation of DEX within the cardiomyocytes. Interestingly, the intermediates B and C were distributed to the heart much easier (233). Unfortunately, there is a lack of information on concentration–time profile of DEX and its metabolites in the heart. The PK/PD investigation with a focus on the heart may shed more light on importance of DEX metabolism for cardioprotective action.
The DEX metabolism has been also studied in vitro using cardiomyocytes, hepatocytes, tissue homogenate fractions, and purified enzymes. The majority of these experiments were performed in the Hasinoff's laboratory, and the authors have proposed that DEX undergoes stepwise hydrolysis by two tissue hydrolases—dihydropyrimidinase and dihydroorotase (87, 94). However, the dihydropyrimidinase, which is expected to hydrolyze DEX in the first step, has been shown absent in the heart, although it is abundant in other tissues, including the kidney and liver. Moreover, in contrast to porcine liver and kidney tissue and purified dihydropyrimidinase, the heart tissue fraction has been shown unable to hydrolyze DEX. In cell culture experiments, Schroeder et al. have demonstrated a rapid and nearly complete decline of DEX concentrations in the medium containing hepatocytes with concomitant, but disproportional, raise of ADR-925 (235). However, the decline of DEX was apparently less profound when isolated neonatal ventricular cardiomyocytes were used. Unfortunately, ADR-925 was not examined in these experiments, and these cell culture studies have not shed light on the intracellular compartment, where the metabolites are formed, and when the active metabolites are thought to exert their chelating or other effects. Hence, the intracellular metabolism of ADR-925 deserves further study with focus on the relative rate of metabolism, subcellular localization, and potential retention of relatively polar metabolites inside the cardiac cells.
Hasinoff's group approached this issue indirectly through measurement of intracellular chelation within the cardiomyocytes after the incubation with DEX using an acetoxymethyl ester of calcein (86). Their data suggest that DEX indeed penetrates into the cardiomyocytes, and is only slowly converted there to the chelating metabolites, which contrasted with fast and significant intracellular chelation induced by 10-fold lower concentration of ADR-925. Although these experiments used rather specific experimental conditions, the results may argue against the role of metabolic bioactivation of DEX to ADR-925 within the cardiomyocytes. Further experiments are needed to clarify this point.
Cardioprotection against ANT cardiotoxicity in vitro
Sawyer et al. have published a seminal study showing that 10 μM DEX protects the neonatal cardiac myocytes from apoptotic cell death induced by clinically relevant DAU concentrations (≤1 μM), while it is unable to protect these cells from necrotic cell death induced by higher concentrations of DAU (≥10 μM) (226). Hasinoff's group has shown that DOX accumulates in the mitochondria and induces depolarization of the mitochondrial membrane potential at clinically relevant concentrations (≤1 μM). DEX preincubation (90 μM) was able to effectively prevent the latter mitochondrial changes. Relatively high DOX concentrations (≥10 μM) were also capable to significantly oxidize reduced dichlorofluorescein in cardiomyocytes, which was preventable by DEX pretreatment (90). However, the same group has shown that unlike the parent DEX molecule, neither the intermediate metabolites B and C nor ADR-925 is able to protect the neonatal cardiomyocytes from toxicity of 1.5 μM DOX (91). Of interest, using the calcein assay, more pronounced intracellular chelation has been found for both metabolites (ADR-925 and C) than for parent DEX. The authors have concluded that either DEX metabolites do not have an access to the critical iron pool within the cell, or other mechanisms might be involved in the cardioprotective effects of DEX (91).
Interestingly, the same group has shown that both DEX and ADR-925 can significantly protect isolated cardiomyocytes from hypoxia–reoxygenation damage, although ADR-925 was less effective (232). Furthermore, others have suggested that DEX may act as a scavenger of ROS without enzymatic hydrolysis to ADR-925, and its antioxidant effect may be iron independent (132). In addition, using H9c2 myoblasts and fibroblasts from TOP2β knockout mice, Lyu et al. have suggested that the parent compound DEX may be protective through inhibition of ANT-induced and TOP2β-mediated DNA damage (173).
Cardioprotection against ANT cardiotoxicity in vivo
In the first in vivo studies, Herman et al. have shown that razoxane pretreatment (100 mg/kg) can partially decrease the mortality rate and ameliorate certain signs of acute cardiovascular toxicity induced by sublethal DAU doses (50 and 25 mg/kg) in Syrian golden hamsters (97, 104).
With recognition of clinical importance of chronic ANT cardiotoxicity, the interest has been shifted toward the animal models receiving repeated cycles of clinically relevant individual ANT doses. Again, Herman's laboratory has made a significant part of this pioneering job. With accumulating body of evidence, it has become firmly established that DEX is able to effectively protect the heart and cardiomyocytes from chronic ANT cardiotoxicity in various animal models, including rabbits, dogs, mice, normotensive and spontaneously hypertensive rats, guinea pigs, and pigs as reviewed elsewhere (101, 124)
Furthermore, it has also become obvious that DEX-afforded cardioprotection is applicable on cardiotoxicity of all studied ANTs, including DOX, DAU, EPI, idarubicin (101, 124), or mitoxantrone (109). Furthermore, using animal models, a dose–response of DEX cardioprotection has been established (125). It has been shown that DEX provides significant cardioprotection when administered at dose ratios (DEX/DOX) 4:1 to 30:1, and the ratio 10:1 to 20:1 has been selected as optimal for clinical studies (124, 125). Regarding the timing, in beagle dogs, it has been found that outcomes of the cardioprotective intervention are significantly better when DEX is administered simultaneously with DOX as compared to administration 2 h after the DOX administration (100). The time intervals between the DEX and DOX administration have been systematically evaluated of in mice (from 2 h before to 1 h after DOX). Both extremes were found inferior to other intervals, whereas administration 30 min before DOX has provided the best results (124). Importantly, Herman and Ferrans have clearly demonstrated that the DEX effect is not a mere delay of cardiotoxicity, but real long-lasting cardioprotection (99), and they have also shown that DEX pretreatment enables applications of much higher cumulative doses of DOX with a bearable risk of cardiotoxicity (103). To our best knowledge, there has been only one study that suggested a failure of DEX in cardioprotection against ANT cardiotoxicity (DEX 60 mg/kg administered to 10 days old rats before single DOX dose—3 mg/kg) (96). Nevertheless, others have demonstrated significant cardioprotection of DEX (20 mg/kg) against chronic DOX-induced cardiotoxicity (1 mg/kg, weekly for 7 weeks) in weanlings and young adults (46).
Interestingly, relatively limited data are available on mechanisms involved in DEX-afforded cardioprotection. An early study of Rajagopalan et al. has shown that 25 μM DEX significantly reduces production of •OH in an isolated heart perfused with 1 μM DOX (211). However, the production of •OH did not show common concentration–response relationship in the studied range (DOX 0.1–25 μM), and it became significant only 10 to 15 min after the beginning of perfusion with the drug. Alderton et al. have not found any changes in total activity of myocardial superoxide dismutase, glutathione peroxidase, catalase, and reduced glutathione after either acute (12 mg/kg) or chronic DOX dosing (1 mg/kg, weekly for 12 weeks), alone or with DEX pretreatment (5). Other authors have found no effect of DEX (40 mg/kg) on myocardial TBARS levels after acute EPI treatment (10 mg/kg) or gamma irradiation of the heart (210). However, the DEX/ANT ratio and dosing interval could have been suboptimal in this study. It has been also demonstrated that DEX pretreatment (20:1) is able to very effectively prevent the onset of both degenerative changes and apoptotic cell death induced by chronic ANT treatment in rabbits (DAU 3 mg/kg, weekly for 10 weeks) (208). DEX treatment rescued cardiomyocytes from complex proapoptotic signaling, and resulting cell death and parameters of apoptosis correlated well with the cardiotoxicity parameters. Surprisingly, effective cardioprotection with DEX was not associated with significant decrease of moderately elevated markers of lipoperoxidation. In addition, no correlation has been found between myocardial malondialdehyde levels and parameters of cardiac function (208).
Using a model of late-onset chronic ANT cardiotoxicity (DOX 0.8 mg/kg, weekly for 7 weeks, and 37-week follow-up), Lebrecht et al. have found that DEX can protect the heart mitochondria from late-onset DOX-induced genetic and functional lesions and raise of TBARS. Interestingly, DEX pretreatment markedly reduced the DOX-induced damage to mtDNA (mtDNA/nDNA copy number and occurrence of common deletions) and overcame DOX-induced decline in expression of mtDNA-encoded subunits of the respiratory chain (150). Another study has shown that DEX is able to prevent complex changes in myocardial metabolism induced by chronic DOX treatment (35).
DEX pretreatment (50 mg/kg) has prevented downregulation of myocardial ryanodine receptor expression induced by chronic DOX treatment (2.5 mg/kg, weekly for 6 weeks) in rats (31). Others have documented that DEX (40 mg/kg) markedly reduces deleterious effects of DOX (2 mg/kg, weekly for 4 weeks) on trabecular actin–myosin crossbridge cycle (45). DEX (60 mg/kg weekly) was also able to prevent an increase in myocardial total calcium content induced by chronic DAU treatment (3 mg/kg, weekly for 10 weeks) (241). Interestingly, in the latter study, the chronic ANT treatment with or without DEX had no impact on total myocardial iron levels.
Cardioprotection against ANT cardiotoxicity in clinical trials
Cardioprotective effects of DEX against ANT-induced cardiotoxicity have been unambiguously demonstrated in numerous clinical trials and in both adults and children (169, 170, 177, 252, 260, 305). Importantly, significant cardioprotection has been achieved in various chemotherapy regimens, using different DEX-to-ANT ratios and all ANTs in clinical use. Cardioprotective potential of DEX has been evaluated by clinical examination (incidence of cardiac events and symptoms of CHF), using cardiac function examinations (echocardiography or radionuclide ventriculography), by analysis of biochemical markers (e.g., plasma concentrations of cardiac troponins) and/or endomyocardial biopsy.
Pioneering randomized controlled trial conducted by Speyer et al. has shown that DEX permits breast cancer patients to receive significantly more cycles of chemotherapeutic regimen (9 vs. 11, p<0.001) and to achieve higher cumulative doses of DOX (441 vs. 500, p<0.01) with much lower rate of DOX-induced cardiac events (252). Swain et al. have revealed that DEX is also effective when launched in patients exceeding a cumulative dose of DOX of 300 mg/m2 without prior protection (259). Other studies have confirmed the cardioprotective significance of DEX in mixed cohorts involving both adults and children (171, 290). Importantly, the randomized prospective trial by Lipshultz et al. has clearly shown that DEX may prevent cardiotoxicity onset in children with ≈2.5-year follow-up after the chemotherapy (169). A nonrandomized prospective trial by Elbl et al. has suggested that DEX affords a long-term cardioprotection from both clinical and subclinical cardiotoxicity in 8-year follow-up after the ANT treatment (57). In addition, a retrospective study in children with a follow-up of 2–20 years (7 years median) suggested a reduction of both clinical and subclinical signs of late-onset cardiotoxicity in children receiving DEX (58). In a recent prospective randomized study, DEX has been demonstrated to provide a long-term cardioprotection (5-year follow-up) in children, and the benefits from DEX treatment seemed to be more significant in girls than in boys (170). Finally, the cardioprotective effectiveness of DEX has been also supported by the recent meta-analysis of all randomized controlled trials of ANT-treated oncologic patients (275).
Question of potential interference with anticancer effect of ANTs
Firstly, the DEX-afforded cardioprotection has been shown to be not due to its impact on ANT pharmacokinetics and metabolization (13, 113, 219). However, as both ANTs and DEX share an important molecular target, TOP2, the question of potential interference of DEX with anticancer effects of these agents is appropriate.
Early preclinical examinations have shown no effect on anticancer effects of DOX (94, 281). Later on, a significant synergism with ANTs has been reported in a leukemia cell line (30), while others have reported the possibility of antagonism on proliferating noncancer Chinese hamster ovary cells (92). The tumor response was also carefully observed during first two stages of clinical evaluation of this drug. No suggestions of significant impairment of anticancer effects of ANTs have been recognized.
Significant, although not well evidenced, suspicion on potential interference of DEX with anticancer effects of ANTs has arisen from one of phase III trials (260). At this stage, two studies with very similar design have been conducted. In the first trial (n=123), no difference in any oncological endpoints has been found (objective response rate was 54% and 49%; p=0.63 in DEX and placebo-treated group, respectively). However, from another trial (n=293), a significant difference in objective response has been reported (47% vs. 61%, respectively, p=0.019) (260). While unusual was the rather high response in the placebo group, and no other endpoints (including survival or time to progression) were affected in either of these studies, DEX became suspected for negative effect on tumor response. This has been reflected in recommendations for DEX use given by various oncological societies, and DEX has not been recommended for use during initial ANT treatment, but only after the cumulative dose of DOX exceeds 300 mg/m2 (95).
The above-described study has been analyzed in depth with conclusion that these data cannot conclusively blame DEX from reduction of tumor response (257). No significant effect on any oncological parameter has been found ever since, and most importantly, careful meta-analyses of all randomized clinical trials have found no evidence for this hypothesis (235c, 275). However, the stain has remained on DEX, and clinical guidelines as well as oncological practice have not been changed in the light of these data.
Adverse effects
DEX has been reported to be relatively a low-toxic drug, well tolerable by both animals of different species as well as cancer patients (41). Leukopenia and myelotoxicity have been established in early phases of clinical evaluation as the dose-limiting toxicities (283), which are likely due to the TOP2 inhibition induced by the parent DEX molecule. Leukopenia has been also reported as the only grade 3/4 adverse event significantly more frequent in DEX-cotreated cancer patients (260). In addition, less-serious adverse effects included mild-to-moderate increase of risk of anemia, nausea, and fatigue, while phlebitis was more frequent (41). One clinical study has suggested that DEX may increase incidence of secondary hematological malignancies (264), whereas another randomized controlled clinical trial designed and powered for this purpose has found no evidence for these potentially serious adverse effects of DEX (12).
DEX-afforded tissue protection beyond the ANT-induced cardiotoxicity
Besides cardioprotection against ANT-induced cardiac injury, DEX has been reported to protect the kidneys from experimental ANT-induced nephrotoxicity (307, 311). Broader cardioprotective potential has been suggested from experimental studies on ischemia/reperfusion injury (212, 310) and catecholamine-induced cardiotoxicity (63, 93, 205, 306). Although many of these results appeared interesting, the only additional clinical indication for DEX becomes a protection of tissue in accidental ANT extravasation—a rare, but potentially serious, adverse effect associated with long-lasting local damage, ulceration, and necrosis. Langer et al. have reported that systemic treatment with DEX (250 mg/kg or triple treatment with 62.5 mg/kg) markedly decreases the area of wound and healing time when started immediately after induction of ANT extravasation in mice (148, 149). However, such effects were not achievable with ADR-925, EDTA, N-acetylcystein, vitamin E, merbarone, or aclarubicin, which does not suggest involvement of iron chelation, oxidative stress, or TOP2 inhibition (149). Conversely, DEX failed to protect the tissues from hydrogen peroxide-induced tissue damage (149). The mechanisms of tissue protective effects of DEX therefore remain undetermined in this setting.
DEX Derivatives As Cardioprotectants
A number of dioxopiperazine derivatives have been studied for their anticancer and TOP2 inhibition effects (40). However, much less information is available regarding their ability to protect the myocardium against ANT-induced damage. An early study performed by Herman et al. (98) has evaluated the protective action of 19 bisdioxopiperazines together with DEX (12.5–200 mg/kg) against acute DAU toxicity (25 mg/kg) in Syrian golden hamsters. Only three compounds were found effective in terms of reducing the mortality rate and body weight loss (DEX, levorazoxane–ICRF-186, and bimolane–AT-1727), whereas a minimal effect was observed in other five compounds. Later on, it has been shown that DEX and its optic isomer levorazoxane have comparable or slightly lower cardioprotective effects on a chronic SHR model (308). Although other bisdioxopiperazine derivatives also yield the iron-chelating hydrolytic products (49, 118), systematic evaluation of DEX derivatives on chronic ANT cardiotoxicity models has not been performed until 1997. Herman et al. have evaluated ADR-925 and four bisdioxopiperazine derivatives (50 mg/kg, intraperitoneally) on a well-established SHR model (DOX 1 mg/kg, weekly for 12 weeks), and the results were compared to DEX. None of tested DEX derivatives was superior to DEX in terms of cardioprotection (evaluated by histopathology) and prevention of premature death. The marked cardioprotection and decreased mortality rate were found in a demethylated derivative of DEX (ICRF-154) and bis(N-morpholinomethyl) derivative (ADR-559) (Fig. 7). Significant cardioprotection has been also found in ICRF-197 and ADR-239 (Fig. 7), whereas no protection was found in ADR-925 and an ethyl analog of DEX (ICRF-192). These observations contrasted with the notion that all examined compounds are well hydrolysable to metal-chelating products that can displace the iron from complexes with DOX. Although the authors suggested an alternative link to ability of these agents to inhibit TOP2, it might be noted that generally a higher response was seen in agents with better solubility. Thus, potential difference in absorption and pharmacokinetic profile may complicate interpretation of the structure–activity pharmacodynamic data.
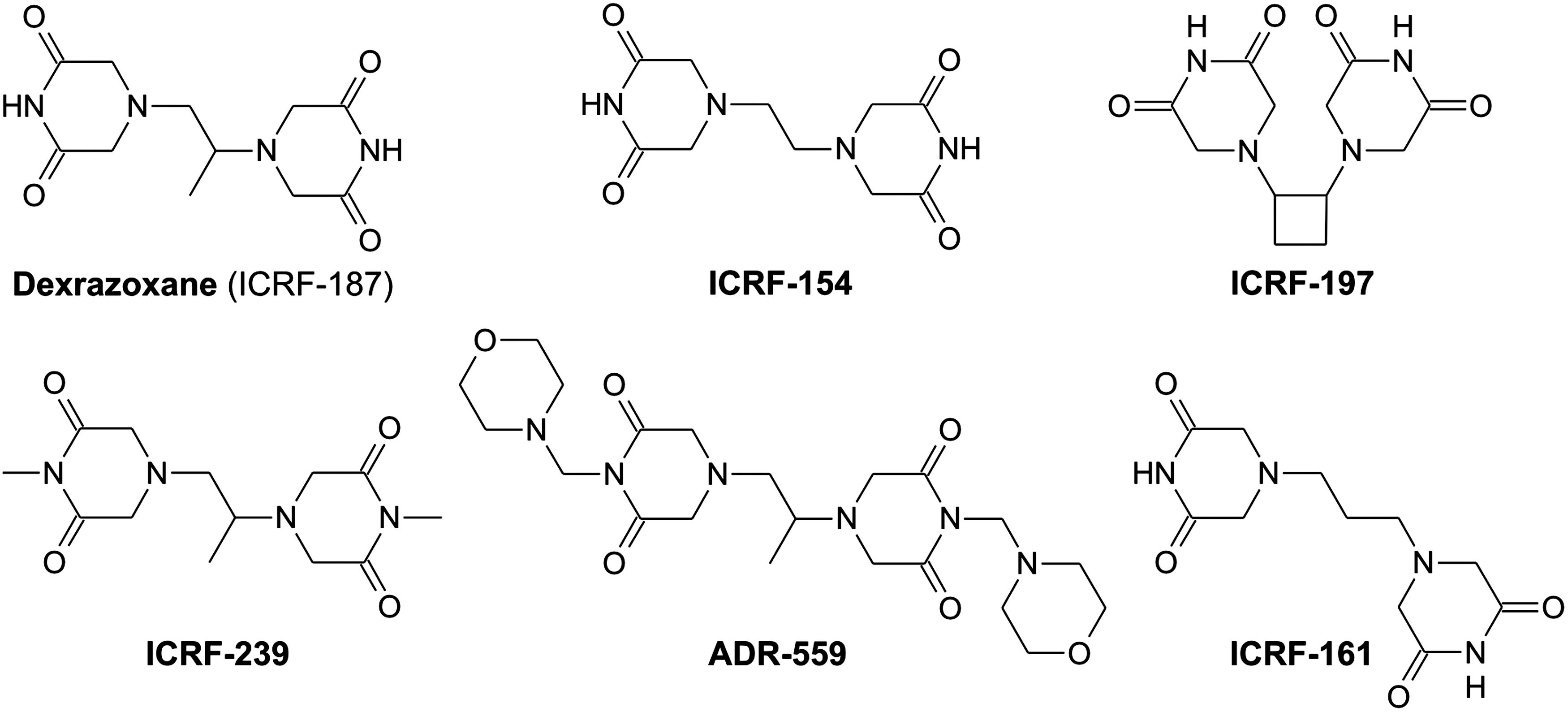
More recently, Martin et al. have performed an elegant study in which the effects of DEX were directly compared with those of ICRF-161 (a close bisdioxopiperazine analog of DEX without TOP2 inhibition properties, Fig. 7) (176). Both compounds were found hydrolyzable to iron-chelating products, and they were comparable in their effectiveness to displace iron from its complex with DOX. Although both compounds inhibited release of LDH from neonatal cardiomyocytes incubated with 1.8 μM DOX, only DEX has been found significantly protective on a well-established chronic ANT model in SHR (DOX 1 mg/kg, weekly for 12 weeks). Unlike DEX, ICRF-161 was unable to significantly decrease DOX-induced premature death and typical histopathological changes in the myocardium despite very similar pharmacokinetic profile of both drugs. These data strongly suggest that (i) mere iron-chelating activity is not a sufficient predictor of cardioprotective effect; (ii) short-term exposure of cardiomyocytes in vitro may not be a reliable predictor of cardioprotective properties of the drug without confirmation on appropriate chronic model, and (iii) it is not sure whether TOP2 effects and effective cardioprotection are dissectible. Hence, further mechanistic studies with focus on structure–activity relationships with employment of DEX-validated chronic ANT models are needed to shed more light on this issue.
Iron Chelators As Cardioprotective Agents
The idea of iron chelation as a means of prevention of the ANT cardiotoxicity arose with the discovery of the cardioprotective effects of DEX. As already mentioned, ADR-925, a DEX hydrolysis product, can chelate free intracellular iron as well as displace Fe3+ from its complex with DOX, and these effects have been long accepted as responsible for cardioprotection (87). Furthermore, it has been demonstrated that in iron-overload conditions, the ANT-induced cardiotoxicity is markedly aggravated (166). Taken together, this provided a strong rationale for studies of ANT cardiotoxicity prevention with iron chelators. While the classical antioxidants scavenge the ROS once they had been formed, chelators are able to prevent the formation of highly toxic •OH through the iron-catalyzed Fenton and Haber–Weiss reactions (68, 84).
An important question regarding any cardioprotective intervention during ANT therapy is whether the prospective cardioprotective drug can decrease the heart damage caused by ANTs without reducing the antitumor efficacy. Despite rare early data, suggesting that DFO might interfere with DOX cytotoxicity in MCF-7 human breast cancer cells (51), it is now generally accepted that iron-mediated ROS formation is not important for anticancer effects of ANTs (297). Nevertheless, different iron chelators, in addition to being examined as protectors against ANT-induced cardiotoxicity in vitro and/or in vivo (see below), have been also shown not to blunt the antiproliferative effects of ANTs in various cancer cell lines (209, 243, 256).
It should be noted that—with the exception of few DEX analogs (108)—no chelator has been specifically designed for use in prevention of ANT cardiotoxicity. Most of the agents studied so far have been primarily developed and/or used for the management of patients with chronic iron overload, such as those with β-thalassemia major (134). Studies on ANT cardiomyopathy prevention with iron chelators are summarized in Table 1.
Improved survival of animals, but cardioprotection did not reach statistical significance.
Partial cytoprotection, but no effect on daunorubicin-induced lipid peroxidation.
PIH, pyridoxal isonicotinoyl hydrazone; o-108, ortho-chlorobenzoyl isonicotinoyl hydrazone; SIH, salicylaldehyde isonicotinoyl hydrazone; Dp44mT, Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone; LDH, lactate dehydrogenase; MDA, malondialdehyde; cTnT, cardiac troponin T; LV EF, left ventricular ejection fraction; GOT, glutamic–oxalacetic transaminase; CPK, creatinine phosphokinase; CK-MB, creatinine kinase (muscle and brain types); AST, aspartate transaminase; GSH, glutathione; ECG, electrocardiography; i.v., intravenously; i.p., intraperitoneally.
Deferoxamine (also desferrioxamine [DFO] Fig. 8) is a well-established hexadentate-chelating agent long used to remove excess iron from the body. In iron overload, it acts by binding free iron in the bloodstream and enhancing its elimination in the urine. By removing excessive iron, DFO significantly reduces the damage to various organs and tissues, such as the liver and heart (134). With respect to ANT cardiotoxicity, DFO received the main attention in the early 1990s. It has been shown to protect the isolated rat neonatal cardiomyocytes against DOX, however, only in the iron-overloaded cells. In contrast, the DFO treatment of normal cardiomyocytes had no measurable cardioprotective effect against DOX toxicity, whether DFO was administered before or simultaneously with DOX (111). Using an acute model of DOX-exposed mouse atria, Voest et al. (282) showed that at the concentration of 200 μM DFO was the most effective protectant of the six various chelators tested, including the DEX. However, with slight increase of DFO concentration to 500 μM, its protective effects completely disappeared. In another acute cardiotoxicity study, DFO was able to protect the heart against lesions induced by single very high DOX (15 and 25 mg/kg) doses (3, 221). In contrast, using a well-established SHR model of chronic ANT cardiomyopathy, Herman et al. (106) have reported a failure of DFO to achieve a meaningful cardioprotection and reduction of DOX-induced death. A conceivable explanation of these findings was a limited penetration of DFO into the cardiomyocytes due to its high molecular weight and hydrophilicity. While DFO is among the fastest and most efficient iron chelators in solution, its access to intracellular labile iron pool is limited. Furthermore, a short plasma half-life of DFO may also reduce its potential. In fact, numerous previous studies have demonstrated that DFO is clearly inferior in protecting cardiac cells from oxidative stress-inducing agents as compared to smaller and more lipophilic ligands (15, 93, 188, 238), and therefore during the last years, the research interest shifted in this direction.

Deferiprone (L1 and CP 20, Fig. 8) is an orally effective α-ketohydroxypyridine iron chelator, effective both in monotherapy or in combination with DFO (134). In contrast to DFO, L1 is a small lipophilic molecule; it penetrates well into various tissues and is able to remove excess iron from the heart (114). It has been also shown to efficiently bind labile cellular iron, both free as well as accumulated within the mitochondria and lysosomes (74). So far, only a limited number of studies have been examining the antioxidant potential of L1 in iron overload-free conditions. L1 protected neurons as well as fibroblasts against H2O2-induced cell death in vitro (165, 189). It also protected an H9c2 cardiomyoblast-derived cell line against model oxidative injury induced by tert-butyl hydroperoxide (15) and rat hearts against the ischemia–reperfusion injury (276). Two in vitro studies demonstrated that treatment with L1 resulted in an inhibition of DOX (0.6–1.8 μM) toxicity in both iron-loaded and normal cardiac culture cells (10, 166). L1 also protected the spontaneously beating isolated rat atria against the contractility impairment induced by acute 30 μM DOX exposure (299). Hence, several lines of evidence indicated that L1, being already a clinically approved drug, might be a promising and readily available option to increase the cardiac safety of cancer patients undergoing treatment with ANTs.
Nevertheless, in a study on a clinically relevant and DEX-validated model of chronic ANT-induced cardiotoxicity in rabbits (239), L1 showed no potential to alleviate either ANT-induced oxidative stress or the LV cardiac damage and CHF, as assessed by both echocardiography and LV catheterization (209). L1 was administered orally in two doses of 10 and 50 mg/kg, 45 min before each DAU injection (3 mg/kg i.v.), and the higher L1 dose (50 mg/kg, well tolerated when administered alone) actually led to earlier animal deaths (209). The doses and timing of L1 administration were specifically designed according to the pharmacokinetic study performed in rabbits (65) to achieve and maintain drug concentrations affording significant protection against ANT cardiotoxicity in vitro (10, 65). Of note, in the most recent in vivo study, oral 10-day administration of L1 (10 mg/kg daily) protected the rat hearts against acute cardiotoxicity induced by a single high DOX dose (15 mg/kg) (6), which again points on the marked difference between acute and chronic ANT cardiotoxicity.
Deferasirox (ICL670A, Fig. 8) is the newest clinically approved orally active iron chelator. It is a synthetic triazole chelator that has been also shown capable to remove cardiac iron and attenuate myocardial oxidative stress in the state of iron overload (4). It has been shown as capable to protect H9c2 cells from tert-butyl hydroperoxide (15), significantly retard the spontaneous catecholamine oxidation, block the intracellular ROS formation, and exert significant protection against the cellular toxicity of catecholamines and their oxidation products with very low (3 μM) effective concentrations (93). ICL670A, tested in the same in vitro model as the L1, did not protect isolated rat cardiomyocytes against DOX (89). Depending upon the concentration, ICL670A either did not affect the DOX toxicity to cardiomyocytes, or even synergistically increased it. Interestingly, this occurred in spite of the fact that ICL670A quickly and efficiently removed Fe3+ from its complex with DOX, rapidly entered myocytes, and displaced iron from an intracellular iron–calcein complex. The fact that the ferric complex of ICL670A was less toxic than ICL670A alone suggested that ICL670A was cytotoxic either by removing or by withholding iron from critical iron-containing proteins (89).
Aroylhydrazones are represented by pyridoxal isonicotinoyl hydrazone (PIH, Fig. 8) and its analogs. This class of chelators has been primarily developed as orally active agents for management of iron overload (33). Previously, micromolar concentrations of PIH were shown to protect plasmid DNA from damage by Fe2+ and H2O2 (110). Salicylaldehyde isonicotinoyl hydrazone (SIH, Fig. 8) was highly effective in the protection of guinea pig-isolated cardiomyocytes, H9c2 cells, as well as other cell types against the H2O2-induced cell injury, apparently due to the preservation of mitochondrial function and/or lysosomal integrity (115, 145, 172, 238). In the in vivo conditions, PIH prevented ischemia–reperfusion injury of retina in a neonatal piglet model where it was markedly more effective than DFO (20). To date, the cardioprotective effects against ANT-induced cardiotoxicity of three aroylhydrazone analogs have been evaluated: PIH, SIH, and o-108 (ortho-chlorobenzoyl isonicotinoyl hydrazone, Fig. 8). Using a rabbit model of chronic DAU cardiomyopathy, firstly PIH was found to reduce the DAU-induced death (240). SIH and o-108 prevented both premature deaths of animals as well as significantly improved the DAU-induced impairment of cardiac function (LV EF, dP/dtmax), biomarkers of cardiac injury (cTnT), as well as histopathological parameters (255, 256). However, dose escalation of all the aroylhydrazone chelators did not provide additional protection, but on the contrary, it hampered virtually all the beneficial effects of the chelators regarding both overall mortality rate and cardioprotection (244, 255, 256). Importantly, these higher chelators' doses were nontoxic and well tolerated when administered to animals alone. Theoretically, the equivocal results of aroylhydrazone chelators in vivo could have pharmacokinetic background. The inability of aroylhydrazone chelators to provide the same degree of protection as DEX could be attributable to their relatively rapid degradation and short plasma half-life, whereas the disappearance of the cardioprotective effect after higher doses might potentially be associated with too high peak plasma concentrations overshooting a desirable degree of iron chelation in ANT-treated cardiomyocytes (140 –142). Therefore, in an attempt to shed light on the results of in vivo experiments, an in vitro study examining protection by SIH against DAU cardiotoxicity and involved mechanisms was conducted, using primary culture of neonatal rat ventricular cardiomyocytes (243). SIH has been shown to protect cardiomyocytes against the 48 h incubation with 10 μM DAU. However, the protection was only partial and with no apparent dose dependency: the reduction of DAU-induced toxicity by 3 μM SIH (22%) was comparable with that of 100 μM SIH (25%; nonsignificant). This sharply contrasted with protection against model oxidative injury by 500 μM H2O2, where SIH pretreatment resulted in dose-dependent protection with EC50 value of 2.1 μM and complete (100%) protection at SIH concentrations higher than 10 μM (243). The observed protection with SIH against both DAU and H2O2 was dependent on iron chelation, as it could be blunted by increasing the concentration of iron salts in the incubation medium. However, while SIH completely abolished the H2O2-induced significant increase of a lipid peroxidation marker, the protection by SIH against DAU cardiotoxicity was not accompanied by any significant change in this parameter (243).
Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT, Fig. 8) is a thiosemicarbazone anticancer iron chelator (291, 305). Dp44mT has been shown as a very effective agent in intracellular iron chelation, and under certain conditions, it was able to protect the cells against oxidative stress—low concentrations of Dp44mT (1 μM) were able to significantly protect H9c2 cardiomyoblasts against 200 μM tert-butyl hydroperoxide (15). Even though Dp44mT has shown some tendency to increase ascorbate oxidation under cell-free conditions, it was the most potent agent in reduction of intracellular ROS production (15). However, Dp44mT has failed to reduce DOX cardiotoxicity both in vitro (88) and in vivo (213). Dp44mT may form redox-active iron or copper complexes (127), and it is therefore possible that the ability of Dp44mT to protect the biomolecules or cells against oxidative injury is concentration-, system-, and/or context-dependent.
To summarize, studies with various iron-specific chelators (both clinically approved drugs as well as experimental agents) have yielded rather mixed results, particularly when evaluated on clinically relevant animal models of chronic ANT-induced toxicity. Clearly, none of the agents reached the high and experimental model-independent protective efficiency of DEX. This has been observed despite the fact that the studied agents are even stronger and more selective intracellular iron chelators being able to displace iron from its complexes with ANTs, and their own complexes with this metal are less likely to redox cycle than those of ADR-925. Furthermore, in some of those studies, the cardioprotective dose range was quite narrow, and even a slight (≈2.5-fold) dose increase resulted in a disappearance of protective activity (244, 255, 256, 282). It should be noted that while DEX is a prodrug that can be bioactivated to the chelating metabolite ADR-925 at the site of its effect, all the above-listed iron chelators are active ligands from the moment of administration. Of note, aroylhydrazone prochelators have been recently developed where the Fe-binding groups are chemically masked, and the active chelators PIH and SIH are generated only at the sites of ROS production (35b, 35c, 304). Unfortunately, to date, none of these agents has been tested so far as a protector against ANT cardiotoxicity. In addition, unlike the chelators discussed above, ADR-925 is far from being selective for iron, and thus it cannot be excluded that chelation of other biometals can be involved in the DEX action.
More recently, it has been shown that ANTs are able to interact with cellular iron in a more complex way than merely producing the toxic free oxygen radicals (Fig. 9). In vitro, ANTs decreased the binding of IRPs to the iron-responsive elements of mRNA, thus modifying expression of the proteins that are critical for maintaining optimal intracellular iron levels (186). Kwok and Richardson have shown that DOX induced an accumulation of iron in ferritin and prevented its mobilization, which might possibly result in functional iron deficit (147). Therefore, iron chelators might also interfere with ANTs and cellular iron in a more complex way than only shielding free iron from being a catalyst in Fenton chemistry. It is possible that an excessive iron binding, such as that apparently induced with ICL670A or Dp44mT treatments (88, 89) and too high doses of DFO (282) or aroylhydrazones (244) might exceed the optimal degree of chelation and rather act synergically with the ANT-induced iron metabolism misbalance. Of note, oxidative stress may not be only caused by iron excess, but oxidant-induced damage to mitochondria has been also described from iron deficiency (287). In fact, there is also considerable uncertainty regarding possible spatiotemporal requirement of metal chelation in ANT cardiotoxicity settings.
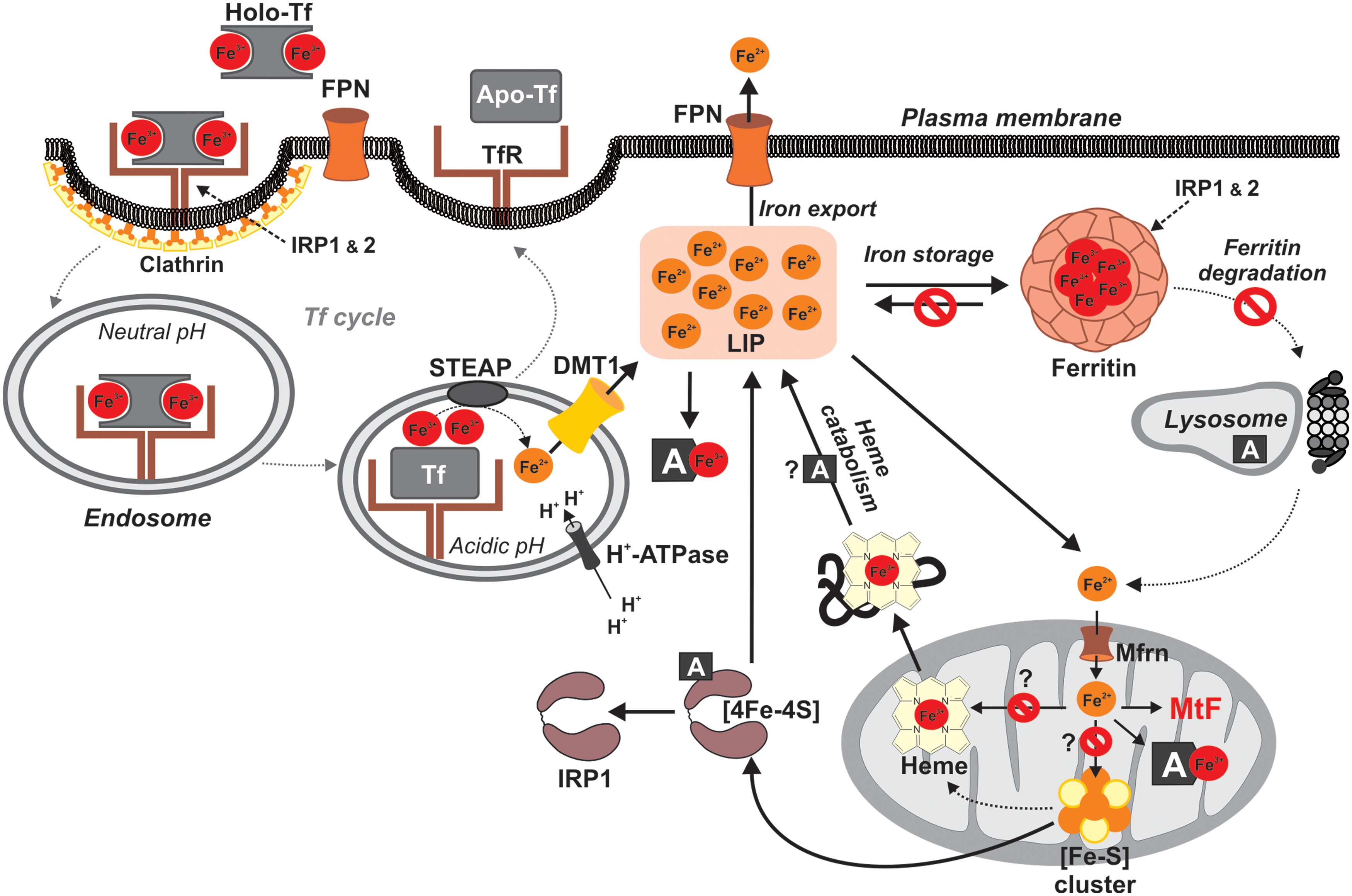
Apart from (relatively) selective chelators of iron, limited attention has been paid to chelators of other metals. Sodium 2,3-dimercaptopropane-1-sulfonate (DMPS) is a dithiol orally active chelating agent that may be used for treatment of poisoning with heavy metals such as mercury or arsenic (130). Tested on the rabbit model of chronic DAU-induced cardiotoxicity, DMPS has not shown any protective effects (117). Interestingly, no study has been published so far with copper chelators, such as D-penicillamine. Given the propensity of copper to catalyze the Fenton reaction, which is even higher than that of iron (84), and in the light of the still-prevailing oxidative stress hypothesis of ANT-induced cardiotoxicity, this approach warrants investigation.
Concluding Remarks
ANTs continue to play an undisputed role in the treatment of many forms of cancer, including hematological malignancies and solid tumors. Their clinical use has resulted in prolongation of lifespan of cancer patients, but also it has brought the risk of cardiotoxicity, which negatively affects the long-term outcomes. Considerable body of evidence demonstrated that DEX markedly reduces or even prevents ANT-induced cardiotoxicity in the clinical settings. However, due to its real or assumed drawbacks, DEX is unfortunately underutilized, which creates a room for alternative cardioprotective agents.
The classical ROS and iron hypothesis of ANT-induced cardiotoxicity is listed on the first place in practically all textbooks and a vast majority of review articles. Indeed, the DEX-afforded effective cardioprotection has been the strongest piece of evidence supporting this theory. However, studies with other iron-chelating agents—often stronger, more selective for iron, and less redox active than ADR-925—have yielded no or only partial response. Furthermore, some recent data cast doubts on the real role of DEX metabolites in the cardioprotective effects seen after DEX administration, and a detailed PK/PD approach focused on the heart may shed more light on this issue. Furthermore, if the metal chelation is indeed involved, its intracellular spatiotemporal characteristic might be critical. In recent years, numerous alternative or complementary theories have been proposed and described for both ANT cardiotoxicity and protection. The critical problem is enormous heterogeneity of the experimental models used and little effort on confirmation of proposed hypotheses using long-term chronic animal models. This may explain many contradictory findings and questionable translatability of some experimental data into the real-world clinical situation. Hence, although huge amounts of data have been published, it is very difficult to arrange them to a clear and cohesive picture. Therefore, continued efforts need to be placed on further research of the molecular pathogenesis of ANT cardiotoxicity as well as search for alternative cardioprotective therapies.
Footnotes
Acknowledgments
The authors' own work has been supported by the Czech Science Foundation (grant GACR 305/09/0416), the Internal Grant Agency of the Ministry of Health of the Czech Republic (grant NT13457-4/2012), and the Charles University in Prague (grants UNCE 33/2012 and SVV 265004, the program PRVOUK P37/5).