
Abstract
Introduction
P
The role of reactive oxygen species (ROS) and their possible sources in pulmonary hypertension (PH) is not fully understood. Mitochondria and NADPH oxidases (Nox) have been suggested as sources of altered ROS generation in PH, and it is unclear whether increased or decreased ROS contributes to the development of PH. Our studies provide evidence for the role of an increased superoxide production derived from Nox1, triggering increased migration and proliferation of pulmonary arterial smooth muscle cells in monocrotaline-induced PH. This finding is in contrast to the role of Nox4 in hypoxia-induced PH. Thus, different Nox isoforms may be targeted in different forms of PH to interfere with the pathophysiological mechanisms.
All Nox are membrane-bound, superoxide (O2
Against this background, we examined the expression of Nox isoforms and their possible role in PASMC pathobiology in MCT-induced pulmonary vascular remodeling in rats.
Results
Single subcutaneous MCT injection induces PH
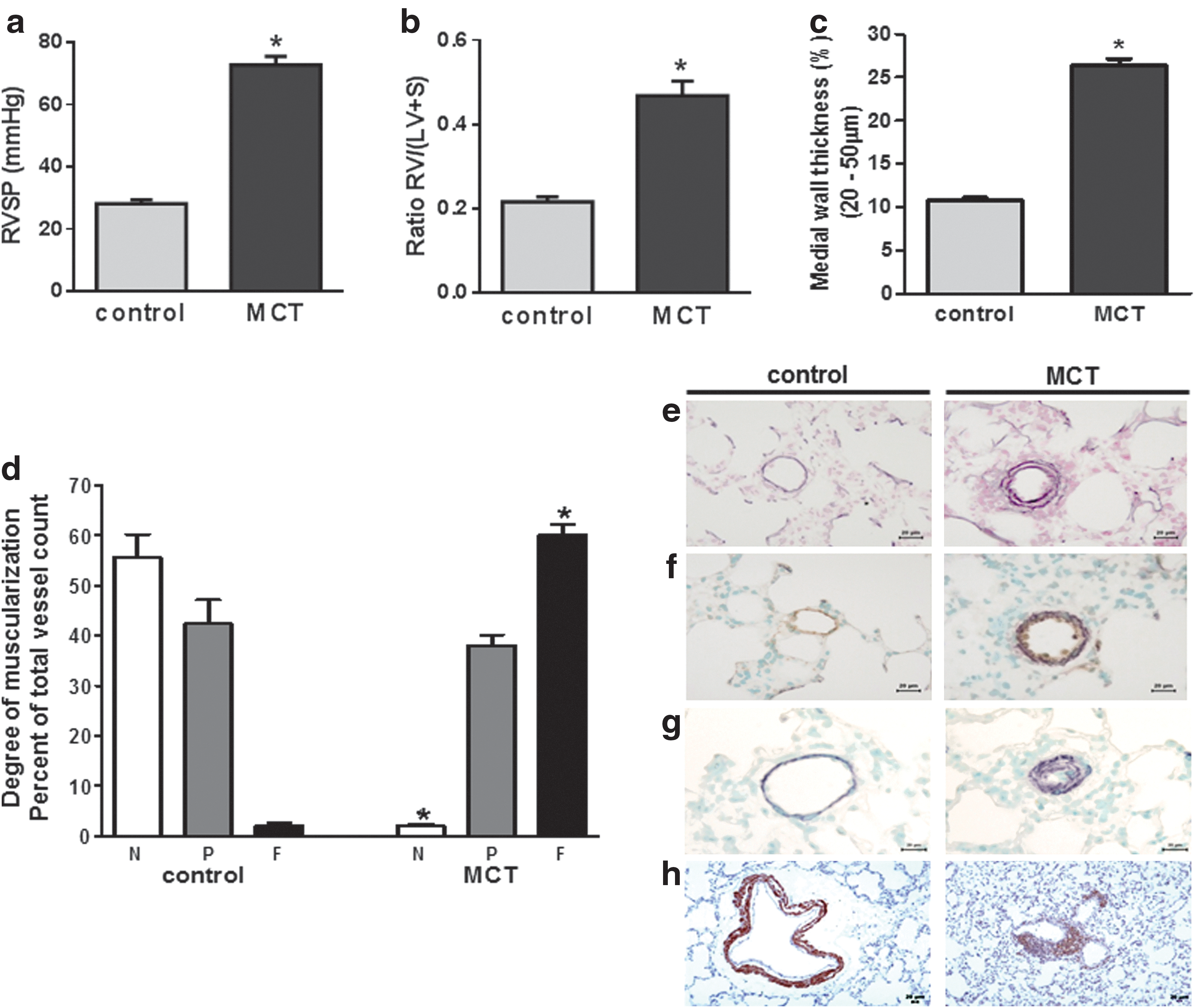
MCT injection led to PH with an increase in right ventricular systolic pressure (RVSP) from 29.3±2.1 mmHg (controls) to 70.0±3.8 mmHg (MCT) (Fig. 1a) and a right heart hypertrophy as depicted by the increment in the ratio of the right ventricle/(left ventricle+septum) from 0.22±0.01 (controls) to 0.47±0.034 (MCT) (Fig. 1b). Furthermore, MCT caused pulmonary vascular remodeling as shown by an increased medial wall thickness (Fig. 1c, e) and increased muscularization (Fig. 1d, f–h) in small precapillary pulmonary arteries, which is concomitant with a reduction of the internal vessel diameter.
MCT-dependent regulation of Nox subunits in PASMC and microdissected pulmonary arteries and localization of Nox1 in rat lungs
As shown in Figure 1, PASMC are a major site of vascular remodeling underlying MCT-induced PH. To address the role of Noxes in PH, we screened for the expression of the Nox subunits Nox1, Nox2, Nox4, Duox1, Duox2, NoxA1, NoxO1, p67phox, and p47phox in PASMC (Fig. 2a) isolated from healthy and MCT-treated rats by RT-PCR (Fig. 2b). PASMC isolated from MCT and control rats were stained for alpha-smooth muscle actin (α-sma) (Fig. 2a), and all Nox subunits except Nox2 were detected in PASMC of both MCT and control rats (Fig. 2b). Among these isoforms, Nox1 and p22phox were significantly up-regulated after MCT treatment (Fig. 2b-i). Interestingly, the control in systemic VSMC revealed that Nox1 was not regulated in smooth muscle cells derived from the aorta of MCT and control rats (aortic smooth muscle cell [ASMC]) (Fig. 2b-ii). The up-regulation of both Nox1 and p22phox was confirmed at the protein level (Fig. 2c, d). The increased expression of p22phox was not further addressed. Since Nox1 has been shown to induce mitogenic activity in nonvascular cells (66), and since mitogenesis is also a feature of PH, we next focused on Nox1. In situ hybridization revealed co-localization of Nox1 with α-sma in small pulmonary arteries (Fig. 2e). Nonvascular areas that stained positive for Nox1 messenger RNA (mRNA) comprised bronchial smooth muscle cells. The up-regulation of Nox1 in the vascular compartments was then confirmed by investigation of microdissected pulmonary arteries from MCT-treated rats and healthy controls (Fig. 2f).

Nox1 causes increased intracellular generation of superoxide and oxidative stress in PASMC after MCT treatment
In comparison to healthy controls, PASMC isolated from MCT-treated animals showed an increased intra- as well as extracellular superoxide concentration under nonstimulated conditions, as well as after stimulation with phorbole myristate acetate (PMA) (Fig. 3a and Supplementary Fig. S2a; Supplementary Data are available online at
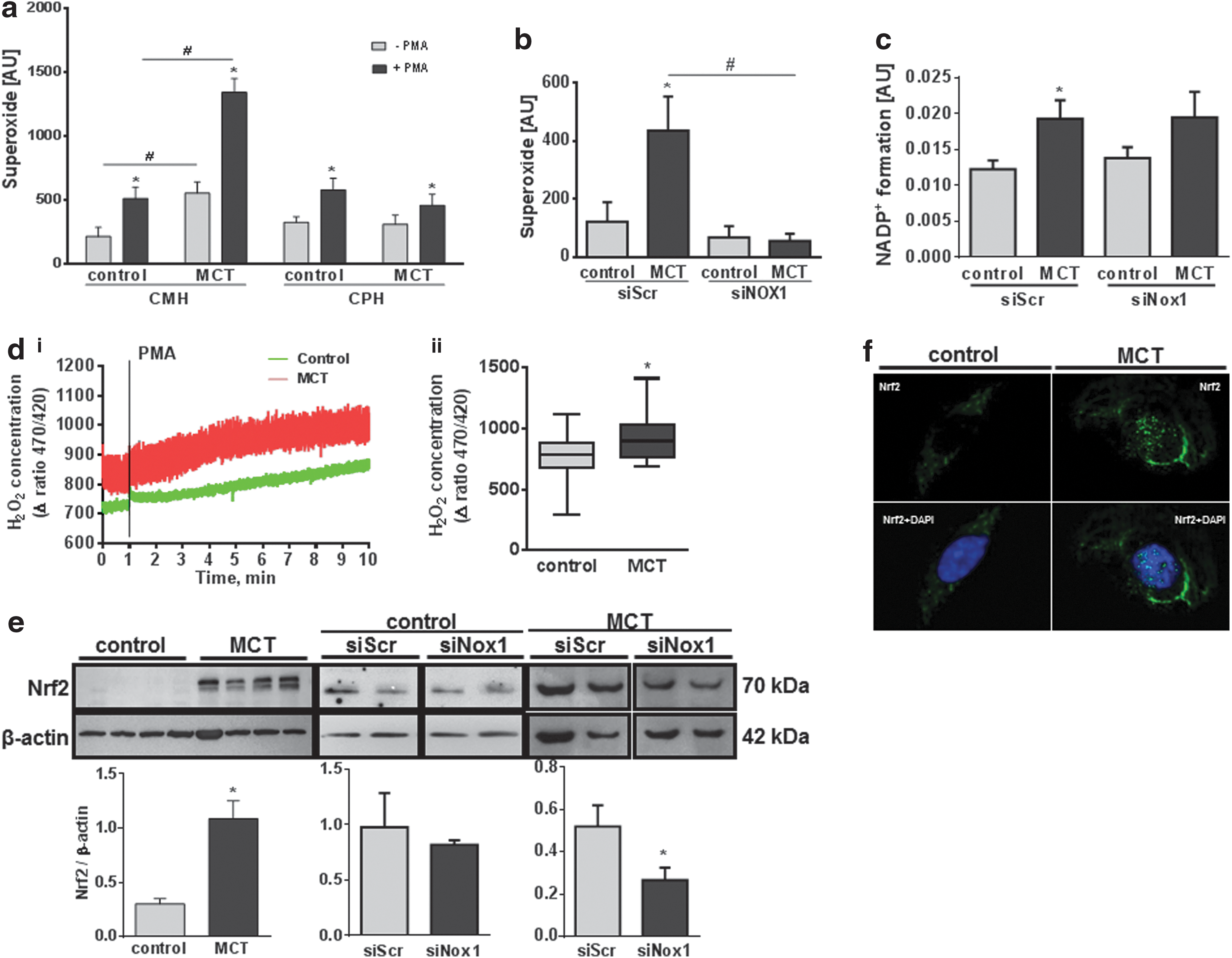
Increased intracellular ROS production does not cause unavoidable oxidative stress. To investigate whether the increased superoxide production by Nox1 in MCT-PASMC caused oxidative stress, we next analyzed the expression levels and localization of NFE2-related factor 2 (Nrf2), one of the most important regulators of cellular redox responses and detoxification (11, 39, 42, 82). MCT-PASMC exhibited an increased expression of Nrf2 and the Nrf2 target gene NAD(P)H quinone oxidoreductase 1 (NQO1) when compared with controls. MCT-induced Nrf2 and NQO1 levels were normalized by knockdown of Nox1 (Fig. 3e and Supplementary Fig. S3). Furthermore, immunostainings showed increased translocation of Nrf2 to the nucleus in MCT-PASMC (Fig. 3f).
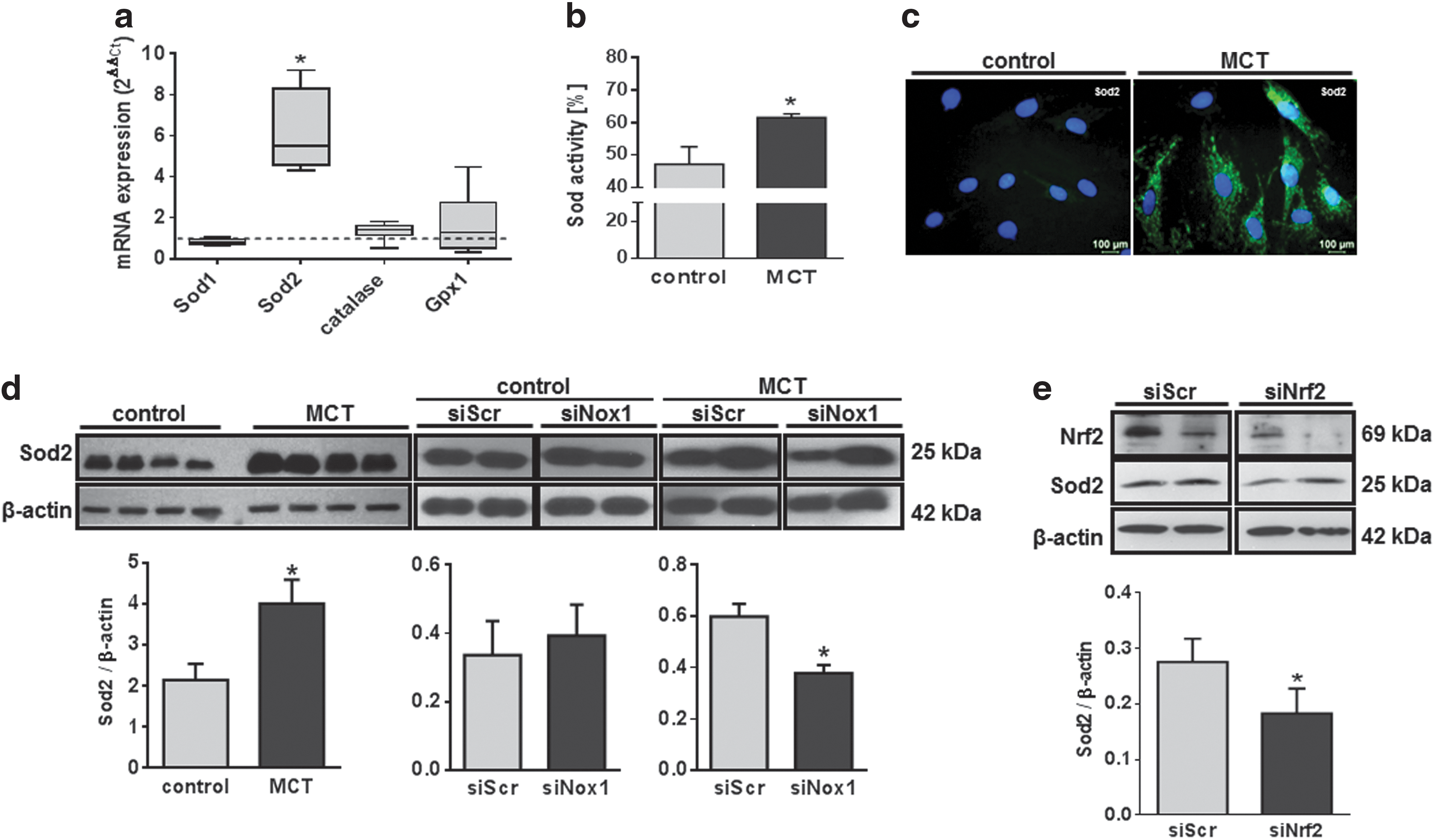
As a response to oxidative stress, we expected changes in the regulation of the antioxidative capacity of the cell. Screening for the expression of the main antioxidative enzymes revealed an increased mRNA expression of Sod2 (mitochondrial superoxide dismutase [Sod]) in MCT-PASMC, whereas the antioxidative enzymes catalase, Gpx1, and Sod1 (cytosolic Sod) were not differentially regulated (Fig. 4a). In line with the increased Sod2 expression, total Sod activity was elevated in MCT-PASMC. Accordingly, immunofluorescence imaging revealed a more intense Sod2 staining (Fig. 4c), and western blotting confirmed the Sod2 up-regulation at the protein level (Fig. 4d). Expression of Sod2 could be reduced by both the suppression of Nox1 (Fig. 4d) and Nrf2 (Fig. 4e), demonstrating clearly that Sod2 expression is dependent on Nox1. The measurements of Sod2 expression after Nrf2 suppression were performed in MCT-PASMC only, as control-PASMC showed very low expression levels of Nrf2 (Fig. 3e).
Nox1 causes increased proliferation and migration of MCT-PASMC
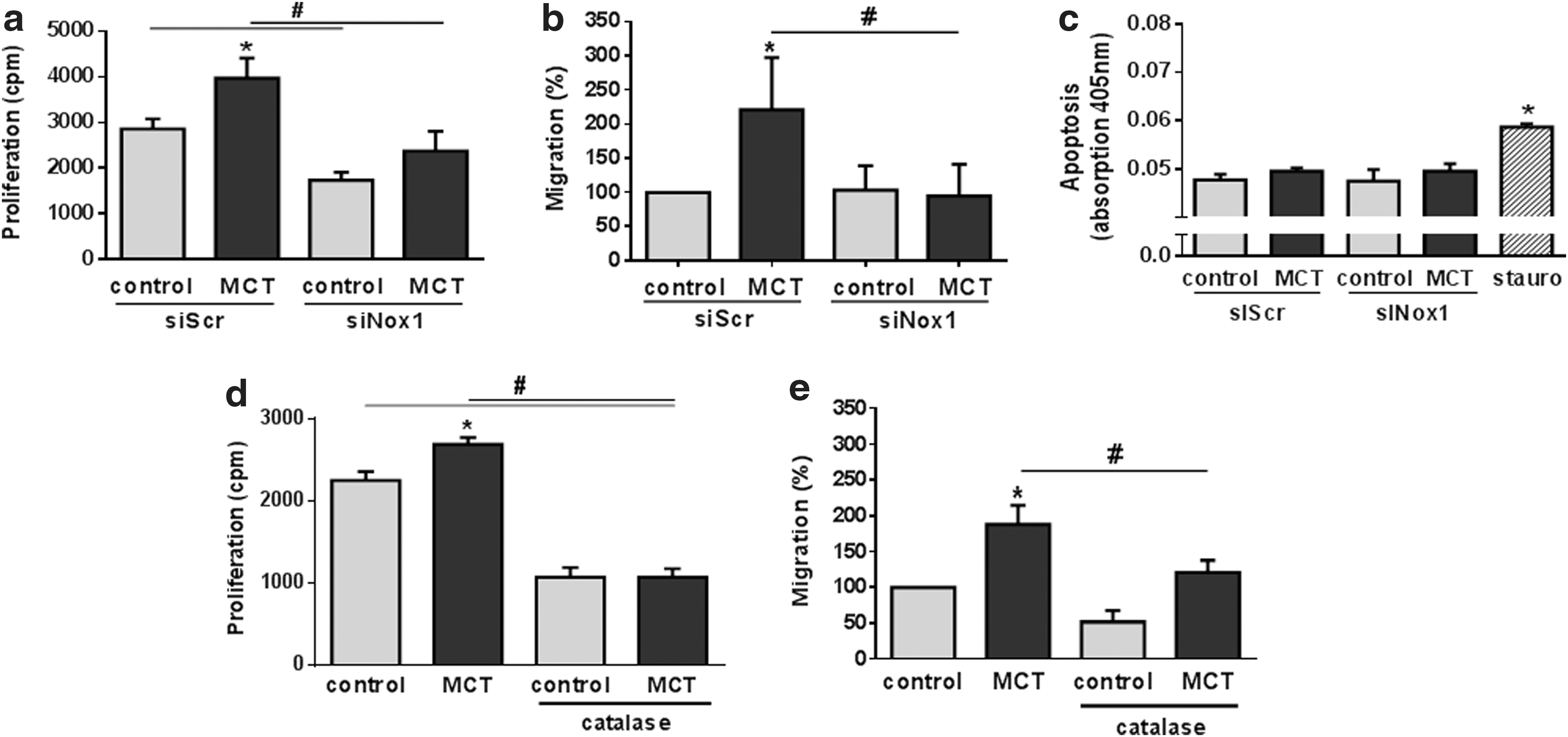
In addition to increased intracellular superoxide production, MCT-PASMC showed an Nox1-dependent increase in proliferation and migration compared with control PASMC (Fig. 5a, b and Supplementary Fig. S4). However, determination of caspase-3 activity revealed that neither MCT treatment nor Nox1 small interference RNA (siRNA) had an effect on apoptosis of rat PASMC (Fig. 5c). Treatment of PASMC with polyethylene glycol (PEG)-conjugated catalase reduced proliferation and migration of PASMC in both groups (Fig. 5d, e).
Nox1 has been implicated in cell death and necrosis by prolonged c-jun N-terminal kinase (JNK) activation and superoxide or H2O2 can induce JNK (41). The fact that we did not observe changes in JNK expression and/or phosphorylation is, thus, in line with our measurements of unchanged apoptosis (Fig. 5c and Supplementary Fig. S5).
Based on the elevated migration and proliferation of MCT-PASMC, we next screened for the expression of possible underlying Nox1 downstream targets. In this regard, we focused on the phosphorylation of extracellular signal regulated kinases (Erk) and the expression of cyclin D1, which have been shown to be primary targets for the mitogenic effects of Nox1 in lung epithelial cells and of reactive oxygen and/or nitrogen species (9, 66). We observed an up-regulation of cyclin D1 as well as an increased phosphorylation of Erk in MCT-PASMC. Both could be reduced by suppression of Nox1 (Fig. 6a, b).

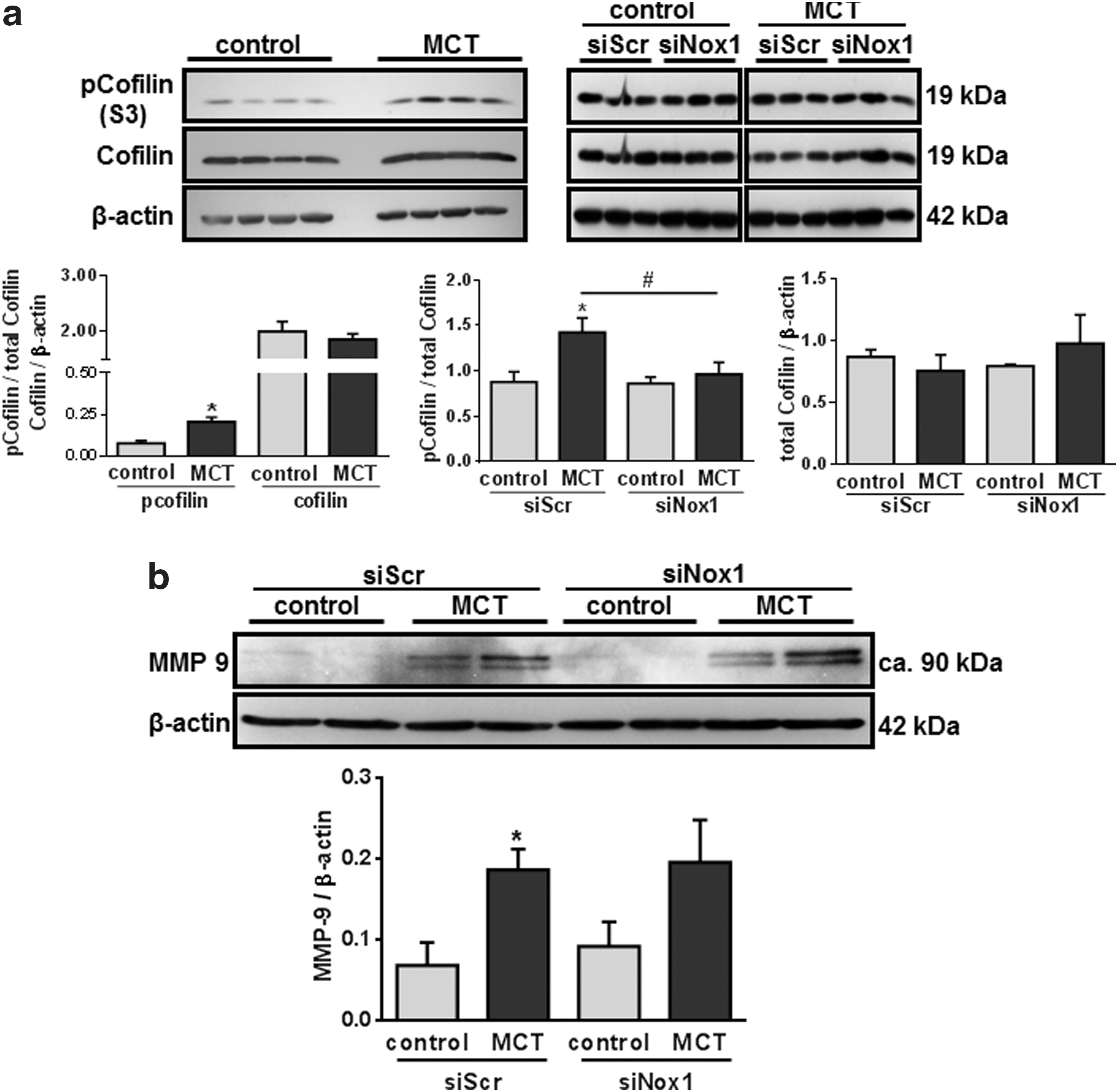
With regard to the increased migration of MCT-PASMC, we screened for the expression and/or phosphorylation of p21-activated protein kinase-1 (PAK1), cofilin, and matrix metalloproteinase-9 (MMP-9), which have been described as potential downstream targets of Nox1 with regard to migration (36, 69, 89, 94, 95). MCT treatment resulted in an increased cofilin phosphorylation (Fig. 7a) and MMP-9 expression (Fig. 7b). Expression of total cofilin (Fig. 6d) as well as the expression and phosphorylation of PAK1 (Supplementary Fig. S6c) were not altered. However, only cofilin phosphorylation, cyclin D1 expression, and Erk phosphorylation were Nox1 dependent (Figs. 6a, b and 7a). Suppression of cyclin D1 by siRNA and inhibition of Erk led to decreased proliferation and migration, respectively (Supplementary Fig. S6). Knockdown of cofilin did not alter migration of PASMC in both groups (Supplementary Fig. S7c). As proof of principle, blockage of ROS signaling using a combination of N-acetylcysteine (NAC) and 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) could reduce cofilin phosphorylation and cyclin D1 expression by trend tendency (Supplementary Fig. S7).
Platelet-derived growth factor causes increased expression of Nox1 in MCT-PASMC
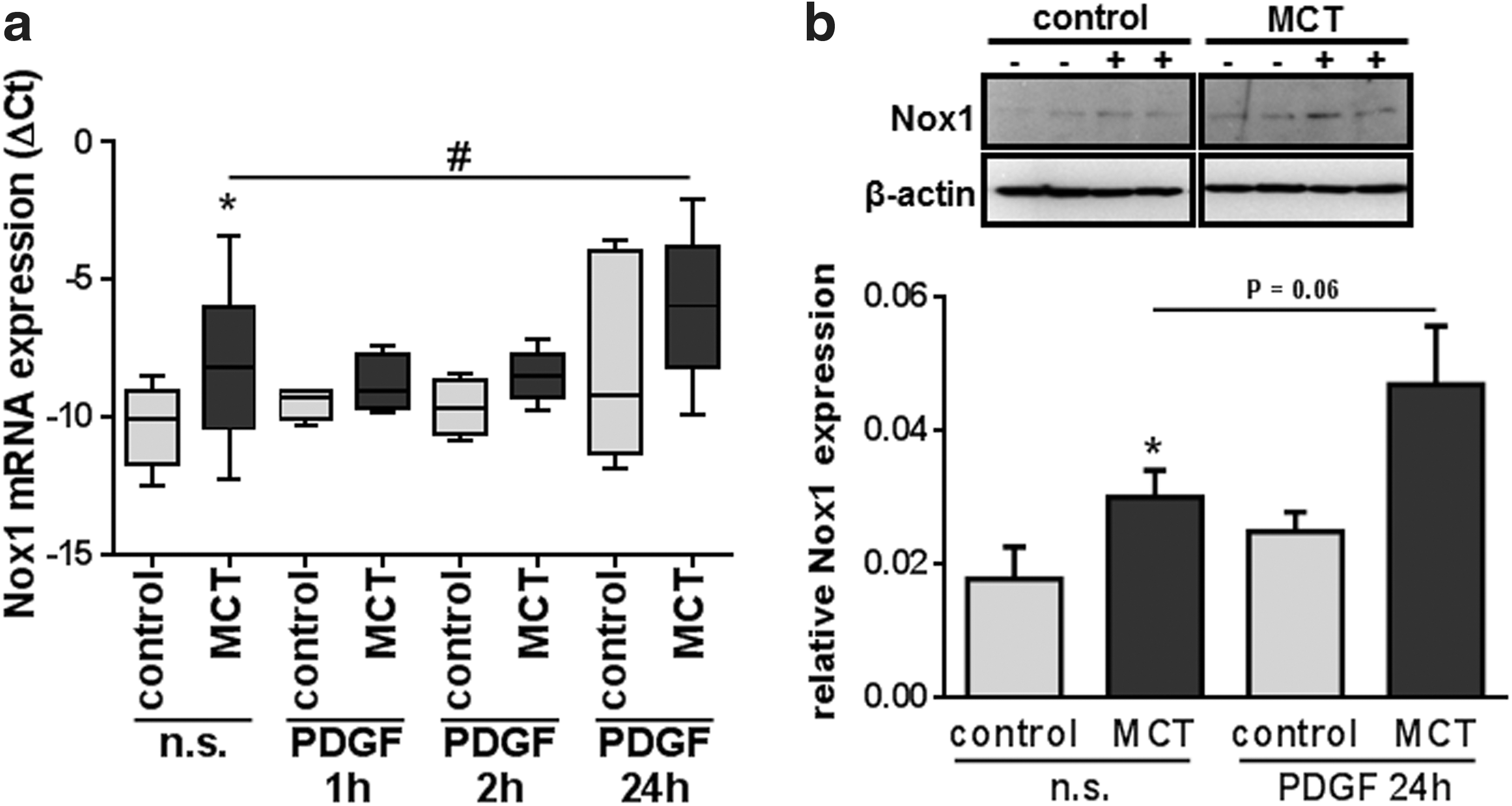
To investigate a possible link between MCT-induced PH and Nox1 up-regulation, we stimulated MCT- and control PASMC using platelet-derived growth factor (PDGF). We choose PDGF as a stimulatory agent, because it is known to induce activity and expression of Nox1 (8, 80). Furthermore, among many other growth factors, PDGF is released after MCT-induced endothelial damage, and Schermuly et al. showed an increased expression and phosphorylation of the PDGF receptor β (PDGFRβ) in MCT-treated rats (70). We stimulated PASMC of both groups with PDGF and quantified mRNA expression after 2, 4, and 24 h. In MCT-PASMC, an increase of Nox1 mRNA expression was observed after 24 h of stimulation (Fig. 8a). This was in contrast to control PASMC, where PDGF had no effect. Both, however, groups showed increased Nox1 expression at the protein level after 24 h of PDGF stimulation (Fig. 8b).
Discussion
Our data provide evidence for an important role of Nox1 and Nox1-derived superoxide release as a trigger for elevated proliferation and migration of PASMC derived from MCT-induced pulmonary hypertensive rats. PASMC migration and proliferation are key features of the vascular remodeling process underlying PH. These findings are in contrast to the role of Nox in hypoxia-induced PH in rats and mice (53, 60), as in our current study, not Nox4, but instead Nox1, was found to be up-regulated during the course of vascular remodeling. Thus, different Nox subunits may be involved in the pathogenic mechanisms of different forms of PH-related vascular remodeling. Moreover, we have demonstrated that Nox1 activates cyclin D1 expression as well as phosphorylation of Erk1/2 and cofilin, leading to increased proliferation and migration of PASMC.
In contrast to hypoxia-induced pulmonary vascular remodeling, MCT injection triggers a severe pulmonary vascular remodeling that was concomitant with an inflammatory response, endothelial cell death, and release of growth factors (52). Increased muscularization and de novo muscularization of small pulmonary arteries caused by proliferation and migration of PASMC are known to be major causes of PH development (55, 56). In this regard, ROS are thought to play a crucial role for migration and proliferation of PASMC. However, the role of ROS and their possible sources in PASMC are still under debate. For example, in chronic hypoxia, some investigators suggested a decrease of ROS, whereas others provided evidence for increased ROS generation (68, 91). Mitochondria and Nox have been suggested as sources of altered ROS generation in hypoxia-induced PH (7, 59, 60, 92). In addition to hypoxia-induced PH, these issues are also not resolved for nonhypoxia-induced forms of PH.
MCT injection was followed by extensive pulmonary vascular remodeling that was accompanied by an increase in RVSP and right heart hypertrophy. After it is metabolized in the liver, MCT is thought to cause endothelial damage mainly in the pulmonary vessels, as it is the first vasculature with small capillary vessels distal of the liver (52). Screening of Nox mRNA expression levels in PASMC after MCT treatment revealed an increased expression of Nox1 and a minor up-regulation of p22phox, but no changes in Nox4 expression. These results were confirmed at the protein level. Independent of the different Nox isoform expressions, the increase in p22phox can contribute to the Nox1 effects, as it is an universal stabilizer of almost all Nox isoforms (2). Since MCT, however, might also act on the systemic vasculature, we assessed Nox1 mRNA expression also in ASMC. Nox1 expression was increased in PASMC only, excluding a systemic up-regulation of Nox1 after MCT treatment, thus confirming the specificity of the observed effects for the pulmonary circulation.
The finding that different Nox isoforms are active in different types of PH or vascular remodeling may be explained by the different underlying pathological mechanisms of PH. In contrast to hypoxia, MCT injection causes a strong inflammatory response with a different profile of the secretion of growth factors and cytokines, and appearance of, for example, PDGF, Angiotensin II (ATII), Thrombin, and TNF-α. Especially PDGF and ATII are known to stimulate Nox1 expression and activity (8, 93), thereby triggering an increased Nox1 expression and activity in microvascular PASMC after MCT injection. On the other hand, in hypoxia-induced PH, the expression and, consequently, the activity of the constitutively active Nox4 appear to be induced by HIF1α stabilization (20). Although they are members of the family of Nox, the functions of Nox4 and Nox1 are quite different. In contrast to Nox1, Nox4 is thought to produce H2O2 as a primary product and seems to be constitutively active independent of the activation by cytosolic subunits, although Poldip2 has been described as a potential regulator (50, 83).
In addition, under certain conditions, Nox4, in contrast to Nox1 and Nox2, may have beneficial effects by protecting mouse lung endothelial cells from ischemic or inflammatory stress (75). Thus, Nox4 and Nox1 can exert different functions. In a contradictory study, Dorfmüller and colleagues reported an increased mRNA expression of the Nox isoforms Nox2 and Nox4 in an MCT-induced PH model (24). However, for their expression studies, lung homogenate was used, which might have masked regional changes in the expression of Nox isoforms in, for example, PASMC or pulmonary vessels, investigated in our study. Indeed, a compartment-specific regulation was shown to not be visible when investigating whole lung tissue due to interference with nonvascular cells (87). Based on the increase in Nox1 and p22phox expression and since Nox1 is known to be a superoxide-generating enzyme (80), we discriminated between intra- and extracellular superoxide production using the highly cell permeable spin probe CMH and the only partially cell permeable CPH (low cell permeability at physiological pH) (23) alone or in combination with PEG-Sod or Sod, respectively. With such an approach, it has previously been shown that a differentiation between extra- and intracellular superoxide can be achieved (44). Moreover, EPR technology is thought to overcome false-positive results of fluorescent dyes for ROS detection (21). Our data revealed that the Nox1-derived superoxide release occurs in the intracellular milieu only. siRNA experiments confirmed the Nox1 dependency of superoxide production in MCT-PASMC. In line with these findings, Nox1 has been shown to produce intracellular superoxide in gut cells (40), and the subcellular localization of Nox1 has been allocated to caveolae and early endosomes. Moreover, Nox1 is known to alter intracellular signaling via generation of superoxide (57). In addition to unstimulated superoxide production, we also analyzed superoxide release after PMA stimulation. PMA has been shown to activate protein kinase C (PKC) and, thus, to cause the assembly of the cytoplasmic and membrane bound subunits of Nox, thereby activating this multiprotein complex (38). PMA exclusively activates Nox isoforms that are activated on by such a mechanism. The stimulating effect of PMA on superoxide release in our experiments, thus, excludes the activation of the constitutively active Nox4 isoform and supports our findings of increased intracellular superoxide production due to increased Nox1 expression and activity (53). We could also detect increased NADPH consumption in MCT-PASMC. However, this could not be linked to Nox1, as the rather small effect of Nox1 on cellular NADPH consumption is probably masked by the contribution of biochemical processes such as the pentose phosphate pathway, regeneration of glutathione, and lipid synthesis.
Besides superoxide, H2O2 has been suggested as being a very important ROS signaling molecule (29). In this regard, we also found that H2O2 generation was increased in MCT-PASMC. We cannot exclude an increased activation of other Nox isoforms beside Nox1, independent of their expression levels. However, related to the fact that we were not able to detect Nox2 in rat PASMC and that Nox4 is constitutively active after expression (53), we speculate that an increased activation of Noxes via increased phosphorylation of the cytosolic subunits would affect Nox1 almost exclusively. In this context, a potential role of the dual oxidase (Duox) enzymes for H2O2 generation can, nevertheless, not be eliminated, but Duox activity cannot contribute to an elevation of superoxide production. Furthermore, Duox is thought to play an important role in host defense, rather than in disease (45). With regard to other contributing effects, we are not aware of any post-translational modifications altering the activity of Nox, except the phosphorylation of the cytosolic subunits (81).
Our findings of elevated Nrf2 levels and nuclear translocation of Nrf2 in MCT-PASMC are in accordance with increased superoxide production, as they indicate a shift of the redox state to a more oxidative environment with oxidative stress (63). In addition, the dependence of Nrf2 on Nox1 was proved by our Nox1 knockdown experiments. As a response to oxidative stress, PASMC may adapt their antioxidative capacity. In this regard, screening for the expression of the main antioxidative enzymes at the mRNA level revealed the mitochondrial Sod (Sod2) to be up-regulated; whereas Gpx1, catalase, and Sod1 were not altered. Increased Sod2 expression could be confirmed at the protein level and was accompanied by an increment of total Sod activity. The induction of Sod2 in response to oxidative stress is well established, whereas the Sod1 gene is often constitutively expressed, not as easily inducible as Sod2, and has been considered a “housekeeper gene” (58). The regulation of Sod2 in different models of PH is controversial. Archer et al. described an epigenetic attenuation of Sod2 in pulmonary arterial hypertension (3). In contrast, Sod2 expression was up-regulated over time in a lamb model of increased postnatal pulmonary blood flow, secondary to in utero aortopulmonary graft placement (77). In human IPAH, Fijalkowska et al. observed a decreased Sod2 expression, but this decrease was shown in endothelial cells only (27). Thus, the regulation of Sod2 seems to be dependent on the model, the cell type, and the time point examined. Nox1 as well as Nrf2 siRNA caused a counter regulation of Sod2. We, thus, propose that Nox1 causes Sod2 up-regulation by increasing the expression of Nrf2.
Nox1 knockdown was followed not only by normalization of PMA-stimulated superoxide production, but also by normalization of migration and proliferation of MCT-PASMC, indicating its essential role in these processes. Along these lines, catalase treatment significantly decreased proliferation and migration of MCT-PASMC, supporting our findings of an elevated H2O2 production after MCT treatment. Not surprisingly, catalase treatment also reduced proliferation of control PASMC, as Nox enzymes generate H2O2 or O2
Focusing on possible downstream targets of Nox1, we investigated the regulation and/or phosphorylation of PAK1, cofilin, cyclin D1, MMP-9, and Erk1/2. These targets were chosen, as (i) it was observed that Nox1 along with hypertrophy has also been implicated in cell migration (74); (ii) the downstream effects of Nox1 with regard to migration can be mediated by activation of PAK1 (89) and dephosphorylation and activation of cofilin (95); and (iii) Nox1 was demonstrated to play an essential role in proliferation, and cyclin D1 as well as Erk have been identified as targets of Nox1 during cell-cycle regulation (4, 9, 66). The increased phosphorylation of Erk and expression of cyclin D1 in MCT-PASMC can account for the observed accelerated proliferation of MCT-PASMC (9). Moreover, its Nox1 dependency and relevance in MCT-PASMC is shown by our data that Nox1 knockdown prevented cyclin D1 up-regulation and Erk phosphorylation in MCT-PASMC and that suppression of cyclin D1 and inhibition of Erk reduced proliferation and migration, respectively. Thus, cyclin D1 and Erk are likely downstream candidates of Nox1. Moreover, cofilin phosphorylation was linked to Nox1 signaling in our study. Interestingly, we did not observe changes in PASMC migration after cofilin suppression, which may be explained by the fact that the activity of cofilin is regulated by phosphorylation and dephosphorylation and not only by its expression (32). Although the achieved knockdown of cofilin was rather low, its mechanism of action is probably based on the circle of phosphorylation and dephosphorylation, rather than total cofilin expression. In its unphosphorylated form, cofilin severs and depolymerizes actin filaments (51), promoting migration of nonpulmonary VSMC. Our study, however, revealed increased cofilin phosphorylation in MCT-PASMC. In this regard, different studies showed that the activation of cofilin is induced by dephosphorylation of the protein (1, 18, 61, 95) or that dephosphorylation of cofilin is not necessarily coupled with activation (62, 79). The phosphorylation of cofilin and the subsequent stabilization of actin filaments might, therefore, be an essential mechanism that is used to control turnover processes of actin filaments and cytoskeletal alterations to protect cells from apoptotic cell death (6, 18, 51). The activation mechanism of cofilin seems to differ in diverse cell types.
A further mechanism explaining how Nox1 can lead to increased migration of VSMC is the promotion of MMP-9 expression via an ROS-dependent phosphorylation of Erk 1/2 (36, 94). Although we found an increased MMP-9 expression in MCT-PASMC, in contrast to cyclin D1 expression, cofilin, and Erk phosphorylation, this increase was not Nox1 dependent. However, further studies may address MMP-9 activity in this context, which speculatively is affected by Nox1-dependent superoxide production.
With regard to Nox1 upstream signaling expression, activity of Nox1 can be induced by a variety of factors. For example, pulmonary circulation MCT-induced endothelial damage causes release of a variety of different growth factors. Among those, PDGF is known to be increased in MCT-induced PH. In addition, the PDGFRβ is phosphorylated on MCT application in pulmonary vessels, and PDGF has been shown to induce expression and activity of Nox1 in nonpulmonary systems (8, 80). To focus on such a possible link between endothelial damage and increased Nox1 expression, we stimulated our PASMC with PDGF. After 24 h of PDGF stimulation, we observed an increased mRNA expression of Nox1 in MCT-PASMC, but not in cells from untreated controls. However, the effect of PDGF treatment on Nox1 expression in MCT-PASMC was rather small. With regard to the interpretation of these data, it has to be taken into account that the basal expression of Nox1 in MCT-PASMC is already elevated, which might weaken the effect of a further PDGF stimulation.
Thus, among many other potential stimulators of Nox1 expression, PDGF might be of special interest, (i) as MCT-PASMC appeared to be more susceptible to PDGF stimulation than control-PASMC (at least at the mRNA level in the investigated time frame) and (ii) the finding that the tyrosine-kinase inhibitor imatinib was able to reverse MCT-induced PH (70).
According to our current and previous (60) investigations, different Nox isoforms may be targeted in different forms of PH to interfere with the pathophysiological in PASMC, leading to alterations in the pulmonary arterial vessel structure. Previously detected up-regulation of Nox4 in hypoxia-induced PH in mice as well as in end-stage human IPAH may be related to the fact that MCT-induced PH in rats is much more severe than hypoxia-induced PH in mice, ultimately leading to death. In addition, the type of vascular remodeling addressed here (e.g., inflammation, contribution of the endothelium) is different than that observed in hypoxia-induced PH. Moreover, the MCT model may represent features of human PH that are different from end-stage IPAH (which was investigated in our previous study), that is, features being active during development of IPAH or in other forms of PH, for example, those in which inflammation plays a major role. Those differences between these models may also explain why we could not detect an up-regulation of Nox1 in end-stage IPAH (data not shown) (12, 73, 86). The authors are aware that there is currently no single model which represents all facets of IPAH or other forms of PH (apart from hypoxia). Nevertheless, animal models can be valuable in terms of identifying new mechanisms of pulmonary vascular remodeling, which, in turn, may to lead to the development of new therapies in humans (13, 30, 33, 72).
With regard to Nox1 as a potential target for therapeutic intervention in MCT-induced PH and other forms of PH, as well as PH in different species, including man, further investigations are needed. With regard to human PH, screening of early-stage IPAH or, for example, HIV-associated PH and those forms with inflammation should be helpful to elucidate this issue. Interestingly, along these lines, antioxidant therapies have been shown to exhibit beneficial effects on MCT-induced PH (14, 67, 78).
In summary, apart from the fact that different Nox isoforms may be active in different species, types and stages of PH, we deciphered in detail the role of superoxide generation by Nox1 and its possible downstream signaling mechanisms with regard to vascular pathobiology. In addition to PH, such mechanisms might be active in other proliferative vascular diseases. Beyond the speculative aspects on human disease, our data and previous investigations (60) support the fact that different Nox in different species and models of PH can contribute to PH development. The mechanism described in the current article would be taken into account for PH development not only in humans, but also in animals such as cattle, horses, sheep, goats, and dogs (26, 34, 37).
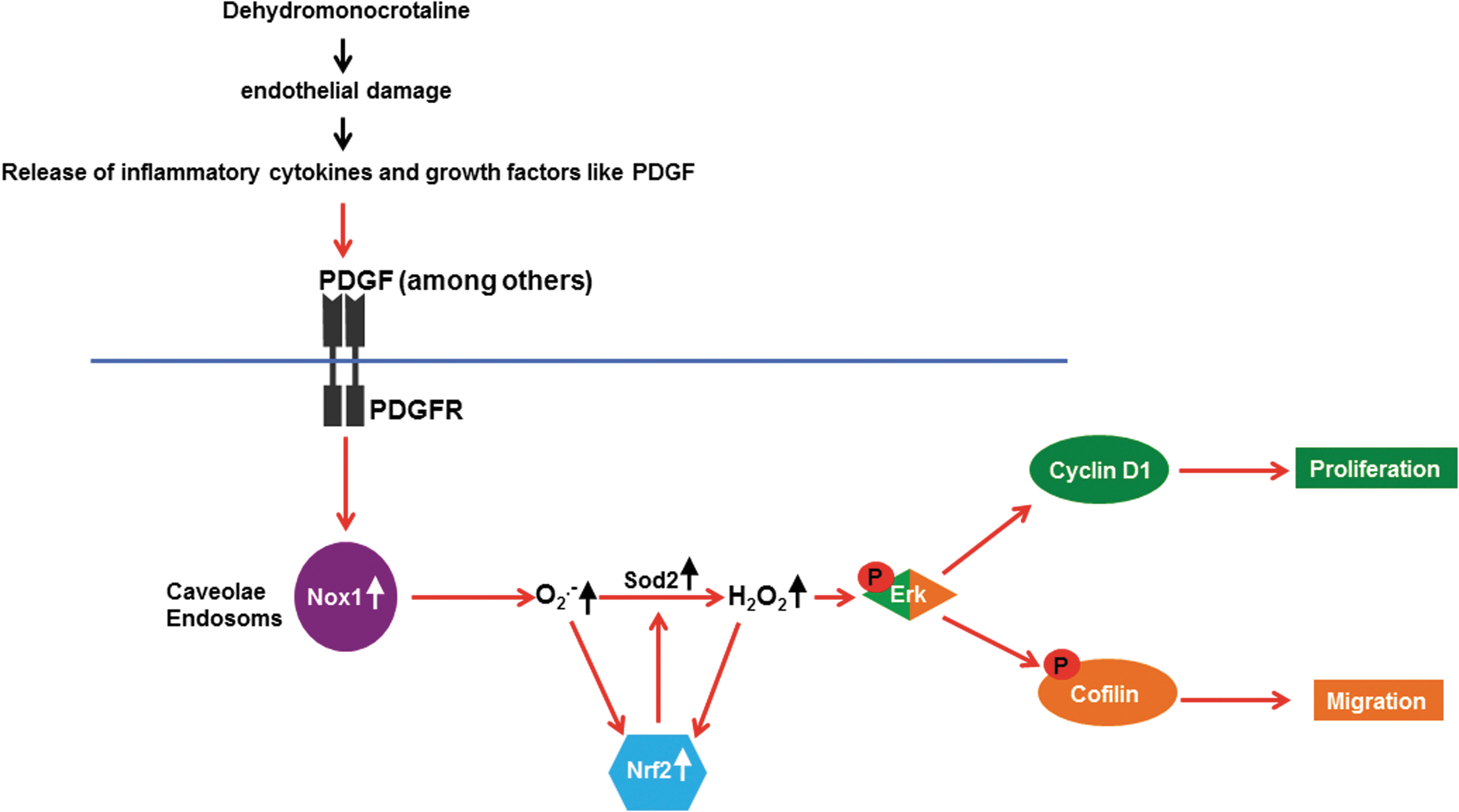
Furthermore, we hypothesize that Nox1 might be of particular importance for pulmonary vascular remodeling which is associated with inflammatory conditions such as in the MCT-induced PH, as Nox1 was shown to be up-regulated in inflammatory diseases such as chronic obstructive pulmonary disease, acute lung injury, and different forms of cancer (12, 73, 86). Figure 9 summarizes our findings in a schematic illustration.
Materials and Methods
Animals
Adult male Sprague–Dawley rats (300–350 g body weight) were obtained from Charles River Laboratories (Sulzfeld, Germany). Animal experiments were approved by the Federal Authorities for Animal Research of the Regierungspräsidium Giessen (Hessen, Germany). A total number of 41 rats has been used.
MCT treatment
MCT (Deishofen) was administered as a single subcutaneous injection in a dose of 60 mg·kg−1 body mass, as described (70). Control rats received an equal volume of isotonic saline. Rats were sacrificed 35 days after MCT injection.
Isolation of pulmonary arterial and aortic smooth muscle cells
PASMC were isolated from healthy rats and animals 35 days after MCT injection, modified from a previously described protocol (88). In brief, rats were anesthetized, and the lung vasculature was flushed via the pulmonary artery with 0.9% NaCl in order to remove the blood. Subsequently, we instilled iron particles (Iron-II, III-oxide, 98% Fe3O4; Sigma-Aldrich, Munich, Germany) into the precapillary vessels of the pulmonary artery. Afterward, lungs were dissected, and the iron particles sticking to pieces of the pulmonary artery were isolated from the mixture using a magnetic concentrator (Invitrogen, Karlsruhe, Germany), washed, and seeded on cell-culture plates in M199+10% FCS+1% penicillin/streptomycin (P/S). Aortic smooth muscle cells (ASMCs) were isolated by scratching the media off the longitudinal opened aorta, followed by chopping the media into small pieces. Those pieces were seeded on cell culture plates in M199+10% FCS+1% P/S. Cells were shock frozen at P0 in liquid nitrogen and stored for later experimental usage or cultured until P2 and used directly.
Cultivation of pulmonary arterial and ASMCs
Cultures were maintained at 37°C in a humidified 5% CO2, 95% O2 atmosphere. For all experiments, cells were used in passage 2. A comparison between freshly isolated (passage 0) and passage 2 and 3 cells revealed that no changes in cell morphology or mRNA expression of Nox1, Nox4, p47phox, p67phox, p22phox, NoxA1, and NoxO1 occurred between these passages (Supplementary Fig. S8).
Immunocytochemistry
Two thousand cells/well were grown on eight-well permanox chamber slides (Nunc GmbH & Co. KG, Wiesbaden, Germany) in 300 μl M199+10% FCS+1% P/S and fixed in ice-cold acetone/methanol (1:1) (Sigma-Aldrich, Steinheim, Germany). Cells were then washed with PBS, and nonspecific binding sites were blocked for 60 min in 3% BSA in DPBS buffer. Incubation of primary antibodies was performed in 0.1% BSA at 4°C overnight. The following antibodies were used: α-smooth muscle actin (α-sma, 1:2500), Superoxide dismutase 2 (Sod2, 1:5000), and Nrf2 (1:100). Antibodies were purchased from Abcam plc, Cambridge, the United Kingdom. Cells were washed in PBS, and incubation of secondary antibodies (Alexa Fluor® 488 F(ab′)2-Fragment goat anti-rabbit IgG [2 mg/ml] and Alexa Fluor 555 goat anti-rabbit IgG; Molecular Probes, Eugene, OR) was performed at room temperature for 1 h. In the case of α-sma, we performed a direct staining. Cells were then washed in PBS again, and nuclei were stained with 4′,6-Diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 10 min.
Immunohistochemistry
Medial vessel wall thickness and degree of muscularization were assessed as previously described (43). In general, the assessment of pulmonary vascular remodeling was done by determination of the degree of muscularization of the rats' small peripheral pulmonary arteries. Intra-acinar arteries in rats were analyzed by categorizing them as fully muscular, partially muscular, and nonmuscular. In addition, the medial wall thickness of the vessels was analyzed. All analyses were done in a blinded fashion.
Medial wall thickness
After dehydration and paraffin embedding, 3 μm sections were stained for Elastin—Nuclear Fast Red to assess the medial wall thickness. Medial wall thickness was defined as the distance between the lamina elastica interna and lamina elastica externa. Percentage of medial wall thickness (% MWT) was examined by light microscopy using a computerized morphometric system (Qwin; Leica, Wetzlar, Germany) and was calculated by a formula:
% MWT=2×medial wall thickness/external diameter)×100
Degree of muscularization and myosin heavy chain staining
Three-micrometer sections of formalin-fixed and paraffin-embedded lung tissues were obtained, and double immunostaining was performed with an anti-α-smooth muscle actin antibody (1:900, clone 1A4; Sigma, Saint Louis, MO) and anti-human von Willebrand factor antibody (vWF, 1:900; Dako, Hamburg, Germany) to assess the degree of muscularization. In addition, we performed myosin heavy chain staining (MHC, 1:100; Abcam plc). The sections were initially maintained for 60 min at 58°C–60°C in the heating chamber. After that, the sections were deparaffinized in xylol and progressively rehydrated in a graded ethanol series. The endogenous peroxidase activity was blocked by using a freshly prepared solution of H2O2 in methanol in the volume ratio of 1:1. Similarly, endogenous biotin and streptavidine were eliminated by using a specific biotin/streptavidine blocking solution. Antigen retrieval was performed by treatment with trypsin for 10 min at 37°C (15 min proteinase k treatment at room temperature in case of MHC). The slides were then incubated with normal horse serum for 30 min to avoid nonspecific binding caused by immunoglobulin cross-reactivity and subsequently incubated with the primary antibodies at 37°C. Both antibodies were diluted 1:900 in 10% BSA and maintained for 30 min. The slides were washed several times with PBS and further incubated with the corresponding biotinylated secondary antibodies. Two different substrates were used for visualization by a reaction with horseradish peroxidise/streptavidine complex coupled to the secondary antibodies: The smooth muscle layer was labeled purple/violet using the VIP substrate, and DAB turned the vascular endothelium brown. Finally, the sections were counterstained with methyl green, then progressively dehydrated, coverslipped using mounting medium, and examined by light microscopy using a computerized morphometric system (Qwin; Leica). Vessels of 20–50 μm outer diameter were used for analysis. Each vessel was categorized as nonmuscularized (N), partially muscularized (P), or fully muscularized (F). The percentage of pulmonary vessels in each category was determined by dividing the number of vessels in that category with the total number counted.
RNA interference and catalase treatment
PASMC from passage 2 were cultured in six-well tissue culture plates, and used for RNA interference. Depending on the experiment, cells were cultured in 6-well, 24-well cell, or migration assay culture plates, and used for RNA interference or treatment with PEG-conjugated catalase. Cultures were maintained at 37°C in a humidified 5% CO2, 95% O2 atmosphere for 1 day. The next day, the medium was changed to low-serum medium containing antibiotics (1% (v/v) FCS, 1% (m/v) penicillin, and streptomycin in M199 (Gibco, Karlsruhe, Germany) and incubated overnight for cellular synchronization. Transfection of siRNA was performed in low-serum medium containing no antibiotics. The medium was changed at least 1 h before transfection. About 100 nM of ON-TARGETplus SMARTpool Nox1, Nox4, Cofilin 1/2, Cyclin D1, and Nrf2 siRNA (Thermo Fisher Scientific, Inc., Waltham, MA) was transfected using 1 μl Lipofectamine RNAiMAX (Invitrogen, Frankfurt, Germany) per cm2 of the well. Both siRNA and the transfection reagent were diluted in OPTI-MEM medium as well. For controls, scrambled siRNA (Thermo Fisher Scientific, Inc.) was employed. After 8 h of transfection, the medium was changed to M199 medium containing antibiotics (10% (v/v) FCS, 1% (m/v) penicillin and streptomycin. Forty-eight to 72 h after transfection, cells were prepared in different ways for respective experiments.
Proliferation assay
PASMC (5000 cells per well) from passage 2 were cultured in 48-well tissue culture plates (Greiner bio-one, Frickenhausen, Germany) in an atmosphere of 37°C, 5% CO2, and 95% O2, and used for proliferation assays. Depending on the experiment, cells were then starved for 24 h and treated with siRNA or PEG catalase, which was again followed by starvation and 5 h of incubation with 3H-thymidine (Amersham, München, Germany). After that, cells were washed with ice-cold PBS and lysed in NaOH at 4°C overnight. 3H-thymidine in the lysate was measured by liquid scintillation counting (Rotiszint® eco plus, Roth, Germany) in a liquid scintillation counter (Packard Bioscience [now Perkinelmer], Meriden, CT).
Migration assay
Migration of PASMC was assayed by transwell in vitro migration assays. Five thousand cells/well in 100 μl Medium+0% (v/v) FCS, 1% (m/v) P/S (Gibco) were added to the upper chamber of the transwell (Becton Dickinson GmbH, Heidelberg, Germany). The pore size of the filters separating the upper and the lower chamber was 8 μm. The lower chamber was filled with 600 μl medium+10% (v/v) FCS, 1% (m/v) P/S. Cells were allowed to migrate for 8 h at 37°C, 5% CO2, and 95% O2 atmosphere. After 8 h, the filters were washed with PBS, and cells were fixed in an ice-cold acetone/methanol mixture (1:1). The upper side of the filter with the nonmigrated cells was wiped with a cotton swap, while the bottom side was stained with crystal violet for 15 min and washed with water. Migrated cells were counted on a glass slide using light microscopy.
Apoptosis assay
For assessment of apoptosis, the CaspACE Assay System, Colorimetric (Promega, Mannheim, Germany), was applied according to the manufacturer's instructions.
Inhibition of extracellular regulated kinase
For the inhibition of extracellular-regulated kinase (Erk), PASMC were treated with the ERK Inhibitor II, FR180204 (1 μM) (Calbiochem; EMD Millipore Corporation, Billerica, MA) for 24 h. Afterward, cells were prepared for migration assays.
N-acetylcysteine and TEMPO treatment
MCT-PASMC were treated with NAC (25 μM) and TEMPO (1 μM) for 24 h. Both substances were added to the complete growth medium. Afterward, cells were prepared for western blotting.
Measurement of Sod activity
Activity of total Sod was measured by an Sod assay (Cell Technology, Mountain View, CA) based on inhibition of formazan formation by Sod. The assay was performed according to the protocol provided by the manufacturer.
Hemodynamic
Measurements were performed as previously described (43). As an index of RV hypertrophy, the ratio of the right ventricle weight to left ventricle plus septum weight (RV/LV+S) was calculated.
Real-Time PCR
Relative quantification of the Nox subunits and the antioxidative enzymes was done using Mx3000P QPCR System (Agilent Technologies, Waldbronn, Germany). Data are shown as fold regulation of the expression in controls. Controls were normalized to 1 (dotted line). All primers used in the real-time PCR experiments were intron-spanning. The Ct values were normalized to the endogenous control HPRT (Hypoxanthine-guanine phosphoribosyltransferase). For cDNA synthesis, reagents and incubation time were applied as described earlier. The fold change 2ΔΔCt was calculated as previously described, and controls were normalized to 1 (dotted line) (28). The following sequences were used as primers in real-time PCR:
HPRT forward: 5′-CTCAGTCCCAGCGTCGTGAT-3′
reverse: 5′-AGCACACAGAGGGCCACAAT-3′
Nox1 forward: 5′-ATGGTCCCTTTGGCACAGTC-3′
reverse: 5′-ATCCCAGCCAGTGAGGAAGA-3′
Nox2 forward: 5′-GCAGTGTCCAAGCTGGAGTG-3′
reverse: 5′-CCAATCCCAGCTCCCACTAA-3′
Nox4 forward: 5′-CCAGTGGTTTGCAGACTTGC-3′
reverse: 5′-CGAGGACGCCCAATAAAAAG-3′
Duox1 forward: 5′-CTGATGAGGGCAGCAGTGAC-3′
reverse: 5′-CCTTGTCGGCCAGAGAAAAC-3′
Duox2 forward: 5′-ATGCTCGCAAGAGGGTCATT-3′
reverse: 5′-CTGGAGAGCTGCTGGAACCT-3′
NoxA1 forward: 5′-GCCATGGACAAAGTGCAGAA-3′
reverse: 5′-TACACACGGGTGAGGACGAC-3′
NoxO1 forward: 5′-CTGGCAACCTCACAGCACAT-3′
reverse: 5′-TCCAGGTAGGGAGCTGGAAA-3′
p67
reverse: 5′-ACTCCCCTTCCAGCCATTCT-3′
p47
reverse: 5′-TGATGTCCCCTTTCCTGACC-3′
Gpx1 forward: 5′-CGTGCAATCAGTTCGGACAT-3′
reverse: 5′-TAAAGAGCGGGTGAGCCTTC-3′
Sod1 forward: 5′-GCGTCATTCACTTCGAGCAG-3′
reverse: 5′-CCTGCAGTGGTACAGCCTTG-3′
Sod2 forward: 5′-GGGCCATATCAATCACAGCA-3′
reverse: 5′-CCAGCCTGAACCTTGGACTC-3′
catalase forward: 5′-CTGGAGCACCATAGCCAGTG-3′
reverse: 5′-TCAGGTGGTTGGCAATGTTC-3′
In situ hybridization
Rat lung preparation for in situ hybridization
35 days after MCT injection, the MCT-treated rats and time-matched controls were euthanized by an intraperitoneal injection of a lethal ketamine and domitor dose, and the lungs were flushed with phosphate-buffered saline (PBS; PAA Laboratories, Cölbe, Germany) through a catheter in the pulmonary artery at a pressure of 20 cm H2O at room temperature. During perfusion of the lungs, the buffer was allowed to drain freely from a catheter in the left ventricle. Once the effluent was clear of blood, 800 μl prewarmed TissueTek® (Sakura Finetek, Zoeterwoude, Netherlands) was instilled into the airways via a tracheal cannula. After ligation of the trachea, the lungs were excised and immediately snap frozen in liquid nitrogen.
Probes for in situ hybridization
Probes were obtained from Exiqon A/S (Vedbaek, Denmark) and were prepared and used according to specifications provided by the manufacturer.
Nox1:/5DigN/TTCACTCATGCTCTCTTCTGTT/3Dig_N/- custom LNA mRNA detection probe 1 umole, RNA Tm: 84°C, DNA Tm: 70°C, Yield: 2.5 OD/13.5 nmole/111.2 μg
Neg. control:/5DigN/GTGTAACACGTCTATACGCCCA, Tm: 78°C (DNA-Tm)/87°C (RNA-Tm)
Pos. control: PolyT(25)Vn Control, LNA™ mRNA in situ hybridization probe, 5 nmol, 5′-DIG labeled, and 3′DIG labeled
Combined approach of nonisotopic in situ hybridization and immunofluorescence in rat lung sections
The nonisotopic in situ hybridization was performed on 8 μm-thick TissueTek-embedded rat lung cryostat sections. The sections were heated at 60°C for 15 min followed by transferring the sections into 2×standard saline citrate (SSC) buffer for 30 min at 70°C, diethylpyrocarbonate-treated water for 1 min, Proteinase K (5 μg/ml) for 10 min at room temperature, 0.2% (m/v) glycine in PBS solution (for the inactivation of Proteinase K) for 30 s, PBS for 30 s, freshly prepared cold 4% (m/v) paraformaldehyde for 20 min, PBS for 5 min, 0.1 M acetylated triethanolamine (0.5 ml of acetic anhydride/200 ml of triethanolamine) on a shaking platform for 10 min, and PBS for 3 min, followed by dehydration of slides by passing through 70% (v/v), 80% (v/v), and 90% (v) ethanol (each for 2 min). The slides were then prehybridized with 2×Prehyb solution (1 M NaCl, 0.02 M Tris (pH 7.5), 2×Denhardt's reagent, 2 mM EDTA, 10 mg/ml salmon sperm DNA, and 0.2 mg/ml yeast tRNA) at 55 °C for 2–3 h in a humidified chamber, followed by hybridization with a denatured antisense Nox1 probe in 2×hybridization solution (1 M NaCl, 0.02 M Tris (pH7.5), 2×Denhardt's reagent, 2 mM EDTA, 2 g dextran sulphate, and 0.2 mg/ml yeast tRNA) at 55°C overnight. The next day, slides were washed from low to very highly stringent conditions as follows: on shaking platform, 2×SSC for 1 h at room temperature; 0.1×SSC at 60°C; and, finally, followed by incubation in preheated 0.1×SSC at room temperature. The sections were treated with blocking buffer (2% Blocking reagent (Roche, Mannheim, Germany), 0.1% (m/v) BSA, 0.1 M Tris (pH 7.5), and 5 M NaCl) for 30 min at room temperature, followed by incubation with a peroxidase-labeled anti-DIG Fab fragment (Roche) in 1:20 dilution for 2 h at room temperature. After antibody incubation, sections were washed in TBT buffer (50 mM, 1 M Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% Triton X-100; 3×15 min). The fluorescent substrate Alexa fluor 555 tyramide (Molecular Probes, Invitrogen, Karlsruhe, Germany) at a dilution of 1:60 in amplification buffer was applied to the sections for 2 h. Subsequently, sections were washed (3×20 min) in PBT buffer (PBS, 0.1% [v/v] Tween 20) and incubated with a mouse monoclonal FITC-labeled α-smooth muscle actin antibody (Sigma-Aldrich Chemie Gmbh, Munich, Germany) at 1:100 in PBS, for 1 h. The sections were washed (3×3 min) in PBS and subsequently incubated with DAPI (1:10,000 in PBS; Invitrogen) for 10 min, washed, and mounted in Dako Fluorescence Mounting Medium (Dako).
Western blot
For western blotting, the following antibodies were used. Nox1 (1:700; Santa Cruz, Santa Cruz Biotechnology, Heidelberg, Germany), Sod2 (1:1000; Abcam plc), Gpx1 (1:1000; Abcam plc), Erk 1/2/phospho Erk 1/2 (T202/Y204) (1:1000; Cell signaling, Denver, MA), Cyclin D1 (1:1000; Cell signaling), MMP-9 (1:750; Abcam plc), PAK1/phospho PAK1 (S144) (1:1000; Abcam plc), JNK1/2/3/phospho JNK (T183/Y185) (1:700; Cell signaling), Cofilin/phospho cofilin (S3) (1:2000; Abcam plc), NQO1 (1:1000; Abcam plc), Nrf2 (1:1000; Abcam plc), and β-actin (1:30,000; Abcam plc). Protein extracts were prepared from isolated PASMC from healthy control rats and MCT-treated rats in RIPA buffer containing 1 mM sodium vanadate, Protease-Inhibitor Mix complete (Roche), and 0.1 mM PMSF. Equivalent amounts of protein were resolved on 12% SDS polyacrylamide gels. Proteins were transferred to polyvinylidene fluoride membranes (Pall Corporation, Dreieich, Germany) by semi-dry electroblotting. Nonspecific antibody binding was blocked by incubation in 6% (m/v) nonfat dry milk powder in T-TBS (20 mM Tris-Cl, pH 7.5, 150 mM NaCl, and 0.1% [v/v] Tween 20) at room temperature for at least 1 h. Incubation with primary antibodies was performed at 4 °C overnight. After washing the membranes in T-TBS buffer, specific immunoreactive signals were detected by enhanced chemiluminescence (ECL; Amersham, Freiburg, Germany) using a secondary antibody coupled to horseradish peroxidase.
Electron paramagnetic resonance measurements
Superoxide release from PASMC was measured, as described, with modifications (21). Briefly, electron paramagnetic resonance measurements were performed at −170°C using an MS 100 ESR spectrometer (Magnettech, Berlin, Germany). We used the spin probes CMH and CPH (500 μM) for the detection of extracellular (CPH) and intra-+extracellular (CMH) superoxide production by PASMC. Since CMH and CPH are known to react with superoxide and peroxynitrite, we ran parallel samples containing either the spin probe alone or in combination with Sod (CPH) or Sod conjugated with polyethyleneglycol (PEG-Sod; CMH), respectively. This setup enabled us to define the superoxide portion of the total CMH or CPH signal. Cells were harvested, normalized to 100,000 cells per sample, and incubated with Sod or PEG-Sod (15 U/ml) for 2 h at 37°C. Afterward, we added CMH (+PEG-Sod) or CPH (+Sod) and incubated the samples again for 20 min. As a positive control, we used PMA (phorbol myristate acetate), which was added along with CMH or CPH. After 20 min, the samples were shock frozen and stored in liquid nitrogen. The spectrometer settings were as follows: g-factor=2.0063, center field=3349.95G, microwave power=200 mW, sweep time=20 s, and sweep number=5.
DHE staining
Briefly, the cells were grown on chamber slides and incubated with 5 μM of DHE for 15 min. Subsequently, the cells were washed in PBS, fixed in acetone and methanol mixture (1:1) for 10 min, and nuclei were stained with Hoechst-33258. Images were quantified using Image J software (U.S. National Institutes of Health, Bethesda, MD).
Detection of NADP+ formation
NADP+ was detected using the Amplite Fluorimetric NADP/NADPH Assay Kit (AAT Bioquest, Inc., Sunnyvale, CA). The assay was performed according to the manufacturer's instructions. Briefly, cell extracts were incubated with the NADP/NADPH reaction mixture provided by the manufacturer and incubated for 2 h at room temperature. Fluorescence intensity was monitored at Ex/Em=540/590 nm.
Determination of intracellular H2O2
For intracellular H2O2 detection, the coding information for the H2O2-sensitive, enhanced yellow fluorescent protein variant HyPer48 was subcloned under the control of the EF-1α enhancer/promoter into the pWPXL plasmid (distributed by Addgene, Boston) and packed with a second-generation lentivirus transduction system (see
PDGF stimulation
For stimulation, PASMC were starved for 24 h and treated with 5 ng/ml PDGF (R&D Systems, Minneapolis, MN) for 1, 2, and 24 h. After stimulation, cells were used for RNA and Protein isolation.
Statistics
Values are given as mean±standard error of mean if not indicated differently. For statistical analysis, a Student's t-test was used for a comparison of two groups. For more than two groups, ANOVA with student-Newman-Keuls posthoc test was performed. A probability value of less than 0.05 was considered significant.
Footnotes
Acknowledgments
The authors thank Dr. Megan Grether for the linguistic and grammatical editing of the article. This work was funded by the Deutsche Forschungsgemeinschaft (DFG 1978/4-1 and IRTG1062).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.