
Abstract
Reactive Oxygen Species
Redox Proteins
The redox environment is modulated by a number of proteins that are able to control the intracellular levels of ROS and regulate redox-sensitive signaling pathways. Superoxide dismutated to hydrogen peroxide can be converted into water by catalase and peroxidases, including glutathione peroxidases and peroxiredoxins (Fig. 1). SOD and catalase do not require co-factors to function, however the peroxidases require serial co-factors and proteins (66, 100).
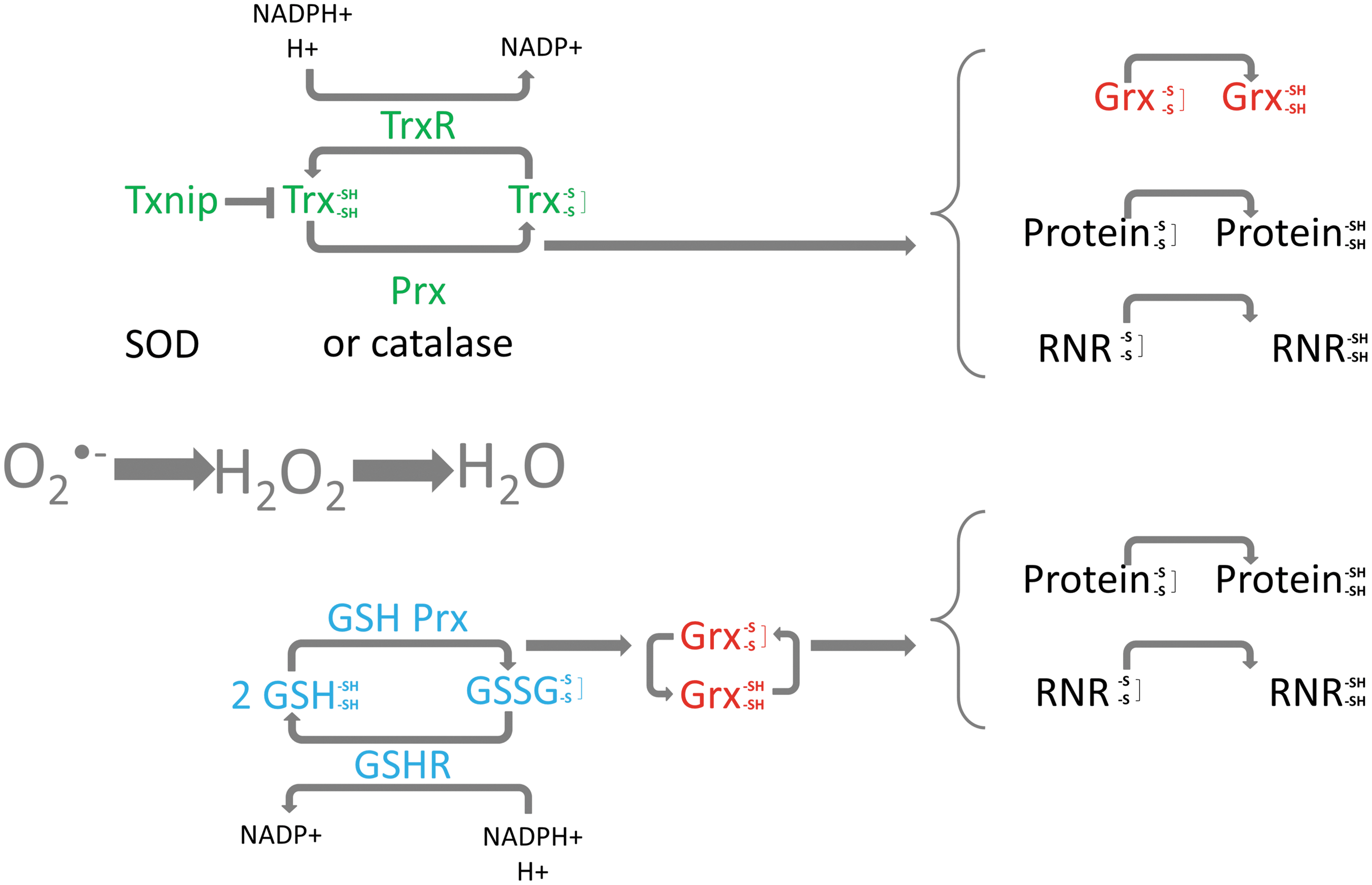
Three primary redox systems involved in the regulation of hydrogen peroxide levels are the glutathione, the glutaredoxin, and the thioredoxin systems (Fig. 1). These systems can reverse the formation of disulfide bonds and counteract potentially damaging ROS levels.
The glutathione system
Glutathione exists in both a reduced (GSH) and oxidized (GSSG) state and can modulate both the glutathione and glutaredoxin systems. GSH functions as a direct electron donor and is present in millimolar concentrations within the cell (reviewed in (67)). As part of the regulation of redox homeostasis, the glutathione redox couple GSH/GSSG can transfer two electrons to downstream factors. Glutathione reductase, another member of the glutathione system, is a dimeric flavoenzyme and functions to reduce oxidized GSSG into GSH at the expense of NADPH (Fig. 1) (98). Glutathione peroxidase is a selenoenzyme that reduces hydrogen peroxide to water via oxidation of selenol to produce selenic acid which can react with two GSH which is oxidized to GSSG.
The glutaredoxin system
The glutaredoxins are oxidoreductases that are able to reduce a wide variety of substrates such as ribonucleotide reductase (RNR), peroxide, and methionine sulfoxide, and are reduced by oxidation of glutathione (reviewed in (57)). In addition, glutaredoxin can be reduced by the thioredoxin system (26, 37) (Fig. 1). Structurally, the glutaredoxins are similar to thioredoxin; however, they are more versatile with respect to substrate (22, 57). Four glutaredoxin proteins are found in human cells; cytosolic dithiol Grx1, the predominantly mitochondrial Grx2 (Grx2a) (two additional cytosolic/nuclear isoforms of Grx2, Grx2b and Grx2c, are expressed in testicular cells and some cancers), the multidomain monothiol Grx3, and the mitochondrial single-domain monothiol Grx5 (46, 58, 101).
The thioredoxin system
The thioredoxin system plays an important role in regulating the intracellular redox state and is principally composed of thioredoxin and thioredoxin reductase (Fig. 1). Thioredoxin is reduced by thioredoxin reductase in an NADPH-dependent manner and in turn reduces oxidized cysteine groups on downstream proteins, such as signal-regulating kinase 1 (ASK-1) and proliferation-associated gene (PAG) (reviewed in (81, 82)). The mammalian thioredoxins include thioredoxin-1 (Trx1), mitochondrial thioredoxin-2 (Trx2), and a larger thioredoxin-like protein, p32TrxL, which have a conserved active site (30). The thioredoxin reductases are selenium-containing flavoproteins that have conserved NADPH binding catalytic sites and C-terminal cysteine-selenocysteine sequences (31, 81). Both TrxR1 and TrxR2 have been cloned, with the former found predominantly in the cytoplasm and the latter containing a mitochondrial import sequence (31, 72). Thioredoxin-Interacting Protein (Txnip) functions as a negative regulator of thioredoxin by directly interacting with its catalytic site (75) and was originally identified as a Vitamin D3 upregulated protein 1 (VDUP1) (also called thioredoxin binding protein 2 (TBP2)). The interaction between Txnip and thioredoxin blocks the reducing activity of thioredoxin by preventing/competing with the interaction between thioredoxin and its downstream factors (47, 111).
The antioxidant activity of the thioredoxin system is primarily exerted by peroxiredoxins (102) (Fig. 1). The peroxiredoxins (Prx) can scavenge intracellular ROS with thioredoxin as their electron donor, and six isoforms are found in mammalian cells; cytosolic Prx I and Prx II, mitochondrial Prx III, secreted Prx IV, mitochondria and peroxisome PrxV, and one-cysteine type PrxVI which differs to the other enzymes that have two conserved cysteine residues. Each isoform contains a conserved cysteine residue located in the N-terminal region which is the active site of catalysis (74, 93).
SOD and catalase
Three SOD enzymes exist, Mn-SOD (SOD-1), CuZn-SOD (SOD-2), and EC-SOD and are localized in different subcellular locations; Mn-SOD is located in the mitochondria, CuZn is located in the nucleus and cytoplasm, and EC-SOD is more abundant in the extracellular matrix and fluids (25). Catalase functions to convert hydrogen peroxide to water and oxygen and is largely located in subcellular organelles known as peroxisomes (100).
Additional important factors
NRF2 and its inhibitor Kelch-like ECG-associated protein-1 (KEAP1) are important in modulating the response to cell stress. ROS can modify cysteine residues within KEAP1 to alter the structure of the protein which results in active NRF2. NRF2 can bind antioxidant response elements (AREs) or electrophile response elements (EpREs) on DNA and can induce the expression of important enzymes such as catalase (reviewed in (89)).
Oxidative DNA Damage and Repair
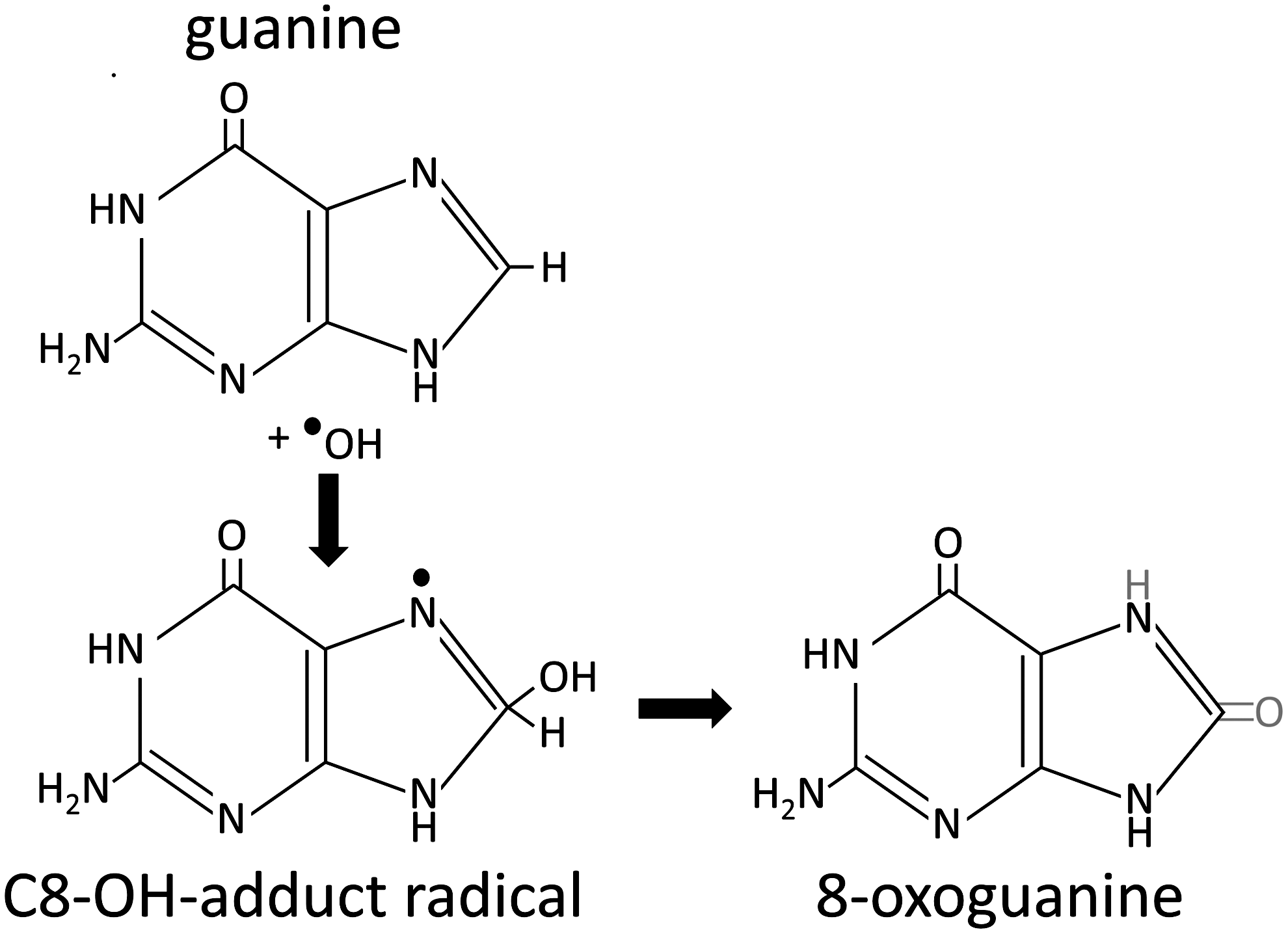
A large proportion of DNA damage can be attributed to ROS, including single- and double-strand breaks and DNA–protein crosslinks (reviewed in (10, 13, 24)) and may partly explain the induction of certain spontaneous cancers (94). Although superoxide and hydrogen peroxide are not directly responsible for DNA damage, they are involved through the accumulation of the hydroxyl radical. Free radicals can cause DNA damage by a variety of mechanisms which include addition to double bonds of DNA bases and removal of hydrogen from both 2’deoxyribose and thymine. Oxidative DNA damage can cause a multitude of base lesions; with guanine preferentially oxidized due to its low redox potential. The most commonly described DNA lesion is 7,8-dihydro-8-oxoguanine (8-oxoguanine; 8-oxoG) (Fig. 2). The 8-oxoG lesion can result in guanine to thymine transversions, as adenine can freely pair with 8-oxoG in the syn conformation during replication, whereas 8-oxoG paired with cytosine is recognized as an aberrant pairing (42, 86). Although 8-oxoG is the most commonly described lesion, the hydroxyl radical can interact with adenine, cytosine, and thymine, and this has been extensively reviewed (24). Major lesions resulting from oxidative DNA damage include: 5-hydroxy-6-hydro-thymine, thymine glycol, cytosine glycol, 5-hydroxycytosine, uracil glycol, 5-hydroxyuracil, 8-hydroxyadenine and 2-hydroxyadenine, amongst a large number of others (24). Although this review focuses on nuclear DNA, it is important to note that mitochondrial DNA is susceptible to oxidative damage, and that a higher mutation rate is observed than that of nuclear DNA.
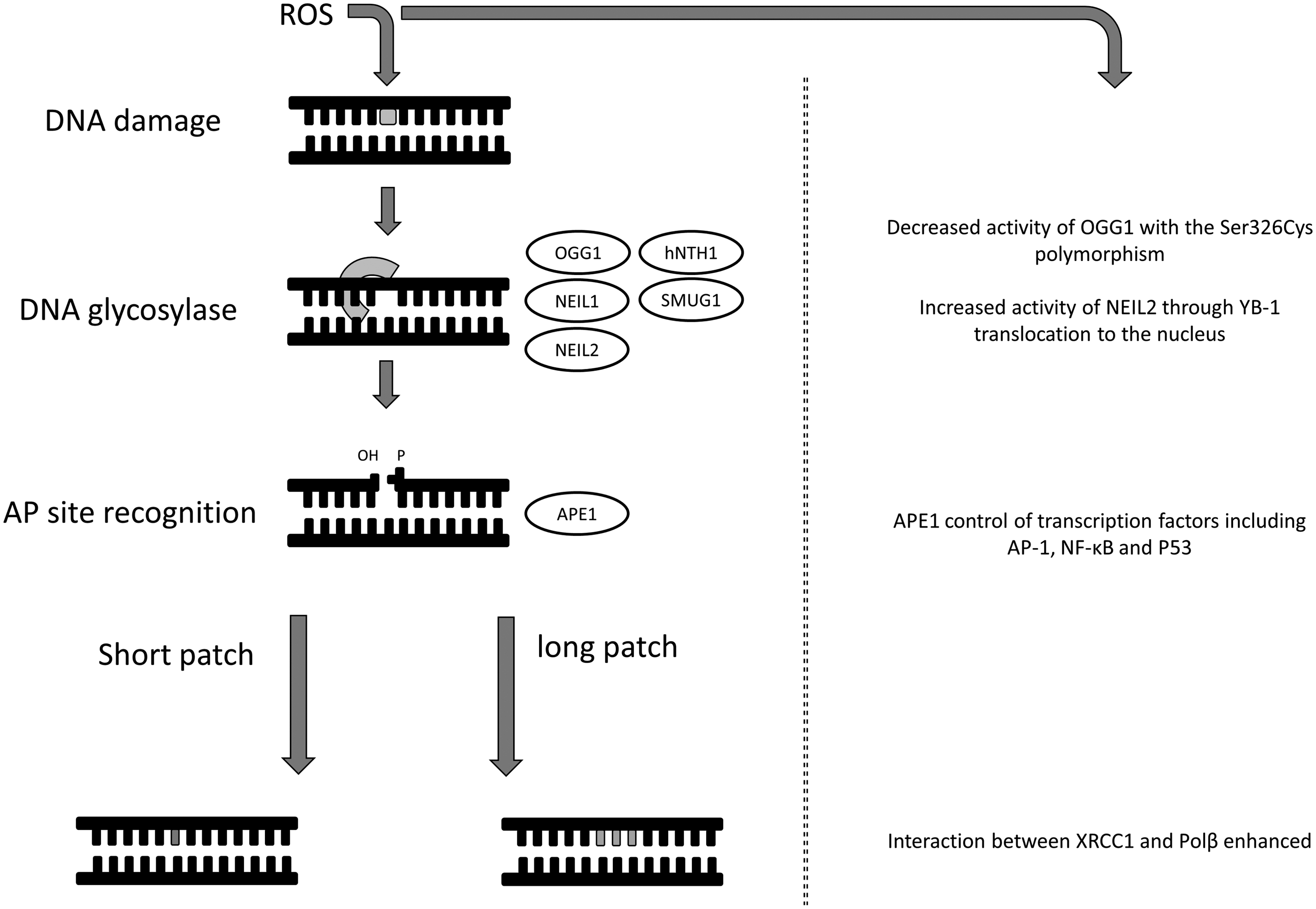
DNA that is damaged by ROS is a direct risk to genomic stability. As DNA is prone to oxidative damage, numerous repair systems have evolved to protect cells from these potentially dangerous lesions. Oxidative DNA damage can be removed by a number of DNA repair pathways, including base excision repair (BER), nucleotide excision repair (NER), and nucleotide incision repair (NIR) (reviewed in (18, 33, 91)), although the BER pathway is generally recognized as the primary mechanism for both nuclear and mitochondrial DNA damage (reviewed in (91)). During the course of DNA repair by the BER pathway, the initial recognition of the lesion is performed by a specific mono or bi-functional glycosylase, which can excise the damaged base. The bi-functional glycosylases have apurinic/apyrimidinic (AP) lyase activity, whereas, in the case of a mono-functional glycosylase, an abasic site is generated (AP-site). This is followed by gap filling and ligation to repair the oxidative damage. The AP site, if left unrepaired, is also mutagenic and can result in DNA strand breaks and apoptosis (reviewed in (59)). Human AP endonuclease, APE-1 (also called APEX, APE, HAP1, and Ref-1), is an important enzyme in BER that cleaves phosphodiester bonds 5’ and adjacent to an AP site. In addition, it has a 3’ phosphoesterase activity, which can be used to remove 3’-blocking damage that can also result from ROS. APE-1 is not always required; NEIL1 can perform βδ-elimination of the AP site (70). BER can be separated into two pathways, short-patch and long-patch, with short-patch BER responsible for the repair of single nucleotides and long-patch BER responsible for the repair of two or more nucleotides. Although recognition of damaged bases in the short- and long-patch BER pathways is similar, different proteins are involved downstream, including ligase 1 (LIG1) for long patch and ligase 3 (LIG3) for short patch. In the case of the 8-oxoG DNA lesion, there is a dedicated 8-oxoG DNA glycosylase (OGG1) which specifically recognizes 8-oxoG paired with cytosine to exclude the oxidized base pair. Interestingly, the recognition capacity of this protein lies in its ability to interact with the proton on N7, rather than recognizing the 8-oxo-carbonyl feature (9). In addition to OGG1, Nei-like glycosylase 1 and 2 (NEIL1 and NEIL2), and OGG2 are all able to recognize 8-oxoG lesions; repair of this lesion is reviewed by David et al. (2007) (17). There is evidence that some degree of cooperation exists between the glycosylases, such as OGG1 and NEIL1 through enhanced turnover (70). Other glycosylases involved in recognizing the plethora of oxidized base lesions, such as human Nth homolog-1 (hNTH1) and single-strand mismatch-specific uracil DNA glycosylase (SMUG1), may also co-operate [reviewed in (33)].
Redox Modulation of Oxidative DNA Damage Repair
The redox environment is capable of directly modulating DNA repair. One of the initial observations that the redox environment could modulate DNA repair showed that both an increase in 8-oxoG and a reduction in DNA repair occurred in vitro following treatment with cadmium (15), this was subsequently shown to be due to cysteine modification of OGG1 following cadmium treatment (7) (Fig. 3). The redox environment has been shown to modulate the activation of OGG1 with the Ser326Cys polymorphism, although this reduction is not apparent in mononuclear blood cells of healthy individuals. The hOGG1 Ser236Cys polymorphism has been implicated in cancer development, however contradictory information exists as to its importance (8, 45, 54, 114). Furthermore, an interaction between OGG1 and poly(ADP-ribose) polymerase 1 (PARP-1), a sensor of DNA damage involved in DNA repair, has recently been described. This interaction is enhanced by oxidative stress to stimulate PARP-1 activity (76). OGG1 is not the only protein in the DNA repair pathway that can be modulated by the redox environment. Oxidative stress causes the translocation of the Y-box binding protein (YB-1) to the nucleus where it has a stable interaction with NEIL2 to increase NEIL2 activity in the BER pathway by seven fold (16) (Fig. 3). As a further example of redox modulation of DNA damage repair, the interaction between oxidized XRCC1 (x-ray repair, cross-complementing defective, in Chinese hamster, 1) and DNA polymerase β (Pol β) is enhanced due to the formation of a disulphide bond (12) (Fig. 3).
APE-1 is a multifunctional enzyme that acts in BER pathways and transcriptional regulation. Human APE-1 was first described and cloned as a DNA repair protein in 1991 and the redox activity of APE-1 was described in 1992 (19, 84, 108). The redox and DNA repair activities of APE-1- are located in different, distinct areas of the protein (108, 109), but they are not independently folded structural domains. APE-1 can be activated by nontoxic levels of ROS which promotes translocation into the nucleus (83) and ROS production following Ca2+ mobilization via extracellular ATP stimulation of purinergic receptors is responsible for the localisation of APE-1 (80). Furthermore APE-1 phosphorylation by protein kinase C (PKC) after an oxidative challenge has been shown to increase the activity of the APE-1 redox domain (41).
In the nonhomologous end joining (NHEJ) double-strand DNA repair pathway, Ku DNA binding is lower in an oxidizing environment, although the mechanism of this alteration is not clear (5). Ku is a heterodimer that encircles broken DNA ends during repair and can recruit the DNA-PK catalytic subunit (DNA-PKcs). The time that Ku is bound to the DNA impacts the likelihood of recruitment of DNA-PKcs to the DNA-PK complex (2). Ku is inactivated during oxidative stress in G6PD null mutant Chinese hamster ovary cells (3). The ataxia-telangiectasia mutated (ATM) protein is activated following DNA damage to sense double-strand breaks, and elevated ROS can activate ATM to activate the tumor sclerosis complex (TSC2) tumor suppressor that represses mTORC1 and induces autophagy (1). ATM was originally identified as the protein lost in the genetic disorder ataxia-telangiectasia and it is thought that loss of ATM results in oxidative stress (50). ATM has subsequently been shown to be activated through ROS oxidation at specific cysteine residues (34). Evidence also shows that ATM can promote an antioxidant response by regulation of the pentose phosphate pathway, one of the main sources of NADPH (11). An additional example of a DNA repair pathway protein affected during oxidative stress is human replication protein A (RPA). RPA is a DNA binding protein implicated in replication, repair, and recombination, and in an oxidizing environment the cysteines in the zinc-finger motif of the p70 subunit can form disulfide bonds that can impair DNA binding (69).
Redox Proteins and DNA Repair Pathways
The redox control of oxidative stress is known to be a critical determinant of all normal cellular functions and signaling processes. The term oxidative stress has been redefined recently as “an imbalance in pro-oxidants and antioxidants with associated disruption of redox circuitry and macromolecular damage” (32). From the numerous DNA repair proteins that are capable of dealing with oxidative DNA damage, only APE-1 is currently known to have an integral redox capability. APE-1 does not contain the C-X-X-C active site motif of thiol-disulfide oxidoreductases, but Cys65 is thought to be a critical residue for its redox activity and may achieve this in a similar manner to the 2-Cys or 1-Cys peroxiredoxin family (36, 62). APE-1 regulates the DNA binding activity of several transcription factors, including activator protein-1 (AP-1), nuclear factor-κB (NF-κB,) and p53, via redox-dependent and independent mechanisms. Transcription factors such as p53 and AP-1 are then capable of regulating a significant number of the DNA repair proteins [i.e., uracil DNA deglycosylase-2 (UNG2), APE-1, polβ in the BER pathway (reviewed in (61)] and excision repair cross complementing-1 (ERCC1) in the NER pathway (55, 56). However, ROS activation of APE-1 may have implications on its DNA repair activity. Immediately adjacent to a crucial histidine in the DNA repair active site is a cysteine that can be redox regulated (52), potentially interfering with the function of the DNA repair domain. In some cancers, APE-1 is upregulated, and in pancreatic cancer inhibition of the redox domain of APE-1 reduces NF-κB, AP-1, and HIF1α activity in vitro and can inhibit tumor growth (28). Following inhibition of the redox domain of APE-1 in pancreatic cancer cells, in vitro increased oxidative stress is observed which results in inhibition of cell growth and migration (116). Furthermore, growth inhibition of endothelial cells has been demonstrated, as a surrogate measure of abrogation of angiogenesis (60).
In situations where a large excess of 8-oxoG lesions are induced by ROS, the activity of the histone acetyltransferase p300 is enhanced, which in turn acetylates and increases the turnover of OGG1 in the presence of APE-1 (6). APE-1 can enhance the activity of OGG1 by sitting 5’ to the OGG1/DNA complex, distorting the DNA which actively displaces OGG1 enabling it to search for new lesion (87). It has also been speculated that the redox domain of APE-1 can influence the activation/deactivation of an XRCC1 disulfide switch. The affinity of XRCC1 for DNA polymerase β can be enhanced via formation of a disulfide bond in its N terminal domain that alters its folding topology (12) (Fig. 4A). This interaction is central to the base excision repair process of oxidative DNA damage repair. In addition to XRCC1 and OGG1, APE-1 can interact with other DNA repair proteins such as flap endonuclease-1 (FEN1) and proliferating cell nuclear antigen (PCNA) (20). There also appears to be cross talk between the breast cancer susceptibility gene, BRCA1 and APE-1, in protecting cells from oxidative stress as APE-1 knockdown increases ROS levels in a manner that can be partly reversed by BRCA1 overexpression and vice versa (85). BRCA1 also plays an important role in redox homeostasis as overexpression can upregulate gene expression of important antioxidant proteins, including glutathione S-transferase theta 1, glutathione peroxidase 3, and oxidoreductases (NAD(P)H dehydrogenase quinone-1 (NQO1) and NAD-dependent malic enzyme, mitochondrial (ME2)). A decreased expression of glutathione S-transferase A2, glutathione peroxidase 3, nuclear factor-like 2, and superoxide dismutase 1 has been demonstrated with BRCA1 deficiency (Fig. 4B). Bae et al. also demonstrated that BRCA1 could influence the cellular redox balance by enhancing the production of glutathione under hydrogen peroxide-induced stress (4).
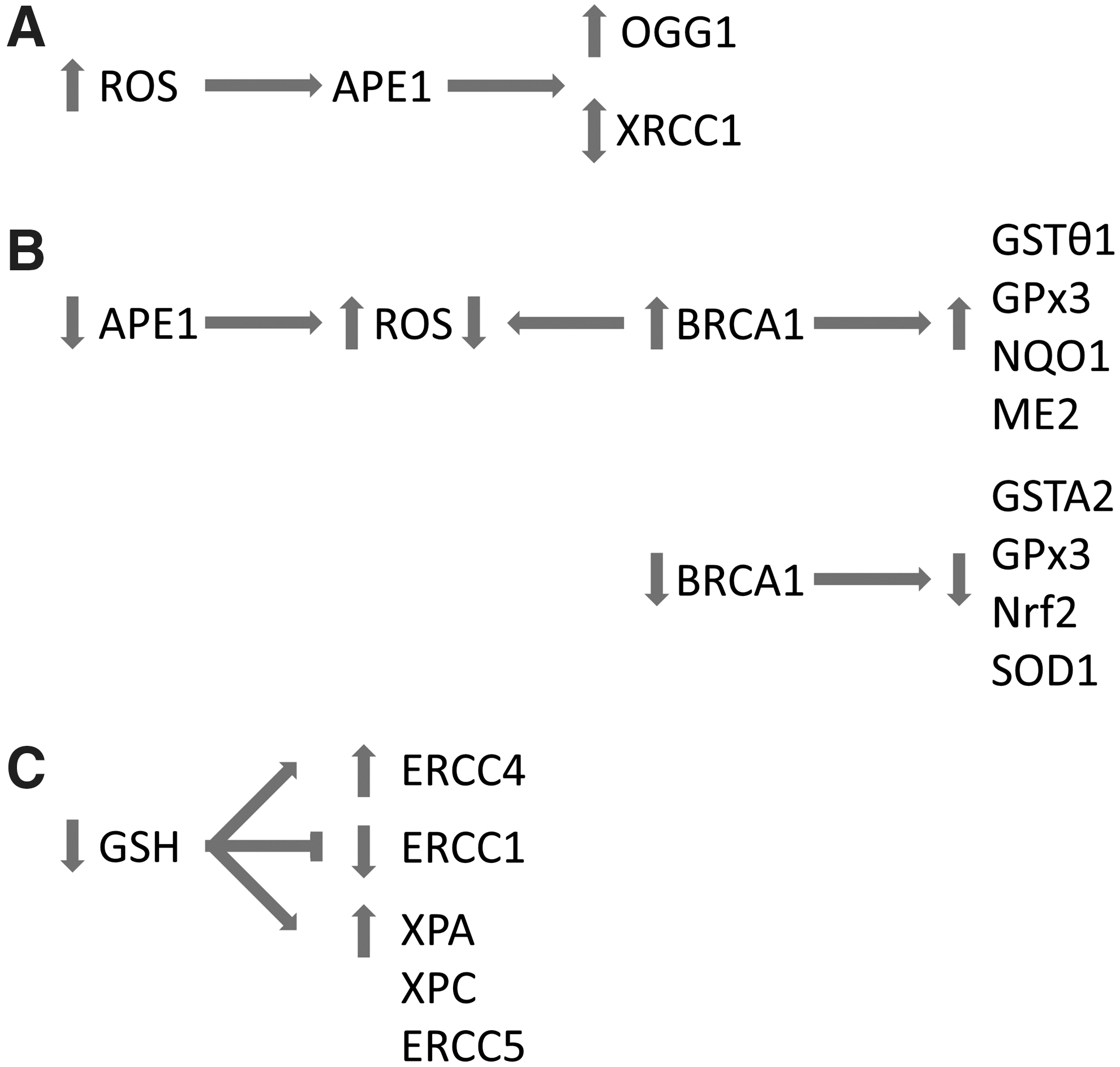
In response to H2O2 exposure, expression of some NER genes can be mediated by glutathione in three ways: either glutathione depletion further enhances the upregulation of genes by H2O2 (i.e., excision repair complementing defective in Chinese hamster 4 (ERCC4)), glutathione depletion inhibits the downregulation of genes by H2O2 (i.e., ERCC1), or the effects are very minimal (i.e., xeroderma pigmentosum group A complementing protein (XPA), xeroderma pigmentosum group C complementing protein (XPC), and xeroderma pigmentosum complementation group G (ERCC5)) (53) (Fig. 4C). However, in XPC-deficient mice exposed to the pro-oxidant Bis(2-ethylhexyl)phthalate (DEHP), the glutathione response is missing which is active in wild-type mice (unpublished results in (68)). Overall, depletion of glutathione resulted in a 50% increase in NER activity (53).
A review of the cellular response to oxidant generation between different cellular compartments demonstrated that the nuclear glutathione and thioredoxin systems can function independently from the cytoplasmic systems (32). In response to oxidative stress, thioredoxin translocates to the nucleus and increases the activity of co-activators such as APE-1 and transcription factors such as NF-κB, AP-1, and p53 (39, 40, 95, 99). In turn, APE-1 can maintain transcription factors in their reduced active state. In addition, the thioredoxin and glutathione systems play an important role in the reduction of ribonucleotide reductase, which is rate limiting in synthesis of deoxyribonucleotides for DNA repair and replication (113).
Redox Proteins and DNA Damage Repair in Disease
The accumulation of oxidative DNA damage is implicated in many diseases, such as cancer, but also occurs in normal conditions such as aging. The process of DNA damage and disease has been discussed in a number of excellent reviews (24, 43, 44, 65). The majority of diseases have some degree of oxidative stress involvement whether in a causative capacity or as a by-product of the disease state. The redox systems normally capable of counteracting these effects are sometimes themselves affected, allowing progression and further damaging effects to occur.
Cancer
If oxidative DNA lesions are not removed quickly enough, they can become self-perpetuating mutations that contribute to the development of disease states, particularly cancer. The importance of redox proteins in cancer is well known (79, 90, 97) and the indirect link between their altered expression and oxidative DNA damage leading to cancer formation is established. A number of studies have examined if the expression of redox proteins are associated with response to ROS inducing DNA damaging agents used therapeutically, such as ionizing radiation and certain chemotherapeutic agents in various tumor types (103 –106); but few have correlated expression with DNA repair proteins or with induction of DNA lesions or their repair.
Through detection of 8-oxoG, glutathione peroxidase 1 null mice that are heterozygous for manganese superoxide dismutase show increased DNA damage and increased incidence of tumours (115). Mice lacking peroxiredoxin 1 had an increased tumor incidence and increased levels of 8-oxoG formation (73). Glutathione peroxidase 7 suppresses ROS and oxidative DNA damage in esophageal epithelial cells and it is downregulated during Barrett's tumorigenesis (78). However, any direct role in oxidative DNA damage and its repair, or lack of, in the cancer setting is less understood. Guo et al., eloquently stated “Most studies addressing thiol/disulfide systems in DNA damage and repair have focused on protection against reactive species and have not distinguished whether separate redox signaling and control events also function in regulation of the repair machinery” (32).
Alterations in APE-1/Ref-1 have been reported in many human tumors (23, 112). In breast cancer, the cellular localization of APE-1 determines whether the immunohistochemical expression associates with good or poor prognostic indicators (49). In lung cancer, levels of APE-1 expression between normal and carcinomas has not been observed (48) but differential patterns of expression were dramatically different in cervical, prostate, and epithelial cancers (51, 71, 110). The mechanistic implications of these changes need to be fully explored and may be tissue type specific.
Diabetes
Dincer et al., (21) hypothesized that oxidative DNA damage may be an important factor in the development of diabetic complications, especially in those with poor control of their disease. They used a comet assay to determine oxidized purines including 8-oxoG via the damage specific-repair endonuclease, formamidopyrimidine DNA glycosylase (Fpg), in type 2 diabetic cases. Fpg-sensitive sites were considerably higher in the poorly controlled diabetics compared to the well-controlled population and controls. The frequency of Fpg-sensitive sites was also negatively correlated with glutathione levels and positively correlated with HbA1c (an index of glycemic control and serum glucose) in control and diabetic patients. Therefore, decreased glutathione levels may be a contributing factor to oxidative DNA damage in type 2 diabetics. Hyperglycemia in diabetics can reduce the cellular glutathione content and the uptake of cystine which limits the maintenance of glutathione levels (92). In a type 1 diabetic rat model, Simone et al., (88) provided a mechanism of oxidative stress mediated DNA damage with hyperglycemia leading to the phosphorylation/inactivation of tuberin and downregulation of the DNA repair protein OGG1 via ROS-dependent activation of Akt.
Cardiovascular disease
In atherosclerotic plaques compared to normal vessels, several repair enzymes were increased, including APE-1, poly(ADP-ribose) polymerase 1, p53, and DNA dependent protein kinase, with constitutive expression of OGG1, polβ, endonuclease III-like 1 (NTH1), and 8-oxo-7,8-dihydroguanosine triphosphatase (8-oxo-dGTPase) (64). APE-1/Ref-1 strong nuclear immunoreactivity was present in the entire plaque (>90% of the total area of macrophages and smooth muscle cells) but the role of APE-1 in atherosclerotic plaques is unclear. In hyperhomocysteinemia-accelerated atherosclerotic lesion formation, ROS upregulates the expression and translocation of APE-1, promoting the binding of NF-κB. This leads to the expression of monocyte chemoattractant protein-1 in macrophage that promotes the development of atherosclerosis (14). However, overexpression of APE-1 was not specifically demonstrated to increase resistance to DNA damage (38).
The overexpression of some BER proteins does not necessarily lead to an increased protection from DNA damaging agents (29). In the case of PARP-1, an imbalanced activation leads to necrotic cell death and may be the contributing factor to the hallmark necrotic core of an atherosclerotic lesion (35, 64).
Neurological diseases
Oxidative stress is a common feature of many acute and chronic neurological disorders such as Alzheimer's disease, Parkinson disease, and amyotrophic lateral sclerosis, but very little is understood about the role redox proteins play. In sporadic and familial Alzheimer's disease, a higher number of nuclei were intensely positive for APE-1 in all cortical layers compared to normal brain. This may be an adaptive consequence of oxidative stress, but whether it reflects DNA repair or transcription factor activation is still unclear (63). The risk of Alzheimer's disease has not been found to be linked to polymorphisms in BER genes such as OGG1, XRCC1, or APE-1 (77).
Conclusion
In this review we have discussed the generation of ROS and their modulation by various enzyme systems; how ROS are able to damage DNA; and the mechanisms to repair this DNA damage that can be modulated by the redox environment. Furthermore, we have described redox proteins that have direct influence over the repair of oxidative DNA damage. Redox homeostasis is extremely important for cellular wellbeing, and a breakdown of this system has significant clinical consequences, with a number of diseases associated with aberrant expression of these systems. Further research into the mechanisms of redox modulation of both DNA damage and repair is essential to fully elucidate the complex role of the redox environment within the cell. Future directions of this complex research area will aim to characterize the role that the redox environment, principally ROS levels, has on influencing both DNA damage and repair; understanding this complex balance is essential to understand many disease pathologies and subsequent disease progression.
Footnotes
Acknowledgment
The authors wish to thank the Breast Cancer Campaign for supporting their research.
Author Disclosure Statement
No competing financial interests exist.