
Abstract
Introduction
The Nrf2 transcription factor is a member of the cap-n-collar (CNC) basic-region leucine-zipper (bZIP) family of proteins (24). It mediates induction of over a hundred genes encoding proteins that are involved in various cytoprotective functions, including synthesis of GSH, maintenance of protein thiols, inactivation of electrophiles, drug efflux, generation of NADPH, and activity of the proteasome (32, 42, 56); each of the genes regulated by Nrf2 contains an antioxidant-response element (ARE, i.e., 5′-A/GTGAC/GNNNGCA/G-3′) † in its regulatory region (7, 17, 53, 58, 75). Induction of ARE-driven genes helps mitigate injury to cells caused by oxidants and electrophiles (25).
Innovation
This study provides evidence that hydrogen sulfide (H2S) activates the Kelch-like ECH-associated protein-1 (Keap1)–NF-E2 p45-related factor-2 (Nrf2)–antioxidant-response element (ARE) cytoprotective pathway. The ubiquitin ligase substrate adaptor Keap1 serves as a versatile stress sensor. When Keap1 is exposed to H2S, an intramolecular disulfide bond is formed between its C226 and C613 residues, which prevent it from repressing Nrf2- and ARE-driven gene expression. Inhibition of Keap1 by H2S involves production of H2O2, which also entails oxidation of C226 and C613 in the adaptor protein. Thus, activation of Nrf2 by H2S partly involves H2O2 signaling. Lastly, Nrf2 regulates genes involved in H2S synthesis and catabolism, suggesting that an inter-relationship exists between Nrf2 and H2S.
The activity of Nrf2 is tightly controlled. It is primarily regulated at the level of protein stability (43, 52) through an interaction with the ubiquitin ligase adaptor Kelch-like ECH-associated protein-1 (Keap1), which allows the Keap1-Cul3-Rbx1 E3 ubiquitin ligase complex (i.e., Keap1CRL) to ubiquitylate Nrf2 under nonstressed conditions (35, 68). Keap1 is essential for maintenance of redox homeostasis, because its substrate adaptor activity ensures that Nrf2 has a short half-life, and the expression of ARE-driven antioxidant genes is restricted under normal homeostatic conditions. However, the activity of Keap1 is redox sensitive. Inactivation of Keap1 by agents that produce ROS or are soft electrophiles increases Nrf2 stability, and this in turn results in enhanced transactivation of ARE-driven gene expression (8, 39, 43, 52, 74). The increase in stability of Nrf2 observed upon stress appears to occur as a consequence of thiol-mediated inactivation of Keap1CRL, which allows the CNC-bZIP factor to evade ubiquitylation.
Keap1 is a dimeric protein that contains five domains: the N-terminal region (residues 1–56) ‡ , the Broad complex, Tramtrack, and Bric-à-Brac (BTB) domain (residues 57–177), the Intervening region (IVR, residues 178–326), the Kelch-repeat domain (Kelch, residues 327–598), and the C-terminal region (CTR, residues 599–624) (54). The BTB domain is responsible for dimerization of Keap1 subunits and recruitment of Cul3, while the Kelch and CTR domains form a six-bladed β-propeller to which substrates bind. Mammalian Keap1 proteins typically contain 25–27 cysteines, many of which are susceptible to adduction by thiol-reactive stressors (12, 27, 41). It is becoming clear that Keap1 contains several distinct thiol-based sensors that exhibit selectivity for different types of stressor. First, for example, Keap1 can be inactivated through covalent modification of C151 within its BTB domain by a diverse range of compounds, including tert-butylhydroquinone (tBHQ), diethylmaleate, Ebselen, nitric oxide (NO), and sulforaphane (SFN) (14, 36, 46, 73). Secondly, Keap1 can be inactivated through adduction of C273 and/or C288 within its IVR by chemicals that contain an α,β-unsaturated carbonyl moiety, such as dexamethasone mesylate, acrolein, 4-hydroxynonenal, and 15-deoxy-Δ12,14-prostaglandin J2 (12, 14, 36, 46). In addition to inactivation of Keap1 by electrophiles through the covalent modification of C151, C273, and C288, the transition metals Zn2+, Cd2+, As3+, and Se4+ can inactivate the substrate adaptor through noncovalent binding by coordination chemistry to both C226 and C613 (46). It therefore seems likely that although the C226 and C613 residues of Keap1 are far removed from each other in the primary structure, they are situated close to each other in the native protein. Weight is given to this view by the finding that the IVR domain of Keap1 partially envelops the six-bladed β-propeller formed by the Kelch and CTR domains in the folded protein (54). The notion that C226 and C613 in Keap1 are in juxtaposition in the native protein is also supported by the finding that ex vivo treatment with H2O2 results in formation of an intramolecular disulfide bridge between C226 and C613 (15), but the functional consequence of this oxidative modification of Keap1 is unknown.
Although xenobiotic-inducing agents are commonly used as a means to activate Nrf2 via modification of Keap1 thiols (41, 62, 64), the ability of endogenous signaling molecules to inhibit Keap1 has attracted little attention. Endogenous molecules that modify Keap1 and stimulate Nrf2 activity include 4-hydroxynonenal, 15-deoxy-Δ12,14-prostaglandin J2, nitrofatty acids, and 8-nitro-cGMP (18, 28, 30, 38), as well as NO and H2O2 (3, 10, 15, 46, 71). An example of a relatively poorly characterized second messenger that may regulate the Keap1-Nrf2 pathway is the gaseous signaling molecule hydrogen sulfide (H2S), which is enzymatically generated in a pyridoxal-5′-phosphate-dependent manner by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) (6, 19, 40) and in a pyridoxal-5′-phosphate-independent manner by 3-mercaptopyruvate sulfur transferase (3MST) (60). Specifically, the H2S gasotransmitter exerts antioxidant properties (21, 33, 34, 67). Evidence has been provided that it stimulates nuclear accumulation of Nrf2, and that preconditioning with H2S confers protection against ischemia (4), but the mechanism underpinning such activity has not been elucidated. It is plausible that H2S modifies and inactivates Keap1, causing Nrf2 accumulation and induction of ARE-driven genes, because the gasotransmitter has been reported to S-sulfhydrate Cys residues in proteins (51), though this has yet to be tested.
Results
The H2S donor NaHS activates Nrf2
H2S is unstable and volatile (70), and it is not practicable to expose cells to a sustained concentration of the gasotransmitter. Therefore, to determine its effect on Nrf2, mouse embryonic fibroblast (MEF) cells were treated with a single 400 μM dose of the H2S donor NaHS before they were lysed 0–120 min later; the isothiocyanate electrophile SFN was used as a positive control. Figure 1A shows that NaHS stimulated a rapid increase in the steady-state level of Nrf2 protein. Using 60 min after administration of NaHS as an end-point, Figure 1B shows that the gasotransmitter elicited a dose-dependent increase in Nrf2 protein. To explore whether H2S can delay destruction of Nrf2 protein, fibroblasts were treated with cyclohexamide (CHX) to block protein translation for various lengths of time after treatment with NaHS; in these experiments, fibroblasts were treated with a single dose of NaHS, and 60 min later, they were treated with CHX for various time intervals before Nrf2 protein was measured by Western blotting. As shown in Figure 1C and D, treatment of MEF cells with NaHS stimulated a delay in the destruction of Nrf2 protein when compared with vehicle-treated fibroblasts.
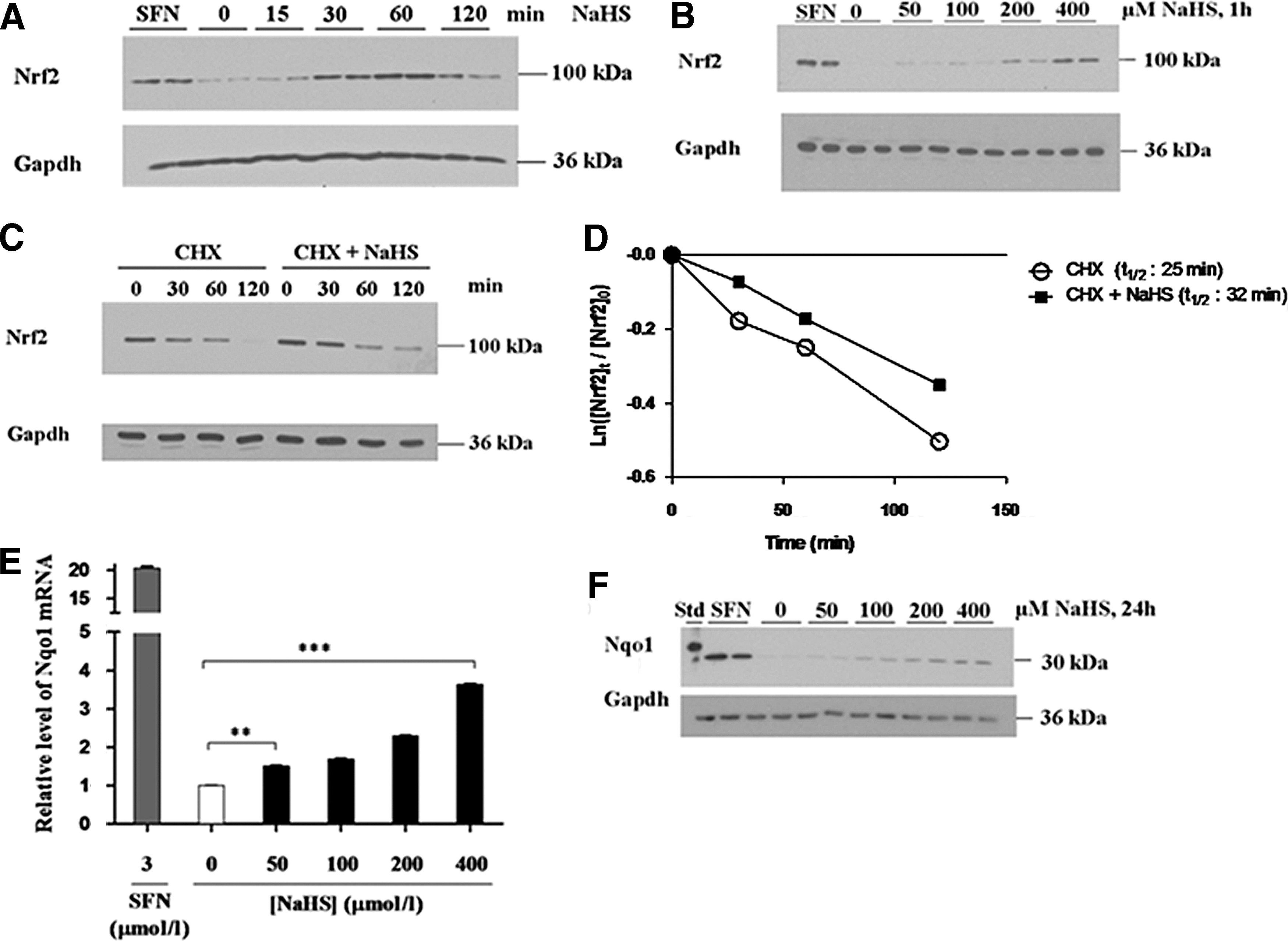
To establish if the increase in Nrf2 protein results in induction of its target genes, fibroblasts were treated with increasing concentrations of a single dose of NaHS before they were lysed 18 h later and the levels of mRNA for NAD(P)H:quinone oxidoreductase-1 (Nqo1) measured by TaqMan™ real-time polymerase chain reaction (RT-PCR). Figure 1E shows that the Nqo1 mRNA levels increased in a dose-dependent manner when wild-type MEF cells were challenged with increasing concentrations of NaHS. Consistent with the TaqMan results, treatment of the fibroblasts with NaHS increased the level of Nqo1 protein, as measured 24 h after administration of the H2S donor (Fig. 1F). Collectively, these data indicate that H2S stabilizes Nrf2 protein and stimulates transactivation of the Nrf2 target gene Nqo1.
H2S protects cells from oxidative stress in an Nrf2-dependent manner
To test whether H2S protects cells from oxidative stress, fibroblasts were pretreated with NaHS, and 24 h later, they were exposed to the redox-cycling compound menadione for a further 24 h. Using the MTT assay, pretreatment of wild-type MEF cells with NaHS increased the lethal dose 50 (LD50) of menadione from 16 to 23 μM, indicating that H2S can protect cells from oxidative stress (Fig. 2A). The role of Nrf2 in mediating protection by H2S against oxidative stress was assessed by pretreating Nrf2−/− MEF cells with NaHS before they were exposed to menadione. This showed that the mutant fibroblasts were substantially more sensitive to menadione than their wild-type counterparts, with an LD50 of ∼3.8 μM, and that pretreatment with NaHS did not influence their sensitivity to the redox-cycling agent (Fig. 2B). Therefore, the cytoprotection conferred by H2S against menadione is dependent on Nrf2.

We next examined whether the ability of H2S to protect against menadione entails suppression of ROS levels. To this end, we pretreated Nrf2+/+ and Nrf2−/− fibroblasts with NaHS before exposing them 24 h later to 23 μM menadione for a further 24 h; the concentration of menadione was chosen as it corresponds to the LD50 of the compound in wild-type fibroblasts that had been pretreated with NaHS. As is evident in Figure 2C, pretreatment of wild-type fibroblasts with NaHS before exposure to menadione for 24 h diminished the amount of ROS generated by the redox-cycling agent to levels that were not statistically different from those under basal conditions (i.e., cells that had been treated with neither NaHS nor menadione). To examine if Nrf2 was involved in the H2S-stimulated decrease in ROS, similar experiments were performed in Nrf2-null fibroblasts. Although pretreatment of the mutant fibroblasts with NaHS decreased modestly the amount of ROS created by menadione, these were substantially higher than the basal levels in Nrf2-null MEFs. By contrast, pretreatment of wild-type fibroblasts with NaHS decreased the amount of ROS created by menadione to levels that were statistically indistinguishable from those observed under basal conditions. Thus, H2S stimulated a reduction in ROS levels and protected cells from oxidative stress in an Nrf2-dependent manner.
Nrf2 regulates the expression of Cbs and Cse that are involved in H2S synthesis
To examine whether Nrf2 contributes to the regulation of murine Cbs, wild-type and Nrf2-null MEFs were grown under normal conditions or were treated with a single dose of 400 μM NaHS 18 h before the Cbs mRNA and 24 h before Cbs protein levels were examined. No significant difference was observed in the basal amount of Cbs mRNA in untreated Nrf2+/+ MEFs and Nrf2−/− MEFs as measured by TaqMan RT-PCR (Fig. 3A). By contrast, treatment of Nrf2+/+ MEFs with NaHS produced a 2.2-fold increase in the Cbs mRNA levels relative to that in untreated wild-type fibroblasts, but this increase was not apparent in the Nrf2-null MEFs. The level of Cbs protein was also increased in Nrf2+/+ fibroblasts treated with NaHS, but this was not observed in the mutant cells (Fig. 3B). Thus, H2S regulates Cbs expression in an Nrf2-dependent manner.
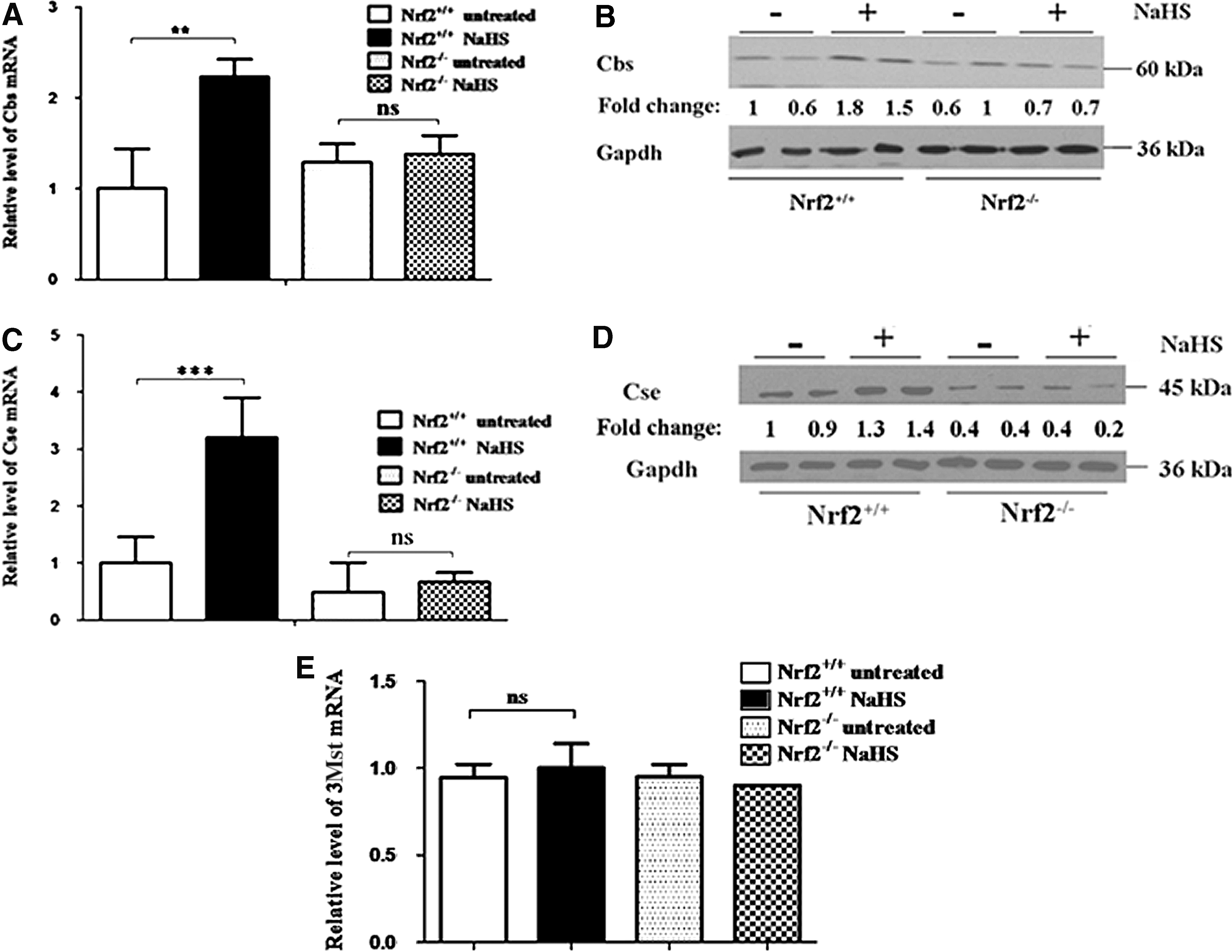
Similar experiments, to those described above, were performed to examine whether Nrf2 regulates Cse. As shown in Figure 3C, the Cse mRNA levels did not differ between untreated Nrf2+/+ and Nrf2−/− MEF cells. However, NaHS increased Cse mRNA 3.2-fold in Nrf2+/+ fibroblasts, but not in their Nrf2−/− counterparts. Western blotting similarly showed that NaHS increased Cse protein modestly in Nrf2+/+ MEFs, but not in Nrf2−/− MEFs (Fig. 3D). These results indicate that induction of Cse by H2S requires Nrf2.
We also measured the levels of mRNA for murine 3Mst in Nrf2+/+ and Nrf2−/− MEFs by TaqMan RT-PCR. This revealed no difference in 3Mst mRNA levels in fibroblasts from wild-type and mutant mice. Moreover, treatment with NaHS did not increase the expression of 3Mst in either wild-type or mutant fibroblasts.
Nrf2 regulates the expression of Sqrdl that contributes to the removal of H2S
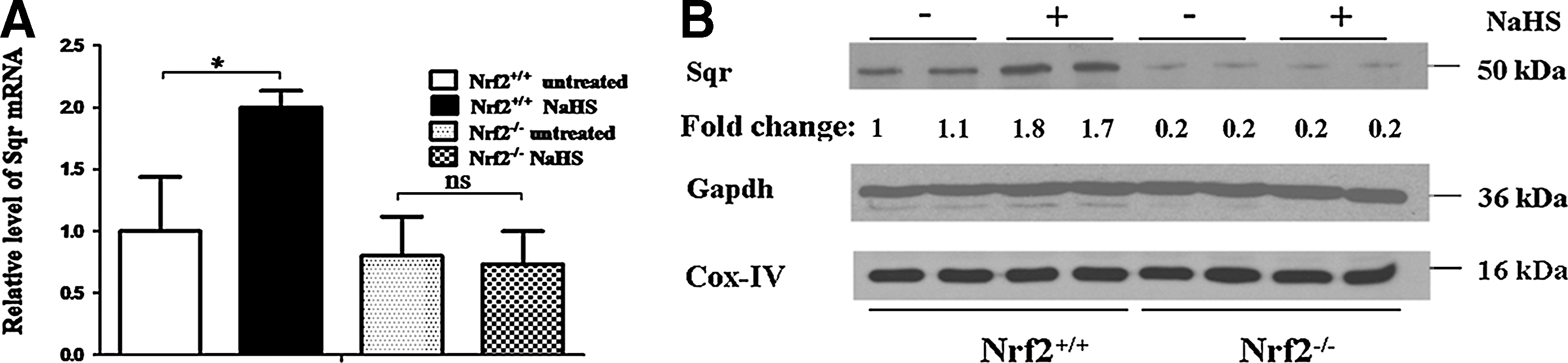
We next examined whether Nrf2 controls the expression of the gene Sqrdl, which encodes the enzyme Sulfide:quinone oxidoreductase (Sqr), which catalyzes the oxidation of H2S. No significant difference in Sqr mRNA was apparent in untreated Nrf2+/+ and Nrf2−/− MEF cells (Fig. 4A). However, treatment of the wild-type fibroblasts with NaHS produced a 2.0-fold increase in Sqr mRNA when compared with untreated fibroblasts, whereas no difference was observed in Nrf2−/− fibroblasts. The level of Sqr protein was higher in wild-type MEFs grown under basal conditions than in their Nrf2 knockout counterparts (Fig. 4B), though this was not evident at an mRNA level. Moreover, treatment with NaHS produced an increase in Sqr protein in Nrf2+/+ MEFs, but not in Nrf2−/− MEFs. These data indicate that H2S regulates Sqrdl expression in an Nrf2-dependent manner.
H2S triggers the formation of disulfide bonds in the dimeric Keap1 protein
We hypothesized that H2S increases the abundance of Nrf2 protein, because it inactivates Keap1 by a mechanism that entails modification of thiols in the BTB-Kelch substrate adaptor. To investigate whether this notion is correct, we used nonreducing sodium dodecyl sulfate–polyacrylamide-gel electrophoresis (SDS-PAGE) to identify oxidized forms of Keap1. Simultaneously, we used conventional SDS-PAGE to analyze the abundance of Nrf2 in the same samples. In our experiments, we cotransfected COS1 cells with expression plasmids encoding untagged Keap1 and V5-tagged Nrf2 for 24 h, after which they were treated with a single dose of 400 μM NaHS and left for a further 0–360 min before examination. At appropriate time intervals after treatment with NaHS, cell extracts were prepared in Redox Lysis Buffer (defined in the Materials and Methods section) to allow subsequent resolution of oxidized and reduced protein by nonreducing SDS-PAGE, followed by identification of the various Keap1 forms through Western blotting. On all occasions, the amount of reduced Nrf2 protein in the same samples was measured by immunoblotting after they had first been treated with 2-mercaptoethanol, to give a final concentration of 6% by volume, before SDS-PAGE.
Nonreducing SDS-PAGE revealed that treatment of cells with NaHS stimulated the formation of several electrophoretically distinct Keap1 protein bands (Fig. 5A). We have labeled these bands Inter-1, Inter-2, Intra-1 and Reduced, based on the fact that a similar Keap1 electrophoretic pattern has been reported previously by Toledano and his colleagues when they used nonreducing SDS-PAGE to analyze ectopic Keap1 in HeLa cells that had been treated with H2O2 (15): our Inter-1, Inter-2, Intra-1, and Reduced Keap1 electrophoretic bands appear, respectively, equivalent to the Oxidized Intermolecular (OxIR) 1, OxIR2, Oxidized Intramolecular (OxIM), and Red bands described by Toledano and colleagues. Specifically, the Inter-1/OxIR1 and Inter-2/OxIR2 electrophoretic bands migrate more slowly during SDS-PAGE than the reduced Keap1 polypeptide, and represent a protein containing an intermolecular disulfide bridge between C151 of two different subunits: the C151 residue is located in the BTB dimerization domain of Keap1, and its involvement in formation of the Inter-1 and Inter-2 bands is concluded from the fact that neither band is observed when the Keap1C151S mutant is subjected to nonreducing SDS-PAGE after treatment with H2O2 [see Ref. (15) and Fig. 5B below]. By contrast, the Intra-1/OxIM electrophoretic band migrates more quickly during SDS-PAGE than the reduced Keap1 polypeptide, and represents a protein containing an intramolecular disulfide bond between the C226 and C613 residues within a Keap1 subunit, as deduced from the fact that the band is not observed when either Keap1C226S or Keap1C613S mutants are examined by nonreducing SDS-PAGE [see Ref. (15) and Fig. 6A below].
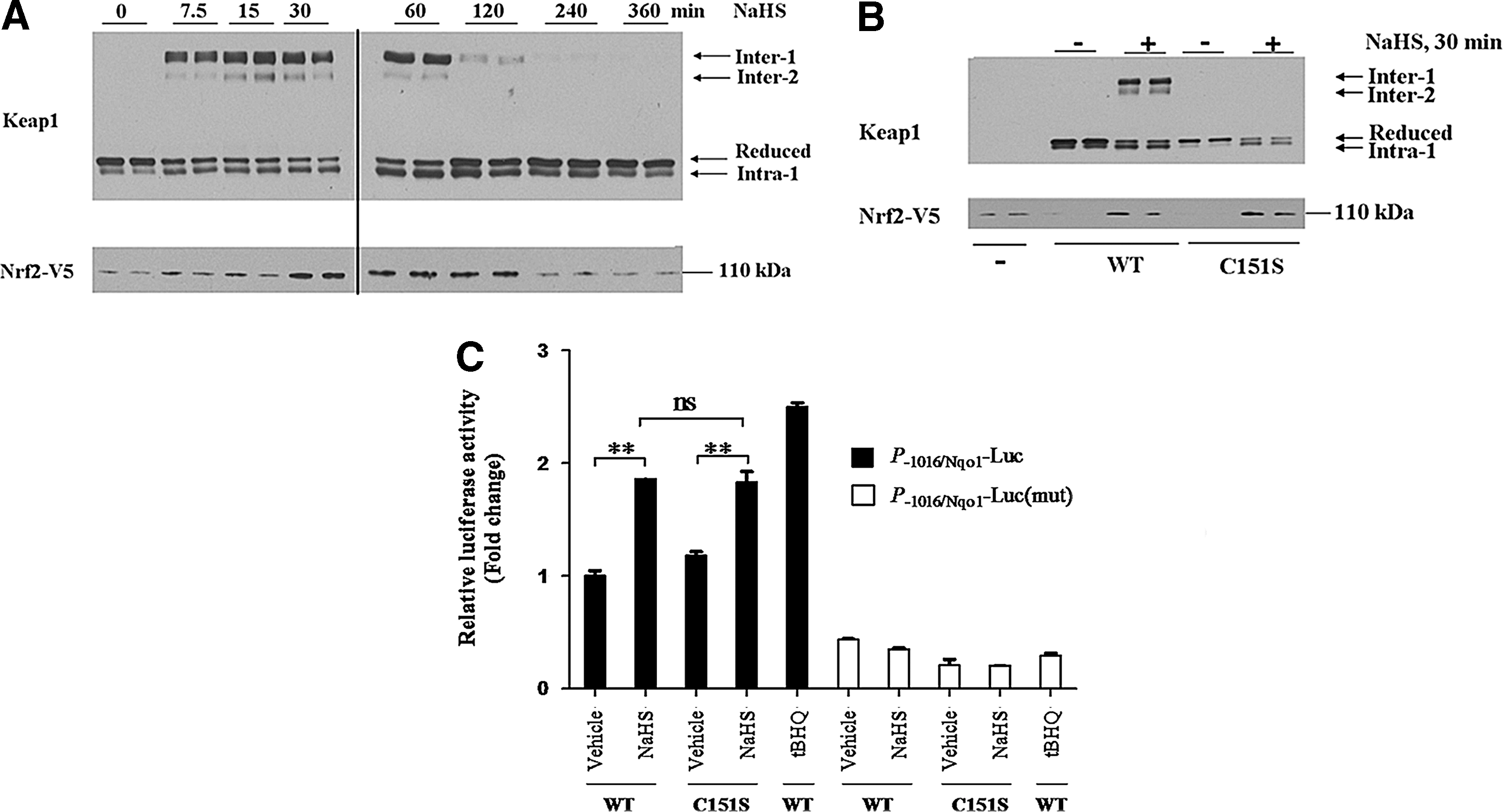
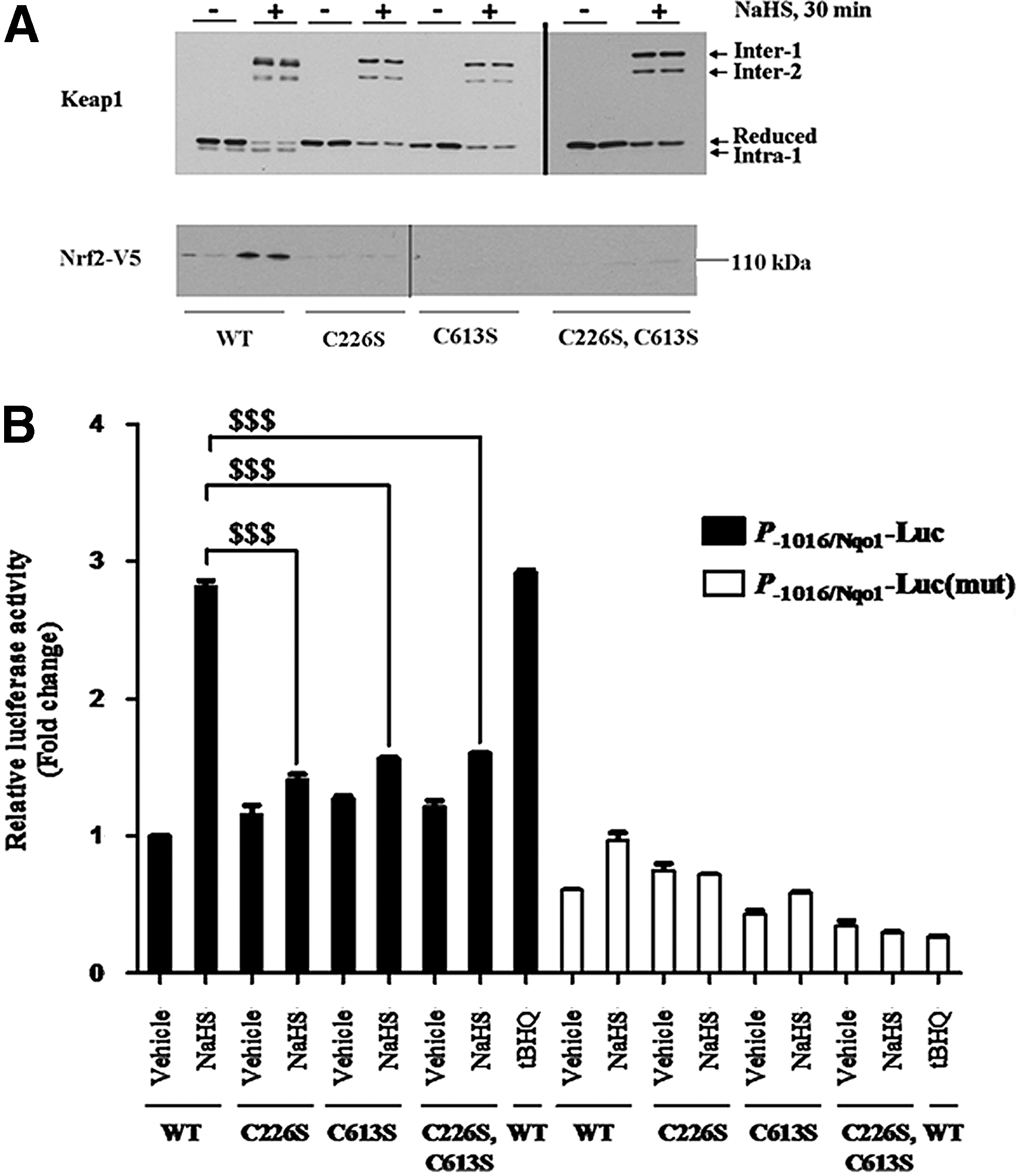
In our experiments, the Inter-1 and Inter-2 Keap1 bands were not present under basal conditions, but appeared within 10 min of treatment with NaHS (Fig. 5A). The Inter-1 and Inter-2 bands were however relatively short-lived and were no longer apparent 240 min after administration of a single dose of NaHS. Furthermore, we found that the Inter-1 Keap1 band was relatively more abundant than the Inter-2 band, which was also the case with the OxIR1 and OxIR2 bands in the experiments described by Fourquet et al. (15). By contrast with the Inter-1 and Inter-2 Keap1 bands, the Intra-1 band was present under basal conditions and was most abundant after treatment with NaHS for between 60 and 120 min. After treatment with NaHS, the increase in the abundance of ectopic Nrf2 protein mirrored most closely the increase in intensity of the Intra-1 band. It was noted that the appearance of Inter-1 and Inter-2 seemed to precede the increase in Nrf2 protein. Moreover, Inter-1 and Inter-2 disappeared 60 min before Nrf2 protein returned to basal levels. These observations suggest that a link exists between the formation of disulfide bonds in Keap1 and inhibition of its substrate adaptor activity toward Nrf2.
Keap1 contains a sensor that responds to H2S and is distinct from that for NO
The gasotransmitter NO has previously been shown to inactivate Keap1 by S-nitrosylation of its C151 residue (46). To determine if the C151 residue in Keap1 represents a general gasotransmitter sensor that also recognizes H2S, we examined whether the Keap1C151S mutant was resistant to inactivation by H2S. Thus, COS1 cells were transfected with Nrf2-V5 alone or jointly with an expression construct for either wild-type Keap1 or Keap1C151S. After recovery from transfection, the COS1 cells were exposed to a single dose of NaHS for 30 min, harvested in Redox Lysis Buffer, and analyzed as described above. The level of ectopic Nrf2 protein was low in samples from untreated COS1 cells, but it was more abundant in samples from cells that had been challenged with NaHS (Fig. 5B). The increase in the amount of Nrf2 upon NaHS treatment was observed in both samples that expressed ectopic wild-type Keap1 and those that expressed Keap1C151S. These results demonstrate that Keap1C151S is inhibited by H2S and therefore indicate that C151 is not required by the BTB-Kelch protein to sense the gasotransmitter. Figure 5B also shows that mutation of C151 in Keap1 prevents formation of the Inter-1 and Inter-2 disulfide bands, which agrees with the findings of Fourquet et al. (15). As nonreducing SDS-PAGE resolves two electrophoretically distinct forms of Keap1 that appear to contain an intermolecular disulfide bond, it is possible that C151 can form a disulfide bridge with another cysteine in the other subunit besides C151. Candidate residues are likely to reside in the BTB domain, and include C77 or C171, but this possibility has not been explored.
To determine whether failure of Keap1 to form Inter-1 and Inter-2 bands influences its ability to repress Nrf2, ARE luciferase reporter assays were performed in RL34 cells. This also allowed us to monitor the ability of Keap1C151S to repress Nrf2 after treatment with H2S; the hydroquinone electrophile tBHQ was used as a positive control. Figure 5C shows that NaHS can induce ARE-driven luciferase activity to a similar extent in RL34 cells expressing wild-type Keap1 and the Keap1C151S mutant protein. Thus, formation of an intermolecular C151–C151 disulfide bond in Keap1 is not required for H2S to inactivate the substrate adaptor protein. The fact that Keap1C151S is resistant to inhibition by NO (46), but can be inactivated by H2S, indicates that Keap1 senses these two gasotransmitters by different mechanisms.
H2S upregulates Nrf2 by inhibiting Keap1 through its C226 and C613 residues
To access whether inactivation of Keap1 by H2S is synonymous with formation of the Intra-1 electrophoretic band, COS1 cells were transfected with an expression vector for Nrf2-V5 along with ones encoding wild-type Keap1, Keap1C226S, Keap1C613S, or Keap1C226S,C613S proteins. Under basal conditions, the level of Nrf2 protein in the transfected COS1 cells was low due to Keap1-mediated ubiquitylation (Fig. 6A), but treatment with NaHS resulted in a significant increase in Nrf2 in some, but not all, cases. Importantly, the accumulation of Nrf2 upon NaHS treatment only occurred in cells that expressed ectopic wild-type Keap1 and was not observed in cells expressing the Keap1C226S, Keap1C613S, or Keap1C226S,C613S mutant proteins. Therefore, H2S did not inactivate Keap1 point mutants that lacked C226 and/or C613 residues (i.e., had been replaced with Ser). Furthermore, mutation of either C226 or C613 in Keap1 prevented the formation of the Intra-1 electrophoretic band. These results indicate that inactivation of Keap1 by H2S requires formation of the C226–C613 disulfide bond.
To examine whether both C226 and C613 in Keap1 are required for H2S to alleviate repression of Nrf2 by the substrate adaptor, a series of ARE-driven luciferase reporter assays were carried out. Figure 6B shows that the ability of NaHS to induce luciferase activity is severely blunted in cells expressing ectopic Keap1C226S, Keap1C613S, or Keap1C226S,C613S proteins. Taken together, the above data are consistent with the hypothesis that the oxidized form of Keap1 that contains the C226–C613 disulfide bridge is inactive and cannot direct ubiquitylation of Nrf2.
The environment of both C226 and C613 contributes to the sensitivity of Keap1 to H2S
A molecular model of Keap1 around C226 predicted that its sulfhydryl group is orientated in close proximity to S224 and H225 (Fig. 7A). It is reasonable to suppose that S224 and H225 may diminish the pK a of the C226 thiol group (46, 59), thereby increasing its reactivity. Furthermore, S224, H225, and C226 are conserved in Keap1 proteins from mammals and fish (Fig. 7B). To determine if S224 and H225 in Keap1 contribute to the reactivity of C226, COS1 cells were transfected with an expression construct for Keap1S224A,H225N. Western blotting showed that mutation of S224 and H225 in Keap1 resulted in the loss of the Intra-1 electrophoretic band, suggesting that these amino acids contribute to the ability of the substrate adaptor to sense H2S (Fig. 7C).
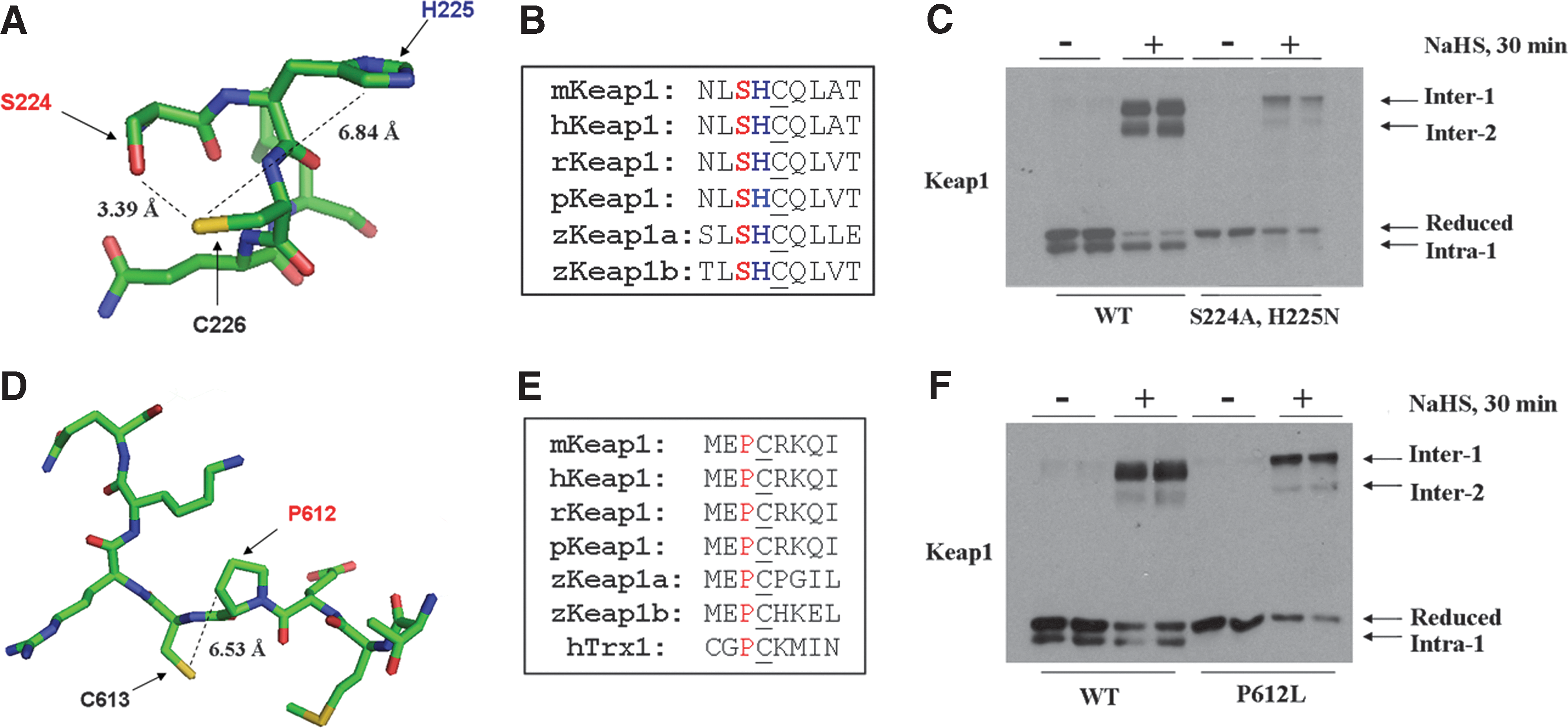
A model for Keap1 around C613 indicated that its sulfhydryl group is not orientated near any hydrogen-bonding or basic amino acids, but is located near the pyrrolidine ring of P612 and may alter its position and/or reactivity (Fig. 7D). Moreover, P612 and C613 are conserved in Keap1 across species (Fig. 7E). To test whether P612 in Keap1 is important for the reactivity of C613 and its ability to form a disulfide bond with C226, COS1 cells were transfected with either expression constructs for the wild-type protein or the Keap1P612L mutant. After overnight recovery, the cells were treated with NaHS and lysates prepared in Redox Lysis Buffer. Immunoblotting showed that mutation of P612 resulted in the loss of the Intra-1 electrophoretic band under basal conditions, and it was still not observed even after treatment with NaHS. These results suggest that P612 influences the ability of C613 to form an intramolecular disulfide bond with C226 (Fig. 7F).
Inhibition of Keap1 by H2S involves H2O2
Recent evidence suggests that H2S may undergo auto-oxidation to form H2O2 (26). To test the notion that H2O2 contributes to the actions of H2S, we examined whether forced expression of catalase might diminish activation of Nrf2 by H2S. Thus, we transfected COS1 cells with an expression vector for Nrf2-V5 alone, or with vectors for Nrf2-V5 and wild-type Keap1, either with or without a construct encoding catalase. After recovery from transfection, cells were treated with NaHS for 30 min before Nrf2 levels were estimated. It is clear from Figure 8A that the increase in Nrf2 protein caused by NaHS is decreased, but not abolished, by the presence of ectopic catalase. These data suggest that the effect of H2S on the Keap1-Nrf2 pathway is mediated in part by H2O2.
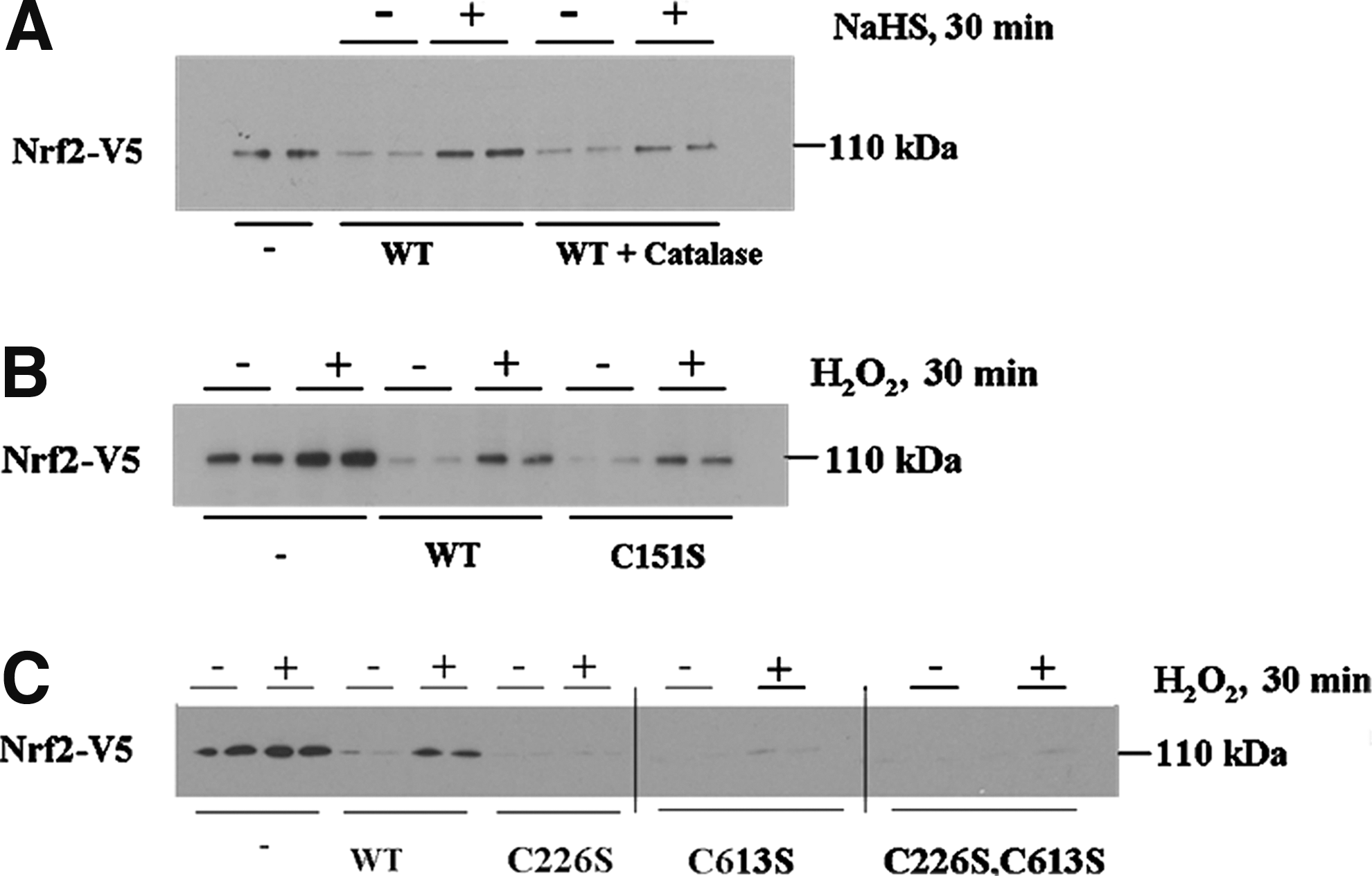
Keap1 senses NO via C151 (15, 46), and Fourquet et al. inferred that the BTB-Kelch protein also senses H2O2 via C151 (15). To evaluate this possibility, we tested whether the Keap1C151S mutant protein is resistant to inactivation by H2O2. COS1 cells were transfected with expression plasmids for Nrf2-V5 alone, or in combination with plasmids for either wild-type Keap1 or Keap1C151S, before they were treated with H2O2 for 30 min, and Nrf2 levels measured. Figure 8B shows that treatment of the transfected cells with H2O2 caused stabilization of Nrf2 to the same extent in both wild-type Keap1-expressing cells and Keap1C151S-expressing cells. Thus, the BTB-Kelch protein does not sense H2O2 via its C151 residue. To determine if Keap1 can be inhibited by H2O2 through triggering the C226/C613 sensor that responds to H2S, the same strategy was used as described above, but in this case, the COS1 cells were transfected with expression plasmids encoding Keap1C226S, Keap1C613S, or Keap1C226S,C613S proteins rather than Keap1C151S. As is clear from Figure 8C, mutation of the Keap1 H2S sensor cysteines to serines prevented H2O2 from stimulating an increase in Nrf2 protein. These results indicate that C226 and C613 in Keap1 sense H2O2 as well as H2S.
Keap1 is S-sulfhydrated by H2S
We used a Biotin-switch technique (BST) method (51) to determine if Keap1 is S-sulfhydrated by H2S. COS1 cells were transfected with an expression vector for wild-type Keap1, and after recovery from transfection, they were treated with NaHS for 30 min before being harvested in radioimmunoprecipitation assay (RIPA) buffer. The cell lysates were subjected to the BST method, and after elution of biotin-labeled proteins from strepavidin beads, the amount of Keap1 protein pulled down was determined by Western blotting. For unknown reasons, the BST method caused Keap1 to migrate during SDS-PAGE as two bands, with the mobility of the slower band corresponding to a protein of 69 kDa (this is equivalent to the molecular weight of Keap1). Figure 9A shows that treatment of cells with NaHS increased the amount of Keap1 protein in the pull-down samples, indicating that the substrate adaptor is S-sulfhydrated. Interestingly, Keap1 was also found to be present at low levels in pull-down samples from lysates of untreated cells, indicating that it is subject to S-sulfhydration under basal conditions.
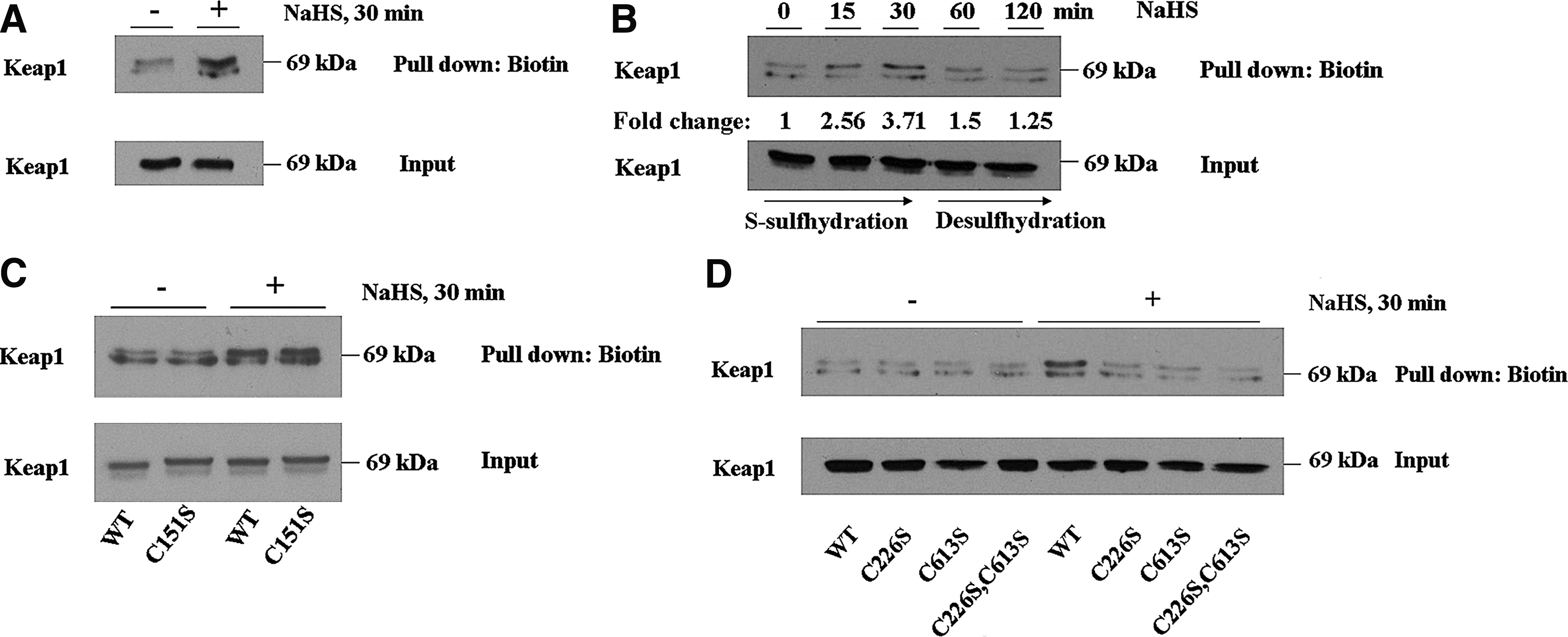
To further examine S-sulfhydration of Keap1, COS1 cells expressing wild-type Keap1 protein were treated with a single dose of 400 μM NaHS, before they were harvested 15, 30, 60, or 120 min later in the RIPA buffer, and the lysates subjected to BST. As shown in Figure 9B, the amount of Keap1 protein in the pull-down samples at the 15- and 30-min time points was greater than that at 0 min, suggesting that S-sulfhydration of the adaptor protein increases acutely upon exposure to H2S. However, 60 min after treatment with a single dose of NaHS, the amount of Keap1 protein in the pull-down samples decreased to the level observed at the 0-min time point. This finding indicates that S-sulfhydration of Keap1 by H2S is a reversible process and suggests that the modified protein is subject to desulfhydration.
To determine whether the NO sensor in Keap1 acts as a site of S-sulfhydration, COS1 cells expressing either wild-type Keap1 protein or Keap1C151S protein were treated with NaHS, and 30 min later, they were harvested in RIPA buffer and the lysates subjected to BST. As shown in Figure 9C, the amount of Keap1 protein in the pull-down samples did not differ between wild-type Keap1-expressing cells and Keap1C151S-expressing cells treated with NaHS, suggesting that C151 is not a major site of S-sulfhydration.
We next examined whether C226 and C613 in Keap1 are S-sulfhydrated, because they comprise the H2S sensor. COS1 cells were transfected with expression vectors for wild-type Keap1, Keap1C226S, Keap1C613S, or Keap1C226S,C613S, and after treatment with NaHS were examined using the BST method. Figure 9D shows that the amount of Keap1 protein in pull-down samples from wild-type Keap1-expressing cells increased upon treatment with NaHS when compared with untreated cells. Importantly, the accumulation of Keap1 protein upon NaHS treatment only occurred in cells that expressed ectopic wild-type Keap1 and was not observed in cells expressing the Keap1C226S, Keap1C613S, or Keap1C226S,C613S mutant proteins. These findings indicate that C226 and C613 are the major sites of S-sulfhydration in Keap1, but that such modification can only occur when both cysteines are present.
Discussion
The results reported in the present article have demonstrated that H2S can induce the classic Nrf2-target gene Nqo1, and that this requires a functional ARE sequence in the Nqo1 gene promoter. We also found that stimulation of ARE-driven gene expression by H2S is associated with stabilization of the Nrf2 protein. Moreover, evidence is provided that cytoprotection afforded by H2S against the redox-cycling agent menadione requires activation of Nrf2.
Regulation by Nrf2 of enzymes involved in the metabolism of H2S and the production of cysteine
It is evident from our in vitro study of MEFs that Nrf2 regulates the expression of Cbs. Consistent with this view, in silico examination of the upstream region of the gene has revealed that it contains the sequence 5′-GTGATCTAGCA-3′ that is likely to function as an ARE to which Nrf2 can bind as a heterodimer with a small musculoaponeurotic fibrosarcoma (Maf) protein. Similarly, we have provided evidence that Nrf2 regulates Cse, and its upstream region contains the sequence 5′-ATGAGGCAGCT-3′ that is likely to serve as an ARE. Our finding that mouse Cse is regulated by Nrf2 is consistent with the recent observations of Hassan et al. (22), who reported that rat CSE is induced by platelet-derived growth factor-BB in an Nrf2-dependent manner. We found no evidence that Nrf2 regulates the levels of mRNA for 3Mst in mouse fibroblasts. However, Nrf2 does regulate human thioredoxin (TRX1) [for a review, see (24)], and as TRX1 is a cofactor for 3MST (48), it is possible that Nrf2 regulates 3MST indirectly.
Our TaqMan and immunoblotting experiments indicate that Cbs and Cse ought to be included as members of the ARE gene battery. In addition to catalyzing the production of H2S, Cbs and Cse probably contribute to cytoprotection by augmenting levels of cysteine and thus aid the synthesis of GSH. In particular, CBS and CSE catalyze the reverse trans-sulfuration pathway that is responsible for production of cysteine from homocysteine (29, 34, 40). The availability of cysteine seems to be a rate-limiting factor for GSH synthesis, and in accordance with this, Cbs−/− and Cse−/− mice have diminished GSH levels and enhanced intracellular levels of ROS (11, 69). We have presented a model that depicts the interplay between Nrf2 and H2S (Fig. 10A); this includes a role for Nrf2 in mediating the cytoprotective effects of H2S and in controlling the expression of Cbs and Cse, which in turn stimulate production of both cysteine and the H2S gasotransmitter.
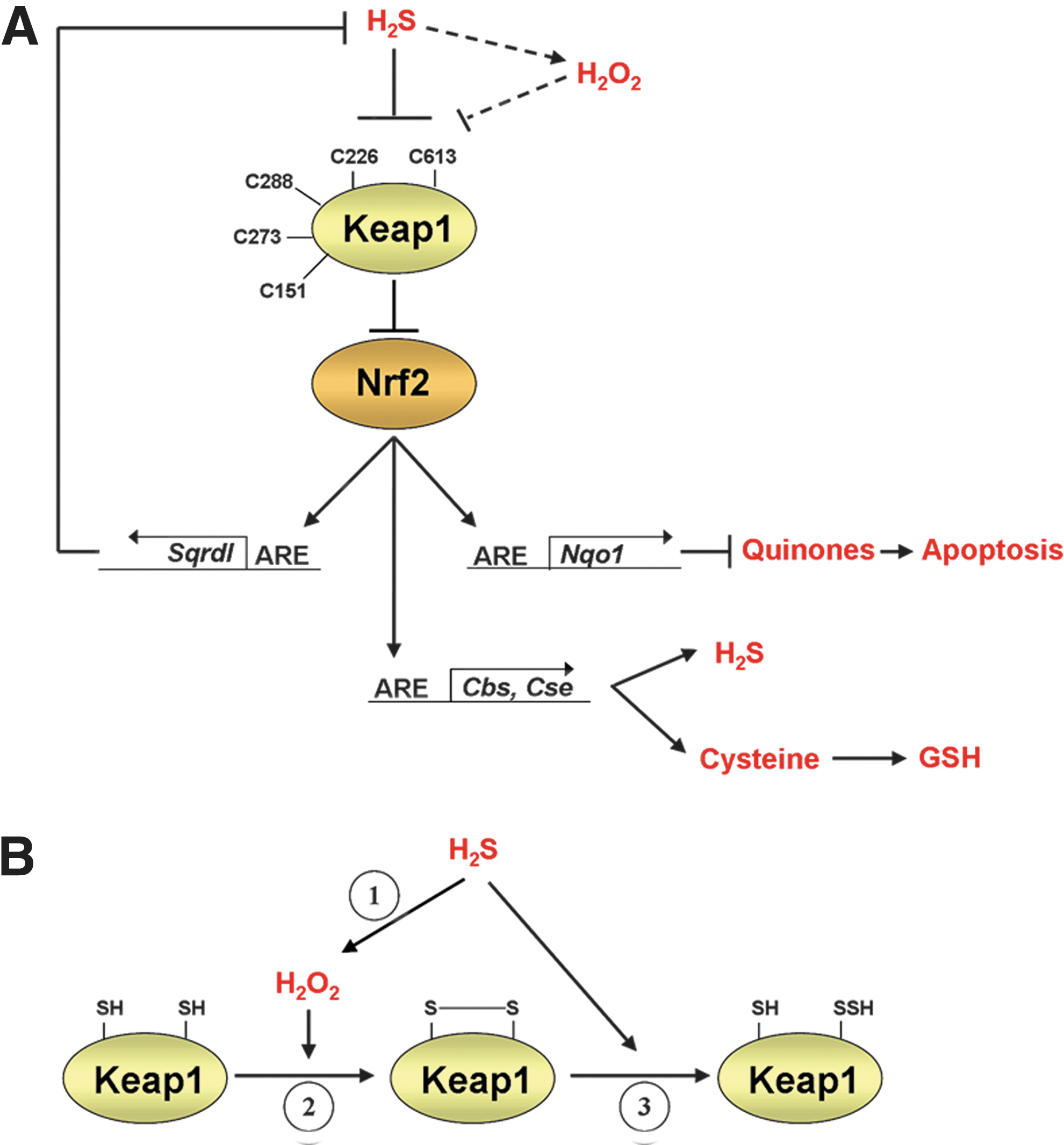
While low levels of H2S confer cytoprotection against oxidative stress, it can cause toxicity when present in high concentrations (66). Excessive levels of H2S can be countered by activation of the sulfide oxidation pathway, which involves the enzyme Sqr (1, 49). As demonstrated in this study, Nrf2 regulates the expression of Sqrdl, which encodes Sqr. In silico analysis of the flanking region of Sqrdl showed that it contains the sequence 5′-ATGACATAGCC-3′, that is likely to function as a binding site for an Nrf2–small Maf heterodimer. Collectively, our data suggest that Nrf2 is a principal regulator of H2S metabolism, as it regulates genes involved in both its synthesis and its elimination.
Modification of Keap1 by H2S
An aim of this project was to identify the mechanism by which H2S increases Nrf2 activity. We have found that the gasotransmitter antagonizes Keap1 activity through modification of reactive cysteines in the substrate adaptor. While our data provide evidence that inactivation of Keap1 by H2S requires the presence of both C226 and C613, we have also identified the amino acids surrounding these two residues that contribute to the sensitivity of Keap1 to inhibition by H2S, presumably through lowering the thiol pK a of C226 and C613 (55, 59). While the reactivity of C226 is dependent upon adjacent basic and hydrogen-bonding amino acids (i.e., S224 and H225), it appears that the reactivity of C613 is dependent upon its adjacent P612 residue. Although, proline is not usually thought to affect redox signaling, it is noteworthy that an active-site proline contributes to the activity of TRX1 (57).
Whether H2S is capable of reacting directly with the reduced thiol groups of C226 and C613 in Keap1 is not known. This uncertainty arises because H2S is a weak reducing agent and exhibits little reactivity toward free thiols (9). Given the fact that H2S readily reacts with oxidized thiols (16), it is possible that the gasotransmitter reacts with C226 and C613, to form S-sulfhydrated Keap1, principally after they have been oxidized to form a disulfide bridge. Consistent with this hypothesis, our experiments involving forced expression of catalase suggest that H2O2 makes a significant contribution to activation of Nrf2 by H2S. In this context, it is notable that H2O2 may be generated directly from H2S by auto-oxidation (26), or indirectly by inhibition of cytochrome c, causing increased mitochondrial production of ROS (66). We therefore speculate that S-sulfhydration of Keap1 may occur by a two-step process in which the C226 and C613 residues are first oxidized by H2O2 to form a disulfide bridge before they are subsequently reduced by H2S (Fig. 10B). Thus, it may be necessary for H2S to stimulate formation of a C226–C613 disulfide bridge in Keap1 before it is able to S-sulfhydrate the BTB-Kelch protein.
H2S can S-sulfhydrate Keap1 at C226 and C613, but it is a reversible process. Removal of the S-sulfhydrated group from the thiol indicates a level of regulation, and it will therefore be important to identify the proteins responsible for desulfhydration. Recent work by Krishnan et al. (37) suggests that TRX1 can desulfhydrate PTP1B in an in vitro setting, but the physiological significance of this remains to be determined. Of particular interest, our data indicate that mutation of either C226 or C613 prevents S-sulfhydration of Keap1, which suggests (i) that the BST is not sensitive enough to detect low levels of S-sulfhydrated protein, or (ii) that S-sulfhydration of one cysteine promotes modification of the second cysteine through direct transfer of the persulfide bond (trans-sulfhydration). In this case, both cysteines in Keap1 are required for the modification to occur, but only one cysteine is ultimately S-sulfhydrated. It remains to be determined whether the putative trans-sulfhydration process, which is analogous to transnitrosylation where the nitro group attached to one cysteine is transferred to a second cysteine, occurs in Keap1.
Mechanism by which Keap1 is inhibited through the C226/C613 sensor
We have established previously that Keap1 is inactivated by the noncovalent binding of Zn2+, Cd2+, As3+, and Se4+ to its C226 and C613 residues through a process that we envisage involves coordination chemistry (46). The present study has demonstrated that Keap1 is inactivated by H2O2 through formation of an intramolecular disulfide bridge between C226 and C613. It could be argued that in both cases (i.e., the binding of transition metals and thiol oxidation), the loss of Keap1 activity is associated with restriction in the freedom of movement of the IVR and CTR domains. If this hypothesis is correct, it follows that the mobility of residues in regions of Keap1 that contain C226 and C613 is essential for its substrate adaptor activity. It is possible that flexibility in this area influences the relative positions of the two β-propeller protein-docking sites in the Keap1 dimer, and this in turn may block binding of Nrf2 through both its DLG and ETGE motifs (45). Formation of the C226–C613 disulfide bridge in Keap1 might either prevent it from mediating the ubiquitylation of Nrf2 by Cul3-Rbx1, or prevent the transfer of ubiquitylated Nrf2 to a receptor involved in transport to the proteasome.
If the prediction that loss of mobility in the IVR and CTR is sufficient to trigger the C226/C613 sensor, it seems unlikely that S-sulfhydration of C226 and C613 by H2S would inhibit Keap1. Indeed, if H2S cleaves the C226–C613 bond in Keap1 formed by H2O2, then H2S might restore substrate adaptor activity, because the reaction would free the IVR and CTR domains from restriction in their mobility caused by the disulfide bridge. In a related vein, electrophiles that form adducts with C226 or C613 might prevent the sensor being triggered by H2O2, because they block formation of the disulfide bridge.
Basal Nrf2 activity and endogenous signaling through the H2O2 sensor
Fourquet et al. (15) were the first group of workers to use nonreducing SDS-PAGE to show that Keap1 forms a C151–C151 intermolecular disulfide bond and a C226–C613 intramolecular disulfide bond upon treatment with H2O2. These workers assumed that NO and H2O2 trigger the same sensor in Keap1, and on the basis of the finding that NO inactivates Keap1 through C151 in the BTB-Kelch protein, they proposed that H2O2 also triggers the C151 sensor; in fact, they did not test whether H2O2 inactivates Keap1 via C151. Herein, we have demonstrated that inactivation of Keap1 by H2O2 occurs independently of C151. Moreover, our study of Keap1 mutants indicates that the BTB-Kelch protein is only sensitive to H2O2 inactivation if it possesses both C226 and C613. Our results are therefore consistent with the hypothesis that H2O2 inactivates Keap1 by formation of the C226–C613 intramolecular disulfide bond, not through formation of the C151–C151 intermolecular disulfide bond.
Nonreducing SDS-PAGE has revealed that the C226–C613 intramolecular disulfide bond in Keap1 is formed in the absence of exogenous stressors. It therefore appears that the C226/C613 sensor is triggered under basal conditions and may thus be responsible for the low Nrf2 activity that occurs under normal homeostatic conditions. This observation suggests that Keap1 may be subject to a modest level of inhibition by the generation of H2O2 that occurs during normal mitochondrial metabolic processes (65). It is notable that in previous cotransfection experiments described by Toledano and his colleagues, knockdown of endogenous thioredoxin reductase (TXNRD1) resulted in a significant increase in the portion of ectopic Keap1 that forms a C226–C613 disulfide bridge, and that this was associated with a marked increase in the relative amount of ectopic Nrf2 (15). These results suggest that inactivation of Keap1 through formation of a C226–C613 disulfide bridge may be rapidly reactivated by reduction of cystine by TRX1, which is in turn reduced by TXNRD1. As TRX1 and TXNRD1 are both members of the ARE gene battery (24), it is evident that an Nrf2-mediated feedback loop may exist between Keap1 and the thioredoxin pathway.
Concluding comments
Transcription factor Nrf2 is a master regulator of cellular redox homeostasis and mediates preconditioning, stimulated by H2S, to confer protection against ischemia. While Nrf2 is of benefit to normal cells, it is frequently upregulated in nonsmall-cell lung cancer, and to a lesser extent in tumors of the head and neck, liver, ovary, and stomach (5, 72). Tumors in which Nrf2 is constitutively active have a poor prognosis, because the CNC-bZIP factor increases resistance to chemotherapeutic drugs and radiotherapy, increases angiogenesis, and allows higher rates of cell proliferation [reviewed in Refs. (23, 63)]. Upregulation of Nrf2 can arise as a consequence of somatic mutations or epigenetic changes that diminish Keap1 activity. Alternatively, Nrf2 may evade repression by Keap1 as a consequence of somatic mutations in the CNC-bZIP factor, or by overexpression of the Nrf2 gene. It is also possible that mutations in enzymes involved in the synthesis or metabolism of endogenous signaling molecules such as H2S, H2O2, or NO that antagonize Keap1 might contribute to the permanent activation of Nrf2 in tumors. This possibility warrants investigation.
Materials and Methods
Chemicals
These were all readily available commercially, and in all cases, the highest grade of purity was used.
Cell culture
The COS1 and RL34 cells were grown in the Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM
Plasmids
Expression constructs encoding V5-tagged mouse Nrf2 (mNrf2-V5) and untagged mouse Keap1, both in pcDNA3.1, have been described previously (43, 44). Also, the expression constructs encoding mouse Keap1C226S, Keap1C613S, and Keap1C226S,C613S have been described previously (46). The β-galactosidase expression plasmid pCMVβgal, used to correct for transfection efficiency, was from Clontech. The expression construct for mouse Keap1S224A,H225N was generated using a Multi-site-directed mutagenesis kit (Stratagene) with the following primer: forward, 5′-GAGGAGTTCTTCAACCTG
Analysis of mRNA levels by real-time quantitative PCR
Total RNA was isolated from cell lines by standard methods, and cDNA was prepared using the Omniscript Reverse Transcription Kit (Qiagen) according to the manufacturer's instructions. The cDNA was diluted 1:7.5 in ddH2O and used as a template for real-time quantitative PCR. ABI PRISM 7700 Sequence Detector (Applied Biosystems) was used for analyses; cycling conditions were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and finally, 60°C for 1 min. All samples were analyzed in triplicate, and reactions containing no template were also carried out as a negative control. Actin served as an internal reference. Fold induction values were normalized to controls using the comparative threshold cycle values. Most probe and primer sets were purchased from Applied Biosystems (ABI) and were supplied in a single-tube format with the exception of the Nqo1 probe/primer set (forward, 5′-GCAGGATTTGCCTACACAATATGC-3′ and reverse, 5′-AGTGGTGATAGAAAGCAAGGTCTTC-3′) described elsewhere (47). The gene expression primer and probes for Cse, Cse, 3Mst, Sqrdl, and actin were Mm00461247_m1, Mm00460654_m1, Mm00460389, Mm00502443_m1, and Mm00607939_s1, respectively.
Luciferase reporter gene assay
ARE-driven luciferase activity was measured using P −1016/Nqo1-Luc, as described previously (53); a mutant form of this plasmid in which point mutations have been introduced into its ARE [i.e., P −1016/Nqo1-Luc(mut)] served as a negative control. The pRL-TK Renilla reporter plasmid was used to correct for transfection efficiency.
Nonreducing SDS-PAGE
To analyze redox-modified Keap1 protein, cells were transfected with expression constructs encoding either the wild-type or the mutant forms of the substrate adaptor. After 18-h recovery, cells were treated with NaHS, or vehicle control, before they were lysed, and the protein was extracted in Redox Lysis Buffer (100 mM Tris, 150 mM NaCl, 0.2% [w/v] deoxycholic acid, 5% [v/v] NP40, 200 μM sodium fluoride, 250 μM EDTA, 100 μM phenylmethylsulfonyl fluoride, and 40 mM N-ethylmaleimide). Proteins from cell lysates were diluted with two volumes of redox sample buffer (200 mM Tris, pH 6.8, 8% [w/v] SDS, 30% [v/v] glycerol, and 0.05% [w/v] bromophenol blue) before being subjected to Tris–glycine SDS-PAGE in 6% (w/v) polyacrylamide gels. For the analysis of reduced Nrf2 that was done in parallel with nonreduced Keap1, the samples prepared in Redox Lysis Buffer were allowed to react with 6% (v/v) 2-mercaptoethanol for 10 min at 95°C before electrophoresis.
Immunoblotting
For most immunoblotting experiments, cells were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% [v/v] NP40, 0.5% [w/v] deoxycholic acid, and 0.1% [w/v] SDS) and allowed to react with 6% (v/v) 2-mercaptoethanol for 10 min at 95°C. Nonreduced proteins (in the Redox Lysis Buffer) and reduced proteins (in the RIPA buffer) that had been resolved by SDS-PAGE were transferred electrophoretically onto polyvinylidene fluoride membranes and probed with antibodies using standard methods. Antibodies against rat NQO1, mouse Nrf2, and mouse Keap1 have been described previously (31, 44, 45). Antibodies against CBS were provided by Dr. Ruma Banerjee (University of Michigan, Ann Arbor, USA) and have been described (29). The other antibodies were obtained commercially as follows: anti-GAPDH (Sigma), anti-CSE (Abnova), and anti-SQR and anti-Cox-IV (Abcam). ImageJ software was used to determine the densitometry of immunblots relative to the loading control. The density of each protein band was expressed as a fold change of the vehicle-treated control.
Detection of ROS
MEF cells, grown until they were 80%–90% confluent, were washed with PBS and incubated with 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) in Hanks Buffered Salt Solution (HBSS; Gibco) for 30 min at 37°C in the dark. After incubation, the fibroblasts were washed 3×5 ml HBSS before 1 ml of HBSS was added to each plate. Fluorescence was measured by excitation at 485 nm and emission at 538 nm. Cells were lysed, and fluorescence was normalized to protein levels.
Cell viability
Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (25, 50). Cells were challenged with xenobiotics in triplicate wells for 24 h, after which 20 μl MTT (5 mg/ml in PBS) was added to the culture medium to achieve a final concentration of 1 mg/ml. Cells were incubated at 37°C for 60 min before being lysed in the MTT lysis buffer and incubated at room temperature for 8 h. The formation of formazan was measured by absorbance at 570 nm.
Determination of the half-life of Nrf2
MEF cells were treated with NaHS for 1 h before treatment with CHX (final concentration 40 μg/ml) for between 0 and 120 min. Cell lysates were prepared, and the amount of Nrf2 protein was determined by Western blot. Densitometry analysis was carried out to calculate band intensities, and the relative amount of Nrf2 was plotted on a semi-log graph as described elsewhere (43).
Biotin-switch technique
To determine the extent of protein S-sulfhydration, the BST method was used (51). Duplicate dishes of COS1 cells that had been transfected with expression vectors for Keap1 were allowed to recover for 18 h before they were treated with NaHS for various periods of time. Thereafter, they were subjected to a modified BTS method. Briefly, cells were lysed in the RIPA buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% [v/v] Nonidet P-40, 0.5% [w/v] deoxycholic acid, 0.1% [w/v] SDS) supplemented with 10 mM S-methyl methanethiosulfonate (MMTS) to alkylate free thiols. After centrifugation of lysates, the supernatant was added to 1.6 ml of HEN buffer (100 mM Hepes, 1 mM EDTA, and 0.1 mM neocuproine [pH 8.0]), supplemented with 2.5% (w/v) SDS and 20 μM MMTS, and incubated at 20°C for 18 min to complete the blocking of free thiols. The MMTS was removed from protein samples by acetone precipitation, and proteins were resuspended in the HEN buffer supplemented with 1% SDS. Thereafter, proteins were incubated with 4 mM Biotin–HPDP (Thermo scientific) for 3 h at room temperature to label S-sulfhydrated thiols. Biotinylated, and thus S-sulfhydrated, proteins were precipitated by streptavidin–agarose beads and eluted in an SDS-PAGE sample buffer.
Bioinformatics and statistical analyses
Swiss-Model software was used for molecular modeling of mKeap1 with 3i3nA.pdb as a template. Throughout this study, GraphPad Prism 4 software was used for statistical analysis. The luciferase data were analyzed using Student's t-test, whereas TaqMan data were analyzed using one-way analysis of variance, followed by Newman–Keuls multiple comparison test. Significant increases are expressed as p≤0.001 (***); p≤0.01 (**); p≤0.05 (*); and not significant. Significant decreases are similarly expressed using the dollar ($) symbol.
Footnotes
Acknowledgments
We thank Dr. Ruma Banerjee for providing antibodies against CBS, and Dr. Nick Tonks for providing an expression vector for catalase. We are most grateful to Dr. Simon Fourquet and Dr. Michel Toledano for helpful comments during the initial stages of this project. We are indebted to Dr. Mike McMahon and Dr. Albena Dinkova-Kostova for their expert help and critical suggestions throughout this project. This work was funded in part by a Biotechnology and Biological Sciences Research Council Ph.D. studentship (BB/E528995/1) under the BBSRC Industrial CASE Partnership scheme and by Cancer Research-UK (C4909/A13786).
Author Disclosure Statement
The authors declare no conflict of interest.
Abbreviations Used
†
The ARE has also been called an electrophile-response element (EpRE).
‡
The numbering of amino acids in Keap1 is based on the human protein.