
Abstract
Introduction
T
The medical application of ionizing radiation was realized early after the discovery of X-rays by Röntgen in 1895, when Emil Grubbé used X-rays to treat an ulcerated breast cancer 60 days after the discovery of X-rays (15). Since that time, efforts have been made to improve the efficacy of radiation therapy, increasing the killing effect on cancer cells while minimizing the detrimental effects on normal tissues. Various drugs have been developed to modulate the DNA damage response in tumor cells, alter the activation of signal transduction pathways activated after irradiation, and control the influence of the tumor microenvironment [reviewed in ref. (12)]. Despite these advances, there is a need for further improvements.
Reactive oxygen species (ROS) are produced as a byproduct of oxygen metabolism (70). ROS, while harmful to cells when produced in excess through oxidative modification of lipids, proteins, and DNA, are also vital mediators of multiple cellular processes, including cell growth and differentiation (18), the immune response, cell adhesion, and apoptosis (47). ROS are also second messengers in cell signaling (69, 81, 181, 210). The rate of ROS production and destruction is carefully maintained in the cell, and interruption of this process contributes to the development of different diseases, including cancer (75, 210, 215).
ROS play a major role in the damaging effects of low linear energy transfer (LET) ionizing radiation on cancer cells. ROS are formed by the radiolysis of water, and these ROS (137), particularly the hydroxyl radical (214), participate in damaging DNA. Roughly two-thirds of radiation-mediated DNA damage is caused by indirect effects from ROS (146). Although radiation is an important treatment for cancer, it can also be harmful to normal tissues (1). Therefore, methods that can simultaneously increase the radiosentivity of cancer cells and radioresistance of normal tissues are needed to improve the treatment outcome in patients.
Mitochondria are the major sites of metabolic ROS production in the cell, with the superoxide radical as the primary ROS generated by the organelle as a byproduct of oxidative phosphorylation (2, 97). Cells are equipped with many systems to scavenge ROS, with the superoxide dismutases (SODs) as the chief ROS scavenging enzymes in the cell (228). Because of the importance of ROS in cancer development, and the role of ROS in the radiation-induced damage, methods to alter the redox environment of cancer cells may enhance the response of cancer cells to ionizing radiation.
In this review, we will discuss the effects of ionizing radiation on the cell. We will also discuss two factors that affect the efficacy of radiation therapy: the bystander effect and the tumor microenvironment. We will also discuss SODs, their role in cancer development, and their importance in the radiation response of both normal tissues and cancer cells. Finally, we will detail different methods to increase SOD expression and/or activity and its effects on simultaneously protecting normal tissues and sensitizing tumor tissues to ionizing radiation.
Effects of Radiation on Biological Tissue
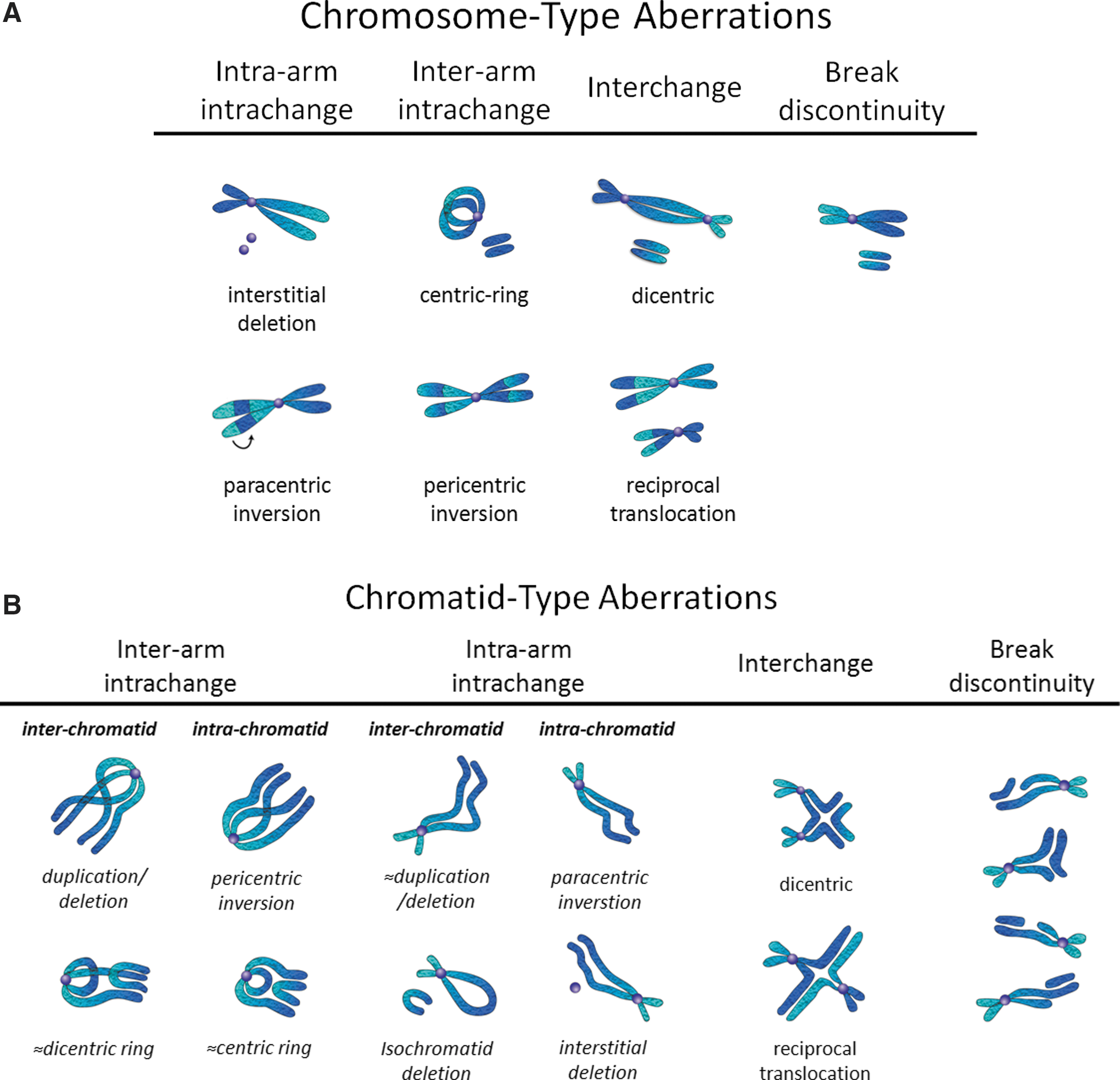
The primary target of ionizing radiation in the cell is chromosomal DNA. Ionizing radiation damages intracellular molecules by direct ionization and through indirect ionizations mediated by water radiolysis products (Fig. 1A), with an estimated two-thirds of radiation-induced DNA damage due to indirect effects (146). This process elicits myriad types of DNA damage, including base and sugar damage, as well as single- and double-strand breaks. Radiolysis of water leads to the formation of a variety of free radicals. For example, water is ionized to form a radical and a free electron. The water radical can either undergo decomposition or interact with a water molecule to form hydroxyl radicals. The free electron can be hydrated by surrounding water molecules, and this stabilized electron can interact with another water molecule to form a hydroxyl radical or a proton to form a hydrogen radical (137) (Fig. 1B). Direct effects of ionizing radiation on DNA lead to a variety of damage products, such as oxidized bases and cleavage of the sugar–phosphate backbone (Fig. 2) (189). Water radiolysis products can damage nucleic acids (Fig. 3) and other cellular molecules, including proteins and lipids (180, 214). Free radical damage of DNA, whether direct or indirect, results in the formation of myriad types of DNA lesions (137, 167, 182). Base lesions may be benign, with no apparent effect on the cell after irradiation, or the base lesions may lead to miscoding of the DNA, resulting in mutation formation. Single-strand breaks can also form, which may result in mutation formation if this damage is not properly repaired. Double-strand breaks also occur, in particular, following the replication of DNA that sustained oxidative damage, resulting in chromosomal abnormalities (137). These double-strand breaks, if not re-annealed at the site of the original break, may re-anneal with other breaks on the same chromosome or a different chromosome to form chromosomal aberrations (88). Chromosomal aberrations are divided into two broad types: chromosome type and chromatid type. Chromosome-type aberrations occur when breaks and rejoining change both sister chromatids at one locus (Fig. 4A). Chromatid-type aberrations occur when breaks and rejoinings alter only one sister chromatid at any one locus (Fig. 4B). The type of damage that occurs after irradiation depends on the stage of the cell cycle at which a cell is irradiated. Chromosome-type aberrations typically occur during G1 of the cell cycle, whereas chromatid-type aberrations typically occur during G2 and S phases of the cell cycle (3).

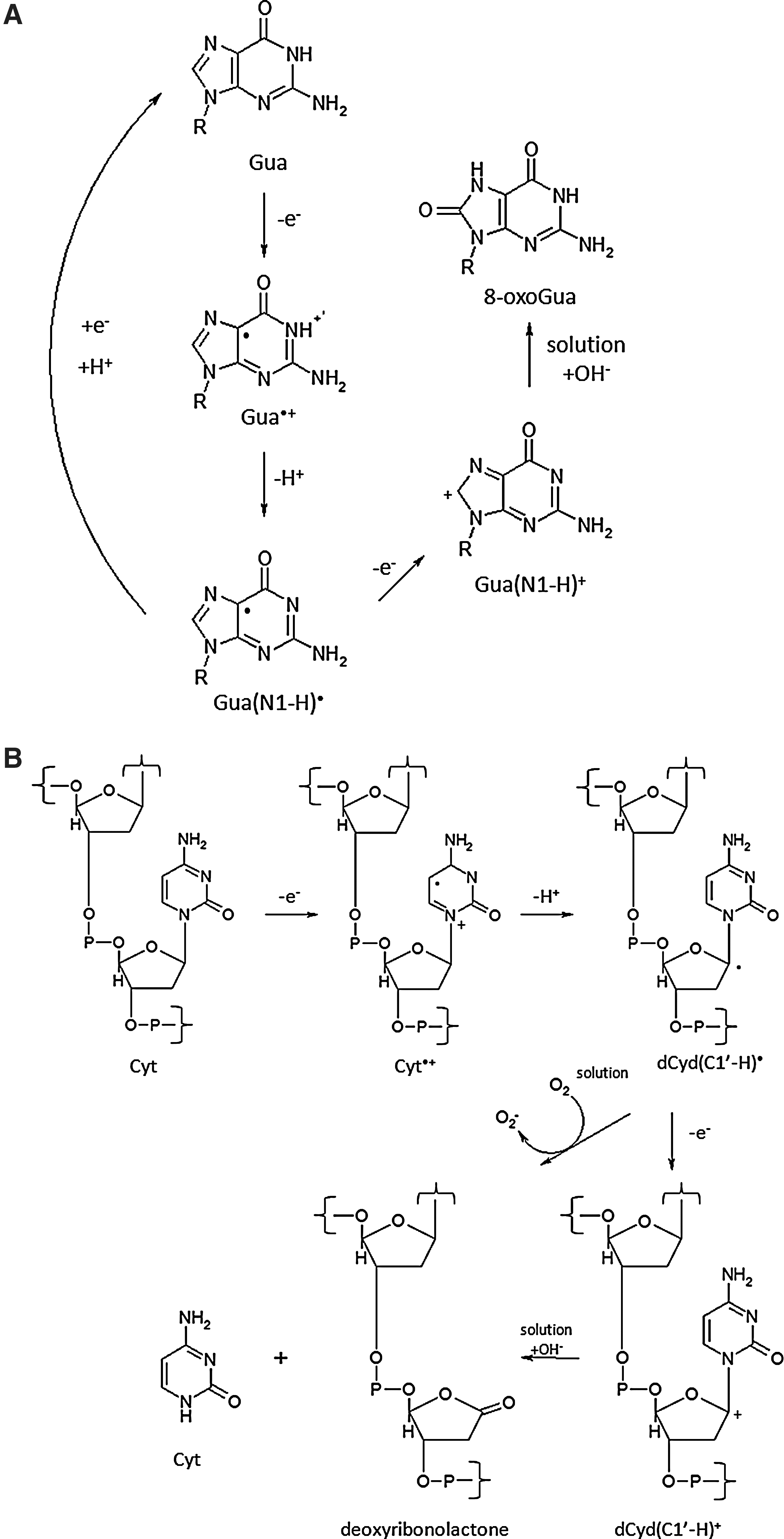
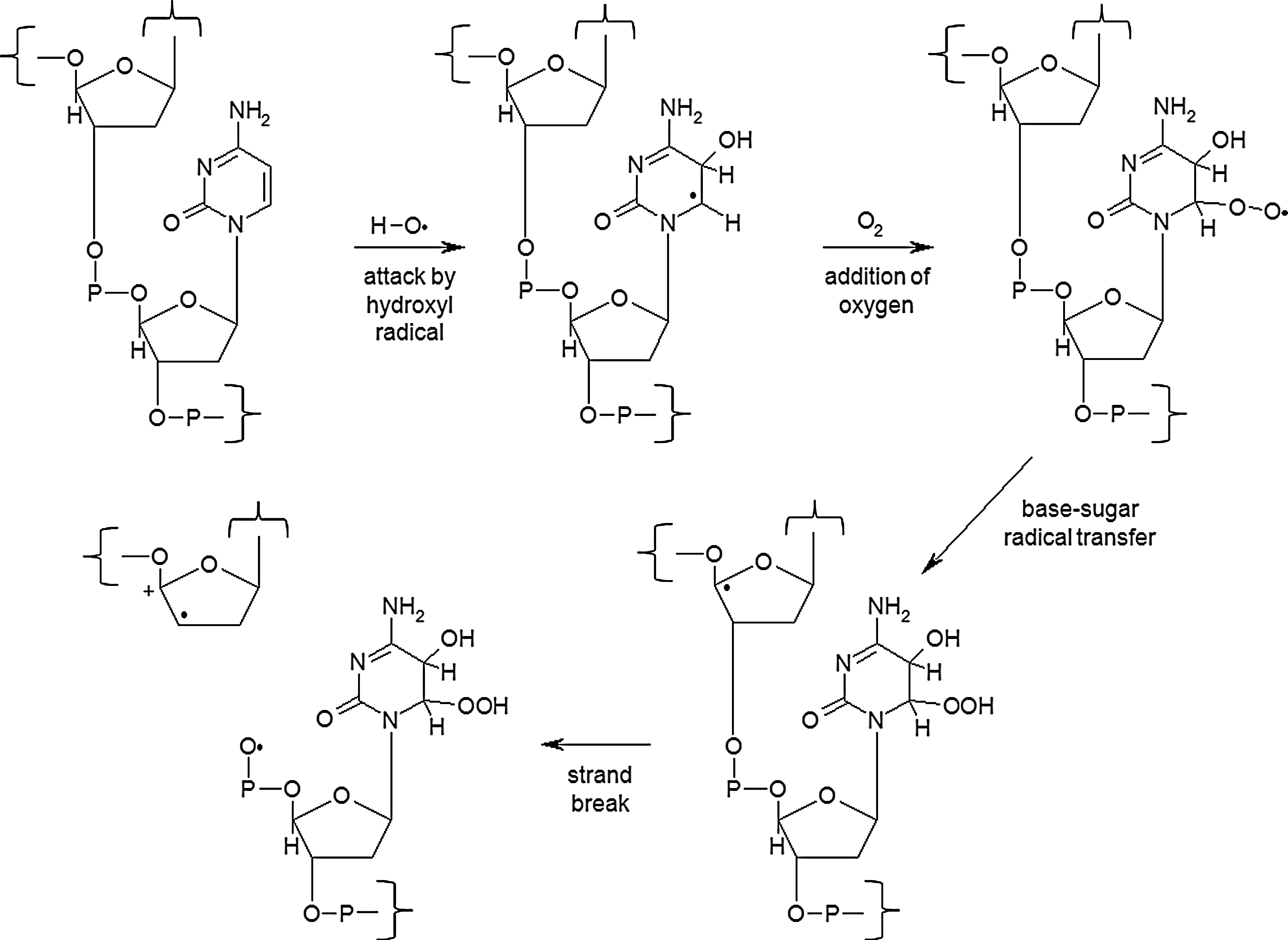
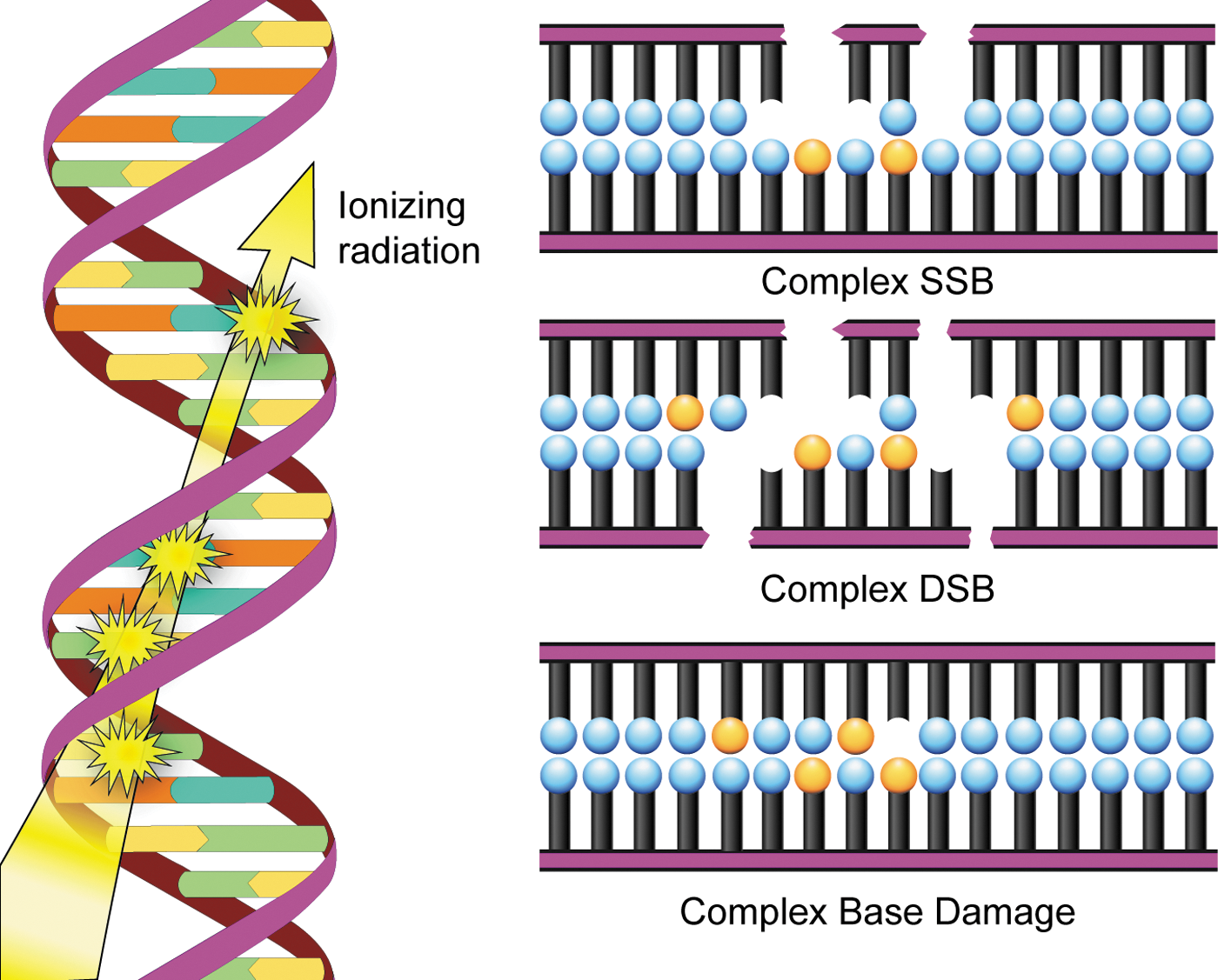
Another important type of radiation-induced DNA damage identified is referred to as clustered damage, where two or more closely spaced types of damage occur, such as abasic sites, oxidized bases, and tandem lesions, contributing to the formation of double-strand breaks (77, 78, 198, 213) (Fig. 5). This clustered damage is thought to have significant biological consequences because it is much more difficult to repair. Clustered damage is longer-lived, increasing the likelihood of incorporation of an inappropriate base, resulting in mutation formation [reviewed in ref. (152)]. Unrepaired clustered damage also leads to a multitude of chromosomal aberrations (5), and attempts to repair clustered damage can result in further double-strand break formation (191). Not only does clustered damage form from the direct interaction of ionizing radiation with DNA, it can also be induced by ROS, including hydroxyl radicals (180).
Although radiation therapy has proven valuable in the treatment of cancer, it can also have detrimental side effects. Tissues most susceptible to ionizing radiation are those with a high replication rate and are least differentiated (1). This characteristic makes cancer cells, with their high proliferation rate, more prone to radiation-induced damage than normal tissues (12), but can also affect normal tissues (1). One example of the detrimental effects of ionizing radiation is the formation of secondary cancers. Diallo et al. studied the frequency of secondary cancer formation in relation to irradiated volume in patients with childhood cancer that received radiation therapy. The researchers found that the majority of secondary tumors (66%) formed in the region bordering planning target volume, and a small percentage of tumors formed in a region >5 cm from the irradiated volume (22%) or within the irradiated volume (12%) (46). Therefore, methods that increase the radiosensitivity of cancer tissues and simultaneously increase the radioresistance of normal tissue are needed.
Radiation-Induced Bystander Effect
Description
The bystander effect is defined as an induction of some biological effect in cells that have not been directly traversed by radiation but are in close proximity to a cell that has received radiation (87). Radiation-mediated changes in unirradiated cells can be divided into three classifications: bystander effects, abscopal effects, and cohort effects. Bystander effects occur in cells in an irradiated tissue that are not directly bombarded by radiation. Abscopal effects occur to unirradiated cells outside the irradiated volume. The cohort effect is an indirect effect within an irradiated cell due to some signal received by another irradiated cell within the same irradiated tissue (17) (Fig. 6). Several biological endpoints have been used to prove the presence of radiation-induced bystander effect, including micronuclei formation, clonogenic survival, mutation formation, apoptosis, and neoplastic transformation, among others [reviewed in refs. (34) and (89)]. Another important consequence of the bystander effect is increased radioresistance in nonirradiated tissue. For example, the exposure of normal human lung fibroblasts (NHLFs) to medium from HLFs irradiated with 1 cGy γ-rays increased clonogenic survival after exposure to 10 and 19 cGy radiation. The increased radioresistance in bystander cells correlated with an increase in the expression of AP-endonuclease (102).
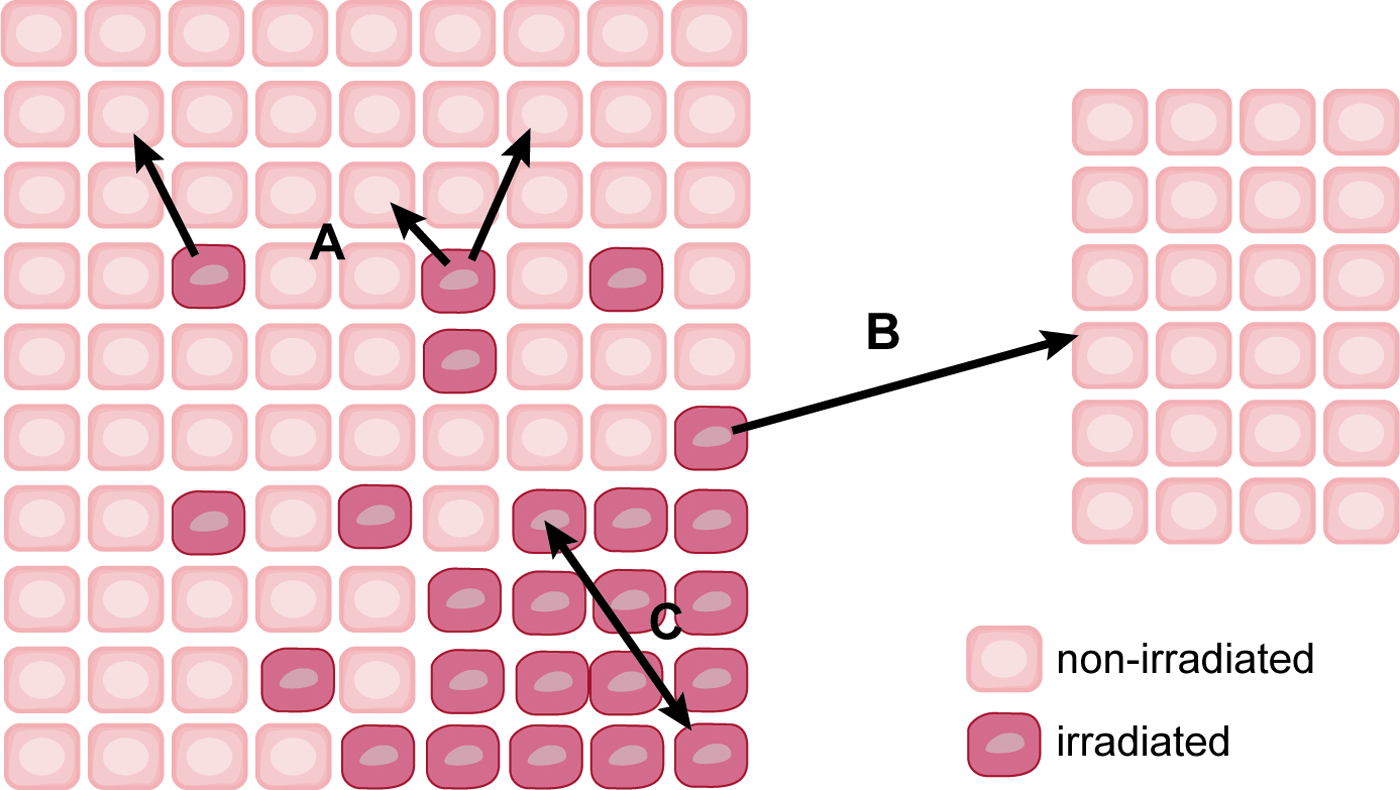
One of the first reports of a radiation-induced bystander effect was a study by Nagasawa and Little (145), in which the researchers reported the induction of sister chromatid exchanges in Chinese hamster ovary (CHO) cell cultures exposed to very low mean doses of α-particles (0.31 mGy). While less than 1% of the cell nuclei were struck by an α-particle, 30% of the cells had increased sister chromatid exchange. Irradiation with X-rays (65, 112, 226) and low LET β-particles (163) also produced stressful bystander effects.
Irradiated cells can induce a bystander effect long after the irradiation event. Lyng et al. (129) found that medium collected from progeny cells of γ-irradiated HPV-G human keratinocytes up to passage 7 after irradiation induced rapid calcium fluxes, loss of mitochondrial membrane potential, and increased ROS production in bystander HPV-G cells. No differences were observed in the bystander effect between medium collected from irradiated cells or from the cells passaged from the irradiated cells (129). A better understanding of the mechanisms of the bystander effect can lead to improvements in cancer therapy that mitigate harmful effects of radiation on nontarget tissue.
Mechanisms of the bystander effect
Direct intercellular communication by junctional channels.
Microbeam technology has proven invaluable for study of the bystander effect, allowing for the delivery of a specific dose of radiation to an individual cell or groups of cells, leaving surrounding cells untouched [reviewed in ref. (170)]. Alpha particle irradiation of 20% of AL human hamster hybrid cells in a microbeam dish resulted in a threefold higher mutation fraction than predicted due to the formation of mutations in nonirradiated cells. When cells were pretreated with the gap junction intercellular communication (GJIC) inhibitor lindane, there was a significant reduction in mutation formation, indicating the importance of cell–cell communication in the induction of the bystander effect (231). Further studies revealed that GJIC is vital for propagating signals involved in the bystander effect. Inhibition of gap junction communication by chemical inhibitors or by expression of a dominant-negative form of connexin-43 (Cx43) resulted in diminution of bystander effect-induced mutation frequency (162, 233). Using various fibroblast and epithelial cell lines, Azzam et al. (8) demonstrated the importance of GJIC in the bystander effect. The researchers found that treatment of the cells with α-particles resulted in the induction of p21Waf1, and pretreatment with the GJIC inhibitor lindane, use of cells that are GJIC incompetent or use of Cx43 knockout cells, resulted in diminished radiation-induced p21Waf1 levels (8).
GJIC is also important in potentiating the cell-killing effects of ionizing radiation due to the propagation of stress signals throughout the irradiated volume. Little effect is seen with low LET radiation, but a significant enhancement is observed for high LET radiation. Using confluent cultures of AG1522 human diploid fibroblasts, Autsavapromporn et al. (6) discovered that incubation of the cells for 3 h after irradiation with α-particles resulted in decreased survival and increased DNA damage compared to cells irradiated with γ-rays. This effect was due to GJIC and was attenuated by pretreatment with the GJIC inhibitor AGA or knockdown of Cx43 by siRNA (7). A similar enhancement of cell killing and DNA damage was observed for high-charge high-energy iron ions compared to protons (6).
Intercellular communication via release of soluble factors
Numerous studies have found that conditioned medium obtained from irradiated cells induces a bystander effect in unirradiated cells, suggesting soluble factors released by irradiated cells are involved in the bystander effect. For example, Mothersill and Seymour (141) found that treatment of various epithelial cell lines with the medium from γ-ray-treated cells led to a decrease in clonogenic survival in both a radiation dose- and irradiated cell number-dependent manner. This effect was only seen in the medium from irradiated cells, and not irradiated medium in the absence of cells (141). Similar results have been reported in various cell types using different methods, including medium transfer (123, 159), double mylar dishes (which allow exchange of medium between irradiated and nonirradiated cells) (232), and microbeam treatment (14, 169), leading to increased apoptosis (14, 123, 159, 169), ROS formation (159), as well as micronuclei formation (14, 169).
Extracellular DNA (ecDNA) has recently been identified as an important soluble factor that stimulates the bystander effect. Ermakov et al. (60) discovered that release of ecDNA from X-ray-treated primary human lymphocytes induced a bystander effect in nonirradiated lymphocytes. Both the release of ecDNA and induction of the bystander effect were blocked by the administration of the antioxidant α-tocopherol. Based on these results, the researchers suggested that ROS are important for both the response to radiation, leading to apoptosis and release of ecDNA, and induction of DNA damage in bystander cells (60). Similar results have been observed in X-ray-treated human umbilical vein endothelial cells (HUVEC) (61, 118). The authors proposed that ecDNA is released due to ROS-mediated apoptosis induced by ionizing radiation. DNA-binding receptors (TLR9) on bystander cells detect these DNA fragments, stimulating ROS production and DNA damage. Inhibition of TLR9 by treatment with an oligonucleotide inhibitor or chloroquine blocked the bystander effect, whereas treatment of unirradiated cells with ecDNA isolated from irradiated cell medium stimulated the bystander effect (61, 118). These results suggest that scavenging of ROS may be a valuable strategy for assuaging the bystander effect.
Altered gene expression and signal transduction
Induction of the bystander effect leads to the activation of various signal transduction pathways and different transcription factors, resulting in changes in gene expression, culminating in the biological endpoints that characterize the bystander effect. Two important genes identified in the bystander effect are cyclooxygenase-2 (COX-2) and insulin growth factor binding protein 3 (IGFBP-3). Zhou et al. (229) using NHLFs found that α-particle irradiation resulted in a bystander effect by a reduction in the surviving fraction and an increased mutagenesis rate. Using a signal transduction gene array, the researchers identified a significant increase in the expression of COX-2 gene and a decrease in IGFBP-3 gene expression. Co-treatment of NHLFs with NS-398 (a COX-2 inhibitor) reduced the mutagenesis rate in bystander cells, and addition of IGFBP-3 increased survival and decreased the mutagenesis rate in bystander cells. Treatment of the cells with either the MAP kinase inhibitor PD98059 or an anti-tumor necrosis factor-alpha (TNF-α) antibody significantly blocked the bystander effect (229).
Bystander effect in vivo
While a multitude of studies demonstrate an in vitro bystander effect, the bystander effect has also been confirmed in vivo in various model systems, such as Caenorhabditis elegans, zebra fish, and Arabidpopsis thaliana [reviewed in ref. (30)]. Studies of partial lung γ-ray treatment in rats revealed that the lower lung is much more sensitive to the in-field effects of radiation (as determined by micronuclei formation) than the upper lung, whereas the upper lung demonstrated greater response to bystander effects after lower lung irradiation. Copper-zinc superoxide dismutase (CuZnSOD), manganese superoxide dismutase (MnSOD), or the nitric oxide synthase inhibitor nitro-
Bystander effect in neoplastic transformation
Not only can the bystander effect result in cellular damage and death in nonirradiated cells, it can also induce neoplastic transformation in nonirradiated cells. Exposure of CGL1 HeLa skin-fibroblast hybrid cells to medium collected from X-ray-treated HeLa-skin fibroblast hybrid (CGL1) cells significantly increased the neoplastic transformation frequency (123). A similar increase in neoplastic transformation has been observed in JB6 mouse epithelial cells co-cultured with JB6 cells treated with γ-rays (216). Bystander effect-mediated neoplastic transformation has also been observed in vivo. Mancuso et al., using Patched-1 (Ptch1) heterozygous knockout mice (Ptch1 +/−), discovered that medulloblastoma formation was significantly greater in Ptch1 +/− mice with their heads shielded (but the remainder of their body receiving radiation treatment) compared to untreated mice but was not as great as mice receiving whole-body radiation (134). A later study investigated the role of GJIC on propagation of bystander effect signaling in vivo and its role in radiation-associated bystander effect cancer formation in a Cx43 heterozygous knockout mouse (Cx43+/− ) model. Radiation-induced medulloblastoma formation was significantly lower in shielded Cx43+/− /Ptch+/− mice compared to shielded Cx43+/+/Ptch+/− mice, confirming the role for GJIC in bystander effect-mediated neoplastic transformation (133).
The bystander effect may also contribute to neoplastic transformation in humans. In a study by Diallo et al. (46) investigating the formation of secondary cancer formation in childhood cancer patients who received radiation therapy, there was a substantially greater number of secondary cancers that developed at distant sites from the irradiated volume (25 neoplasms) than what was predicted (nine neoplasms). In this study, the authors defined distant as any area farther than 5 cm outward from the edge of the irradiated volume (46). Given the effects of the bystander effect on neoplastic transformation discussed above, these results suggest that an important bystander effect may also be occurring in humans that may contribute to the secondary cancer formation after radiation treatment. Therefore, adjuvant therapies that can mitigate these unwanted effects of radiation therapy would prove valuable in the clinic.
Although irradiated cells can communicate to unirradiated cells through the bystander effect, bystander cells can communicate back to the irradiated cells. Chen et al. found that incubation of α-particle-irradiated NHLFs with bystander NHLFs significantly reduced 53BP1 foci formation (a marker of double-strand breaks), as well as diminished micronuclei formation and apoptosis, in the irradiated cells compared to irradiated cells not incubated with bystander cells (36). Similar reductions in micronuclei formation have been observed in α-particle-irradiated HeLa incubated with NHLFs (36) and incubation of X-irradiated Me45 human melanoma cells with normal human dermal fibroblasts (219), and the reduction in micronuclei formation corresponded with reduced ROS formation in the irradiated cells (219). These reports suggest that the bystander effect and reciprocal communication from bystander cells back to irradiated cells is an important component in the complex response of an organism to ionizing radiation. This type of communication may have important implications in radiation resistance in cancer cells and may provide an important target to increase radiosensitivity in cancer, especially within the context of therapies that exploit oxidative stress.
ROS in the bystander effect
ROS are important mediators of the bystander effect by myriad mechanisms. Incubation of bystander cells with the conditioned medium from γ-ray-treated human keratinocytes in the presence of the antioxidant N-acetylcysteine (NAC) or the caspase-9 inhibitor Z-LEHD-FMK abrogated the effects of the conditioned medium on bystander keratinocytes (130). Increased lipid oxidation after irradiation with γ-rays (as measured by malondialdehyde formation) has also been observed in both irradiated and bystander Me45 cells and correlated with a significant decrease in the MnSOD activity (171). ROS formation was associated with micronuclei formation and γ-H2AX foci formation in bystander AG01522 normal human fibroblasts, which was inhibited by the addition of either CuZnSOD or catalase (final concentration 500 U/ml and 8×103 U/ml, respectively) directly into the culture medium immediately after irradiation (226). Similar results have been reported in human peripheral blood lymphocytes (13). In HaCaT human keratinocytes, viability of bystander cells was rescued by incubation of either the bystander cells or the γ-ray-treated cells with SOD or catalase (added directly to the cell culture medium), suggesting ROS are both a source of bystander signals and carry out the bystander effect in recipient cells (127). ROS also initiate the bystander effect by stimulating ecDNA release from irradiated cells and response of bystander cells (see discussion above). Therefore, methods that increase ROS scavenging ability in cells may simultaneously abrogate both the generation of bystander signals and the response to bystander signals.
Mitochondria and the bystander effect
Mitochondria are the primary source of ROS in the cell due to oxygen metabolism through oxidative phosphorylation (22, 122, 144), with complexes I (83, 200) and III (205) as major contributors of superoxide produced by mitochondria. Because mitochondria produce ROS, and the role of ROS in propagation of the bystander effect (discussed above), logic dictates that mitochondria may be important in the bystander effect. Rajendran et al. (178) used various human lymphoblastoid cell lines treated with X-rays to study the effects of mitochondrial DNA (mtDNA) mutations on the bystander effect. The researchers found that medium from irradiated cell lines with normal mtDNA stimulated a bystander effect (micronucleus formation) in the same cell line or other cell lines with normal mtDNA, but not in cell lines harboring mtDNA mutations. Medium from irradiated cell lines with mtDNA mutations was unable to stimulate a bystander effect in either normal cell lines or cell lines with mtDNA mutations. These results suggest that functioning mitochondria are vital for both generation and reception of bystander signals (178).
Mitochondria as sources of the bystander effect
Studies using AL human-hamster hybrid cells with either normal mtDNA (ρ+) or depleted of mtDNA (ρ0) have established the contribution of mitochondria in generation of bystander signals from irradiated cells. Either depletion of mtDNA or inhibition of the electron transport chain, as well as inhibition of NO production or calcium uptake, significantly reduced γ-H2AX foci, a marker of DNA damage, in bystander cells (37). A reduction in the bystander effect has also been observed in AG1522 cells incubated with the medium from α-particle-irradiated ρ0 AL cells (38). Depletion of mtDNA had similar results in α-particle-irradiated HeLa cells (202) and human skin fibroblasts (230).
Mitochondria as targets of the bystander effect
Not only are mitochondria important initiators of the bystander effect, they are also recipients of bystander signals. For example, treatment of human keratinocytes with the medium from keratinocytes treated with γ-rays resulted in rapid calcium flux, increased ROS production, and decreased mitochondrial membrane potential, as well as increased apoptosis and decreased clonogenic survival (128). Direct irradiation of Chinese hamster ovary K1 cells or HPV-G cells, or exposure of these cells to irradiated cell-conditioned medium, significantly altered mitochondrial oxygen consumption and increased mitochondrial mass (151), as well as decreased expression of mtDNA-associated genes (150).
mtDNA is an important target of the bystander effect. Several reports indicate that mtDNA mutations (79) and deletions (79, 143, 185) occur upon exposure to the conditioned medium from irradiated cells, with these mutations correlating with a decrease in mitochondrial membrane potential (79). The bystander effect can also affect mtDNA gene expression (35, 150). For instance, Chaudhry and Omaruddin (35) found that in both X-irradiated and bystander TK6 human lymphoblast cells, there was a statistically significant change in the expression of various mtDNA encoded genes for different components of the electron transport chain. MT-ND1, MT-ND5, and MT-ND6 (components of NADH dehydrogenase) were upregulated in directly-irradiated cells but were downregulated in bystander cells, whereas MT-ATP6 and MT-ATP8 were upregulated in both cell types (35).
Although cytochrome c (cytc) does not affect the direct response of cells to ionizing radiation or the generation of bystander signals, it has a significant impact on response to bystander signals from irradiated cells. Co-culture of mouse embryonic fibroblasts (MEFs) that are cytc-null (cytc −/−) with α-particle-irradiated cytc −/− or cytc +/+ cells resulted in no significant induction of micronuclei formation in cytc −/− bystander cells. The conditioned medium from irradiated cytc +/+ or cytc −/− cells stimulated ROS production in cytc +/+ bystander cells, but not cytc −/− bystander cells (225).
Genomic DNA damage occurs in bystander cells as a result of signaling events modulated by mitochondria. Telomere shortening and anaphase bridge formation caused by incubation of bystander SW480 cells with the medium from explanted tumor tissue exposed to γ-rays correlated with a decrease in mitochondrial membrane potential and an increase in ROS formation. Overexpression of MnSOD in the SW480 cells not only diminished the effects of the conditioned medium on mitochondrial membrane potential and ROS formation, but also significantly reduced the effects of the conditioned medium on telomere shortening and anaphase bridge formation. These results suggest that MnSOD may be important for inhibition of the bystander effect in nontarget cells (80).
Tumor Architecture and Its Effects on Radiation Medicine
Tumors as organs
Long thought to be only a clonal expansion of mutated cells, tumors are actually complex mixtures of various cell types organized as abnormal organs. A variety of cells have been identified in tumors, many of which contribute to tumor progression, including adipocytes, fibroblasts, endothelial cells, and myriad immune cells [reviewed in ref. (49)]. During tumor progression, changes in the interactions between the cancer cells and supporting cells, including cancer stem cells [recently reviewed in ref. (26)], as well as the differences in the interaction between cancer cells with extracellular matrix (and the composition of the extracellular matrix) occur, which can contribute to radiation resistance in tumors. For example, Josson et al. (107) found that coculture of ARCaPE (with an epithelial morphology and phenotype) human prostate cancer cells with either bone fibroblasts or stromal fibroblasts isolated from normal or cancer-bearing prostate tissue significantly enhanced radioresistance of ARCaPE cells. This radioprotective effect was mediated by both E-cadherin and integrin signaling. Interestingly, coculture of ARCaPM (with a mesenchymal morphology and phenotype) human prostate cancer cells with bone or prostate fibroblasts did not affect radioresistance (107), indicating an important role for phenotype on cancer cell responsiveness. Coculture of Suit-2 human pancreatic cancer cells with γ-ray-treated MRC5 human fibroblast cells or primary pancreatic fibroblasts significantly enhanced invasiveness both in vitro and in vivo. Enhancement of Suit-2 invasiveness and cell scattering was induced by incubation with the conditioned medium from irradiated MRC5, coinciding with an increase in p44/p42 MAPK and c-Met activation, and was blocked by pretreatment with the hepatocyte growth factor inhibitor NK4 (156). In a study by Tsai et al., (206) coculture of MCF-7 or MDA-MB-231 human breast cancer cells with senescent primary human mammary fibroblasts (induced by X-ray treatment) significantly enhanced cell growth due to the stimulation of the expression of mitotic genes. Senescent fibroblasts, as well as senescent fibroblast-conditioned medium, also conferred radioresistance to the breast cancer cells, which was partially attenuated by expression of dominant-negative AKT in the breast cancer cells (206). These results demonstrate the importance of supporting cells for the response of cancer cells to ionizing radiation. Because of the role of soluble factors secreted by the supporting cells in conferring radioprotection and stimulation of aggressiveness of cancer cells, the bystander effect may be vital contributor of this process. Methods that target the bystander effect between cancer cells and the supporting cells within the tumor may prove attractive for enhancing the effectiveness of radiation therapy.
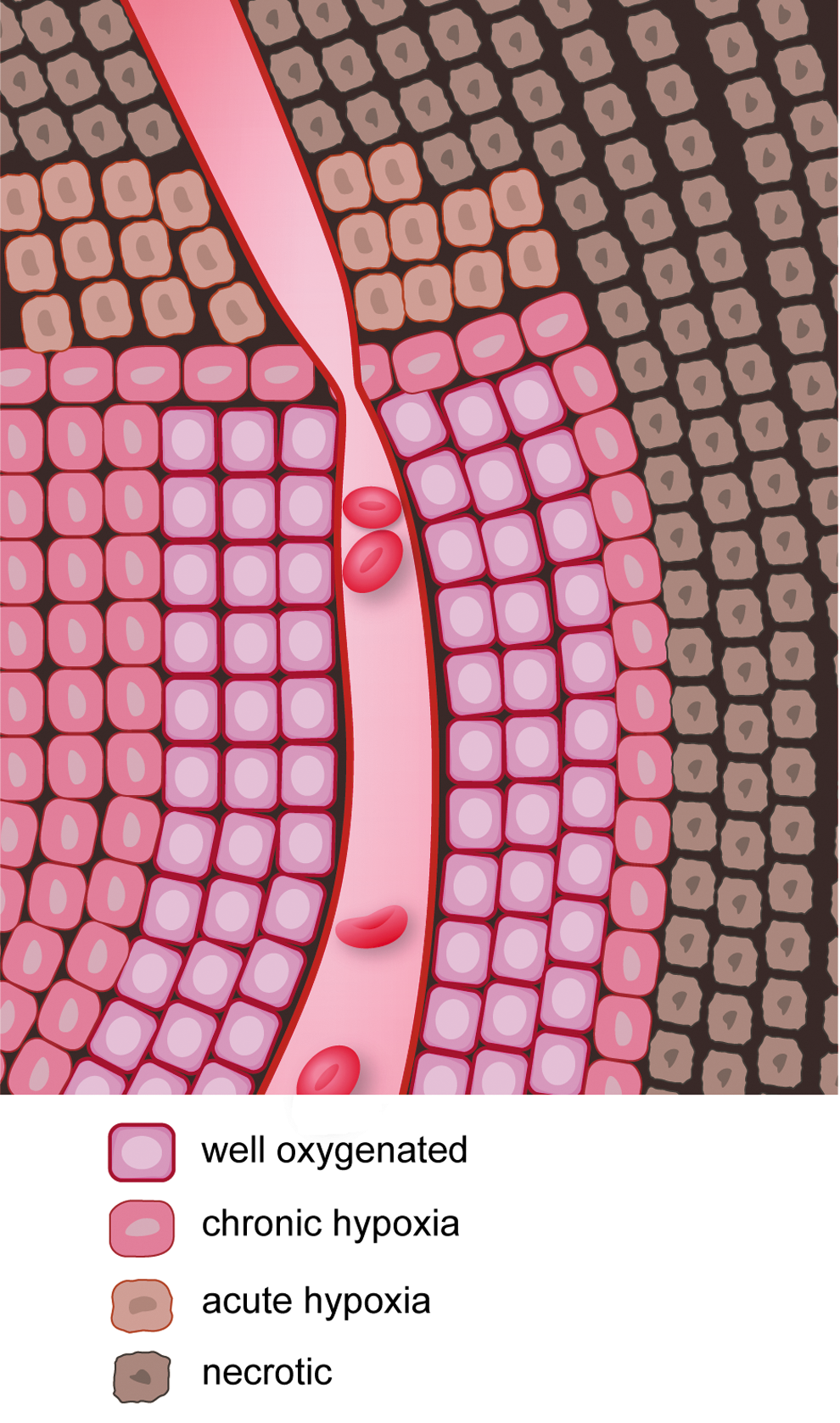
Tumor hypoxia and radiation treatment
Given the importance of oxygen in the response of cells to radiation (23, 82, 106), hypoxia becomes a major problem in the treatment of tumors by radiation. Two types of hypoxia exist in tumors: chronic and acute (Fig. 7). Chronic hypoxia occurs due to the diffusion limits of oxygen through tissues, whereas acute hypoxia occurs due to temporary blockage of a tumor blood vessel (25). Chronic hypoxia was first identified by Deschner and Gray, who found that oxygen supply to cells within a tumor is limited to a region ∼150 μm from the vasculature (44). Acute hypoxia was first identified by Brown (24) and can have important implications in the response of tumors to radiation (32). Several reports show that radiosensitivity of cancer cells is inversely proportional to the distance from the blood supply (142, 201). Chaplin et al. discovered that tumor cells closest to the blood vessels of the tumor were most sensitive to ionizing radiation (as measured by clonogenic survival), whereas the tumor cells farthest from the blood vessel were most resistant to ionizing radiation. Ionizing radiation resulted in acute hypoxia in the tumor due to transient changes in blood flow, and this acute hypoxia increased radioresistance in the tumor (33). Hsieh et al. compared the influence of either cycling hypoxia or uninterrupted hypoxia on radiation resistance of U87 glioma cells and found that cycling hypoxia greatly increased U87 glioma cell radiation resistance compared to uninterrupted hypoxia due to increased ROS production, leading to greater stabilization, expression, and transcriptional activity of hypoxia-inducible factor-1α (HIF-1α). Suppression of HIF-1α induction by siRNA resulted in radiosensitization of U87 cells both in vitro and in vivo (98).
Because hypoxia diminishes the effectiveness of radiation therapy, several methods have been devised to increase oxygen concentrations at the tumor. These methods include increasing blood oxygen saturation, diminishing tumor oxygen consumption (106), and normalization of tumor vasculature (76). Secomb et al. (186) studied the effects of blood flow, arterial pO2, and oxygen consumption on tumor cell hypoxia using a simulation based on observations from a transplanted mammary adenocarcinoma. While hypoxia was reduced with increasing blood flow and arterial pO2 and decreased oxygen consumption, tumors were much more sensitive to changes in oxygen consumption, with a 30% reduction in oxygen consumption, compared to controls, completely eliminating tumor hypoxia (186). McGee et al. (136) studied the effects of interferon-β (IFN-β) or the monoclonal human vascular endothelial growth factor (VEGF) antibody bevacizumab, on the radiation response of orthotopic U87 glioma xenografts. IFN-β or bevacizumab decreased tumor hypoxia by normalization of tumor vasculature. Combination of radiation with either IFN-β or bevacizumab synergistically reduced tumor size compared to any individual therapy alone, demonstrating the importance of tumor oxygenation by normalization of vasculature in radiation therapy (136).
Another important method to treat tumors that can affect tumor oxygenation is fractionated radiotherapy. By applying radiation in a fractionated regimen, normal tissues can recover through repair of sublethal damage and repopulation of normal cells (10). Fractionated therapy takes advantage of the increased proliferation rate in cancer cells compared to normal tissues (15). Fractionation, especially hyperfractionation, permits a higher total radiation dose to be delivered. Fractionated therapy also allows for the reoxygenation of tumors and increases their radiosensitivity (Fig. 8) (137). The combination of radiation with chemotherapy postradiotherapy can enhance drug delivery and improve patient outcome.

SODs in Normal and Tumor Tissues
General description of SODs
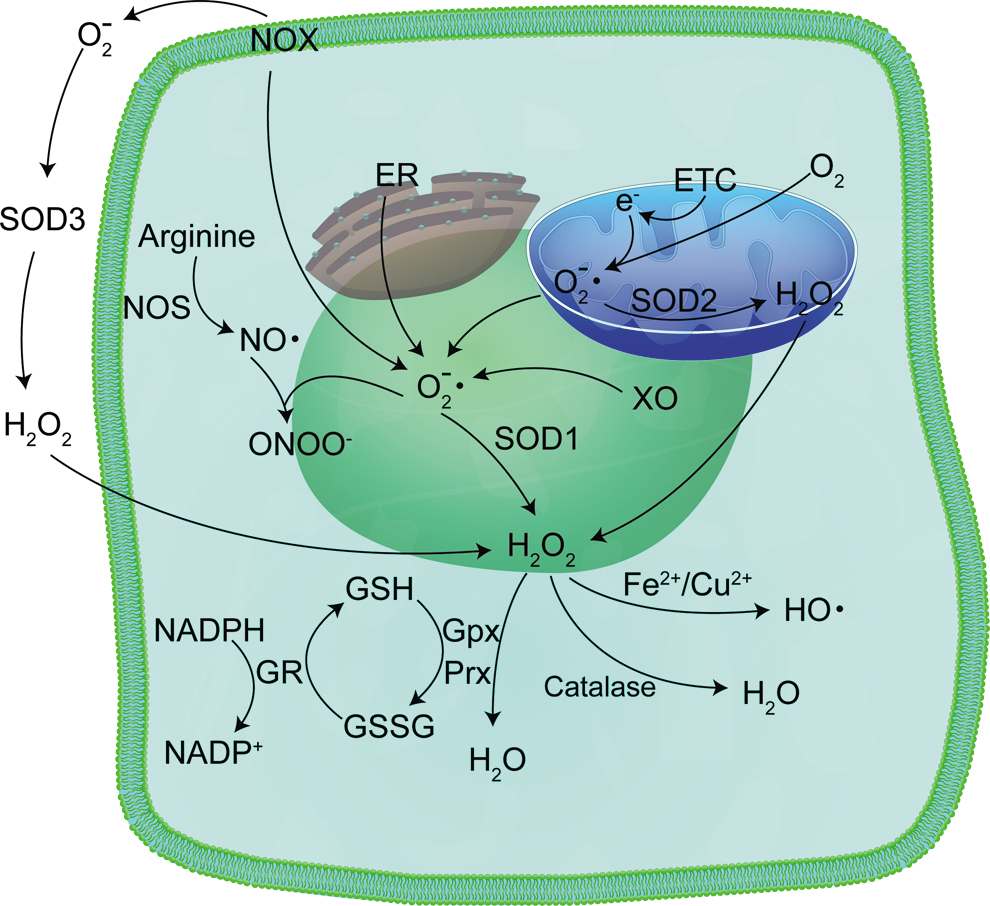
Because SODs are discussed in much greater detail in other review articles in this Forum, we will only briefly describe the SODs. SODs are the chief superoxide-scavenging enzymes in a cell (71), and three types exist in cells, with each form encoded by a separate gene [reviewed in ref. (228)]: CuZnSOD (SOD1), MnSOD (SOD2), and extracellular superoxide dismutase (SOD3, ECSOD). CuZnSOD is a cytoplasmic homodimer (113, 135), although small amounts have been identified in the intermembrane space of mitochondria (157, 217). ECSOD shares 40–60% amino acid homology with CuZnSOD, and like CuZnSOD, contains both copper and zinc in its active site. However, ECSOD is a homotetramer found in the extracellular region of the cell (68, 92). MnSOD is a homotetramer found exclusively in the mitochondrial matrix (19, 157, 179, 217, 220). All three enzymes catalyze the dismutation of superoxide radicals to hydrogen peroxide and molecular oxygen. Hydrogen peroxide, itself a ROS, is decomposed to water by myriad enzyme systems, including peroxiredoxins, glutathione peroxidase (GPx), and catalase (4). All three enzymes have forms localized to mitochondria (31, 62, 131, 155, 160, 177, 183, 188, 190) (Fig. 9).
SOD in cancer development
Given the role of ionizing radiation in the development of cancer (46) and the significance of ROS in the development and progression of cancer (75, 209, 215), the importance of MnSOD in cancer development becomes quite evident due to its ability to scavenge ROS. However, a careful examination of the literature reveals a dual function for MnSOD in cancer [recently reviewed in ref. (94)]. Many studies show a reduction in MnSOD expression in various types of cancer compared to normal tissues (39, 42, 100, 153, 192), suggesting MnSOD acts a tumor suppressor. Conversely, other studies report an elevation in MnSOD expression in cancer (93, 99, 103, 104, 132, 203, 207) and its association with cancer aggressiveness, growth, survival (140, 158), and metastatic potential (95, 132, 149, 184), implying that MnSOD supports progression of tumors to a more aggressive stage.
Recent work by this laboratory has begun to shed light on the dual role of MnSOD in cancer development. Using the 7,12-dimethylbenz(a)-anthracene (DMBA)/12-O-tetradecanoylphorbol-13-acetate (TPA) two-stage model of skin cancer development in a unique mouse model expressing a MnSOD promoter-linked luciferase reporter gene (45), this laboratory discovered that treatment with the tumor initiator DMBA, followed by repeated exposure to TPA over 25 weeks, resulted in a significant reduction in MnSOD luciferase reporter gene activity, MnSOD mRNA, protein, and enzyme activity in both DMBA/TPA-treated skin and papillomas compared to vehicle controls. When the observation period was extended to allow squamous cell carcinoma (SCC) formation, MnSOD expression significantly increased at the reporter gene activity, mRNA, protein, and enzyme activity levels during the transition from the relatively nonaggressive papilloma to the more aggressive SCC. These differences in MnSOD expression between papillomas and SCCs are due, in part, to changes in Sp1 and p53 transcription factor binding activity on the Sod2 promoter (45).
One potential mechanism by which MnSOD is involved in tumor suppression and tumor aggressiveness is altered hydrogen peroxide flux with changes in MnSOD levels. Buettner et al. (27) showed that steady-state levels of hydrogen peroxide are affected by MnSOD where the equilibrium constant for superoxide production (K) is less than 1. Under these conditions, the rate constant for the back reaction (conversion of superoxide back to molecular oxygen) is greater than the rate constant for the forward reaction (conversion of molecular oxygen to superoxide). This condition is seen for superoxide production by the electron transport chain. When MnSOD is present, there is a proportional increase in hydrogen peroxide production with increasing MnSOD levels. The greatest effect occurs when MnSOD levels are low, but at sufficiently high levels of MnSOD, there is only a modest further increase in hydrogen peroxide production. Increased hydrogen peroxide production occurs because MnSOD is driving the equilibrium of the system to the right, resulting in increased superoxide production. A small amount of superoxide is consumed for the production of hydrogen peroxide and does not participate in the reverse reaction to regenerate molecular oxygen, in agreement with Le Chatelier's principle (27).
Changes in hydrogen peroxide flux with modifications in MnSOD levels can have broad repercussions in cancer progression. During early stages of cancer, when MnSOD levels are low (153), MnSOD overexpression may suppress cancer growth through various mechanisms because of greater hydrogen peroxide flux (126). At later stages of cancer progression, when cancer cells experience persistent oxidative stress (16, 161, 168), increased MnSOD expression may benefit cancer cells by the stimulation of metastasis (40, 90, 95, 147).
SOD in radiation-induced neoplastic transformation
Two important papers established the importance of SODs in radiation-induced neoplastic transformation of normal tissues. This laboratory was the first to demonstrate the protective effects of MnSOD overexpression in the protection of normal tissue from ionizing radiation-induced neoplastic transformation. In C3H 10T1/2 MEFs expressing either an empty vector or a construct containing the Sod2 gene, St. Clair et al. discovered that overexpression of MnSOD did not protect the cells from transformation by the DNA intercalating agent 3-methylcholanthrene but did protect the cells from transformation by ionizing radiation (194). Du et al., (48) using MEFs derived from MnSOD+/+, MnSOD+/−, and MnSOD−/− mice, found a fivefold increase in transformation frequency in MnSOD−/− MEFs compared to MnSOD+/+ MEFs. The lack of MnSOD in MnSOD−/− MEFs enhanced the formation of late ROS, micronuclei (a marker of DNA damage), and binucleated bearing micronuclei cells at 72 h post-irradiation compared to MnSOD+/− and MnSOD+/+ MEFs. MnSOD+/− and MnSOD−/− MEFs also demonstrated decreased exit from the G2 cell cycle checkpoint compared to MnSOD+/+ MEFs, which was reversed in MnSOD−/− cells by overexpression of MnSOD (48). These results suggest that mitochondrial ROS generation is an important component of ionizing radiation-mediated transformation and the vital role for MnSOD for the prevention of cancer formation.
SOD-mediated radioprotection
Many studies have revealed the importance of SOD in protection of normal tissue from the harmful effects of ionizing radiation. Lee et al. (121) used a yeast model with knockdown of MnSOD, CuZnSOD, or both MnSOD and CuZnSOD, to study radiation response. The researchers found that in wild-type cells, there was a dose-dependent increase in the activity of different antioxidant enzymes (catalase, glutathione reductase, and glucose 6-phosphate dehydrogenase), whereas knockdown of either SOD alone or the combination of MnSOD and CuZnSOD resulted in a much smaller increase in the activities of these enzymes. The researchers also found a radiation dose-dependent increase in the levels of ROS and ROS-mediated protein and lipid damage (protein carbonyl formation and thiobarbituric acid-reactive substances, respectively) (121).
Overexpression of MnSOD alone is enough to bestow radioprotection in normal cells. In CHO cells, overexpression of MnSOD, but not CuZnSOD, protected the cells from radiation-induced cell death. Overexpression of GPx was only partially protective against radiation compared to MnSOD (195). Overexpression of MnSOD by retroviral transduction in either K562 human erythroleukemic cells or primary mouse bone marrow-derived myeloid progenitor cells resulted in a significant increase in radioresistance (193). MnSOD is also important for radiation-induced adaptive response in normal cells. In JB6 mouse epidermal cells, exposure to low-dose ionizing radiation significantly increased the clonogenic survival of cells subsequently challenged to 2 Gy radiation due to activation of several NF-κB target genes, including MnSOD (63). An important mechanism by which MnSOD confers radioprotection of normal tissues is to prevent apoptosis (50, 56), in part, by maintaining mitochondrial integrity (56). These results suggest that mitochondria are major targets of ionizing radiation and mitochondria-specific scavenging of ROS by MnSOD may be an important mechanism to safeguard cells against radiation-induced damage.
CuZnSOD also has a radioprotective effect in normal tissue. Petkau et al. found that the administration of mice with bovine SOD by intravenous injection before and after ionizing radiation treatment significantly increased the LD50 dose (164, 165), and exogenous SOD protected the proliferative capacity of bone marrow stem cells from ionizing radiation, indicating a protective effect of SOD (164). The addition of CuZnSOD either before irradiation or after irradiation also had a protective effect in HUVEC, although pretreatment with CuZnSOD gave the greatest level of radioprotection. CuZnSOD pretreatment also partially attenuated the antiangiogenic effects of radiation, as determined by tube formation of HUVEC cells in vitro. The radioprotective effect of CuZnSOD leading to increased angiogenesis required the activation of the mitogen-activated protein/extracellular signal-regulated kinase1/2 pathway, and the pretreatment of HUVECs with the MEK inhibitor PD98059 suppressed the effects of CuZnSOD (204).
ECSOD, like MnSOD and CuZnSOD, confers protection from the harmful effects of ionizing radiation. Lung-specific overexpression of ECSOD in B6C3 mice resulted in a significant reduction in radiation-induced lung damage compared to wild-type mice. For example, ECSOD overexpression delayed the onset of increased breathing frequency and significantly reduced breathing rate compared to irradiated wild-type mice. ECSOD also reduced radiation-induced increases in macrophage and lymphocyte lung infiltration compared to irradiated wild-type mice, as well as decreased TGFβ activation, Smad3 expression, and Smad2/3 activation and reduced lipid peroxidation (111, 174).
While a multitude of studies show a role for SOD in radioprotection of normal tissue, other studies suggest an apparent radiosensitization effect. Fishman et al. (67) discovered that knockdown of either MnSOD or CuZnSOD in C57BL/6J mice attenuated radiation-induced reductions in neuronal cells and cells differentiated into either neurons, astrocytes, or microglia in the dentate subgranular zone of the hippocampus compared to wild-type mice. The authors suggest that one potential mechanism of the protective effect of MnSOD or CuZnSOD knockdown may be increased superoxide levels. This hypothesis was supported by experiments using xanthine or xanthine plus xanthine oxidase. Incubation of neural precursor cells with either xanthine alone or xanthine/xanthine oxidase had a protective effect against radiation-induced diminution of cell numbers. Although the authors did not address the specific mechanisms by which decreased SOD, whether MnSOD or CuZnSOD, imparted a radioprotective effect on neuronal cells, they suggested that the effect may be similar to an adaptive response for radiation, whereby low doses of ionizing radiation are protective against later, high doses of ionizing radiation (67). It would be interesting to determine whether the observed effect of SOD knockdown can occur in other normal cells and tumor cells.
SOD and Radiation Response in Neoplastic Cells
Radiosensitization
Using the MnSOD-overexpressing Fsa-II cells implanted in mice, this laboratory found that MnSOD expression resulted in a significant reduction in the radiation dose required to control one-half of the irradiated tumors compared to control mice (208). Overexpression of MnSOD by transfection with a MnSOD cDNA-expressing plasmid/liposome complex (MnSOD-PL) proved effective in increasing radiosensitivity of SCC-VII mouse SCC cells (D 0=1.244 Gy compared to 3.246 Gy for control cells), and the combination of MnSOD-PL with the EGFR inhibitor gefitinib further increased radiosensitization (D 0=0.785 Gy) (54). The administration of recombinant MnSOD (rMnSOD) was also effective in enhancing radiosensitivity of cancer cells (20).
Radioresistance
Although some studies suggest a radiosensitizing role for MnSOD, other studies show that MnSOD is important in radioresistance of cancer cells. Qu et al. (172, 173) found that the CNE1 human nasopharyngeal cell line was more radioresistant than the CNE2 cell line, which correlated with increased expression and activity of MnSOD in the CNE1 cell line. Knockdown of MnSOD in CNE1 cells decreased radioresistance (172, 173). Feng et al. generated a radioresistant CNE2 cell line (CNE2-IR) by treating parental CNE2 cells with five rounds of sublethal IR. Gene expression between CNE2 and CNE2-IR cells was compared, and there was an increase in MnSOD expression, among other genes, in the radioresistant cell line (64). MnSOD expression correlated well with radioresistance in nasopharyngeal tumors (64, 172), suggesting that MnSOD may be predictive in determining radioresistance of tumors. An important mechanism of radioresistance is cell cycle arrest at the G2 phase of the cell cycle after exposure (66, 109). Scavenging of hydrogen peroxide by catalase expression or treatment with NAC abolished MnSOD-induced radioresistance (199). Using a systems biology approach, Niciforovic et al. discovered that radioresistance was associated with a positive feed-forward cycle with hydrogen peroxide-induced elevation of MnSOD expression (148).
MnSOD is a major target gene for the NF-κB pathway (116, 117, 223). The NF-κB pathway is vital for conferring radiation resistance in cancer cells (124), and a major mechanism of NF-κB-dependent radioresistance is induction of MnSOD [reviewed in ref. (96)]. For example, Guo et al. (84) discovered that MnSOD expression was significantly increased as part of a radioadaptive response in MCF-7 human breast cancer cells. This radioresistance was replicated by stable overexpression of MnSOD, and several NF-κB target genes important for survival were expressed in both radioadapted cells and cells overexpressing MnSOD (84). Work by this laboratory has focused on the mechanisms by which the alternative NF-κB pathway increases radioresistance in prostate cancer cells. In a study by Josson et al., (108) nuclear localization of RelB (a component of the alternative NF-κB pathway) (187) was significantly higher in aggressive PC-3 human prostate cells compared to LNCaP cells and correlated with increased MnSOD expression and radioresistance (108). Inhibition of the alternative NF-κB pathway by expression of RelB-specific siRNA, overexpression of a dominant-negative p100 (108), treatment with the peptide inhibitor SN52 (to prevent nuclear translocation of RelB) (222), or administration of 1α,25-dihyroxyvitamin D3 (to inhibit RelB expression) (221), all resulted in a decrease in radiation-induced MnSOD expression and an increase in radiosensitivity in PC-3 cells.
CuZnSOD can also confer radioresistance in cancer cells. Gao et al. (72) reported that the overexpression of CuZnSOD in U118-9 human glioma cells increased radioresistance compared to vector control and parental cells. ROS accumulation occurred in parental and vector control cells starting at 2 days postirradiation and remained elevated up to 8 days after radiation. This late accumulation of ROS was suppressed in CuZnSOD-overexpressing cells. CuZnSOD overexpression also increased the accumulation of the cells at the G2 phase of the cell cycle, as well as decreased cyclin B1 expression (72).
SODs are a double-edged sword in the radiation response of cancer cells
The studies highlighted above suggest a more complicated role for SODs in the response of cancer cells to ionizing radiation. This dual role for SODs may be due to differences in the expression and/or activity of other antioxidant enzymes in various types of cancer cells. For example, well-differentiated hepatocellular carcinoma cell lines have higher activity or expression levels of catalase, glutathione reductase, CuZnSOD, and MnSOD compared to poorly differentiated cell lines (227). In patients with renal cell carcinoma, there was a significant reduction in the activity of GPx and catalase in cancer tissues compared to normal tissues (166). On the other hand, other studies have demonstrated an elevation in the expression and activity of some antioxidant enzymes in cancer cells. Increased expression of MnSOD and catalase were observed in gastric carcinoma cells compared to noncancerous cells (101), and increased MnSOD expression was associated with lymph node metastasis in gastric cancer patients (132). Overexpression of SODs in cancer cells expressing low levels of other antioxidant enzymes that remove hydrogen peroxide may have a radiosensitization effect, perhaps because of increased hydrogen peroxide levels after irradiation. Conversely, overexpression of SODs in cancer cells with high levels of hydrogen peroxide-scavenging enzymes may have a radioresistant effect because of increased dismutation of superoxide radicals generated after irradiation and the ability to handle the resultant increase in hydrogen peroxide produced.
Given the dual effect of SODs on the response of cancer cells to ionizing radiation, careful screening of the antioxidant status of tumor tissues should be considered before embarking on the use of SOD therapy in parallel with radiation therapy. This screening of tumor tissues may include expression of different antioxidant enzymes alone, or even ratios of different enzymes, as well as a comparison of antioxidant enzyme expression between tumor and normal tissues. This approach would better tailor the therapy to each patient and maximize the effectiveness of the therapy to destroy cancer tissue and mitigate normal tissue injury.
Methods Exploiting SOD for Radiation Response
Because of the detrimental effects of ionizing radiation on normal tissues, the resistance of tumor tissues to radiation therapy, and the role of ROS in these processes, methods that can increase SOD expression and/or activity may prove to be important adjuvants to radiotherapy to increase radiosensitivity in tumor tissues and protect normal tissues. An important mechanism for the effects of SOD overexpression on the simultaneous radioprotection of normal tissues and radiosensitization of tumor tissues may be diminished expression of other antioxidant enzymes, especially catalase, in most cancer tissues compared to normal tissues (9, 20, 21, 41, 154, 196, 197). These enzymes catalyze the decomposition of hydrogen peroxide to molecular oxygen and water. Cancer cells are under higher levels of oxidative stress than nontumorigenic cells (91, 153). Therefore, overexpression of SODs in cancer cells may set up a condition whereby exposure to ionizing radiation results in a greater ROS insult than in normal tissues, resulting in greater cell death and cell cycle arrest. In contrast, overexpression of SODs in normal tissues is radioprotective because of the increased antioxidant capacity in normal tissues to handle the ROS burst resulting from radiation exposure. Discussed below are several strategies that have been devised to increase SOD activity and/or expression in vivo for modulation of the radiation response in normal and tumor tissues.
Overexpression of SOD
Plasmid/liposome therapy
MnSOD-PL are useful for increasing the expression of MnSOD in tissues. This particular approach has proven effective in protecting lung tissue (29, 53, 57), the oral cavity (52, 85), and the esophagus (55, 58) from ionizing radiation-induced damage. Interestingly, this therapy also simultaneously sensitizes cancer cells to ionizing radiation. For example, Epperly et al. (50) discovered that the overexpression of MnSOD using MnSOD-PL inhibited radiation-induced GPx expression in SCC-VII murine and OC-19 human SCC cells and 3LL murine lung carcinoma cells and increased radiosensitization of SCC-VII and OC-19 cells, but did not affect the radiosensitivity of 3LL cells, in vitro. In MEFs, radiation had no effect on GPx activity, but significantly reduced reduced glutathione (GSH) levels, and the administration of MnSOD-PL significantly increased GSH levels after irradiation, suggesting a radioprotective effect on normal cells in vitro. In vivo, the administration of MnSOD-PL had no effect on radiation-induced reduction in GSH in oral cavity SCC-VII orthotopic tumor tissue, but significantly reduced GPx activity in conjunction with radiation. In adjacent normal oral mucosal tissue, MnSOD-PL protected from radiation-induced changes in GSH levels and GPx activity (59). Intraoral administration of the MnSOD-PL therapy significantly decreased the number of ulcerations 5 days after irradiation compared to irradiated control mice. Interestingly, the administration of the MnSOD-PL therapy did not protect SCC-VII tumors from the effects of radiation (85). These results suggest a potential for radioprotection of normal tissue and radiosensitization of tumor tissue by MnSOD-PL.
Virus-mediated overexpression
The use of viruses is another efficient method for overexpression of MnSOD to alter radiation response. Adenovirus-mediated overexpression of MnSOD, both in vitro (234) and in vivo (51), confers protection to lung tissue and lung epithelial cells against radiation-induced damage. For example, intratracheal injection of MnSOD adenovirus significantly decreased alveolitis, in part, by suppressing radiation-induced expression of different inflammatory cytokines (transforming growth factor-beta [TGF-β], TNF-α, and interleukin-1) (51). Retroviral transduction of a MnSOD construct in hematopoietic cells protected the cells from ionizing radiation-induced DNA damage both in vitro and in vivo (193). Intraluminal administration of a herpes simplex virus-MnSOD virus had a radioprotective effect on the small intestine (86). Transfection of urothelia (bladder) with MnSOD transgene did not protect bladders from the acute effects of radiation early after exposure (as measured by transepithelial resistance and permeabilities to water and urea) but did enhance recovery at 4 weeks after irradiation compared to controls (110). These results suggest a persistent effect of ionizing radiation on oxidative metabolism lasting for weeks after radiotherapy and the ability of SOD overexpression to ameliorate this effect.
Recombinant protein
Administration of a recombinant form of MnSOD can also be radioprotective. Borrelli et al. (20) found that treatment with rMnSOD sensitized cancer cells to ionizing radiation and protected normal cells from ionizing radiation in vitro. In vivo, intraperitoneal administration of rMnSOD after irradiation, with subsequent rMnSOD treatments for an additional 6 days, significantly diminished radiation-induced organ damage and increased survival compared to animals receiving phosphate-buffered saline only (20).
SOD mimetics
Another attractive approach to increase the SOD activity in normal tissue and cancer is the use of SOD mimetics. Several manganese porphyrin compounds, as well as non-metal-based compounds, have been developed with variable superoxide-scavenging properties, pharmacokinetics and tissue and subcellular localization (11, 138), and several of these porphyrin-based mimetics (Fig. 10) are effective in decreasing radiation-induced damage in normal tissue and radiosensitizing tumor tissue.
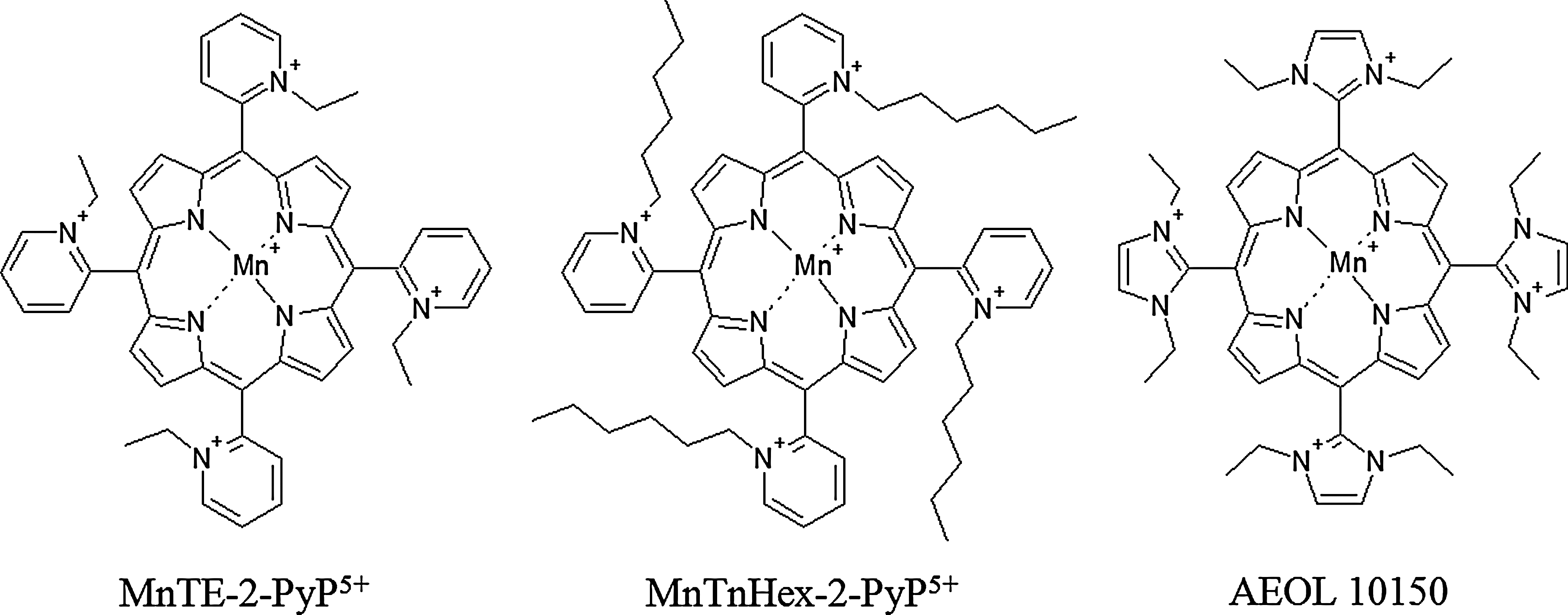
Mn(III) 5,10,15,20-tetrakis(N-ethylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) protects from bone marrow suppression induced by total body irradiation by inhibiting oxidative damage induced by ionizing radiation in hematopoietic stem cells and progenitor cells (125). MnTE-2-PyP5+ has also proven effective in delaying the onset of ionizing radiation-induced lung injury in rats, as indicated by diminished breathing frequency, reduced lung fibrosis, and less hydroxyproline content (indicating collagen deposition) compared to irradiated rats receiving no mimetic (212). MnTE-2-PyP5+ decreased oxidative stress, blocked the activation of HIF-1α and TGF-β, and suppressed the upregulation of carbonic anhydrase-IX and VEGF expression. The protective effect of MnTE-2-PyP5+ can also occur long after the irradiation event. The administration of MnTE-2-PyP5+ at 3 days and 8 weeks postirradiation, while not affecting radiation-induced oxidative DNA damage, significantly reduced histopathological damage in the lungs compared to animals not receiving mimetic (74).
While MnTE-2-PyP5+ is effective in radioprotection of normal tissues, Mn(III) 5,10,15,20-tetrakis(N-hexylpyridinium-2-yl)porphyrin (MnTnHex-2-PyP5+) has been proven to be even more efficacious than MnTE-2-PyP5+ due to its greater lipophilicity and higher biodistribution (218). At high concentrations, MnTnHex-2-PyP5+ can cause toxicity, which may be due to its micellar properties (73). Another SOD mimetic useful in suppressing radiation-induced lung damage after both single-dose (175) and fractionated radiotherapy (176) is Mn(III) tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin (AEOL 10150). Potential mechanism of diminution of radiation-induced lung damage include reduction in inflammatory cytokines in both plasma (212) and lung tissue (74), suppression of oxidative DNA and protein damage (176), as well as a decrease in radiation-induced apoptosis (224).
Not only do SOD mimetics have radioprotective effects on normal tissues, but they can also have radiosensitization effects on cancerous tissues. Moeller et al. (139) found that the SOD mimetic MnTE-2-PyP5+, at all doses tested, was not cytotoxic on the different tumor and endothelial cell lines tested and neither sensitized nor protected the cells from radiation treatment in vitro. Interestingly, MnTE-2-PyP5+ inhibited the radioprotective effects of tumor cell-conditioned medium on endothelial cells in vitro and enhanced radiation-induced damage to tumor vasculature and delayed tumor growth in vivo (139), suggesting that SOD mimetics may disrupt communication between tumor and endothelial cells, leading to breakdown of tumor vasculature after radiation and disruption of the tumor microenvironment.
Concluding Remarks
Ionizing radiation is an efficient method for the treatment of cancer. However, the development of normal tissue injury can limit the effectiveness of radiotherapy. ROS are important mediators of radiation therapy and can also participate in the differential effect of radiation in cancer cells and in normal cells due to differences in the antioxidant potential between tumor and normal tissues. Bystander effects play an important role in the response of cancerous and normal tissues to ionizing radiation, potentially leading to radioresistance of cancer cells and radiosensitization of normal cells. ROS are key mediators of radiation-induced bystander effects. Bystander effects are also important for the effects of tumor microenvironment on radiation responsiveness, with communication between tumor and stromal cells conferring radioprotection and increased aggressiveness of tumor cells. SODs, the main ROS scavengers of the cell, play a complex role in the response of cancer cells to ionizing radiation. Increasing the expression and/or activity of SODs, especially MnSOD, through the use of gene-expressing plasmid delivery systems, viral vectors, administration of recombinant protein, or the use of SOD mimetics, can increase the radiosensitivity of cancer cells while simultaneously increasing the resistance of normal tissues to radiation-induced injury in vivo. Development of these techniques for use in humans may provide unique tools for the radiation oncologist to improve treatment efficacy and enhance the quality of life for cancer patients in the future.
Footnotes
Acknowledgments
The authors thank Mr. Tom Dolan and Mr. Matt Hazzard of the Graphics and Multimedia/Academic Technology Group at the University of Kentucky for their assistance in generating the figures used in this article. This work was supported by NIH grants 2T32ES007266 and CA143428.