
Abstract
Protein and amine halogenation is a type of oxidative stress induced by phagocytic overstimulation, and its role in Parkinson's disease (PD) has not been discerned. We have detected that advanced oxidized protein products, markers of protein halogenation, are reliably enhanced in serum of patients with PD (n=60) relative to control subjects (n=45, p<0.012), and to a lesser extent in the cerebrospinal fluid. Amine halogenation, as evaluated through 3-chlorotyrosine, is not affected. Mieloperoxidase and hydrogen peroxide levels, halogenative factors of phagocytes, are devoid of changes. Levels of advanced oxidized protein products are progressively reduced over time, and the duration of PD is larger in the Hoehn-Yahr-stage-2/3 patients (n=34) with low serum levels (R 2=0.0145, p<0.003). Levodopa treatment contributes to this reduction (R 2=0.259, p<0.001). These protein products are not cytotoxic, unlike 3-chlorotyrosine, but they are known to form inflammatory mediators after conjugation with serum albumin. Our observations lead to the hypothesis that the serum level of advanced oxidized protein products is a prognostic marker of PD duration, and these oxidized proteins could participate in the development of parkinsonian neurodegeneration. Antioxid. Redox Signal. 18, 1296–1302.
Introduction
Innovations
We report for the first time to our knowledge the presence of halogenative oxidation stress in serum and, to a lesser extent, in the cerebrospinal fluid of parkinsonian patients, leading to excess of advanced oxidized protein products, without changes in halogenated amines such as 3-chlorotyrosine. Serum concentration of these protein products is progressively reduced over time, and levodopa treatment contributes to this reduction. These protein products are not cytotoxic, unlike 3-chlorotyrosine, but they form inflammatory mediators after conjugation with serum albumin. Our observations lead to the hypothesis that the serum level of advanced oxidized protein products is a prognostic marker of duration of Parkinson's disease, and these oxidized proteins could participate in the development of neurodegeneration or contribute to the cascade of events leading to neuronal degeneration.
Results and Discussion
First, we verified that main clinical characteristics were similar between the PD and control groups, except for hypertension (p<0.01), as shown in Table 1. We quantified AOPP levels through enzyme-linked immunosorbent assay (ELISA), and they were found to be higher in serum and CSF of patients with PD relative to control subjects, but only serum AOPP values were significantly different (t=2.58, p<0.012; see Table 1). Taking into account the Hoehn-Yahr stages of the disease to measure AOPP levels at the four disease stages, one-way analysis of variance (ANOVA) indicated a Hoehn-Yahr-stage effect on serum AOPP levels (Table 2). Thus, AOPP levels were progressively reduced in serum (F3,56=4.6, p<0.05), and stage-3 and 4 patients showed significantly lower levels of AOPP (p<0.05 vs. stage-1 and 2 patients, Newman–Keuls test), and these values were found not to be different from controls. The lower AOPP levels the higher the Hoehn-Yahr stage. Regarding CSF, AOPP levels were also progressively reduced, but differences were not found to be significant, because CSF AOPP are not detectable in a large group of patients. CSF AOPP levels are quite low in stage-4 patients, similar to controls. The number of years of duration of each stage was observed to be progressively enhanced as expected (F3,56=6.6, p<0.001; see Table 2); hence, AOPP changes decreased over time.
Mean±SEM. Statistical comparisons were carried out with the χ 2 test (qualitative variables) or the Student t-test (quantitative variables).
AOPP, advanced oxidized protein products; PD, Parkinson's disease; nd, nondetectable; NS, nonsignificant; UPDRS, unified Parkinson's disease rating scale.
Mean±SEM, a p<0.05 versus controls; b p<0.05 versus stage-1 and 2 patients (Newman-Keuls).
H-Y, Hoehn and Yahr; CSF, cerebrospinal fluid; AOPP, advanced oxidized protein products.
Since halogenation is related to overstimulation of phagocytes, which release hydrogen peroxide, and myeloperoxidase is the main phagocytic enzyme leading to the formation of halogenated proteins, both hydrogen peroxide and this peroxidase were quantified in serum and CSF. The findings indicated that levels of this enzyme in serum and CSF were not found to be different between patients and control subjects, as shown in Table 1. Besides, hydrogen peroxide was undetectable in both fluids.
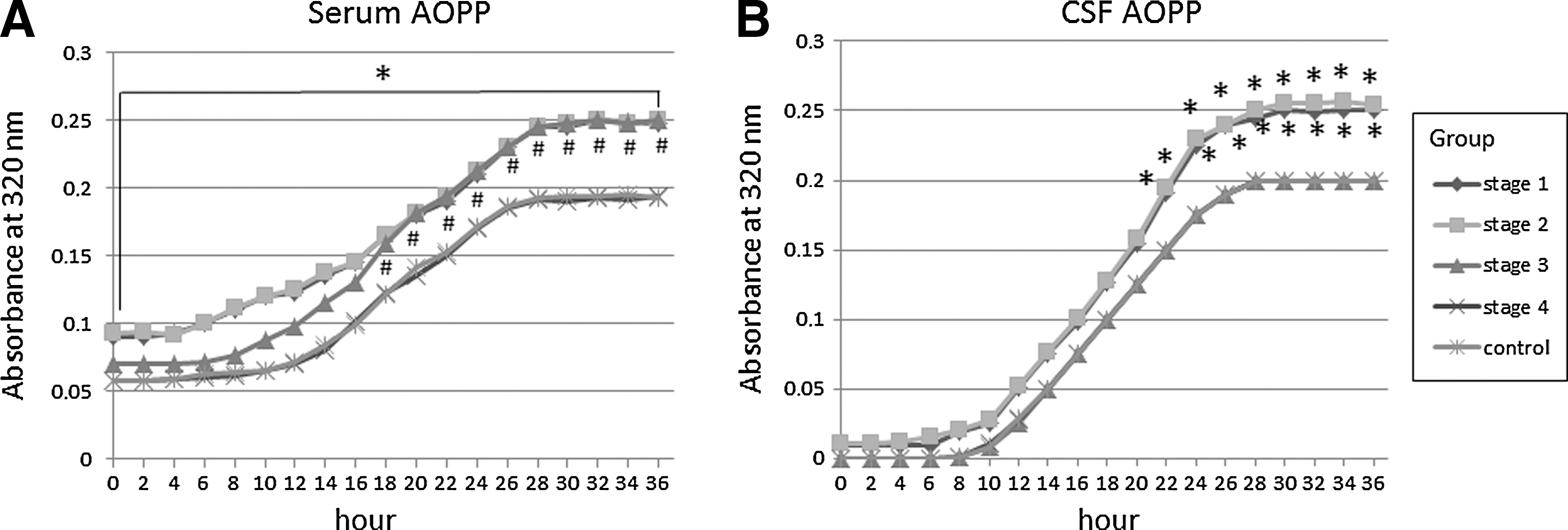
To further characterize AOPP-related effects, we studied the kinetics of self-oxidation of AOPP in serum and CSF. This spectrometric method was based on that previously used for measuring lipoprotein oxidation in plasma and CSF (4). It was observed again that basal AOPP levels were significantly higher in serum of stage-1 and 2 patients versus controls (p<0.05), but not stage-3 and 4 patients. Self-oxidation yielded sigmoid kinetic profiles in every case, as shown in Figure 1. The lag phase was significantly shorter in patients in stage-1, 2, and 3 PD relative to controls (stage 1, 245±30 min; stage 2, 250±37 min; stage 3, 342±35 min; controls, 496±45 min; p<0.01), but it was found to be similar to controls in stage-4 patients (482±33 min). AOPP levels remained significantly enhanced in stage-1 and 2 patients up to 36 h (p<0.05 vs. controls at every time point, Student's t-test), and AOPP levels of stage-3 patients were found to be enhanced relative to controls from 18 h on (p<0.05 vs. controls at every time point, Student's t-test), as shown in Figure 1A. These data indicate that basal AOPP levels are reliably enhanced in stage-1 and 2 patients due to basal halogenative oxidation, and all patients' sera, except stage 4 ones, possess a reduced antihalogenative capacity relative to controls, because their lag phase was significantly shortened. Regarding the CSF, basal AOPP levels were very low in every group, as shown in Figure 1B. Self-oxidation yielded sigmoid kinetic profiles in all cases. The lag phase was observed to be significantly shorter in stage-1 and 2 patients relative to controls (p<0.05), stage-3, and 4 subjects (stage 1, 275±35 min; stage 2, 282±37 min; stage 3, 485±44 min; stage 4, 488±35 min; controls, 496±45 min). AOPP levels of stage-1 and 2 patients were found to be enhanced relative to those of controls and stage-3 and 4 patients from 22 h onward (p<0.05 at every time point, Student's t-test). These data indicate that basal AOPP levels are very low in all patients, and CSF of stage-1 and 2 patients, but not stage 3 and 4 ones, presents a reduced antihalogenative capacity, because the lag phase was significantly shortened, and 36-h AOPP levels were significantly enhanced.
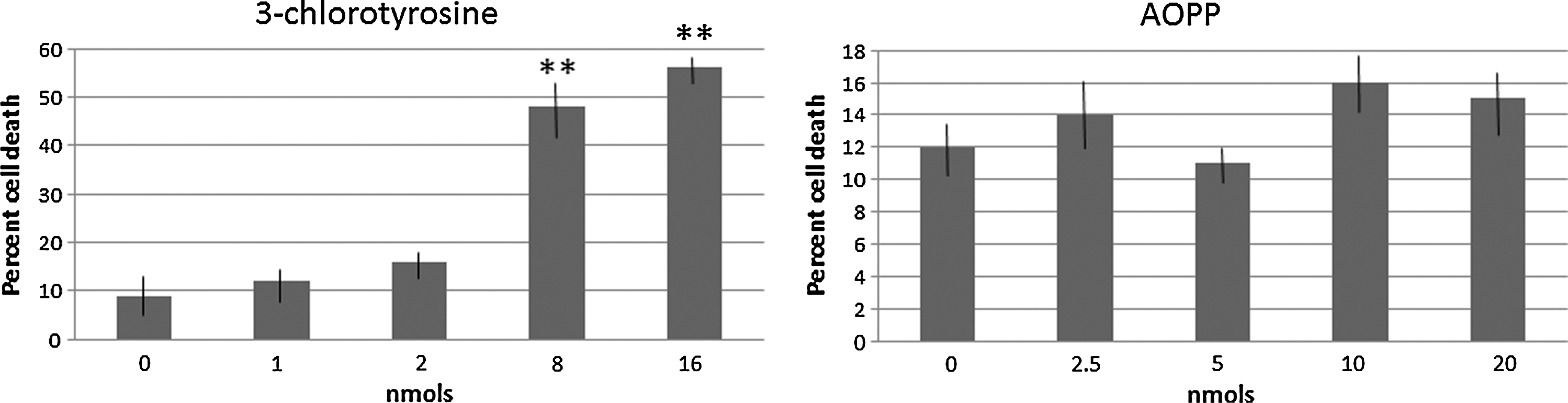
Next, since halogenative oxidation can also yield halogenated amines, we evaluated the presence of 3-chlorotyrosine in serum and CSF from patients with PD with mass spectrometry. 3-chlorotyrosine (retention time=9.17 to 10.20 min) was not detected in any fluid, and only several unknown compounds with different retention times were observed in the MS spectra, as shown in Figure 2. Considering their potentially cytotoxic effect, we were interested to know if halogenated proteins and amines are neurotoxic for dopamine neurons, which are those cells mostly affected in PD. LDH assays of cultured dopamine neurons from substantia nigra of rat pups were carried out, and this study revealed that LDH release was dose-dependently enhanced after 3-chlorotyrosine (dose effect, F4, 25=927, p<0.001; n=6 per group), as shown in Figure 3, but not AOPP, which were devoid of effects. Doses of 8 and 16 nmol chlorotyrosine enhanced cell death significantly (p<0.01 vs. control; Newman–Keuls). Taken together, these data indicate that halogenative oxidation modifies proteins, not free amines. Although 3-chlorotyrosine, unlike AOPP, was observed to be cytotoxic of dopamine neurons, it seems that it does not contribute to in vivo PD damage.

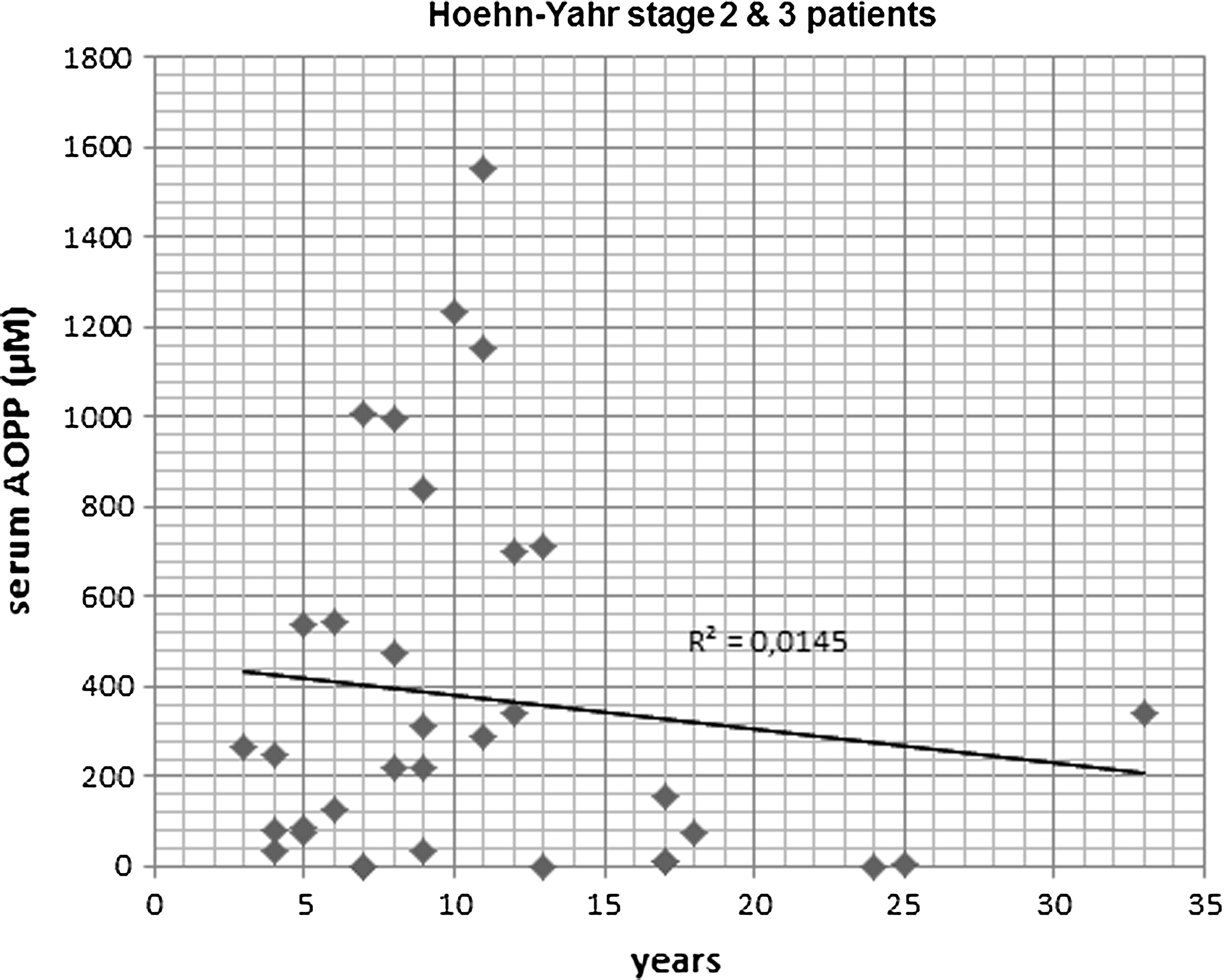
To further analyze AOPP properties, simple linear regression analyses between AOPP levels and clinical characteristics were carried out to discern clinical correlations. No significant regressions were observed in controls, and AOPP levels were not affected by any clinical feature. However, in patients with PD, serum AOPP levels were found to be related to the Hoehn-Yahr stage (R 2=0.114, T=4.4, p<0.01), and to levodopa dosage (R 2=0.259, T=6.4, p<0.001). The higher the Hoehn-Yahr stage or levodopa dose, the lower serum AOPP levels. Besides, considering individual duration of disease and AOPP levels in stage-2/3 patients, a significant regression was observed between this group of patients and serum AOPP levels (R 2=0.0145, T=3.28, p<0.003), as shown in Figure 4. It seems that serum AOPP levels predict how large the disease is in this group of patients. Thus, 9 patients (out of 13) with disease duration longer than 10 years showed serum AOPP levels lower than 350 μM. This effect was not found to be related to levodopa dosage, although treatment with high levodopa doses contributed to global reduction of serum AOPP levels as explained. Hoehn-Yahr stages 2 and 3 were selected, because patients are not at either initial or very advanced timepoint of the disease. Finally, the Tibbling-Link index was quantified to confirm that CSF protein levels were not influenced by blood levels. The Tibbling-Link index in the PD group was lower than 0.7 in every case, except for one patient, and it was observed not to be different relative to the control group (0.46±0.01 PD; 0.41±0.01 controls). Hence, no inflammation of the blood–brain barrier was detected in patients with PD, discarding any meningeal inflammatory process.
In summary, our results in a random sample of patients with PD show that there are indicators of halogenative oxidation stress and lower antihalogenative capacity in serum and, to a lesser extent, CSF of PD, characterized by enhanced levels of AOPP without changes in free 3-chlorotyrosine. In other words, PD serum and CSF show excess of protein halogenation without chloramines. The findings also indicate that serum and CSF AOPP levels are progressively reduced over time, and antihalogenative capacity recovered too. Levodopa contributes to these changes.
Metabolism of AOPP has been associated to several diseases previously (8), where blood phagocytes are over-stimulated, and the formation of HOCl and chloramines from chloride and released hydrogen peroxide is enhanced. Myeloperoxidase catalyzes this process and, as a consequence, AOPP are formed due to oxidative stress. Since AOPP can conjugate with human serum albumin (HSA) giving AOPP-HSA conjugates, inflammatory mediators (2), it can be hypothesized that AOPP could be linked to inflammatory processes in PD. Prior studies have indicated that the other type of advanced oxidized products, advanced glycosylation-end products, are involved in neurodegenerative diseases (6). Our study is the first one, to our knowledge, reporting enhanced formation of AOPP in PD, at early stages of the disease. Of note is that the duration of disease in stage-2 and 3 patients is statistically correlated to serum AOPP levels, and low AOPP levels predict a larger duration of PD. It seems that the AOPP level in serum, not CSF, is a prognostic marker of disease duration, likely because it is a factor contributing to neuroinflammation or that halogenated proteins in blood contribute to the cascade of events leading to neuronal degeneration. Finally, our findings revealed that levodopa treatment was negatively correlated with serum AOPP levels. The contribution of levodopa therapy to oxidative damage is still debated, but our results are in accordance with Prigione et al., who found that levodopa daily dosage is inversely correlated with ROS levels in PD (7). Thus, it seems that levodopa exerts an antihalogenative effect as far as serum AOPP levels are concerned. Such a correlation was not found for the remainder pharmacological treatments based on dopamine receptor agonists or rasagiline, or for the progressive reduction of AOPP levels in CSF. In blood, levodopa is known to give several metabolites with a strong antioxidant capacity such as hydroxytyrosol, and it is possible that a levodopa metabolite rather than levodopa by itself would be the antihalogenative compound.
Notes
Participants
Patients suffering from PD, clinically and single-photon-emission computerized tomography (SPECT)-based diagnosed, were included in the study. Patients were classified according to the Hoehn-Yahr stages, UPDR scales, and duration of PD in years. Duration of PD was calculated on the basis of the year when first symptoms were reported by the patient. All participants were nonsmokers or nonalcohol drinkers. Smoking was defined as a current smoker who consumed cigarettes on a daily basis or occasional smokers who consumed cigarettes less than on a daily basis. Alcohol use was defined as drinking >210 g per week. Control subjects were recruited from either patients' relatives or volunteers subjected to intradural anesthesia for traumatologic surgery in the Surgery Unit of Hospital Macarena (without any neurological disorder). Individuals presenting with any of liver, renal, and cardiac dysfunction, malabsorption, autoimmune diseases, diabetes mellitus, rheumatoid arthritis, AIDS, and infectious conditions (oxidative stress markers in peripheral blood may be altered in such conditions) were excluded from the both PD and control groups.
Clinical information was gathered from each patient: age, gender, body weight, hypertension, dyslipidemia, fasting blood sugar, coffee drinking, smoking, taking of vitamin A/E supplement, statins and aspirins, daily levodopa dose, type and dose of dopamine agonists, and rasagiline. Hypertension was diagnosed when blood pressure repeatedly exceeded 140 mmHg (systolic) and/or 90 mmHg (diastolic) or when a subject was taking antihypertensive medication to control hypertension. Use of vitamin A/E was defined as daily intake. Drinking coffee was defined as daily intake of at least 300 ml of coffee.
CSF collection
CSF was collected by using lumbar puncture. Five ml of CSF was collected and stored in polypropylene tubes (Eurotube) protected by the light, and rapidly aliquoted, coded, and frozen at −80°C for further analyses. A 1 ml collection in a glass tube was employed to observe the absence of traumatic puncture and to quantify red cells before storing. CSF with excess of red cells was discarded (>500 red cells/μl).
Blood collection
Blood was collected by cephalic vein puncture. Five ml of blood was collected in gel-coated tubes to induce blood coagulation, and to obtain serum (BD Vacuotainer). Serum was centrifuged at 2500 rpm during 10 min to separate clot and trapped cells, and then it was immediately frozen at −80°C.
Tibbling-link index
CSF was collected in polypropylene tubes. Simultaneously, blood sample was obtained. CSF samples were centrifuged at 450 g for 10 min, and the supernatant was recovered. Then, paired CSF and serum were aliquoted and stored under biobanking conditions until studies were performed. Albumin and IgG were quantified by standard immunochemical nephelometry in serum and CSF samples (SIEMENS BN II system). IgG indices were calculated as previously described (5). It has been defined that IgG oligoclonal bands are not detected in patients with IgG indices smaller than 0.45; however, indices higher than 0.77 reflect intrathecal IgG synthesis.
Biochemical measures
Serum and CSF aliquots were unfrozen and sonicated with a buffer solution. For measuring AOPP, myeloperoxidase, and hydrogen peroxide, commercial kits were used (AOPP; OxiSelect AOPP Assay kit, Cell Biolabs; myeloperoxidase, Human mieloperoxidase (MPO) Instant ELISA; eBioscience, hydrogen peroxide; Thermo Scientific Pierce Quantitative Peroxide Assay Kit). Other aliquots were used for measuring levels of self-oxidized AOPP through ELISA. These samples were incubated at 37°C in quartz cuvettes and protected against light, and allowed to self-oxidize in contact with the atmospheric air during 36 h. The absorbance was measured at 320 nm in cuvettes (10-mm optical pathway). ELISA measures were performed at the beginning and the end of the incubation period. In every case, serum was diluted 150-fold with phosphate-buffered saline (PBS), and CSF was not diluted.
Self-oxidation kinetics
This method was based on that previously used for measuring lipoprotein oxidation in plasma and CSF (4), and taken into account that most AOPP present a maximum absorbance at 320 nm (9). Thus, previous studies have demonstrated that chromophores detected at 320 nm are mostly related to the presence of modified tyrosines (AOPP) induced by the activity of MPO in the presence of hydrogen peroxide and chloride (23). To register the oxidation rate, serum was diluted 150-fold with PBS that contained 0.6 M NaCl, pH 7.4, treated with Chelex 100 ion-exchange resin (Bio-Rad) for 1 h. The sample was incubated at 37°C for 6 h in the absence of exogenous oxidant and in contact with atmospheric air. The absorbance was measured at 320 nm in quartz cuvettes (10-mm optical pathway), and registered at 30-min intervals up to 36 h. CSF was not diluted, and maintained at 37°C. Absorbance was measured every 30 min up to 36 h too. In both serum and CSF, sigmoid kinetic curves are obtained, with three phases: the lag phase, during which the oxidation rate was close to zero; the propagation phase, presenting a fast accumulation of AOPP; and the plateau phase with an oxidation rate close to zero again.
Mass spectrometry
Chromatographic separation was performed using a PelkinElmer Series 200 HPLC system coupled to an Applied Biosystems QTRAP LC/MS/MS system consisting of an hybrid triple quadruple linear ion-trap (QqQ
In vitro study
Primary cultures of substantia nigra neurons were established as previously described (1). Postnatal pups (P0) were killed by decapitation under aseptic conditions, and the brains were removed and placed in cold Hank's balanced salt solution (HBSS; GIBCO, Invitrogen Corp.). Under a dissecting microscope, a 0.8–1.0-mm-thick coronal section of the mesencephalon was made using a scalpel, and the regions containing substantia nigra were carefully isolated. Tissue was digested in a solution of papain (20 units/ml; Worthington) with 0.2 mg/ml
Exposure to 3-chlorotyrosine (Sigma-Aldrich) and AOPP (AOPP-HSA, 0.14 μmol AOPP/mg protein; Cell Biolabs) was initiated after 4–5 days in vitro. The medium was removed and changed to Neurobasal without B27 for 2 h. 3-chlorotyrosine (0, 1, 2, 8, and 16 nmol in Neurobasal) and AOPP (0, 2.5, 5, 10, and 20 nmol in Neurobasal) were added to neurons. After 30 min, this culture medium was removed, and cultures were gently washed twice with Neurobasal and then further incubated for 24 h, to carry out the lactate dehydrogenase (LDH) assay (Cytotoxicity Detection kit; Roche). Total LDH release was calculated by incubating untreated cells with 0.5% Triton X-100 for 1 h to induce maximal cell lysis. Basal death was calculated from untreated wells without B27. Treatment values were then expressed as the percent of the maximal LDH release. Background LDH release (medium alone) was subtracted from the experimental values.
Statistics and ethics
We enrolled 60 patients, comprising four Hoehn-Yahr stages (stage 1, n=16; stage 2, n=24; stage 3, n=10; stage 4, n=10) and 45 controls. Differences in halogenation markers and clinical characteristics between the PD and control groups were analyzed by the χ 2 test or Student's t-test (independent samples). In patients and controls, simple regression analysis was used for searching for correlations between halogenation markers and clinical factors. Comparisons between clinical characteristics and oxidative markers within patients at different PD stages were analyzed by the χ 2 test and one-way ANOVA followed by Newman–Keuls' test or Student's t-test where appropriate. When two factors were considered (e.g., AOPP levels in all groups before and after self-oxidation), two-way ANOVA was used, followed by Bonferroni correction and Newman–Keuls' test. If needed, normalization was verified with the Shapiro–Wilk test. Informed consent forms under a protocol approved by the University of Seville and Macarena Hospital internal ethics and scientific boards were obtained from all the subjects, and the subjects' consent was obtained according to the Declaration of Helsinki (BMJ 1991; 302: 1194).
Footnotes
Acknowledgments
The authors thank Mara Guerra, Silvia Castellano and Beatriz Galán (University of Seville) for their excellent technical assistance; Dr. Guillermo Izquierdo for allowing the use of the facilities of the Service of Neurology of Hospital Macarena; the Biobanco Hospitalario Virgen Macarena (National Biobank Network, Carlos III Health Institute RD09/0076/00080) for its help and support in the sample collection procedures and storage; Dr. Cintia Calvo and the Service of Nuclear Medicine (Hospital Macarena, Seville) for SPECT analyses; Dr. Eugenia Soria and Servicio de Espectometria de Masas-CITIUS (University of Seville) for mass spectrometry analyses; and Dr. Rodriguez Quecedo (Centro de Salud Mairena, Seville) and Lola Rivero for their medical help. The authors are most grateful to all patients and their partners as well as control subjects who participated in this study.