
Abstract
Introduction: Immunogenic Cancer Cell Death in Cancer?
S
For many decades, most anticancer therapies, especially radiotherapy and chemotherapy, were considered direct tumoricidal compounds, killing cancer cells or inducing a permanent arrest in their cell cycle. Chemotherapies target without any distinction tumor cells and rapidly proliferating host's immune cells, leading to the assumption that cytotoxic chemotherapeutics would induce immunosuppressive rather than immunostimulatory off-target effects (167, 171). This belief has largely discouraged oncologists from performing extensive studies on the contribution of the immune system in anticancer therapies. Indeed, drug discovery programs have neglected the possibility that host immune responses may account for - at least part of - the efficacy of cytotoxic drugs. For example, the guidelines for drug screening and validation provided by the National Cancer Institute recommended the (xeno-) grafting of human cancer cell lines into immunodeficient hosts (168). Moreover, arguing against a dominant role of the immune system in the antitumor effects mediated by select chemotherapies, the fact that most treatment modalities induce “apoptosis,” a cell demise is considered “immunologically silent,” if not “tolerogenic” (67, 89, 131).
Despite these early observations, recent experimental evidence from several groups challenged this notion that necrosis would be immunogenic while apoptosis would not (50), indicating that the nature of cell death defined by distinct molecular, biochemical, and metabolic hallmarks would convey the mandatory links between dying cells, innate and cognate immunity. Hence, while many chemotherapeutic agents (e.g., etoposide, mitomycin C, and cisplatin) trigger a cell death modality that is not immunogenic, distinct cytotoxic compounds such as doxorubicin, oxaliplatin, cyclophosphamide, or ionizing radiation could favor the activation of immune effectors cells while inducing apoptotic tumor cell death (17, 102). This means that (i) cancer cells dying after anthracyclines, oxaliplatin, or irradiation can mount an anti-cancer immune response in the absence of adjuvants and immunize the host against a subsequent tumor rechallenge (17, 102); (ii) that established tumors which regressed postchemotherapy are massively invaded by cytotoxic T cells; and (iii) that chemotherapy-induced control of transplantable tumors depends on T lymphocytes. Indeed, we reported that these anti-cancer treatments were more efficient in immunocompetent than in immunocompromised animals (101, 102, 147).
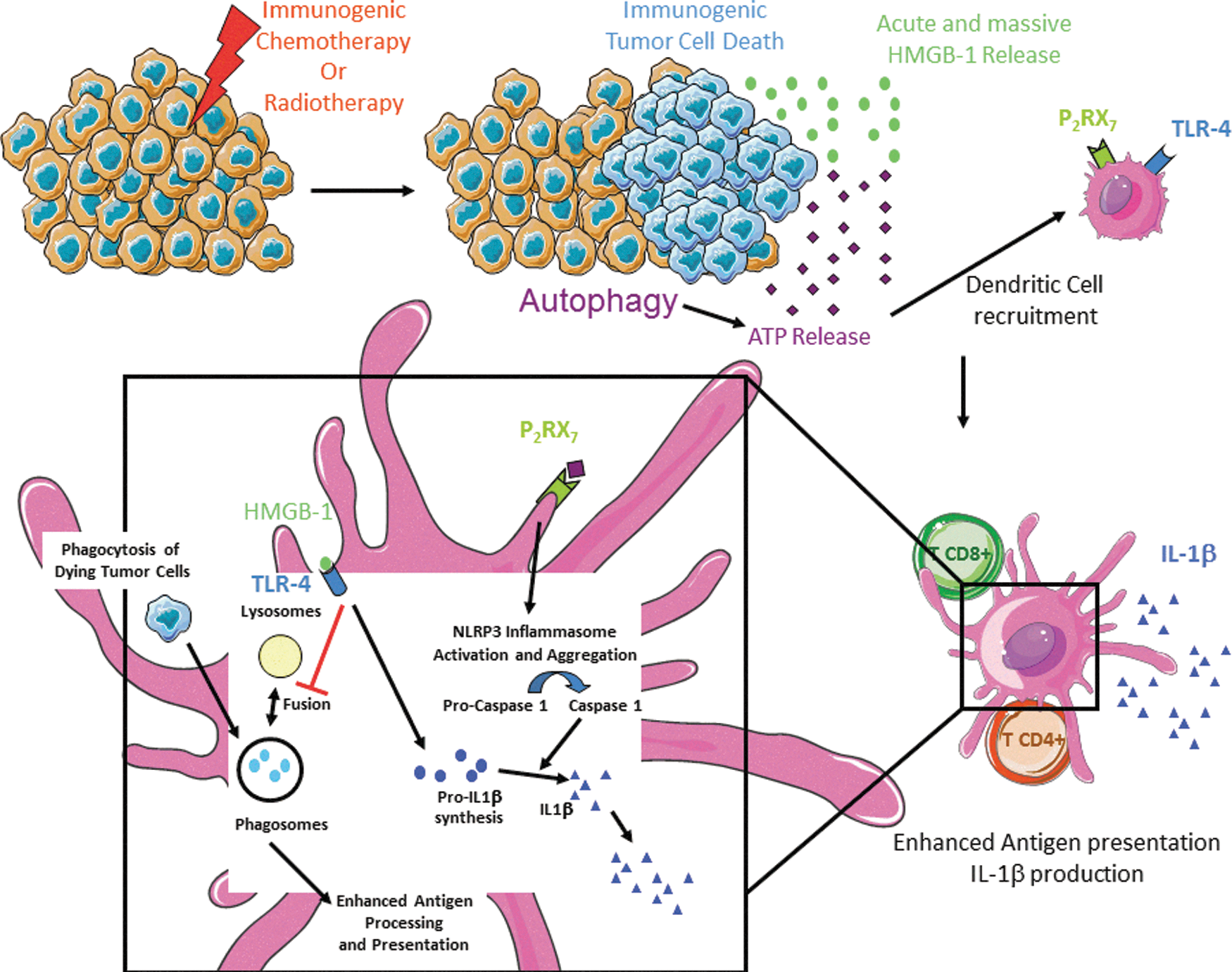
We showed a hierarchy of cytokine-associated events that can be summarized as follows. First, the triggering of the autophagy machinery in chemotherapy-exposed tumor cells promotes adenosine tri-phosphate (ATP) release, which is indispensable for the recruitment of inflammatory monocytes-derived dendritic cells (in a P2Y2-dependent manner) (95). The ATP/P2RX7/Nlrp3/Caspase-1 axis in local dendritic cells (DCs) exposed to dying tumor cells promotes IL-1β release by DCs and activation of γ4/γ6 γδT cells that secrete IL-17 and IL-22 three days after chemotherapeutic treatment. Those γδT17 cells markedly correlate with the brisk recruitment and accumulation of interferon-γ (IFNγ)-producing CD8+ CTLs in tumor beds by day 8 postsystemic chemotherapy. Hence, using neutralizing antibodies and loss-of-function mice targeting IL-17/IL-17RA, IL-1β/IL-1R1, and IFNγ/IFNRII, we showed that these three cytokines are crucial for the immunogenicity of cell death and anticancer efficacy mediated by chemotherapy.
These data generated in transplantable tumor models as well as in methylcholanthrene-induced sarcoma have been extended to humans. Anthracycline-killed tumor cells also appear particularly immunogenic (45). After anthracyline-based chemotherapy, the intratumoral leukocyte infiltrate changes its composition in cancer patients. Accordingly, after exposure to neoadjuvant chemotherapy, one third of stage II/III BCs appear highly infiltrated by cytotoxic T cells with a concomitant reduction of B lymphocytes and Th2-CD4+ T cells (127). Moreover, the ratio of macrophages to CD8+ T cells in primary BCs not only predicts overall survival, but also predicts a pathologic response to chemotherapy in chemo-naïve patients (24). An increased abundance of T lymphocytes, as well as an ameliorated ratio of CD8+ T cells over Foxp3+ regulatory cells postchemotherapy, also predicted favorable therapeutic responses to combination therapy with anthracyclines/taxanes in human BC, and so does invasion by CD8+ T cells in colorectal cancer liver metastases (24, 52, 74, 161).
This review will summarize the mandatory cell-death-associated molecular patterns (CDAMP) for immunogenic cell death (ICD) to occur and will discuss the clinical implementations of this concept.
Early Signaling During ICD: Role of Calreticuline in Antigen Uptake
The uptake of dead cell-derived antigens by DCs constitutes an early step in the sequence of events that leads to anticancer immune responses. One of the first and cardinal molecular events during ICD is the rapid exposure of molecules on the outer leaflet of the plasma membrane that act as “eat-me” signals for antigen-presenting cells such as DCs (47). Interestingly, the analysis of surface proteome changes in anthracycline-treated tumor cells revealed that ICD is associated with the ectopic exposure of the endoplasmic reticulum (ER) chaperone calreticuline (CRT) (102, 109). In homeostatic conditions, CRT is mainly located in the lumen of the ER, in which it functions as a molecular chaperone and modulates calcium signaling and homeostasis (65). In response to anthracyclines or ionizing radiation, CRT exposure occurs before the first morphological signs of apoptosis and before the translocation of phosphatidylserine from the inner to the outer leaflet of the plasma membrane. Another prerequisite for surface CRT exposure is the activation of caspases; the Z-VAD fmk caspase inhibitor abolishing anthracycline-mediated CRT exposure to the cell surface (17). This process is accompanied by other hallmarks of the ER stress response such as phosphorylation of the eukaryotic initiation factor 2α (Fig. 1) and co-trafficking with CRT of another ER-resident protein, the disulfide isomerase ERp57. ERp57 knockdown tumor cells fail to elicit immune responses and to respond to chemotherapy. This can be overcome by exogenous recombinant CRT protein in immunocompetent but not nu/nu mice, indicating that tumors with an intrinsic defect in the CRT-translocation machinery become unable to elicit an efficient anticancer immune response, contributing to tumor regression (109). The presence of CRT on the surface of tumor cells that succumb to distinct anticancer agents correlates with their uptake by DCs (102). Ecto-CRT appears to be indispensable for phagocytosis and ICD in that neutralizing CRT-specific antibodies or silencing of CRT by RNA interference abolished phagocyte engulfment of dying bodies and the immunogenicity of tumor cell death (102).

While we described an ER stress-response-dependent CRT exposure in the context of anthracyclines, cyclphosphamide, oxaliplatin, and irradiation, other reports confirmed the cardinal function of CRT in the recognition of dying cells and the elimination of nascent tumors.
CRT translocation as triggered by hypericin-based photodynamic therapy was independent of ERp57, increased cytosolic Ca2+ concentrations, eIF2α phosphorylation, caspase-8 activation, and lipid rafts (48, 69). Conversely, hypericin-based photodynamic therapy promoted ICD via a molecular cascade that relies on reactive oxygen species (ROS) overgeneration, PERK, BAX, and BAK, PI3K activity, and the ER-to-Golgi transport apparatus (48, 69).
Interestingly, Beneteau et al. just reported that inhibition of glycolysis converted etoposide-mediated non-ICD into an efficient vaccine procedure in mice. They demonstrated that this effect was at least partially mediated through ERp57/CRT exposure on the plasma membrane (12).
Chao et al. demonstrated that CRT is a dominant pro-phagocytic signal in several human cancers (such as leukemia, neuroblastoma, bladder cancers, and lymphoma) binding to CD91 on macrophages and counterbalancing the CD47/SIRPα-mediated anti-phagocytic signal. Interfering in the delicate balance between pro- and anti-phagocytic signals by selectively targeting tumor cells by anti-CD47 antibody effectively prevents immune evasion (18).
Hence, the ER stress response constitutes a central hub in the signaling cascades, leading to the emission of ICD-promoting signals. The accumulation of misfolded proteins, the inhibition of protein glycosylation, and the deregulation of redox and Ca2+ homeostasis can disrupt the delicate homeostasis of the ER-Golgi network, resulting in the activation of adaptive cell responses (120). More work is required to establish which transcription factors of the unfolded protein response are mandatory for the ICD and how the ER stress co-opts the pattern recognition receptor (TLR4 in this case) signaling pathway to ignite innate immunity (84).
Late Signaling During ICD: HMGB1 and Cross-Presentation of Tumor Antigens
The CRT-driven uptake of tumor antigens by DCs is per se insufficient to elicit an antitumor immune response, as these tumor antigens should be also efficiently processed by DCs and exposed at their surface within MHC complexes for the cross-priming of CD4+ and CD8+ T lymphocytes. This implies that, during ICD, other signaling pathways involved in antigen processing, presentation, and polarization are required after antigen uptake. We will now deal in detail with how high mobility group box 1 protein (HMGB1) and ATP can also determine the immunological fate of cell death.
HMGB1 in cancer
The HMGB1 is ubiquitously present in the nucleus of almost all mammalian cells, in which it stabilizes nucleosomes and regulates the transcription of many genes, promoting access to DNA to transcriptional factors (111, 143). Within the nucleus, it binds to the minor groove of DNA and enables interaction between nucleic acids and a variety of molecules, including p53, NF-kB, and steroid hormone receptors. Besides its nuclear role, HMGB1 also functions as an extracellular cytokine-like molecule during various physiological or pathological processes, such as inflammation, cell differentiation, cell migration, or tumor metastasis (31, 36, 77, 98). Indeed, HMGB1 is secreted by macrophages (13, 158), mature DCs (32), and natural killer (NK) cells (139) in response to injury, infection, or other inflammatory stimuli. When released outside the cell, HMGB1 can bind with a high affinity to several receptors, including the receptor for advanced glycation end products (RAGE), TLR-2, TLR-4, TLR-9, and, as a negative signaling molecule, CD24, mediating the response to infection and injury, thereby igniting inflammation (31, 77, 132, 148, 158). For a long time, HMGB1 has been thought to exert a proinflammatory function exclusively during necrosis (132), but recent evidence indicates that it also gets released by cells undergoing the late stages of apoptosis (11, 148). Indeed, many investigators have found that nuclear DNA is released in a time-dependent manner during apoptosis (21). Moreover, binding of HMGB1 to DNA is increased during apoptosis, in line with the fact that late-stage apoptotic cells can release both DNA and HMGB1. Thus, genotoxic agents that activate polyadenosylribosyl polymerase (PARP), including chemotherapeutic agents, can stimulate the release of HMGB1 from its association with chromatin, presumably as a result of the direct PARP-mediated polyadenosylribosylation of HMGB1 (29). Outside the cell, HMGB1 could also favor the establishment of T-cell-dependent, protective immune responses (64, 70, 106), acting as “alarmin” or damaged-associated molecular pattern (DAMP) during cell death. Once passively released from necrotic cells in the extracellular space, HMGB1 acts as a chemoattractant for inflammatory leukocytes and stem cells (19, 108), and is able to trigger the functional maturation of DCs and to support the clonal expansion of IFNγ-producing Th1 cells (32, 97). It also influences the migration of DCs in secondary lymphoid organs (33, 81). Besides the roles of HMGB1 within the nucleus and in the extracellular environment, a third life as a cytosolic chaperon has been recently described, modulating the ability of cancer cells to die via apoptosis or to activate the autophagic machinery. For instance, it has been recently demonstrated that HMGB1/p53 complexes are able to regulate the cytoplasmic localization of the reciprocal protein, and subsequent levels of autophagy and apoptosis (76).
HMGB1 appears to have an alternate paradoxical role during tumorigenesis. Indeed, HMGB1 recognition can favor tumor progression through neoangiogenesis by attracting pro-angiogenic macrophages and endothelial cells in tumor beds (155) (Fig. 2A). Extracellular HMGB1 can trigger cell proliferation, migration, and sprouting of endothelial cells (96, 136). In addition, other studies have suggested a direct role of HMGB1 in tumor cell motility and invasion (39, 114, 115, 121). Altogether, HMGB1 was considered a factor of tumor progression that was associated with a dismal prognosis in cancer (100, 117). Accordingly, HMGB1-RAGE interactions mediated invasion, migration, and the growth of implanted tumors in glioma models (165). In most of the studies, HMGB1 and/or RAGE expression was studied at protein or mRNA levels. Overexpression (compared with healthy tissues) as well as cytoplasmic localization of HMGB1 have been reported in many cancer types, including gastrointestinal stromal tumors (22), colon (157), gastric (72), pancreatic (144), melanoma, prostate, and cervical carcinomas (53, 57, 59, 116, 117, 157). Moreover, in prostate cancer, HMGB1 and RAGE expressions were increased in metastatic sites compared with primary tumors of nonmetastatic patients (71). Enhanced expression of HMGB1 and RAGE is associated with aggressive tumor phenotype and poor prognosis at any stage in colon carcinoma (130). One possible scenario is that neoplastic cells expressing higher amounts of HMGB1 also release the pro-metastatic and pro-angiogenic cytokine in the tumor microenvironment (8, 36). A recent and elegant study demonstrated that HMGB1 can bind to TIM3 in tumor DCs, thereby suppressing the immunostimulatory pathway in DC mediated through nucleic acids released by dying tumor cells (20) (Fig. 2B).

Surprisingly, the relevance of HMGB1 expression has been poorly investigated in BC. In one study, an increased expression of HMGB1 proteins in the tumor beds compared with healthy breast tissues was reported (42). Of note, nuclear HMGB1 markedly enhances DNA binding and transcriptional activity of the estrogen receptors in the estrogen responsive elements (23, 42, 92), suggesting its potential relevance in the biology of estrogen-sensitive BC. Northern blots analyses of the 1.4 kb and the 2.4 kb transcripts of HMGB1 in 13 BC samples revealed strong variations that could potentially account for the response to endocrine therapy in estrogen receptor-positive BC (42). Since a few data were available on HMGB1 expression in BC and normal breast tissues using immunohistochemistry (IHC), we carried out the IHC analysis of the pattern of HMGB1 expression in a series of BC patients from Institut Gustave Roussy, Villejuif, France, and Center George François Leclerc, Dijon, France. We observed that this pattern of expression is variable among tumors and peculiar in cancer versus healthy cells. In our hands, in the vast majority of cases, normal mammary glands and stromal cells harbor a strong nuclear staining (Fig. 3A–C). On the contrary, approximately one-third of breast tumors exhibit a similar nuclear staining (Fig. 4A). The vast majority of BC has lost this nuclear expression and only harbors a weak cytoplasmic or perinuclear staining (Fig. 4B), while in situ carcinoma presents with a mixed heterogeneous pattern of nuclear expression (Fig. 3D). We are currently addressing the prognosis value of HMGB1 nuclear expression in various molecular subtypes of BC and its relevance for the response to conventional therapies.

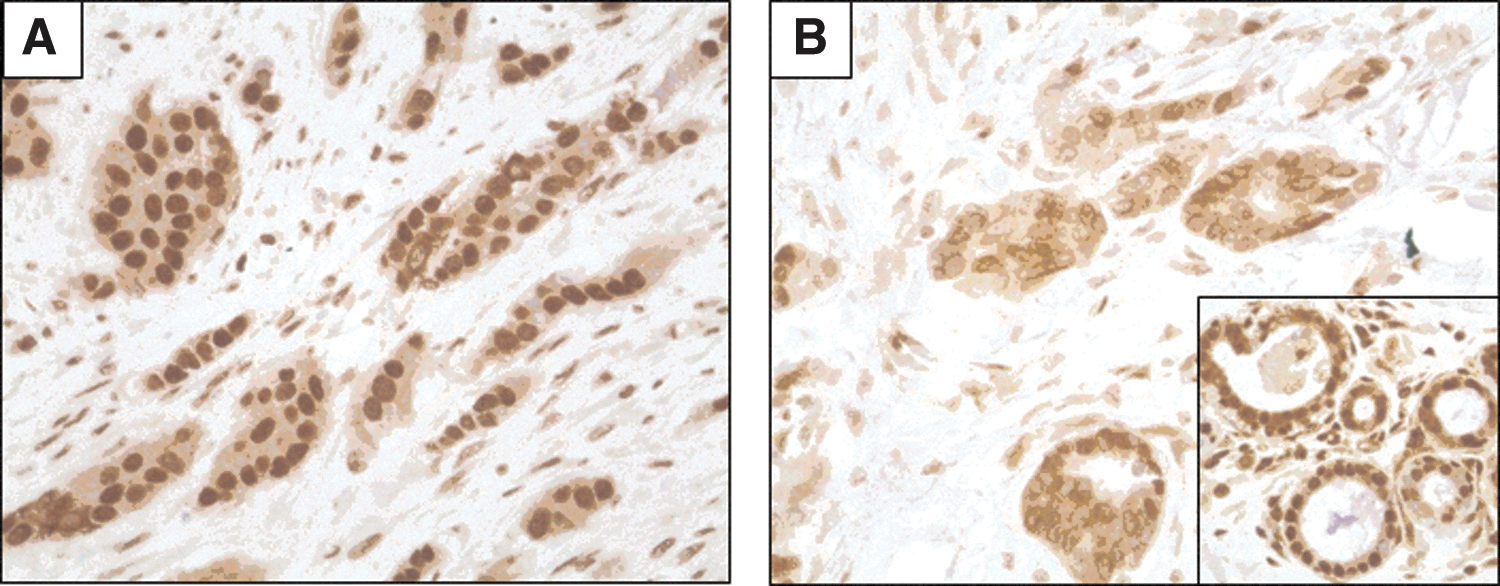
However, HMGB1/RAGE expression was not found to be elevated in all histological types, especially in lung carcinoma in which RAGE and HMGB1 levels are down-regulated compared with benign tumors and normal control tissues (9, 30, 55). Besides its putative role in the promotion of tumor progression, recent preclinical and clinical data revealed that HMGB1 seems to trigger protective anti-cancer T-cell responses. Pioneering studies have shown that this extracellular HMGB1 release could activate DCs (94, 125) and promote the induction of protective immune responses against poorly immunogenic apoptotic lymphoma cells (125). Therefore, when an appropriate antigenic transfer to DCs has occurred, the acute release of HMGB1 may contribute to elicit immune responses in cancer-bearing patients (4, 124, 126). Moreover, chemotherapeutic agents such as alkylating agents and taxanes appear to be more effective than others to trigger the translocation from the nucleus and the subsequent exodus of HMGB1 in the extracellular milieu (31, 43), suggesting that HMGB1 could behave as a DAMP or a CDAMP during ICD.
The multiple and controversial biological functions of HMGB1 are context dependent and could be explained by the redox modifications induced by the cell or the extracellular milieu. Redox-associated modulations of HMGB1 bioactivity oscillate between a chemokine-like molecule (fully reduced HMGB1), a pro-inflammatory cytokine (in case of disulfide-bond possessing HMGB1), and inactivated DAMP (fully oxidized HMGB1). Activation of caspases during apoptosis can amplify mitochondrial ROS production, which may neutralize the DAMP activity of HMGB1 (Fig. 2). Of note, recent studies suggest that the redox modulation might finely control the DAMP activity of HMGB1, regulating leukocyte recruitment or their induction to produce inflammatory cytokines by adopting mutually exclusive redox states (154).
The second face of HMGB1 in cancer: role in processing and cross-presentation during ICD
The fact that some anticancer treatments appear to be more efficient in immunocompetent rather than in immunodeficient mice prompted the search for a link between innate and adaptive immune responses during ICD and led to the systematic screening for a role of toll-like receptors (TLRs) in the efficacy of chemotherapy or radiotherapy. Indeed, tumor cell death triggered by cytotoxic agents may lead to the release of danger signals that favor the development of anticancer immunity (90, 151). Although TLRs were originally described to detect microbial products (145), it has also been shown that endogenous danger signals from dying cells can bind TLRs and, thus, modulate adaptive immune responses (83). Indeed, when TLRs on the surface of DCs recognize pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), signaling pathways are ignited that eventually inhibit the fusion between phagosomes and lysosomes, thereby facilitating antigen processing and the initiation of cognate immunity (141).
To address whether TLRs could be involved in ICD, the immunogenicity of oxaliplatin-exposed tumor cells was tested in TLR-deficient mice. Only those tlr4 deficient mice failed to mount a tumor-specific protective immunity. Next, we tested the efficacy of chemotherapy (anthracyclines or oxaliplatin) and radiotherapy in wild-type versus TLR4-deficient animals in 4 different tumor models (transplantable CT26, EG7, TS/A cell lines, and the graft of an osteosarcoma GOS). Although these cytotoxic compounds significantly reduced tumor growth in wild-type (WT) hosts, they failed to do so in tlr4 −/− mice, underlining the absolute requirement for TLR4 and its downstream effector MyD88 in the chemotherapy-induced control of tumor progression (5, 6). T cells were also mandatory for the observed antitumor effects, as athymic nu/nu mice failed to mount an efficient antitumor response after local treatment with radiotherapy or systemic chemotherapy (6). Interestingly, TLR4 appears to be required for efficient DC-mediated antigen presentation (5, 6). In fact, TLR4 has been shown to inhibit the lysosome-dependent degradation of phagosomes in macrophages (141). Accordingly, the kinetics of fusion between phagosomes and lysosomes was slower in WT DCs than in TLR4-deficient DCs loaded with dying tumor cells, which suggests that the TLR4 defect causes accelerated lysosomal degradation of the phagocytic cargo (Fig. 5). Thus, TLR4-deficient bone marrow-derived DCs were unable to present dying-cell-associated tumor antigens (engulfed by phagocytosis), although they retained the capacity to present antigens from soluble proteins (taken up by pinocytosis) (5, 6). Accordingly, the priming of CD8+T lymphocytes by DCs pulsed with cancer cells undergoing ICD was markedly compromised in tlr4 −/− DC. Altogether, these data suggest that the TLR4/MyD88 pathway contributes to the success of ICD-mediated anticancer therapy.
Identification of the danger molecules that contribute to the immunogenicity of dying tumor cells was performed using the systematic comparison of putative TLR4 ligands released by oxaliplatin or anthracycline-treated tumor cells. These experiments revealed that HMGB1 molecules but not other known TLR4 ligands (such as heat shock proteins, fibrinogen, fibronectin, and beta-defensin 2) selectively accumulated at late time points in the supernatant of tumor cells undergoing ICD (as shown in Western Blotting and ELISA) (5). The release of HMGB1 from cancer cells that succumb to anthracycline or ionizing radiation was measurable within 18h after ICD induction and resulted, similar to CRT exposure, from a caspase-dependent process (5). HMGB1 first translocates from the nucleus to the cytoplasm and then, after the disruption of the plasma membrane, gets released into the extracellular space (147). The binding of HMGB1 to TLR4 was previously shown using fluorescence resonance energy transfer analyses and immunoprecipitation studies (112).
The release of HMGB1 and its interaction with TLR4 on the surface of DCs appears to be mandatory to prevent the fusion of phagosomes with lysosomes and the rapid degradation of tumor antigens (6). Hence, further experiments clearly showed that the ligation of TLR4 by HMGB1 is crucial for the priming of T cells by dying tumor cells. When either HMGB1 or TLR4 was absent from the system, dying tumor cells lost their immunogenicity in vivo. While usually engulfed by DCs, tumor cells were genetically modified to express an siRNA HMGB1 that exhibited reduced immunogenic potential on exposure to doxorubicine. Moreover, a neutralizing antibody directed against HMGB1 blunted antigen presentation of OVA antigens from dying EG7 cells to T cells in vivo (6).
The clinical relevance of the HMGB1/TLR4 pathway as a key determinant for the immunogenicity of dying tumor cells has been analyzed in retrospective clinical studies in N+ BC patients treated with adjuvant anthracyclines. Indeed, a TLR4 single nucleotide polymorphism (SNP) rs4986790 has been associated with decreased responses to lipopolysaccharide (LPS: a well-known ligand of TLR4) (7). The single nucleotide substitution (+896A/G) in the tlr4 gene leads to the replacement of an aspartic acid by a glycine (Asp299Gly) in the extracellular domain of TLR4 and is present in 12%–15% of Caucasians. Hence, alveolar epithelial cells from individuals bearing the Asp299Gly variant form of TLR4 lost their capacity to produce IL-1α after LPS stimulation (7). Coimmunoprecipitation experiments showed that the expression of the variant form of TLR4 significantly decreases the binding of recombinant HMGB1 to TLR4 (6). Moreover, although monocyte-derived DCs from individuals bearing the normal allele of TLR4 could cross-present antigens from dying melanoma cells to T cells in an HMGB1-dependent manner, DCs from individuals bearing the TLR4 Asp299Gly mutation failed to do so (6). In a retrospective analysis of the outcome of 280 localized-BC patients treated with local radiotherapy followed by adjuvant anthracycline-based chemotherapy, patients bearing the Asp299Gly variant TLR4 SNP exhibited a markedly increased incidence of metastatic relapse, when compared with patients bearing the normal TLR4 allele (6). In a multivariate analysis, carrying the TLR4 variant remained independently associated with poorer metastasis-free survival (6). The relevance of the TLR4 Asp299Gly SNP was corroborated in metastatic colon cancers treated with oxaliplatin-based chemotherapy (147).
Altogether, these results identified the interaction of HMGB1 released by dying tumor cells with the TLR4 receptor present in DCs as a major determinant for the cross-presentation of tumor antigens and the promotion of tumor-specific Tc1/Th1-cell responses, culminating in the success of some anticancer treatments (such as anthracyclines and oxaliplatin). It is interesting to note that other investigators have identified alternate DAMP which could mediate antigen processing of cell-associated antigens. Hence, Reis E Sousa's team demonstrated the critical role of DNGR1/CLEC9A expressing CD8a DC in the cross-presentation of necrotic tumor cells to T cells. Their team identified F-actin as the pivotal CLEC9A ligand promoting the processing of the phagocytic cargo by DC, highlighting how an evolutionary conserved cytoskeletal component can represent a relevant DAMP/CDAMP emanating from necrotic cells (1, 128, 129).
However, neither the addition of recombinant CRT nor HMGB1 as a stand-alone DAMP to live cancer cells is sufficient to induce an efficient presentation of tumor antigens by DCs, suggesting that additional factors should be exchanged between dying cells and DCs to induce a full-blown anticancer immune response.
ATP and Inflammasomes in Carcinogenesis and Cancer Immunogenicity
The possible contribution of receptors for DAMPs other than TLRs, which could account for the priming of IFNγ-producing T cells during anticancer chemotherapy, led to an investigation of the putative role of NOD-like receptors (NLRs) that could recognize putative CDAMP and link inflammation to innate immunity (44, 91). Indeed, some of the NLRs also sense nonmicrobial danger signals and form large cytoplasmic complexes called the inflammasome platforms (reviewed in (85, 138)). Inflammasomes are multiprotein complexes that operate as platforms activating caspase-1 and mediating the proteolytic maturation and secretion of pro-inflammatory cytokines (such as IL-1 and IL-18) (61, 138). Inflammasomes can be categorized according to their main constituent: NLRP1, NLRP3, NLRC4, NLRP6, and AIM2. In response to danger signals, the NLR, such as NLRP3, interact with the adaptor molecule apoptosis-associated speck like protein (ASC) to form the inflammasome, the principal caspase-1 activation complex.
Similar to HMGB1, these caspase-1 activation complexes have contrasting roles in the setting of cancer [reviewed in (169)]. First, inflammasomes were associated with pro-carcinogenic functions, especially in human cancers linked to chronic inflammatory processes such as gastric cancers (caused by Helicobacter pylori infection), liver carcinoma (resulting from viral infection), and colorectal cancer (favored by inflammatory bowel diseases) (51, 133). In these contexts, preclinical and clinical data indicate that IL-1β can directly drive oncogenesis (66, 123, 150) or suppress immunosurveillance mechanisms through the recruitment and the activation of myeloid-derived suppressor cells (MDSCs) (15, 150, 152), thus facilitating tumor development. In melanoma or multiple myeloma, tumor cells are able to produce and secrete IL-1β that confers a proliferative advantage to cancer cells through autocrine or paracrine mechanisms (79, 105, 119). Similarly, IL-1β can also convert fibroblasts into tumor-associated fibroblasts, generating a tumor microenvironment that favors cancer progression (134). IL-1 may also favor tumor invasiveness, metastatic spreading, and tumor-driven angiogenesis (35, 99, 135, 156, 160). Moreover, IL-18 secreted by tumor cells can suppress the antimetastatic functions of NK cells in mice (146).
Conversely, in the setting of cancer cell demise, there is growing evidence which suggests that the inflammasome or its products (particularly IL-1β) can also sustain immune response against dying tumor cells, especially in chemotherapy-elicited anticancer immune responses. Indeed, the inflammasome NLRP3 appeared mandatory for the efficacy of some chemotherapies (49). In oxaliplatin-treated mouse tumor models, each individual player of this complex (NLRP3, its adaptor molecule ASC, or caspase-1 or IL-1β/IL-1R1), or its final product IL-1β, was required for the immunogenicity of cell death as well as for the success of chemotherapy (5). Similar results were obtained in other tumor models treated with anthracyclines (49). Hence, caspase-1-deficiency resulted in reduced IFN-γ production by tumor antigen-specific CD8+ T cells in tumor draining lymph nodes (49). IL-1β appears to be a key contributor for the full differentiation of IFN-γ polarized CD8+ T cells during the priming phase of antitumor immune responses in these models. Indeed, in the absence of the IL-1R1 or in the presence of IL-1 receptor antagonist, dying tumor cells failed to prime cancer-specific IFN-γ-producing CD8+ T cells (49).
Multiple distinct bacterial products or endogenous damage signals (such as toxins, ATP, uric acid crystals, alum, and silica) are able to stimulate the NLRP3 inflammasome, resulting in the proteolytic auto-activation of caspase-1 (60, 82, 103). One of the most abundant activators of the NLRP3 inflammasome is extracellular ATP, which is released from stressed cells and acts on purinergic receptors (26). ATP has an important pro-inflammatory role as a danger signal, especially when released in the extracellular environment in response to tissue damage and cellular stress. Through the activation of P2× and P2Y receptors, extracellular ATP enhances tissue repair, promotes the recruitment of immune phagocytes and DCs, and acts as a co-activator of NLRP3 inflammasome. ATP may be released from the mitochondria of necrotic cells (58) or can be secreted in an active, caspase- and autophagy-dependent way by apoptotic cells (87, 95). Once in the extracellular space, ATP may attract and activate innate effector cells of the immune system into the tumor bed (56), at least in part by binding P2Y2 purinergic receptors (37), thus also constituting a “find me” signal (37). Accordingly, expression of the ectonucleoside triphosphate diphosphohydrolase (ENTPD1, an ATP-degrading enzyme) reduced the chemoattractiveness of apoptotic cells (37).
Recent findings indicate that ATP is released by dying tumor cells succumbing to many chemotherapeutic agents (such as cadmium, etoposide, mitomycin C, oxaliplatin, cis-platin, staurosporine, thapsigargin, mitoxanthrone, or doxorubicin) (87, 95). This ATP release became undetectable when cells were incubated with the ATP-degrading enzyme apyrase or with inhibitors of ATP synthesis such as antimycin A plus deoxyglucose (A/D) or the oxidative phosphorylation uncoupler 2,4-dinitrophenol. When oxaliplatin- or anthracycline-treated cancer cell lines were admixed with these ATP scavengers or nonselective P2× receptor antagonists (iso-pyridoxalphosphate-6-azophenyl-2′,5′-disulphonate or oxidized ATP), they lost their capacity to elicit protective antitumor immune responses on subcutaneous inoculation into immunocompetent mice (49). Thus, ATP appears to be mandatory for the efficacy of some chemotherapeutic agents, especially immunogenic treatments (radiotherapy, anthracyclines, and oxaliplatin). Tumor cells manipulated to express the ATP ectonucleotidase CD39 on their surface or to be deficient in essential proteins of the autophagic machinery can usually undergo apoptosis in vitro and in vivo, yet failed to trigger an ICD pattern and to respond to chemotherapy (95). Importantly, the immunogenicity of autophagy-deficient or CD39-overexpressing tumor cells could be restored by the administration of inhibitors of extracellular ATPases (which increase pericellular ATP concentrations) or by the administration of recombinant IL-1β (170).
The high-affinity receptor for ATP present in DCs is the purinergic P2RX7 receptor (40). ATP stimulates P2RX7, thus activating the NLRP3/ASC/caspase-1 inflammasome and driving the secretion of IL-1β (49) (Fig. 5). The IFN-γ-polarized CD8+ T-cell response of P2RX7 −/− mice to oxaliplatin is reduced, and these animals fail to control the progression of established tumors under oxaliplatin treatment (49). Functional P2RX7 receptor on host DCs was mandatory for the chemotherapy-elicited anticancer immune response, as the adoptive transfer of bone marrow-derived WT (but not in P2RX7 −/− ) DCs loaded with dying tumor cells restored the tumor antigen-specific CD8+ T-cell response that had been originally impaired in P2RX7 −/− mice (49).
Altogether, these results are compatible with a scenario in which a danger signal, ATP, activates P2RX7 receptors on DCs, thereby stimulating the aggregation and/or activation of the NLRP3/ASC/Casp-1 inflammasome; then the proteolytic maturation of caspase-1, pro-IL-1β cleavage, and consequent IL-1β release (Fig. 5). Then, IL-1β is required for the priming of IFNγ-producing tumor antigen-specific CD8+ T cells (Th1/Tc1), and, thus, for the chemotherapy-elicited anticancer immune response (49). These data suggest that ICD induced by chemotherapy could influence the composition of the immune infiltrate present in tumors, which, in turn, could affect the therapeutic response. Supporting this assumption, we reported that autophagy-deficient tumors failed to attract DC and to accumulate IL-17-producing γδ T cells as well as IFN-γ-producing CD8+ T cells in tumor beds postimmunogenic chemotherapy, unless apyrase inhibitors be supplied to the tumor microenvironment (Fig. 6) (80, 88). When binding to IL-1R1, IL-1β (most likely produced by antigen-presenting cells exposed to dying bodies) attracts γδ T cells to the tumor site, leading to local production of IL-17 and the secondary recruitment of IFN-γ-producing CD8+ αβ T cells, eventually causing the lysis of tumor cells. Although the molecular links between γδ T17 and cytotoxic T lymphocytes (CTLs) were not elucidated, the percentage of γδ T17 cells appeared closely correlated with the frequency of IFN-γ-producing CD8+ T lymphocytes in the tumor microenvironment. Accordingly, in the absence of γδ T17 or IL-17, the priming of adaptive anticancer Th1/Tc1 responses was hampered, suggesting a helper role of γδ T17 cells in the context of ICD (80, 88).

Accordingly, the TLR4 ligand HMGB1 and ATP, both of which are released by tumor cells exposed to cytotoxic agents, act in concert to promote IL-1β secretion by DCs. The treatment of DCs with oxidized ATP or anti-HMGB1-neutralizing antibodies before loading with dying tumor cells completely abolished IL-1β secretion (49). As for the HMGB1/TLR4 interaction, the integrity of this ATP/P2RX7 pathway appeared to be clinically relevant, at least in N+ BCs, as loss-of-function alleles of P2RX7, which confer a reduced affinity of P2RX7 receptor for ATP, compromised the efficacy of anthracycline-based chemotherapy (49). Supporting this notion, relative elevated expression of ENTPD1 mRNA (which encodes the ectonucleotidase CD39) was associated with a poor outcome in colorectal cancer (73). Similarly, CD73, an ecto-5′ nucleotidase that acts downstream of CD39 to convert AMP into adenosine, was overexpressed in some human tumors (Table 1). CD73-deficient mice were protected against metastatic spreading, and harbor increased antitumor immunity (142). Together, these observations suggest that ATP-dependent, P2RX7-mediated activation of the inflammasome might also be clinically relevant for the efficacy of anticancer therapies in patients (49).
ALL, acute lymphoblastic leukemia; T-ALL, T-lineage ALL; C-ALL, common ALL; CR, complete response; M1, distant metastases; M0, no distant metastases; R, relapse within a 10 year period; NR, without relapse within a 10 year period; CLL, chronic lymphoblastic leukemia; CRC, colorectal cancer; OS, overall survival; EOC, epithelial ovarian carcinoma; GBM, glioblastoma; PTC, papillary thyroid carcinoma.
Perspectives
ICD has now entered the field of cancer immunization and vaccines (69). The comprehensive understanding of the mechanisms governing the immunogenicity of cancer cell death could have a profound impact on the current management of cancer patients. It pertains not only to the use of distinct chemotherapy compounds or various regimens of radiotherapy but also to the ear of “targeted” drugs. As an example, trastuzumab efficacy stems from a functional TLR4 pathway and is boosted by TLR4 agonists (113, 159, 164).
The first obvious and direct clinical implementation of these observations is the subversion of an “immune checkpoint blockade.” Tumors have evolved many immunosuppressive mechanisms to turn down the innate and the effector arms of the immune system. After the pioneering success of Ipilimumab in metastatic melanoma, investigators have conducted trials that are aimed at curtailing the other immune checkpoints. Both CTLA4 and the B7-H1 (PD-L1)/PD-1/B7.1 axis are best understood among the inhibitory B7-H family members featuring in the relevant “immune checkpoint blockers.” The therapeutic potential of novel immune checkpoint blockers (14, 149) has been recently highlighted in advanced cancer patients, including patients harboring lung or brain metastases. ICD may lead to anticancer immune responses that will be unleashed by such immune checkpoint blockers. The current issue is to analyze which regimen of combinatorial therapies (dose and scheduling) will be most suitable to achieve a synergistic anticancer efficacy.
Second, since human cancers carry hundreds of nonsynonymous mutations (several dozens among which lead to the generation of tumor-specific MHC class I-restricted epitopes), every patient's tumor harbors a highly specific mutational and antigenic signature. This “mutanome” can be identified by deep sequencing and can lead to individualized immunotherapies by means of ICD-inducing compounds and/or tailored vaccines (68). Clinical trials are ongoing and are aimed at combining anthracyclines or oxaliplatin and mutanome-derived mRNA epitopes in BCs (Biontech.de).
As already demonstrated, the propensity of tumor cells to undergo ICD in vitro can be exploited toward the therapeutic success of DC-based anticancer vaccines. In indolent B-cell lymphoma patients, antitumor responses were not observed when autologous tumor vaccines failed to harbor the ICD-related hallmarks (166). As mentioned earlier, in cohorts of breast or colon cancer patients treated with adjuvant anthracycline and local radiotherapy or oxaliplatin, respectively, genetic defects in the TLR4 or P2RX7 axis compromised the efficacy of these treatments (6, 49, 147).
However, it is anticipated that exploiting the ICD concept will require a “complete profiling” of the drug-tumor-host putative interactions (Fig. 7). First, compounds should be screened for their capacity to trigger an ICD pattern. The most prominent bottleneck is the failure of most anticancer drugs to trigger an ER stress response, culminating in CRT exposure (93). Hence, although cisplatin alone does not induce the preapoptotic exposure of CRT, it can do so in combination with the ER stressor thapsigargin, de facto rendering cisplatin-induced cell death immunogenic (86). Many novel ER stressors are currently developed and may enter the clinical armamentarium (137). Otherwise, recombinant CRT directly supplied into the tumor bed can compensate for the lack of ER stress response of a given pro-apoptotic compound (101). Second, tumors should exhibit the appropriate machinery to undergo proper ER stress, autophagy, and apoptosis responses when subjected to a tumoricidal compound. Hence, tumors defective in ERp57, HMGB1, or Atg5 will not die in an immunogenic manner and need appropriate compensatory therapies (6, 95, 110) (Fig. 7). We could speculate that failure of HMGB1 release could be compensated by administration of other potent TLR4/Myd88 agonists. Pharmacological inhibitors of extracellular ATPases, which can increase ATP concentration during ICD, could favor an efficient IFN-γ-polarized T-cell response against tumor cells (95). Novel antibodies targeting CD39 (10) could be of particular interest should the safety profile be ascertained. Third, hosts presenting with genetic defects compromising sensing of danger signals may fail to optimally respond to cell death in an immunogenic fashion. Compensation of TLR4 deficiency may rely on the administration of other TLR ligands to boost antitumor immune responses. Thus, administration of specific ligands of TLR3 or TLR9 after irradiation could restore potent antitumor effects in TLR4-deficient mice (5).

It is our hope that future anticancer strategies could be tailored to each patient and a tumor's genetic background, taking into account not only the cytotoxic properties of anticancer agents, but also their capacity to stimulate a host's innate and acquired immune responses. Hence, taking into account these parameters will lead either to a stratification of patients according to various categories pertaining to the tumor profile for ICD hallmarks only or to a personalization according to a complex algorithm retaining in the model the host genetic defects, the resistance to apoptosis, the lack of ICD, and the mutational status of the cancer.
Footnotes
Acknowledgments
This study was supported by the Ligue Nationale Contre le Cancer (Equipes Labellisée; L.Z. and G.K.), Agence Nationale pour la Recherche (L.Z. and G.K.), Fondation pour la Recherche Médicale (L.Z. and G.K.), Institut National du Cancer (L.Z. and G.K.), Cancéropôle Ile-de-France (L.Z. and G.K.), the European Commission (Apo-Sys (L.Z., G.K.), ArtForce (L.Z. and G.K.), ChemoRes (L.Z. and G.K.) and Death-Train (L.Z. and G.K.)), LabEx OncoImmunology (L.Z. and G.K.) and Institut National de la Santé et de la Recherche Médicale (O.K.), and Association pour la recherche sur le cancer (D.H.).