
Abstract
Introduction
W
Inside-out signaling is an important process coordinating intracellular changes in response to stress from cell surface signaling to maintain cellular homeostasis. Mitochondria are vital organelles that are involved in the orchestration of inside-out signaling, with mitochondrial reactive oxygen species (ROS) acting as a key player in this method of signaling. The results presented confirm the importance of mitochondrial ROS in the regulation of inside-out signaling and extend to demonstrate the importance of the ROS-scavenging ability of mitochondria, through manganese superoxide dismutase (MnSOD) expression and mitochondrial localization, in regulating inside-out signaling and the potential importance for pharmacological intervention of this vital pathway by Mn-containing SOD mimetics.
Aberrant mitochondrial function can also affect cell surface signaling in a process known as inside-out signaling. Lim et al. found that inhibition of mitochondrial function in C2C12 myotube cells by treatment with either ethidium bromide (to inhibit mtDNA synthesis) or oligomycin (to inhibit mitochondrial adenosine triphosphate production) resulted in reduced insulin signaling and glucose uptake through changes in the expression of IRS1 and the glucose transporter GLUT4 (32). An informative review by Valerie et al. discussed the importance of ROS in inside-out signaling, especially mitochondria-generated ROS, leading to early activation of different receptors on the cell surface after exposure to ionizing radiation, followed by autocrine/paracrine-dependent activation of cell surface receptor signaling through shedding of proligands on the cell surface (54). While mitochondrial ROS are key mediators of this inside-out signaling, the significance of the ROS-scavenging ability of mitochondria, through expression and mitochondrial localization of MnSOD, in regulation of this type of interorganellar communication, and its implications for disease development, is not well elucidated. Investigations focusing on the role of MnSOD in inside-out signaling will provide important insights into normal cellular function and disease states.
Epidermal growth factor receptor (EGFR) is a part of a family of four receptor tyrosine kinases (ErbB1-ErbB4) and is an important signaling protein in a variety of tissues. The EGFR family regulates diverse cellular functions, such as proliferation, differentiation, migration, and apoptosis (33, 45). EGFR is an important contributor to ultraviolet (UV)-induced skin cancer development. EGFR is a factor in UV-induced cytokine production and inflammation in the skin (15). Activation of EGFR by UV leads to increased keratinocyte proliferation and plays a role in epidermal hyperplasia after UV exposure (14, 17). Inhibition of EGFR has proved effective in suppression of UV-induced skin carcinogenesis (14). ROS are important contributors of EGFR activation, particularly UV-induced ROS. However, the effect of MnSOD on UV-induced EGFR activation in the context of inside-out signaling is not well known. A deeper understanding of the mechanisms of UV-regulated EGFR signaling, particularly ROS-mediated mechanisms, may lead to novel therapies for the treatment or prevention of carcinogenesis.
The purpose of this study was to investigate the hypothesis that changes in MnSOD expression affect inside-out signaling, and we studied how differences in MnSOD expression alter UV-induced EGFR activation. We report here that a decrease in MnSOD expression enhanced UV-induced phosphorylation of EGFR at Tyr1068, while increased MnSOD levels abrogated EGFR phosphorylation, both in vitro in HaCaT human keratinocytes and in vivo in SKH-1 mouse epidermis. This enhanced activation correlated with increased activation of the nonreceptor tyrosine kinase Src and expression of the NADPH oxidase Nox4. Inhibition of Src kinase activity or knockdown of Nox4 by lentiviral transduction of Nox4-specific shRNA blocked EGFR activation. Treatment with the SOD mimetic Mn(III) meso-tetrakis(N-n-butoxyethylpyridinium-2-yl)porphyrin (MnTnBuOE-2-PyP5+) inhibited UV-induced EGFR activation in vitro in MnSOD knockdown HaCaT cells. These results suggest a signaling axis between MnSOD, Src, and Nox4 for UV-induced activation of EGFR and may provide potential targets for early intervention in cancer treatment and prevention.
Results
MnSOD regulates UV-induced EGFR activation in vitro and in vivo
Both in vivo and in vitro, an inverse correlation occurred between MnSOD expression level and UV-induced activation of EGFR (Fig. 1). In MnSOD+/− (MnSOD deficient) epidermis, a significant increase in EGFR phosphorylation occurred at Tyr1068 [a common marker of UV-induced activation (14, 17)] compared with the corresponding untreated control. In MnSOD+/+ (wild-type) mice, an increase in Tyr1068 phosphorylation on EGFR also occurred, but the fold change compared with the corresponding control was much lower than that observed for MnSOD+/− mouse epidermis. In MnSOD+++ (MnSOD TgH overexpressing) mice, no UV-induced change was present in EGFR phosphorylation (Fig. 1A). Interestingly, total EGFR levels were not significantly altered in MnSOD+/− epidermis, while total EGFR levels were significantly reduced in both MnSOD wild-type and MnSOD TgH epidermis (Fig. 1B). Similar results for the effects of MnSOD on UV-induced EGFR activation were observed in vitro in HaCaT human keratinocytes. Knockdown of MnSOD by stable expression of shRNA targeted against MnSOD resulted in increased UV-induced EGFR activation (Fig. 1C), while overexpression of MnSOD suppressed UV-induced EGFR expression (Fig. 1D). These results suggest that changes in MnSOD expression alter the response of the epidermis to UV radiation by regulating EGFR activation.
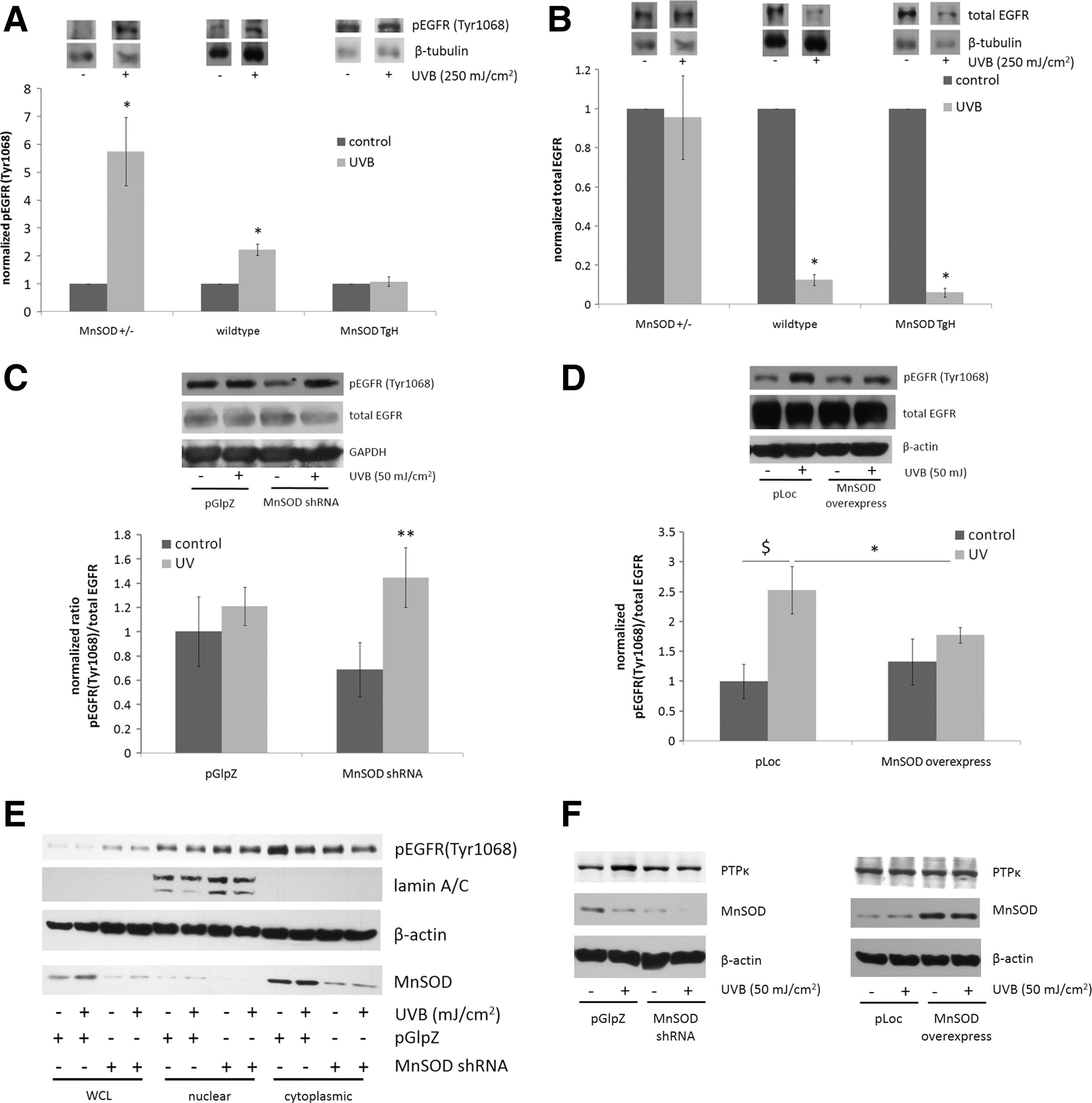
Nuclear translocation of EGFR is a key component of UV-induced signaling (63). To test whether changes in MnSOD expression affect EGFR nuclear localization, nuclear and cytoplasmic fractions were isolated from mock and UV-treated pGlpZ and MnSOD shRNA HaCaT cells and tested for the presence of EGFR. Knockdown of MnSOD resulted in an increase in the nuclear localization, and a decrease in cytoplasmic localization, of Tyr1068 phosphorylated EGFR (Fig. 1E), suggesting that changes in MnSOD expression affect subcellular localization of EGFR after UV exposure.
Protein tyrosine phosphatases are vital regulators of signaling, and receptor protein tyrosine phosphatase κ (PTPκ) has been identified as an important negative regulator of EGFR function. PTPκ particularly targets tyrosines 1068 and 1173 of EGFR, and it regulates UV-induced EGFR activation (62, 64). To test whether alterations in MnSOD expression affect PTPκ, Western blots were performed on mock and UV-treated HaCaT cells and tested for PTPκ protein levels. Knockdown of MnSOD resulted in an inhibition of UV-induced PTPκ protein levels compared with an empty vector control (Fig. 1F, left panel), while overexpression of MnSOD had no effect compared with an empty vector control (Fig. 1F, right panel). These results suggest that decreased MnSOD may enhance UV-induced EGFR activation, in part, by modulation of PTPκ protein levels.
MnSOD scavenges UV-induced ROS in vitro
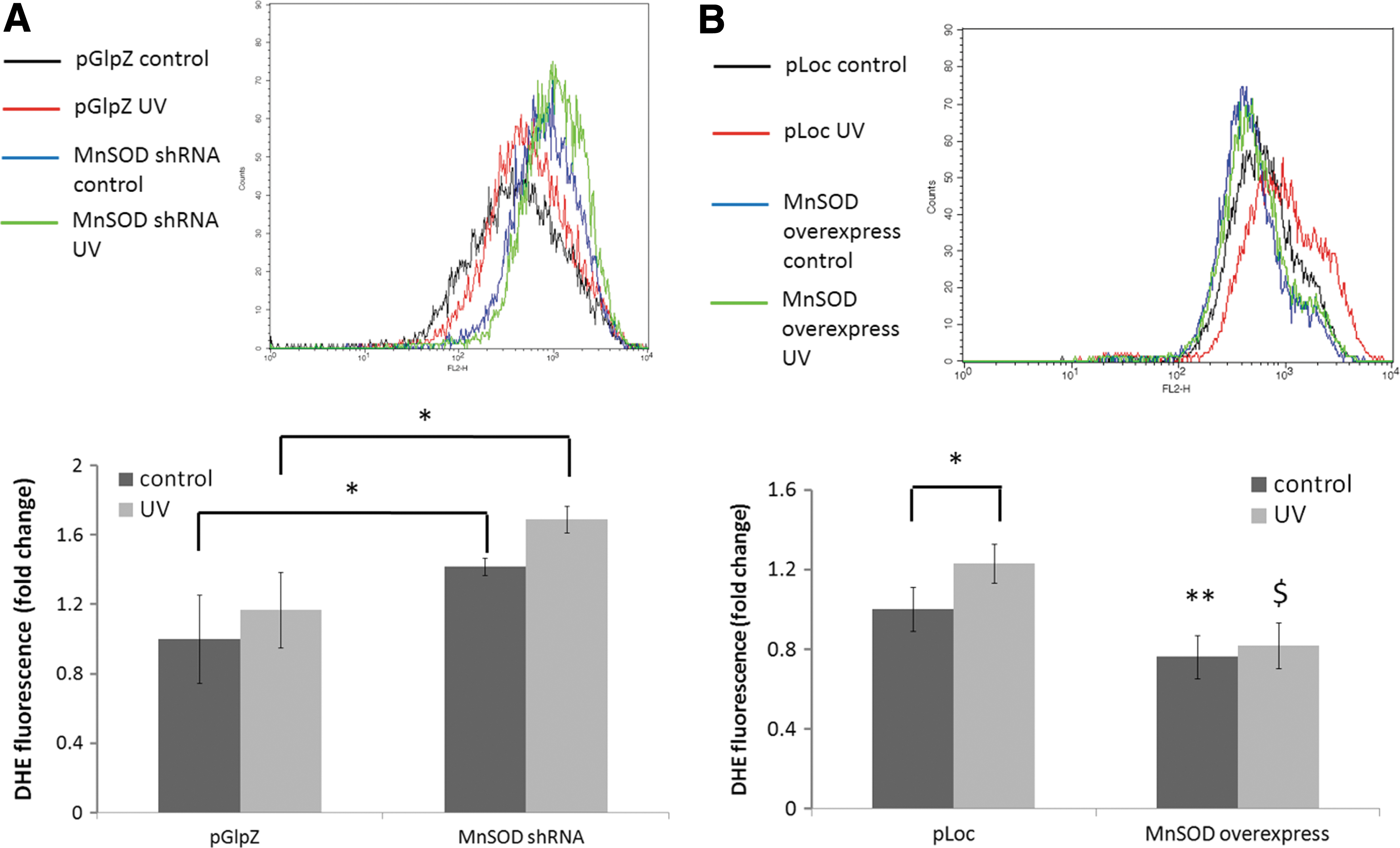
ROS are important mediators for the activation of EGFR by UV (23, 62). Owing to the importance of mitochondria in scavenging ROS, because of the presence of MnSOD, we hypothesized that changes in the level of MnSOD would affect UV-induced ROS production, which would contribute to MnSOD-mediated regulation of EGFR activation. ROS measurements were made using dihydroethidium (DHE) staining and quantification by flow cytometry. Knockdown of MnSOD led to an increase in UV-induced ROS generation at 24 h after UV (Fig. 2A), while overexpression of MnSOD suppressed UV-induced ROS generation (Fig. 2B). To determine whether hydrogen peroxide levels were also increased, an Amplex Red hydrogen peroxide assay was performed on the pGlpZ and MnSOD knockdown HaCaT cells. Interestingly, there was no detectable level of hydrogen peroxide in any of the samples (data not shown). These results demonstrate that MnSOD is important in the regulation of EGFR activation, in part, by scavenging UV-induced superoxide radicals.
Identification of NADPH oxidase 4 as an ROS generator in the effects of MnSOD on UV-induced EGFR activation
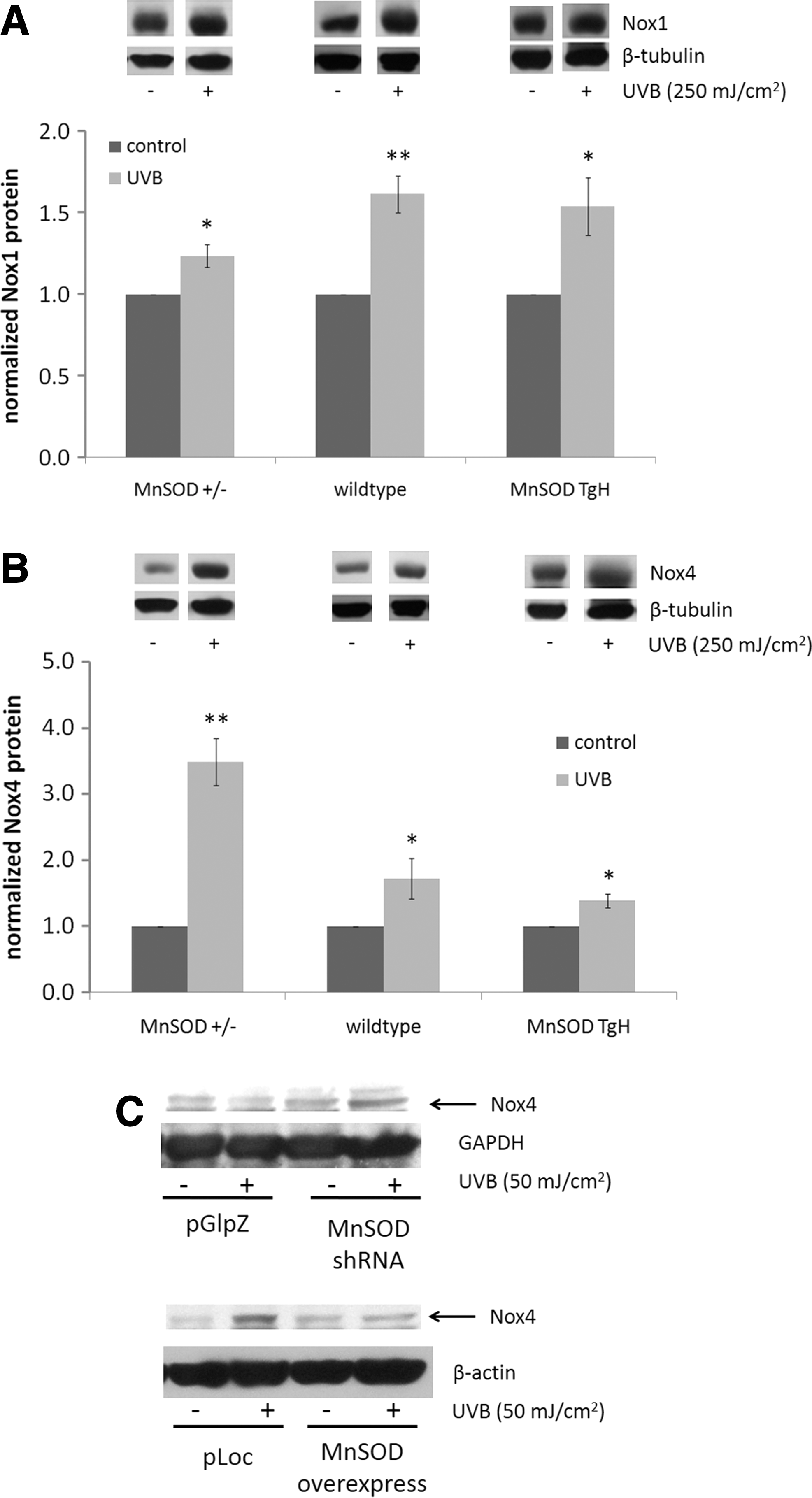
Since increased superoxide production was detected at 24 h postirradiation, we hypothesized that an internal ROS generator was the source of ROS which was expressed long after UV exposure that contributed to the overall pool of ROS. NADPH oxidases (Nox) are a large family of ROS-generating enzymes that are expressed in myriad tissues, and they play important roles in normal cellular function, including immune response and cardiovascular function (7). Nox is important in the activation of EGFR (25, 43); therefore, we hypothesized that changes in MnSOD expression may affect UV-induced ROS generation, in part, by altering the expression of Nox. Two forms of Nox have been identified in the skin: Nox1 and Nox4 (10). In vivo, there was a small, but significant, increase in Nox1 expression in all three genotypes of the epidermis after UV exposure (Fig. 3A). Interestingly, Nox4 protein levels in MnSOD+/− mouse epidermis were substantially and significantly increased after UV irradiation, while a much smaller fold increase was observed in MnSOD wild-type and MnSOD TgH mouse epidermis (Fig. 3B), indicating an inverse correlation between MnSOD expression and Nox4 expression after UV exposure. Similar results were observed in vitro: MnSOD knockdown enhanced, while MnSOD overexpression suppressed, UV-induced Nox4 expression (Fig. 3C). These results suggest a correlation between Nox4 expression and UV-induced EGFR activation with changes in MnSOD expression levels.
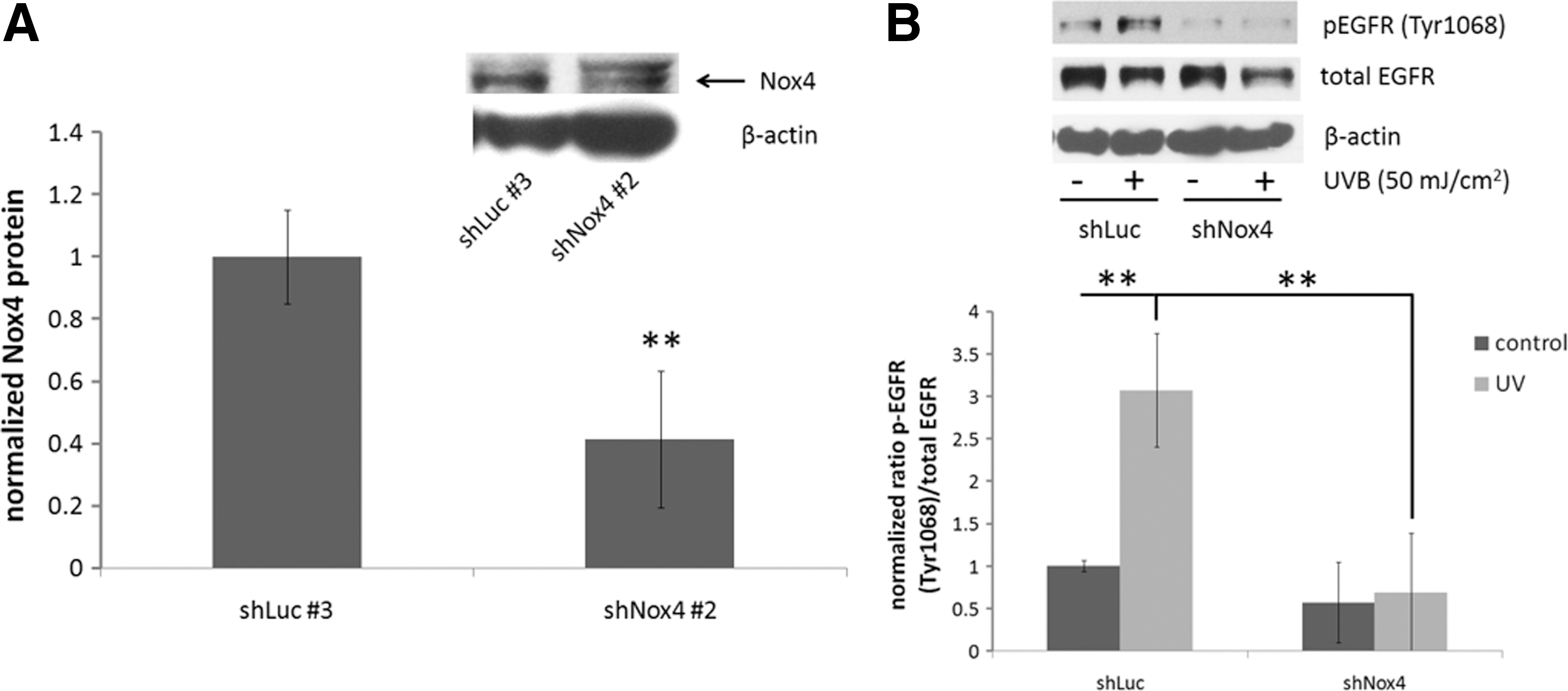
To determine whether Nox4 played a role in UV-induced EGFR activation, HaCaT human keratinocytes were generated by lentiviral transduction with an empty vector or an shRNA targeted against Nox4 and selection of individual clones. A 50% reduction in Nox4 expression was achieved using this method (Fig. 4A). Knockdown of Nox4 resulted in a complete abrogation of UV-induced stimulation of EGFR phosphorylation, while a statistically significant increase in EGFR phosphorylation was observed in vector control cells (Fig. 4B). These results indicate that Nox4 is important for EGFR activation after UV exposure and may play a role in the effects of MnSOD on EGFR activation.
Src kinase is upstream of Nox4 in the effects of MnSOD on EGFR activation by UV
The nonreceptor tyrosine kinase Src is another signaling protein that is vital for UV responsiveness (13), as it participates in cancer development and progression (11, 60). Crosstalk between EGFR and Src exists in cells (27, 44, 48). Therefore, we hypothesized that changes in MnSOD expression alter Src kinase activation, which may affect EGFR activation after UV. In HaCaT human keratinocytes, knockdown of MnSOD resulted in a substantial activation of Src (as measured by Tyr416 phosphorylation) after UV exposure compared with empty vector control cells (Fig. 5A). To ascertain whether Src is involved in EGFR activation after UV exposure in our system, cells were pretreated with dimethyl sulfoxide (DMSO) (vehicle) or the Src kinase inhibitor PP2 (1 μM) and exposed to UVB. PP2 completely inhibited UV-induced EGFR activation (Fig. 5B) and Nox4 expression (Fig. 5C) in both pGlpZ (left panel) and MnSOD shRNA (right panel) HaCaT cells, suggesting that Src activation is involved in the effects of decreased MnSOD on UV-induced EGFR activation, in part, through regulation of Nox4 expression.
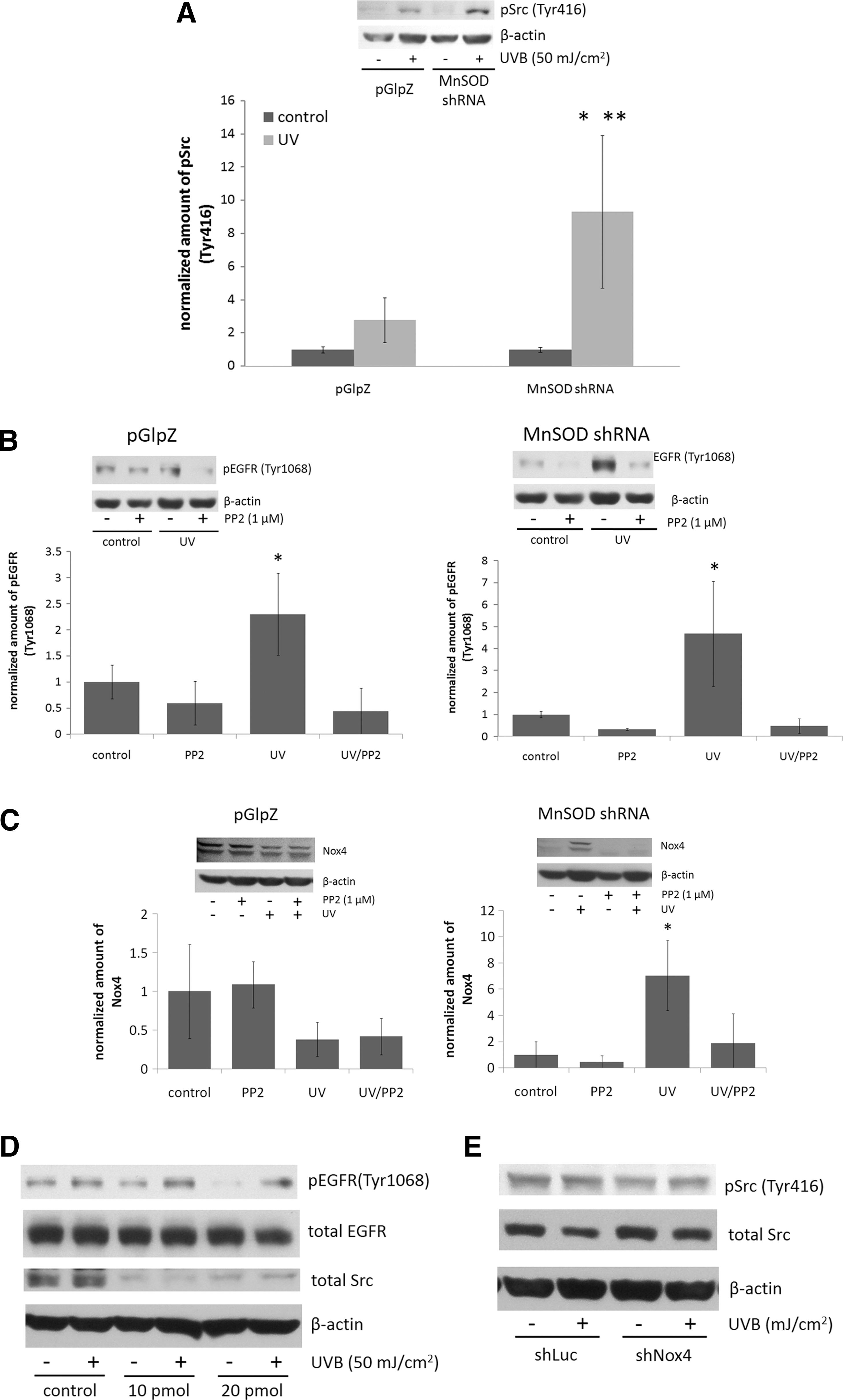
To confirm the role of Src in UV-induced EGFR activation, MnSOD shRNA HaCaT cells were transfected with Src-specific siRNA or control siRNA. There was a substantial knockdown of Src kinase protein levels after 4 days of transfection, and knockdown partially abrogated UV-induced EGFR activation (Fig. 5D) Knockdown of Nox4 had no apparent effect on UV-induced Src kinase activation (Fig. 5E), suggesting that Src activation may be upstream of Nox4 expression.
EGFR activation occurs independent of EGF secretion
Shedding of ligand from the cell surface is an important mechanism by which EGFR is activated (18, 28, 49). A variety of ligands bind to EGFR, including EGF, transforming growth factor α (TGFα), and heparin-binding EGF-like growth factor (HB-EGF) (21). To test the hypothesis that changes in MnSOD expression affect UV-induced secretion of EGFR ligand, we performed ELISAs for EGF on conditioned cell culture medium. Surprisingly, we were unable to detect EGF at 24, 48, and 72 h postirradiation, in spite of a good standard curve and measurement of EGF in control samples within the manufacturer's stated ranges (Supplementary Fig. S1; Supplementary Data are available online at
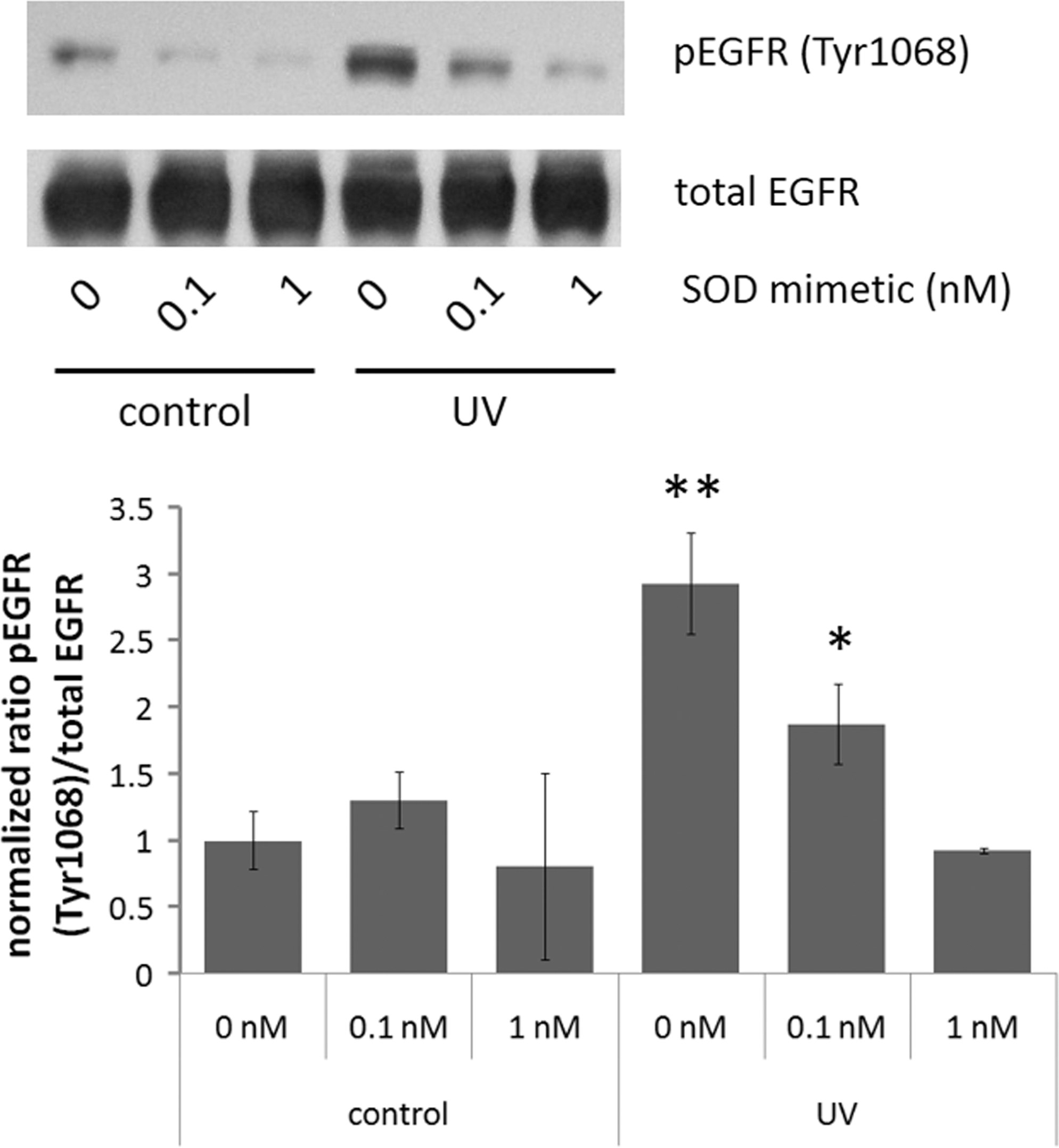
The effects of MnSOD on EGFR activation are replicated by an SOD mimetic
Since our in vitro and in vivo data demonstrated that overexpression of MnSOD suppressed UV-induced EGFR activation, we hypothesized that EGFR activation may be susceptible to inhibition by SOD mimetics. MnTnBuOE-2-PyP5+ is an SOD mimetic with a high lipophilicity and SOD-like activity but with significantly reduced toxicity relative to other Mn(III) N-substituted pyridylporphyrins as a consequence of insertion of one oxygen atom deep into each of N-alkylpyridyl substituents of Mn(III) N-alkylpyridylporphyrin-based mimetics (42, 52). The compound accumulates at a higher level in mitochondria than the cytosol, similar to equally lipophilic but more toxic MnTnHex-2-PyP5+ [(58) and Weitner et al. personal communication]. Its high mitochondrial accumulation is particularly relevant to this study, where MnTnBuOE-2-PyP5+ is used to mimic MnSOD. Pretreatment of HaCaT cells expressing MnSOD shRNA with different concentrations of MnTnBuOE-2-PyP5+ resulted in a concentration-dependent decrease in UV-induced EGFR activation (Fig. 6). MnTnBuOE-2-PyP5+ was well tolerated by the cells, with doses much higher than what were used in the UV experiments having no effect on cell viability (as determined by MTT assay, Supplementary Fig. S2). These results suggest that MnTnBuOE-2-PyP5+, similar to MnSOD overexpression, is capable of suppressing UV-induced EGFR activation, and it may provide an important pharmacological intervention in UV-induced EGFR activation.
Discussion
This study demonstrates a role for MnSOD in the regulation of UV radiation-induced inside-out signaling both in vitro and in vivo. Knockdown of MnSOD enhances, while MnSOD overexpression or treatment with SOD mimetic suppresses, UV-induced EGFR activation (Figs. 1 and 6). Interestingly, we observed a small, but not statistically significant, decrease in total EGFR levels in MnSOD+/− mice compared with MnSOD wild-type and MnSOD TgH mice. However, no change in total EGFR was observed in any HaCaT human keratinocyte cell lines tested. Possible explanations for this observation may include differences in keratinocyte proliferation among the three genotypes or, perhaps, decreased EGFR degradation in the MnSOD heterozygous knockout mice that results in no apparent change in total EGFR after UV. Knockdown of MnSOD also enhanced UV-induced nuclear translocation of EGFR and suppressed UV-induced increases in PTPκ protein. EGFR is vital for the normal response of skin to UV exposure (17) and is an important contributor to UV-induced carcinogenesis by stimulation of pro-survival and anti-apoptotic signaling pathways (16). To our knowledge, our study is the first report that MnSOD has an effect on the regulation of inside-out signaling. This study establishes the importance of mitochondrial ROS in UV-mediated inside-out signaling and extends to demonstrate the significance of the ROS-scavenging ability of mitochondria, through MnSOD, in regulating inside-out signaling.
Mitochondrial ROS are involved in inside-out signaling after radiation exposure by various mechanisms, with different cell surface receptors, including EGFR, being activated through inside-out signaling [reviewed in Ref. (54)]. Our data provide evidence that changes in the ROS-scavenging ability of mitochondria, due to changes in MnSOD expression, result in altered activation of inside-out signaling pathways. ROS play an important role in UV-induced activation of EGFR (23, 40), and our data demonstrate the effects of changes in MnSOD expression on EGFR activation as a part of an inside-out signaling mechanism. We used DHE staining and flow cytometry to monitor changes in superoxide production after UV exposure in MnSOD knockdown and MnSOD overexpressing HaCaT cells, and found that diminished MnSOD increased, while MnSOD overexpression suppressed, ROS production at 24 h after irradiation (Fig. 2). This change in ROS highly correlates with the changes in EGFR activation after UV exposure in MnSOD knockdown and MnSOD overexpressing keratinocytes and mouse epidermis. Our data also reveal that decreased MnSOD enhanced UV-induced expression of the ROS-generating enzyme complex Nox4 both in vitro and in vivo (Fig. 3), which is consistent with the effects of UV on EGFR activation. Using shRNA targeted against Nox4, we demonstrate that Nox4 knockdown in HaCaT keratinocytes results in complete abrogation of UV-induced EGFR activation compared with an empty vector control, supporting a role for Nox4 in UV-induced activation of EGFR (Fig. 4). Nox4 is a member of a large family of NADPH oxidases involved in a multitude of normal cellular activities (7); it is expressed in the skin (10); and its aberrant expression is associated with cancer. For example, increased expression of Nox4 has been identified in thyroid cancer (59), pancreatic cancer (30, 36, 56), as well as colon cancer (57), and a recent study reported that Nox4 localizes to mitochondria and its expression is increased in both breast and ovarian cancer patient samples (20). The fact that decreased expression of an antioxidant enzyme can lead to increased expression of an ROS-generating enzyme may seem counterintuitive. These data suggest an interesting interplay between antioxidant and pro-oxidant proteins that requires further exploration.
Our data also show a role for the nonreceptor protein tyrosine kinase Src in the regulation of UV-induced inside-out signaling by MnSOD. A substantial and significant increase in Src activation (as measured by Tyr416 phosphorylation) occurs in MnSOD knockdown HaCaT cells compared with empty vector controls. Inhibition of Src activation by the chemical inhibitor PP2 or knockdown of Src by siRNA results in the suppression of UV-induced Nox4 expression and EGFR activation (Fig. 5), suggesting that Src activation is an upstream event of Nox4 expression and confirming the importance of Src in MnSOD-regulated EGFR activation. Src activation is an early event in response to UV (13). Crosstalk exists between Src and EGFR, with Src as a target of EGFR (44); while other studies demonstrate that Src activates EGFR (48), especially after UV exposure (27). Src expression and/or activity is elevated in many types of cancer (11, 60), including skin cancers (2, 5, 29). Therefore, UV-induced activation of Src in cells with diminished levels of MnSOD may be considered an important early event in cancer development.
EGFR can undergo autocrine, paracrine, or juxtacrine activation by proteolytic cleavage of pro-ligand on the surface of the same cell or neighboring cells (18, 28, 49). A variety of ligands bind to EGFR, including EGF, TGFα, and HB-EGF (21). Cleavage of pro-ligands is an important component of inside-out signaling, especially long after the irradiation event occurs (54), and it is a major player in UV-induced EGFR activation (46, 47, 50). Interestingly, we were unable to detect any EGF in the conditioned medium in any of the samples at all time points tested (Supplementary Fig. S1), and may suggest a strictly ROS-mediated activation of the receptor independent of EGFR ligand secretion.
Our data also demonstrate that pretreatment of MnSOD knockdown HaCaT cells with the SOD mimetic MnTnBuOE-2-PyP5+ prevents UV-induced EGFR activation in a concentration-dependent manner (Fig. 6), indicating that enhanced UV-induced EGFR activation caused by diminished MnSOD expression is preventable by pharmacological intervention. SOD mimetics, in particular Mn-containing SOD mimetics, have proved attractive pharmacological agents for the modulation of oxidative stress that is associated with a multitude of diseases, including neurological and cardiovascular disorders, diabetes, and cancer. Mn-containing SOD mimetics are also effective for protection of normal tissues and sensitization of cancer tissues to ionizing radiation [reviewed in Ref. (6)]. This laboratory recently demonstrated that in MnSOD+/− C57BL/6 mice, there was an increase in UV-induced mtDNA damage due to oxidative inactivation of DNA polymerase γ (Pol γ) by tyrosine nitration. Pretreatment of the mice with the SOD mimetic MnIIITE-2-PyP prevented UV-mediated inactivation of Pol γ and protected mtDNA from damage resulting from UV (4). Previous work by this laboratory demonstrated that overexpression of MnSOD suppressed skin cancer formation in the DMBA/TPA two-stage model of skin carcinogenesis (67). Furthermore, treatment with MnIIITE-2-PyP SOD mimetic had a similar suppressive effect on DMBA/TPA skin cancer formation (66).
Multiple mechanisms contribute to UV-induced skin carcinogenesis, including epidermal hyperplasia, immune suppression, and DNA damage (34). EGFR contributes to UV-induced skin cancer development, with activation of EGFR by UV leading to increased keratinocyte proliferation, contributing to epidermal hyperplasia after UV exposure (17). ROS are important mediators of UV-induced EGFR activation (23). Mitochondria are major targets of UV radiation and they play a role in the response of skin to UV exposure (8). Mitochondria contribute to oxidative stress in keratinocytes after exposure to both UVA (19) and UVB (39) due to mitochondrial dysfunction.
mtDNA seems to be a particularly important target of UV radiation. Work by Jandova et al. identified a mtDNA mutation hotspot in the gene encoding tRNAArg in SKH-1 mice with UV-induced skin tumors (26). Takai et al. reported that in the SK-HEP-1 hepatoma cell, UV exposure led to increased mtDNA damage through hydrogen peroxide-induced oxidation of mtDNA (51). However, recent work from this laboratory found that MnSOD is vital in protecting DNA polymerase γ [the sole nuclease response for replication and repair of mtDNA (9)] from oxidative inhibition by UV radiation to prevent mtDNA damage (4). The apparent discrepancy between the studies by Takai et al. and this laboratory may be explained by the difference in the ROS balance between normal and cancer cells (37), resulting in the differential response to MnSOD expression and UV exposure. While abrogation of UV-induced mitochondrial ROS may not have a possible effect on nuclear DNA mutations caused by UV exposure, impeding the activation of EGFR may have an impact on the potential for epidermal hyperplasia and clonal expansion that contribute to skin carcinogenesis. This is an avenue of study worthy of further investigation.
Based on our in vitro and in vivo data, we propose the following model (Fig. 7). When MnSOD protein levels are low, UV exposure results in an increase in mitochondrial ROS generation, which leads to increased Src kinase activation. Src activation, in turn, leads to increased Nox4 expression, which contributes to increased ROS generation and EGFR activation long after UV irradiation. When MnSOD is overexpressed, or when an SOD mimetic is present, UV-induced mitochondrial ROS production is suppressed, preventing the downstream effects observed when MnSOD levels are low. This model is consistent with the notion of altering the balance of different ROS in the suppression of cancer development and progression (37). Combined with the mutagenic effects of UV radiation (12, 24, 55), a decrease in MnSOD expression may contribute to UV-induced carcinogenesis, in part, by enhancing UV-mediated activation of the important oncogenic pathway EGFR, via Src kinase, providing a unique and potentially important target for the pharmacological intervention of cancer by SOD mimetics.

Materials and Methods
Chemicals and antibodies
The chemicals used in this study are as follows: PP2 (Abcam Biochemicals; ab120308); DHE (Molecular Probes; D1168); blasticidin (Invitrogen; R21001); puromycin (Clontech; 631305); polybrene (Sigma; H9268); sodium pyruvate (Sigma; S8636); protease inhibitor cocktail III (Calbiochem; 539134); phosphatase inhibitor cocktail 2 (Sigma; P5726); phenylmethanesulfonyl fluoride (Sigma; P7626); dithiothreitol (BioRad; 161-0610), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma; M5655). Nox4 (sc-30141), β-tubulin (sc-9104), lamin A/C (sc-7293), and GAPDH (sc-47724) antibodies were purchased from Santa Cruz. EGFR (2646S), phospho-EGFR (Tyr1068, 2234S), Src (2109S), phospho-Src (Tyr416, 2113S), and cleaved poly(ADP-ribose) polymerase (5625P) antibodies were purchased from Cell Signaling. β-actin (A5316) antibody was purchased from Sigma. The SOD mimetic Mn(III) meso-tetrakis(N-n-butoxyethylpyridinium-2-yl)porphyrin (MnTnBuOE-2-PyP5+) was kindly provided by Dr. Ines Batinic-Haberle (Duke University Medical School). The following were obtained from Dharmacon: Dharmafect 1 transfection reagent (T-2001-01); ON-TARGET plus human Src siRNA (L-003175-00-0005); ON_TERGET plus nontargeting siRNA (D-001810-10-05), 5×siRNA buffer (B-002000-UB-100); molecular-grade RNase-free water (B-00300-WB-100).
Development of MnSOD knockdown and transgenic mice
SKH-1 is a hairless, albino mouse strain that is commonly used for the study of the effects of UV on the skin (3, 35). To study the effects of MnSOD on UV-induced changes in skin, we generated MnSOD heterozygous knockout (MnSOD+/−) and MnSOD transgenic (MnSOD+++) SKH-1 mice by crossing wild-type SKH-1 mice (Charles River) with MnSOD+/− C57BL/6 mice or MnSOD+++ B6C3 mice, respectively, followed by successive backcrossing with wild-type SKH-1 mice. Experiments were conducted with mice of generation 7 and later. Genotyping was performed on DNA isolated from tails as described in (31) and (65) (Supplementary Fig. S3). The animal experimental procedures performed in this study were approved by the Institutional Animal Care and Use Committee of the University of Kentucky.
Cell culture
HaCaT human keratinocytes (a kind gift from Holly Swanson, University of Kentucky) were maintained at 37°C in a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and 1×antibiotic/antimycotic solution (Sigma; A5955) and either 1 μg/ml puromycin (for MnSOD knockdown cells) or blasticidin (for MnSOD overexpressing cells).
Generation of lentiviral vectors and cell lines
Lentiviral constructs expressing SOD2 and its shRNA were obtained from Open Biosystems to manipulate the level of MnSOD in HaCaT human keratinocyte cells. The lentiviral constructs were packaged using the manufacturer-provided translentiviral packaging system before transduction into HEK293T cells, and lentiviruses were titered in accordance with the manufacturer's protocol. For stably expressing MnSOD and silencing MnSOD in HaCaT cells, a pLOC-SOD2ORF lentivirus or a pGIPZ-SOD2 shRNA lentivirus was transduced into the cells and stable pooled clones were selected using 1 μg/ml blasticidin or 2.5 μg/ml puromycin, respectively. Stable pools of cells were verified by green fluorescent protein (GFP) screening and further confirmed by Western blots (Supplementary Fig. S4).
Lentiviral constructs expressing an empty vector and shRNA targeting Nox4 were kindly provided by Dr. Lance Terada (University of Texas, Southwestern). The lentiviral construct was packaged and tittered as described earlier. For stable Nox4 knockdown in HaCaT cells, shNox4 and shLuc empty vector lentiviruses were transduced into parental HaCaT cells. Individual stable clones were selected based on GFP fluorescence and further screened by Western blotting (Fig. 4a).
UV treatment
UV treatments were conducted in a Plexiglas cabinet (Plastic Design Corporation). Mice or cells were exposed to UV radiation generated by UVB lamps (Black light blue lamp; Sankyo Denki Ltd.). UVB output was measured using a UV radiometer (International Light Technologies) that was equipped with a UVB measuring head.
For in vivo UV treatments, the mice were anesthetized and irradiated with 250 mJ/cm2 of UVB radiation (290–320 nm). Twenty-four hours postirradiation, the mice were sacrificed by cervical dislocation. Whole skin was collected, and epidermis was isolated by floating the skin (dermis side down) on filter paper in a 0.5 M EDTA solution (in 1×PBS) for 1.5 h at 37°C. The epidermis was physically separated from the dermis by scraping using forceps. The isolated epidermis was washed twice in ice-cold 1×PBS and flash frozen in liquid nitrogen. Whole tissue lysates were made using a 1×lysis buffer (described later in the Western blotting section).
For in vitro UV treatments, HaCaT cells were washed with 1×PBS, switched to serum-free DMEM (SF-DMEM), and incubated at 37°C for 4 h. After 4 h, the cells were washed, then covered with 1×PBS, and irradiated with 50 mJ/cm2 UVB radiation. The cells were then switched back to SF-DMEM and collected at 24 h postirradiation. The dose 50 mJ/cm2 UVB is a commonly used dose reported in the literature and did not cause any substantial increase in apoptosis in all four HaCaT cell lines used in this study (Supplementary Fig. S5).
Western blotting
Whole cell lysates were made by incubation of the cells in 1×cell lysis buffer (25 mM TRIS (pH=7.8), 2 mM dithiothreitol, 2 mM 1,2-diaminocyclohexane-N,N,N′N′-tetracetic acid, 10% glycerol, 1% Triton X-100, 1:100 dilution of protease inhibitor cocktail III, and 1:100 dilution of phosphatase inhibitor) on ice for 30 min, followed by centrifugation at 2.0 g and 4°C for 10 min. Protein estimation of the supernatant was determined by Bradford assay. Proteins were resolved using 8% or 10% polyacrylamide gels and transferred to nitrocellulose membrane by electroblotting. Membranes were blocked in 1×Tris-buffered saline (with either 0.05% or 0.1% NP-40) containing 5% milk or 5% bovine serum albumin, followed by incubation with primary antibodies (1:1000–1:10,000) overnight at 4°C. Membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies (1:1000–1:2500) for 2–3 h at room temperature. Membranes were treated with enhanced chemiluminescence reagents (GE Healthcare), and films were obtained. Images of the films were obtained, and densitometry of bands was determined by Chemidoc software (BioRad).
Isolation of nuclear and cytoplasmic fractions
Cells were plated at 1.5×106 cells per p100 plate (12 per cell type) and incubated at 37°C overnight. The next day, the cells were washed with 1×PBS, switched to SF-DMEM, and incubated at 37°C for 4 h. After 4 h, the cells were washed with 1×PBS, covered with 1×PBS, and mock or UV-irradiated (50 mJ/cm2). The cells were switched back to SF-DMEM and incubated at 37°C. The cells were collected at 24 h postirradiation. The cells were combined (an aliquot reserved for whole cell lysate), resuspended in buffer A (10 mM HEPES-KOH buffer, 1.5 mM MgCl2, 10 mM KCl, 200 μM PMSF, 0.77 mg DTT, and 1:100 dilutions of protease and phosphatase inhibitor cocktails), and incubated on ice for 15 min. After 15 min, 12.5 μl of 10% NP-40 was added, and the mixture was vortexed for 15 s. The samples were then spun down at 14,000 rpm for 30 s in a microcentrifuge. The supernatant (cytoplasmic fraction) was removed and stored at−80°C, and the resulting pellet was resuspended in buffer B (1.5 mM MgCl2, 420 mM NaCl, 35% glycerol, 200 μM PMSF, 200 μM EDTA, and 1:100 dilutions of protease and phosphatase inhibitor cocktails). The samples were incubated on ice for 20 min, then centrifuged at 12,000 rpm for 2 min in a microcentrifuge. The resulting supernatant (nuclear fraction) was removed and stored at −80°C. Whole cell lysates were made as described earlier. Whole cell lysates, nuclear fractions, and cytoplasmic fractions were resolved on 10% polyacrylamide gels, transferred to a nitrocellulose membrane, and Western blot was performed. Lamin A/C was used as a nuclear marker, and MnSOD was used as a cytoplasmic marker.
ROS detection and flow cytometry
ROS was detected using DHE staining and flow cytometry using a protocol modified from (1). Briefly, cells were plated at a density of 100,000 cells per well of 12-well plates. Twenty-four hours later, the cells were washed with 1×PBS containing 5 mM pyruvate and trypsinized. The cells were resuspended in 1×PBS (5 mM pyruvate) containing either DMSO (for background fluorescence) or DHE (final concentration of 10 μM) and incubated at 37°C for 40 min. After 40 min, the samples were placed on ice and then transferred to Falcon 12×75 mm round-bottom tubes by filtering through a cell strainer. The samples were kept on ice, and flow cytometry was conducted using an FACSCalibur flow cytometer (Becton Dickinson).
Hydrogen peroxide was measured using an Amplex Red hydrogen peroxide/peroxidase assay kit (Life Technologies; A22188). Cells were plated at 200,000 cells/well in six-well plates and incubated overnight at 37°C. The next day, the cells were washed with 1×PBS, switched to SF-DMEM, and incubated at 37°C for 4 h. After 4 h, the cells were washed with 1×PBS, covered with 1×PBS, and mock or UV-irradiated (50 mJ/cm2). The cells were switched back to SF-DMEM and incubated at 37°C. Twenty-four hours postirradiation, the cells were washed with 1×PBS and trypsinized, followed by one additional PBS wash. The cells were lysed in 0.1% triton X-100 (in 1×PBS) using a hand homogenizer. Cell lysate (20 μl) was diluted to 50 μl using 1×reaction buffer and added to separate wells of a 96-well plate. Serial dilutions of a 10 μM hydrogen peroxide stock solution were made and added to the plate. The reaction was started by adding 50 μl of the Amplex Red working solution to each well (100 μM Amplex Red and 0.2 U/ml horseradish peroxidase in 1×reaction buffer). The plate was incubated at room temperature for 30 min protected from light. After 30 min, the plate was read on a fluorescent microplate reader (excitation 544 nm, emission 590 nm). Protein concentration was determined by Bradford assay.
Knockdown of Src expression
Src siRNA experiments were conducted according to Dharmacon's protocol. Briefly, cells were plated at 100,000 cells per well of six-well plates in serum-containing DMEM (without antibiotics) and incubated at 37°C overnight. The next day, the cells were transfected with nontargeting control siRNA (20 pmol) or human Src siRNA (10 or 20 pmol). The next day, the cells were switched to complete DMEM containing both serum and antibiotics/antimycotics and incubated at 37°C for an additional 2 days. The cells were then treated according to the procedure described earlier (UV Treatment, 50 mJ) and collected at 24 h postirradiation. Src knockdown and EGFR phosphorylation were determined by Western blotting.
ELISA for determination of secreted EGF
Cells (pGlpZ and MnSOD shRNA) were plated at 1.5×106 cells per p100 plate (six plates per group). The next day, the cells were treated according to UV treatment protocol stated earlier. Cell culture supernatant was collected at 0, 24, 48, and 72 h postirradiation and spun down for 5 min to remove any debris, aliquoted into 1.5 ml microcentrifuge tubes, and frozen at −80°C. Secreted EGF was measured using an EGF ELISA kit (R&D Systems; DEG00) according to R&D Systems protocol.
MTT assay for cell viability
HaCaT cells (pGlpZ and MnSOD shRNA) were plated at 37,500 cells per well of 24-well plates and incubated at 37°C overnight. The next day, the cells were treated with different concentrations of MnTnBuOE-2-PyP5+ (0, 0.1, 0.5, 1, and 5 μM) and incubated at 37°C. MTT assay was performed after 24 h of mimetic treatment. The cells were washed with 1×PBS and incubated with MTT working solution (5 mg/ml in PBS diluted 1:10 in complete DMEM, 0.5 ml per well) for 1 h at 37°C. After 1 h, 0.5 ml of DMSO was added to each well, and cells were incubated at 37°C for 1 h to dissolve the formazan produced by the cells. Two hundred microliters of the resulting solution was transferred to a 96-well plate, and absorbance was measured at 540 nm.
Statistical analysis
Unless otherwise stated, all data are graphed as the average±standard deviation. Statistical significance was determined by Student's unpaired t-test or either one-way or two-way analysis of variance followed by Bonferroni post-hoc tests using GraphPad Prism software. p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
The authors wish to thank Lance Terada (University of Texas, Southwestern) for the kind gift of the shLuc and shNox4 constructs and Holly Swanson (University of Kentucky) for the kind gift of the HaCaT cell line. The authors also wish to thank Greg Bauman and Jennifer Strange of the Flow Cytometry Core at the University of Kentucky for conducting the flow cytometry experiments. This study is supported by NIH grants CA 49797, CA 073599, T32 ES007266, and the Edward P. Evans Foundation.
Author Disclosure Statement
The authors declare that they have no conflicts of interest to disclose.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.