
Abstract
Introduction
M
Epithelial-mesenchymal transition (EMT), a motogen and redox-dependent program used by cancer cells to escape the hostile primary tumor milieu, is engaged in response to activation of cancer-associated fibroblasts (CAFs) and/or incipient hypoxia. Here, we identify miR-205 as a mandatory molecular player of CAF-driven EMT, acting downstream to cycloxygenase-2-mediated oxidative stress and stabilization of hypoxia-inducible factor-1α and affecting stemness of metastatic cells. Noteworthy, ectopic overexpression of miR-205 can both prevent and rescue stromal reactivity and cancer aggressiveness, in addition to survival and growth of metastatic colonies, thereby representing a novel and promising tool for therapeutic approaches aimed at regulating epithelial/mesenchymal cell plasticity.
In addition to tumor growth factor-β (TGF-β), we recently acknowledged interleukin-6 (IL-6) as the main factor secreted by aggressive prostate cancer (PCa) cells, which elicits reactivity of stromal fibroblasts and converts them into CAFs (23). In turn, activated CAFs secrete MMP-2 and MMP-9, which induce epithelial-mesenchymal transition (EMT) in PCa cells, thus ultimately enhancing their aggressiveness (18,23). Indeed, EMT has been associated with increase in proteolytic motility of cancer cells, enhancement of anoikis resistance, and achievement of stem-like traits (7,24,34). In keeping with such observations, PCa cells experiencing EMT upon CAF contact enhance their invasiveness, self-renewal ability, capacity to grow as adherence-independent prostaspheres, expression of stemness markers, and ability to spread as spontaneous lung metastases. CAF-induced EMT of PCa cells is driven by a pro-oxidant pathway involving activation of Rac1b and leading to delivery of reactive oxygen species (ROS), through the modulation of cycloxygenase-2 (COX-2) (22,50). Oxidative stress leads to activation of two redox-sensitive transcription factors, hypoxia-inducible factor-1α (HIF-1α) and nuclear factor-κB (NF-κB), which start the EMT transcriptional program (22,40,46).
We identify in microRNAs (miRNAs), endogenous small non-coding RNAs that negatively regulate gene expression during key cellular processes (2), potential candidate mediators of CAF-induced EMT. A few miRNAs have been involved in ROS handling by cancer cells and in EMT regulation. For example, members of the miR-200 family have been shown to regulate EMT through the control of key transcription factors like ZEBs and Snails (25,28,37,56), although a direct regulation by elements of the tumor microenvironment has not been reported. In addition, miR-141 and miR-200a have been involved in Nrf2/Keap1 oxidative stress and response to paclitaxel therapy of ovarian cancers (36).
The aim of this study was to identify miRNAs specifically involved in EMT engaged by CAFs in PCa cells and establish their hierarchy with the already accredited signaling pathways involving redox regulation of HIF-1α and NF-κB, to develop possible tools to target EMT and prevent metastasis dissemination.
Results
miR-205 is repressed in PC-3 cells upon CAF stimulation
To verify whether specific miRNAs are involved in CAF-induced EMT in PCa cells, PC-3 cells were subjected to a variety of stimuli [known to induce or not the acquisition of a mesenchymal phenotype (22,23)] and profiled for miRNA expression on a microarray platform. Unsupervised hierarchical clustering showed that miRNA profiles could distinguish cells that underwent EMT following stimulation with activated fibroblasts (including CAFs and human prostate fibroblasts—here referred to as HPFs—activated in vitro with IL-6 or TGF-β) from cells that did not, such as unstimulated or HPF-stimulated cells (Fig. 1A). PC-3 cells pre-incubated with siRNA against HIF-1α or COX-2 (assessment of their knockdown is reported in Supplementary Fig. S1; Supplementary Data are available online at
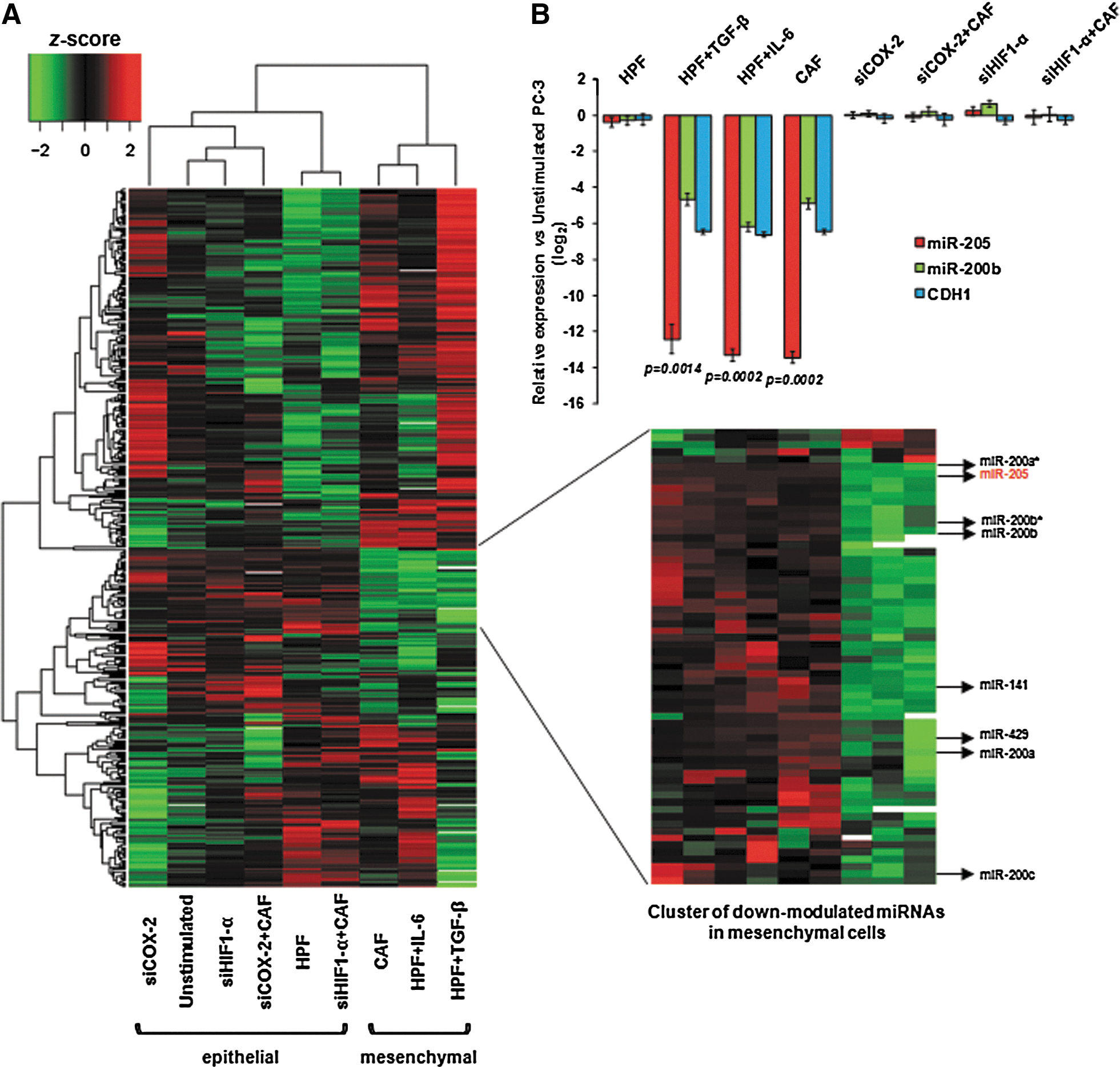
miR-205 loss is associated with PCa progression and metastasis
Downregulation of miR-205 in PCa compared to normal tissues was initially reported by us (19) and confirmed later by a number of miRNA expression profiling studies [reviewed in Gandellini et al. (20)]. To specifically assess the impact of miR-205 downmodulation on PCa progression and metastasis, we analyzed the largest available set of miRNA and gene expression profiling data (52). MiR-205 expression levels were globally reduced in primary carcinomas compared with normal tissues and further diminished in metastases (Fig. 2A). The miRNA level distribution across primary tumors, all obtained from treatment-naive patients subjected to radical prostatectomy, highlighted a subset of cases (about 17%) with expression levels comparable to those of metastases (Fig. 2A, B). Strikingly, such cases derived from patients a significantly increased risk (hazard ratio, 0.28; 95% confidence limits, 0.1–0.77; P-value=0.014) of experiencing biochemical relapse within 5 years (Fig. 2C).
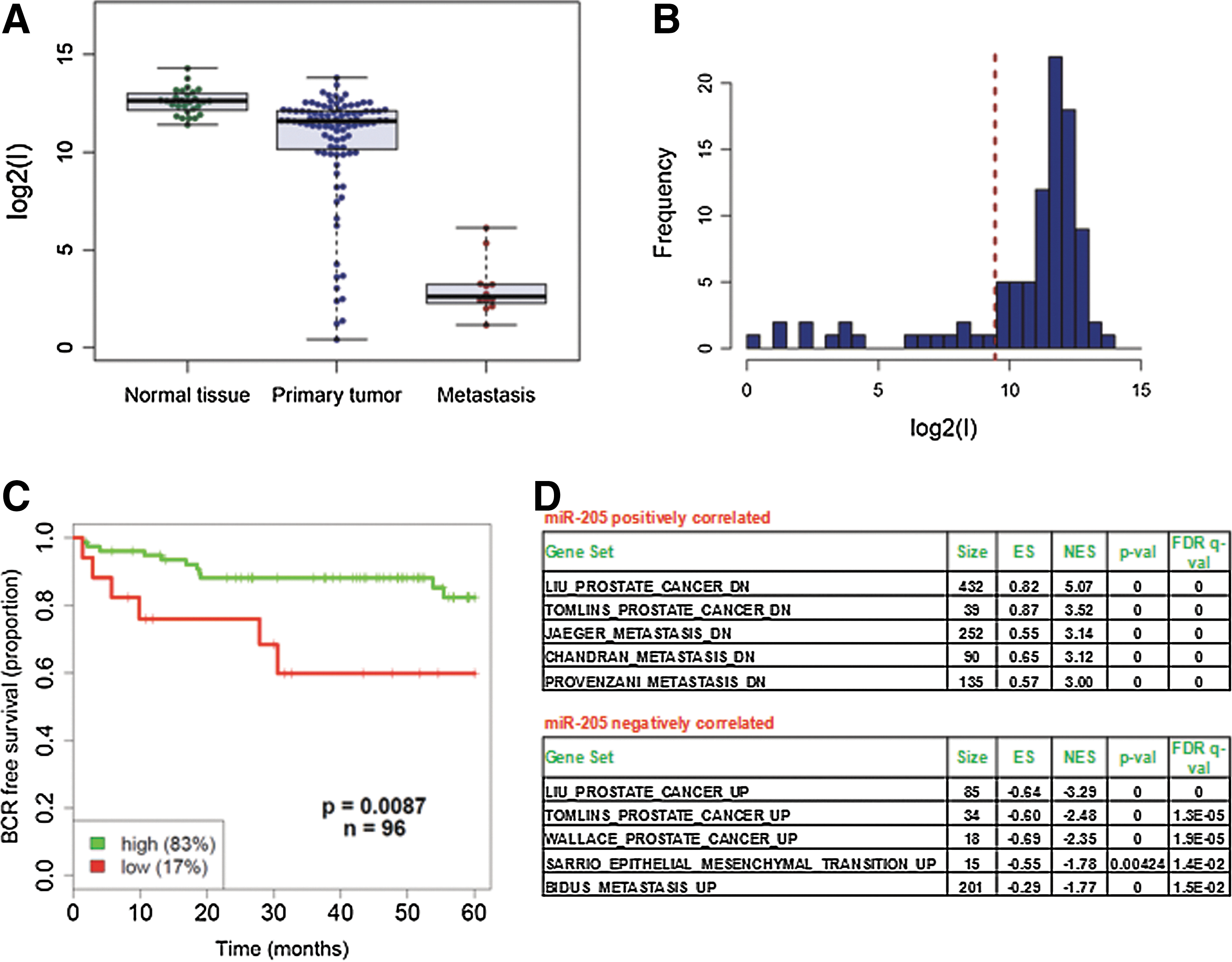
To further elucidate the role of miR-205 in PCa, gene set enrichment analysis (GSEA) was performed on genes correlated with miR-205 expression in the aforementioned dataset. Among positively correlated genes, we found significant enrichment of gene sets reported to be downmodulated in PCa by different groups (Liu, Tomlins datasets) or downregulated in metastates (Jaeger, Chandran, Provenzani datasets) (Fig. 2D and Supplementary Table S2). Among negatively correlated genes were instead those upregulated in PCa (Liu, Tomlins, Wallace datasets), in metastases (Bidus dataset), or during EMT (Sarrio dataset), thus highlighting, in the clinical setting, the possible antagonistic effect of miR-205 on the development and progression of the disease (Fig. 2D and Supplementary Table S3).
A hierarchy exists between miR-205 downregulation and the proinflammatory pathway leading to EMT
To evaluate the kinetics of miR-205 downmodulation induced by tumor stroma and to assess whether reduction of miR-205 was either due to increased degradation or reduced transcription/processing, the levels of the primary transcript (pri-miR-205) and the mature form of the miRNA were measured at different time points after stimulation of PC-3 cells with CAF-conditioned medium (CM), obtained by treating CAFs with serum-free medium for 48 h. Serum-starved cells were used as control. We found that expression of both pri-miR-205 and mature miR-205 started to decrease from day 2 and returned to normal levels from day 5 (Fig. 3A), thus completely matching the derepression of miR-205 direct targets ZEB1 and ZEB2 (Fig. 3A). Accordingly, overlap between miR-205 reduction and appearance of EMT traits, such as downmodulation of E-cadherin and upregulation of vimentin, was observed (Fig. 3B). The expression of other EMT-related transcription factors (i.e., SNAI1, SNAI2, and TWIST1) was not substantially modulated compared to changes in miR-205, ZEB1, and ZEB2 (Supplementary Fig. S3). Notably, activation of HIF-1α, COX-2, and NF-κB pathways peaked at day 1 and became negligible from day 4 (Fig. 3B). Taken together, these data suggested that miR-205 is mainly repressed at the transcriptional level, hypothesis strengthened by the observation that the expression levels of miR-205 primary transcript are lowered despite potential reduced processing to pre-miRNA due to Drosha downregulation (Supplementary Fig. S2). In addition, HIF-1α and NF-κB may be potential direct transcriptional repressors, since their activation immediately precedes reduction of miR-205 expression levels and modulation of ZEB1/2. Again, CAF-induced EMT appears reversible, likely due to depletion of specific factors produced by CAFs. Actually, reversion of EMT is accelerated by removing CAF-CM (Fig. 3C), suggesting that CAF-induced EMT is an epigenetic phenomenon that is not genetically fixed.

To assess whether miR-205 is transcriptionally repressed by HIF-1α, which we have acknowledged to be redox-targeted by the COX-2-dependent EMT activation (22), its promoter (3) was scanned for potential binding sites. We found at least six highly predicted consensus sequences for the transcription factor, three of which on the sense strand (Fig. 4A). Chromatin immunoprecipitation (ChIP) experiments performed using primers to amplify regions containing the three sites (Fig. 4A) evidenced increased PCR signal for all the regions in anti-HIF-1α versus IgG immunoprecipitants from CAF-stimulated cells, especially for site 1 (132-fold increase), thus confirming enriched binding of HIF-1α to such consensus sequences (Fig. 4B, CAF lanes). All three regions appeared to be essentially unbound in HPF-stimulated cells but showed 7.7-, 3.2-, and 2.8-fold enriched binding, respectively, upon CAF stimulation (Fig. 4B). Such findings suggest that changes in HIF-1α steady-state levels, such as those deriving from CAF stimulation, may alter miR-205 promoter occupancy by HIF-1α and, consequently, affect miRNA transcription. To further prove that miR-205 is subjected to direct transcriptional repression, we obtained a plasmid containing a portion of 2 kb of miR-205 promoter, which contains two of the three HIF-1α binding sites, cloned upstream to firefly luciferase (49). PC-3 cells were transfected for 4 h with such a construct, together with pRL-TK renilla luciferase vector, then stimulated with the CM from either CAFs or HPFs. Dual luciferase assay was then performed at different time points after transfection to calculate the firefly/renilla ratio. Results showed a significant decrease in the ratio in CAF-treated compared to HPF-treated cells at all time points considered (24, 48, and 72 h), suggesting that the 2 kb portion of miR-205 promoter is directly responsive to factors produced by CAFs (Supplementary Fig. S4), such as HIF-1α.
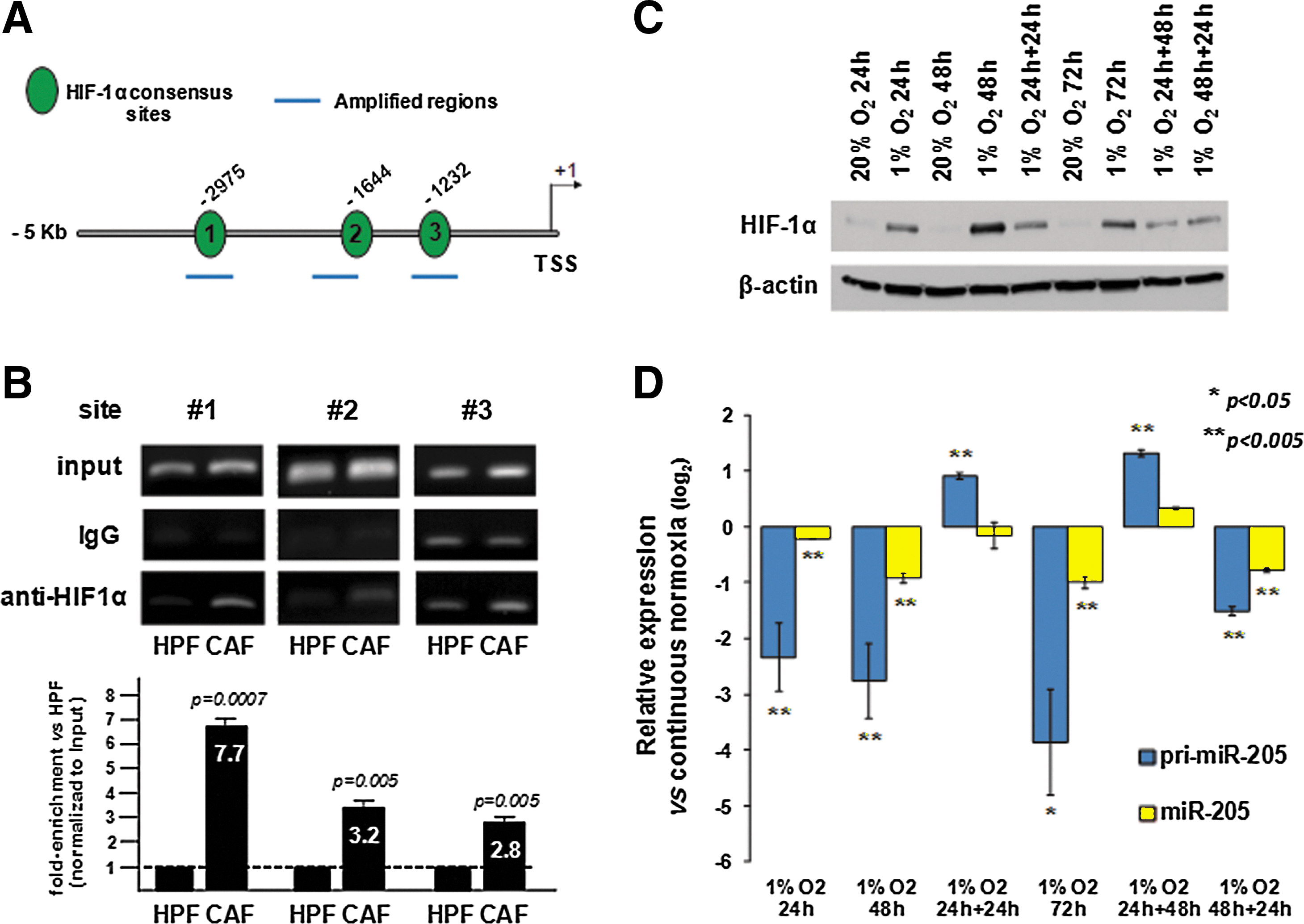
In keeping with these data, culturing of PC-3 cells under hypoxic conditions, which also increase HIF-1α levels (Fig. 4C), caused a time-dependent downmodulation of both primary and mature miR-205 transcripts (Fig. 4D). Interestingly, downmodulation of miR-205 after HIF-1α activation by hypoxia again seemed to be a reversible phenomenon, as recovery of cells in normoxia restored or even increased the levels of primary and mature miR-205 (Fig. 4D). Similar results were obtained by culturing cells in the presence of cobalt chloride (CoCl2), which stabilizes HIF-1α protein by inhibiting prolyl hydroxylase activity (51), for different time intervals and recovering them in CoCl2-free medium (Supplementary Fig. S5). Again, HIF-1α activation induced a reversible downregulation of pri-miR-205 levels, which was, however, insufficient to induce robust modulations of mature miR-205 within the experimental time frame (Supplementary Fig. S5).
miR-205 prevents CAF-induced EMT and fibroblast activation by PCa cells
To investigate whether miR-205 downmodulation is functionally involved in CAF-induced EMT, a rescue experiment, where reduction of miRNA levels was prevented by transfecting PC-3 cells with synthetic miR-205 precursor prior to CAF stimulation, was carried out. Transfection efficiency was checked using a fluorescein-labeled oligomer and found to be ∼90% after a 4-h transfection (Supplementary Fig. S6A). Moreover, transfection of PC-3 cells with synthetic miRNA precursor resulted in a marked overexpression (387- and 179-fold at days 1 and 2 post-transfection, respectively) of mature miR-205, as assessed by qRT-PCR (Supplementary Fig. S6B). Conspicuous overexpression of miR-205 was found to persist in miR-205-transfected cells until 9 days after transfection, when a 17-fold overexpression of the miRNA was observed (Supplementary Fig. S6B).
Ectopic miR-205 expression efficiently prevented loss of E-cadherin expression (Fig. 5A), attenuated the increase in cell invasion (Fig. 5B), and reduced anoikis resistance (Fig. 5C) of PC-3 cells stimulated with CAFs. Further, in keeping with the strict link between EMT and the achievement of stem-like traits (23,34,44), ectopic miR-205 overexpression in PCa cells counteracted the acquisition of stem-like properties, in terms of prostasphere formation (Fig. 5D) and presence of CD44high/CD24low (Fig. 5E) and CD133-positive subpopulations (Fig. 5F), acknowledged markers of PCa stem cells (8,31,48). Interestingly, silencing of the only miR-205 in PC-3 cells by the use of locked nucleic acid (LNA)-modified antisense oligonucleotide phenocopied CAF-induced EMT, in terms of E-cadherin and Vimentin expression pattern, and the CAF-induced pro-invasive effect and acquisition of stem cell traits (Supplementary Fig. S7). Notably, the ability to undergo CAF-mediated EMT is a peculiar property of castration-resistant PCa cells, such as PC-3 and DU145, which express low levels of androgen receptor (AR), and 22Rv-1, which express high levels of AR but are androgen refractory (Supplementary Fig. S8). On the contrary, the androgen-sensitive LNCaP cells are unable to undergo CAF-mediated EMT, while remaining responsive to E-Cadherin upregulation promoted by the ectopic expression of miR-205 (Supplementary Fig. S8).

Once ascertained the capability of miR-205 to interfere with the afferent pathway [i.e., CAF-promoted EMT in PCa cells (23)], we verified whether the miRNA also affects the efferent pathway, that is the tumor-induced activation of fibroblasts. Given that IL-6 is the main soluble factor produced by PCa cells to activate the surrounding stroma (23), cytokine amounts were measured in the CM of PC-3 cells exposed to different stimuli (Fig. 6A). Ectopic miR-205 overexpression was found to counteract the enhancement of IL-6 secretion triggered by CAFs, as assessed by western immunoblotting (Fig. 6A, top) and enzyme-linked immunosorbent assay (ELISA) (Fig. 6A, bottom). As a consequence, media from miR-205-expressing PC-3 cells stimulated with CAFs were unable to promote activation of normal prostatic fibroblasts, as assessed by measuring fibroblast activation protein expression levels (Fig. 6B). Accordingly, such fibroblasts could not enhance PCa cell invasion (Fig. 6C).
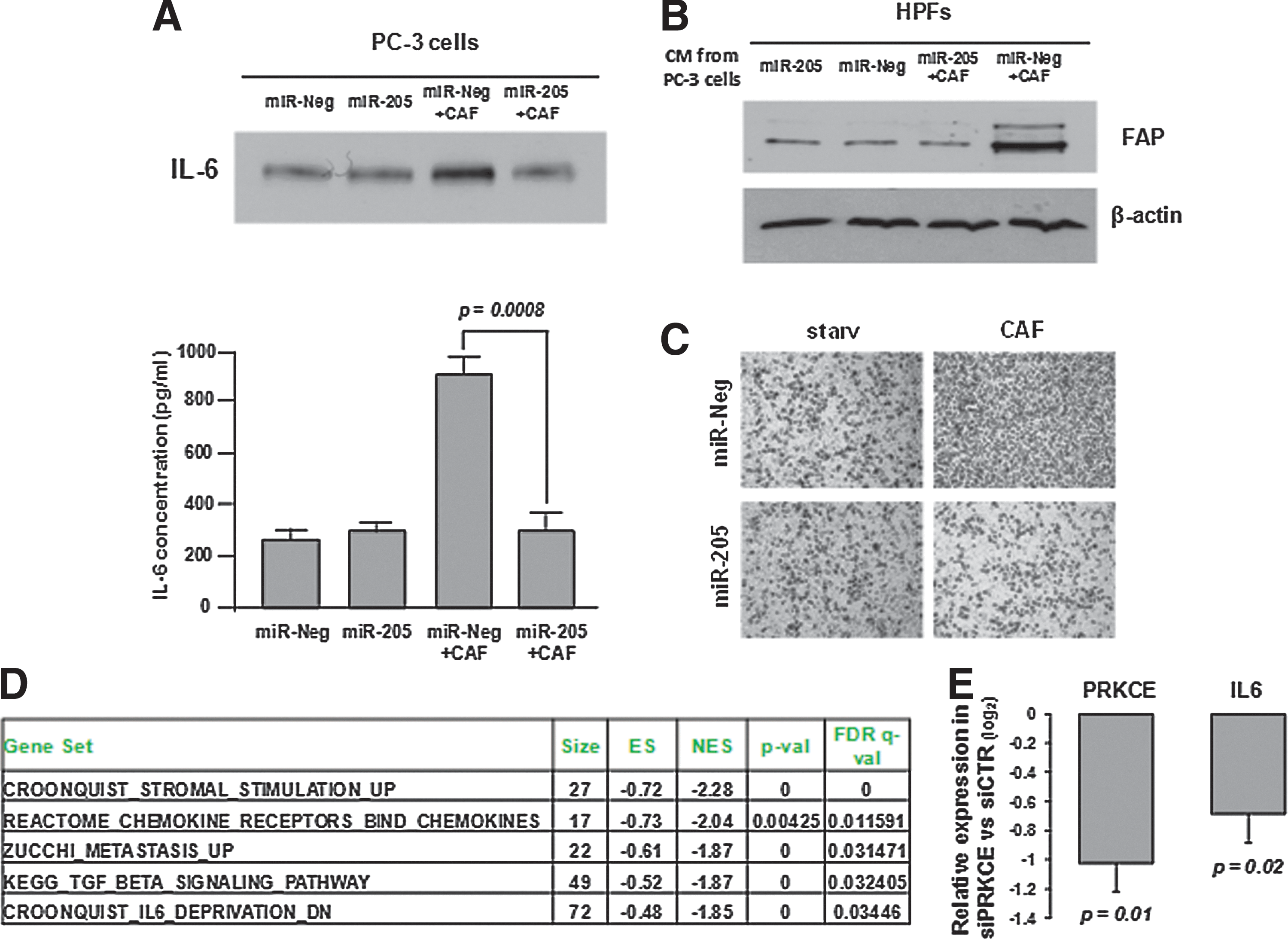
By analyzing gene expression profiles obtained for DU145 PCa cells restored in miR-205 expression (GSE11701 dataset) (19), enrichment of gene sets related to cytokine and chemokine activity (including IL-6) was observed among genes downmodulated by the miRNA (Fig. 6D and Supplementary Table S4), thus confirming the ability of miR-205 to repress IL-6 production by PCa cells. On the same dataset, and consistent with the role of miR-205 in counteracting the EMT program, inverse expression patterns were identified between miR-205 and genes involved in metastasis or TGF-β signaling (Fig. 6D and Supplementary Table S4). Enrichment of gene sets related to the IL-6 pathway was also found among genes negatively correlated with miR-205 expression in Taylor's set of clinical samples (highlighted in red in Supplementary Table S3). Modulation of IL-6 by miR-205 may be the result of the simultaneous suppression of a number of oncogenes that we previously showed to be potentially targeted by the miRNA (19). Among these, protein kinase C epsilon (PKCɛ) has been specifically shown to enhance STAT3 localization onto the IL-6 promoter and thereby increase IL-6 expression (30). It is hence very likely that suppression of PKCɛ by miR-205 may result in reduced transcription and secretion of IL-6. To further support this hypothesis we measured IL-6 expression levels in PC-3 cells silenced for PKCɛ and found that they were downmodulated, thus phenocopying miR-205 effect (Fig. 6E).
Altogether, such findings suggest that miR-205 downmodulation is a prerequisite for the completion of CAF-induced EMT in PCa cells and for the acquisition of activating properties toward surrounding fibroblasts.
To validate this finding in vivo, we analyzed the effects of miR-205 modulation on tumor growth and lung colonization of PCa cells. As previously reported, prostate CAFs powerfully prompt tumor growth and metastatic spread to lungs (23). miR-205 overexpressing PC-3 cells were s.c. co-inoculated with CAFs in the lateral flanks of SCID bg/bg mice, under conditions (i.e., 1×106/flank and in the absence of Matrigel) that prevent their in vivo growth in the absence of activated fibroblasts (5). As shown in Figure 7A, ectopic miR-205 expression significantly impaired tumor growth of PC-3 cells co-injected with CAFs. In addition, an experimental metastasis assay in which PCa cells (22), modulated or not for miR-205 expression, were first exposed in vitro to CAFs to induce EMT and then injected into the tail vein of SCID bg/bg mice showed that miR-205 upregulation markedly inhibited the ability of PCa cells to colonize lungs (as detected by number of micrometastases in animals sacrificed after 8 weeks), highlighting the key role of the miRNA in PCa metastatic dissemination (Fig. 7B, C).

miR-205 reverts CAF-induced EMT
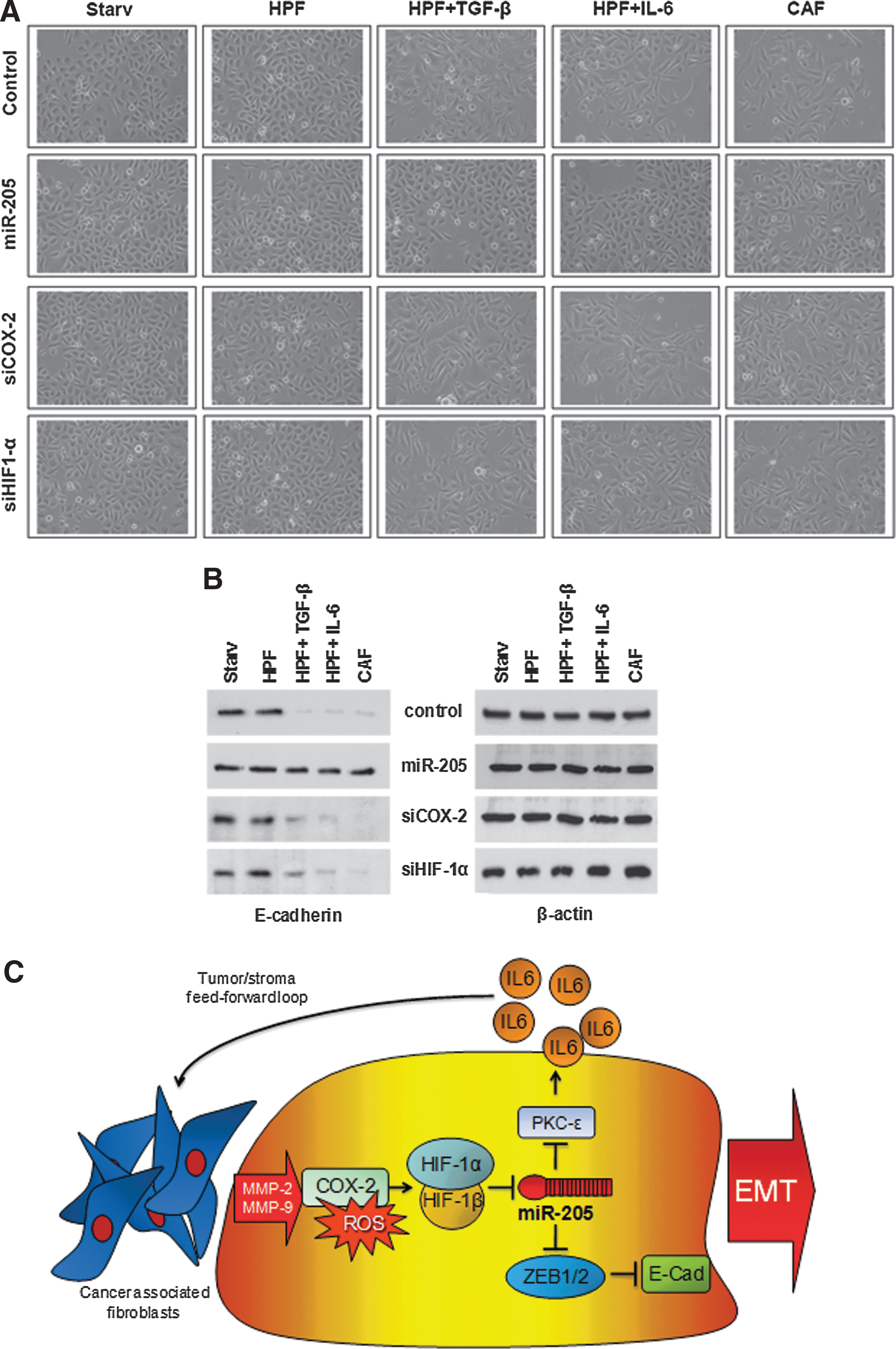
To assess the therapeutic potential of miR-205 reconstitution approach, we investigated whether restoring expression of the miRNA was able to revert the mesenchymal phenotype of PCa cells that had already undergone CAF-induced EMT. Specifically, PC-3 cells were stimulated with the CM from in vitro-activated HPFs or CAFs for 48 h to induce EMT and subsequently transfected with miR-205 precursor or, for comparative purposes, with siRNA against COX-2 or HIF-1α. The replacement of miR-205 was found to successfully revert EMT induced by activated fibroblasts, as detected by changes in cell morphology (Fig. 8A) and increased E-cadherin expression (Fig. 8B), whereas silencing of HIF-1α or COX-2 (assessment of their knockdown is reported in Supplementary Fig. S9) did not (Fig. 8A, B). Such findings confirm the hierarchy between miRNA downregulation and activation of the COX-2/HIF-1α-dependent pro-inflammatory axis (Fig. 8C) and suggest that compounds targeting such a pathway may be ineffective to treat cancer cells that have already undergone EMT once they have experienced miRNA downregulation.
Discussion
The impact of miRNAs on tumor microenvironment has been addressed thus far by only few studies. The first evidence of their involvement in the dynamic crosstalk between the tumor and the surrounding stroma was provided by Aprelikova et al. (1), who identified miR-31 as the most downregulated miRNA in CAFs isolated from endometrial cancers compared with fibroblasts derived from normal adjacent tissues. Functionally, CM from CAFs ectopically overexpressing miR-31 reduced migration and invasion of EC1 endometrial cancer cells, suggesting that miR-31 may target genes responsible for the secretion of soluble factors implicated in promoting tumor cell sprouting (1). Concerning PCa, the only available information indicates that miR-15 and miR-16 downregulation in CAFs promotes tumor growth and progression through reduced post-transcriptional repression of fibroblast growth factor-2 and its receptor FGFR1, which act on both stromal and tumor cells to enhance cancer cell survival, proliferation, and migration (41). However, no evidence is currently available on miRNAs that either regulate pathways activated in tumor cells upon CAF contact or influence the capability of tumor cells to activate surrounding stroma. The identification of such miRNAs might set the rationale for developing specific therapeutic approaches.
In the present study, we explored the modulation of miRNA expression in PCa cells upon stimulation with patient-derived CAFs. Consistent with induction of an EMT phenotype, miRNAs known to be associated with the process, such as miR-200 family members, were markedly downmodulated. However, changes in the expression of other miRNAs with unknown function or at least with undocumented participation in EMT were observed, suggesting that we are still far from understanding which pathways are activated in cancer cells upon interaction with their microenvironment. The most downmodulated miRNA was miR-205. We previously reported that miR-205 exerts a tumor-suppressive effect in PCa cells by inducing a mesenchymal-epithelial transition (MET) and the concomitant downregulation of oncogenes involved in disease progression (i.e., IL-6, PKCɛ, caveolin-1, and EZH2) (19).
Consistent with such tumor-suppressive function, miR-205 expression levels appear reduced in tumors, especially those from patients with disease spread to regional lymph nodes, suggesting a functional role of miR-205 loss in PCa dissemination (19,20,57). In keeping with these findings, here we show that miR-205 expression is almost completely lost in PCa metastases, which emerged from the analysis of a large publicly available dataset. In addition, reduced miR-205 levels in primary tumors correlated with increased risk for patients experiencing biochemical recurrence after radical prostatectomy. Recently, we also showed that miR-205 can enhance basement membrane deposition both by normal and tumor cells, thus conferring to the miRNA a further repressive function against metastatic dissemination (21).
Marked downregulation of miR-205 in cells undergoing EMT upon CAF stimulation is consistent with the aforementioned roles played by the master regulator miRNA. Noteworthy is the observation that miR-205 can be responsive to stromal signals, in addition to the reported regulation by TGF-β in Madin–Darby canine kidney cells (25). The participation of miR-205 in EMT induced by stimuli other than TGF-β is not trivial, considering that signals relevant for the metastatic spur induced by stroma interactions have not yet been identified. Notably, only androgen-refractory PCa cells undergo EMT in response to CAFs, and they do it in a miR-205-dependent manner. This finding would suggest that the malignant interplay between cancer and stromal cells is mainly effective in aggressive and hormonal therapy-resistant PCas.
MiR-205 downmodulation in clinical PCa may hence be the result of crosstalk between epithelial cells and fibroblasts. Obviously, it is hard to say whether mutations in epithelial cells converting them into tumor cells precede activation of fibroblasts, or whether epigenetic changes within the microenvironment can per se transform epithelial cells. An established finding is that activated stroma can exacerbate miR-205 downmodulation in cancer cells, thus prompting EMT and metastasis. In this regard, stimulation of PCa cells by CAFs needs to be continuous to maintain EMT features. It is possible that PCa cells may leave the primary tumor together with associated fibroblasts to maintain their mesenchymal traits, consistent to what was observed for Lewis lung carcinoma cell model (15), where stromal cells from the primary tumor site were detected in metastatic nodules spontaneously formed in mouse lungs after resection of subcutaneously grown xenografts. The data highlight how metastatic cancer cells may bring their own soil, including activated fibroblasts, from the primary site to the colonized organs.
In the present study, HIF-1α has been acknowledged as a direct transcriptional repressor of miR-205, corroborating previous data showing miRNA downmodulation in human placental trophoblasts under hypoxic conditions (39). The capability of HIF-1α to function as a transcriptional repressor, besides being a well-known transcriptional activator, has been recently demonstrated in different cell models (9,16,17,38,58). Several mechanisms have been proposed to mediate the transcriptional repression activity of HIF-1α. Specifically, in addition to a direct transcriptional repressor activity (9,43), indirect mechanisms involving the recruitment of already acknowledged HIF-induced transcriptional co-repressors (17,33) have been suggested. Although at present we do not have a comprehensive knowledge of the molecular events engaged by CAFs to convert HIF-1α into a transcriptional repressor, we obtained several lines of evidence suggesting HIF-1α repressive role on miR-205 transcription. First, silencing of HIF-1α severely prevented miR-205 downregulation induced by CAF treatment. Second, ChIP analysis confirmed an increase in miR-205 promoter occupancy by HIF-1α upon CAF stimulation, by binding to three HIF-1-responsive elements. Third, the modulation of HIF-1α levels by culturing PC-3 cells under hypoxic conditions or in the presence of CoCl2 induced a clear downregulation of miR-205 primary transcript, thus confirming that HIF-1α can ultimately repress the transcription of the miRNA.
The key upstream regulator of HIF-1α is confirmed as COX-2, which we already acknowledged as a source of oxidants essential to stabilize the HIF-1α subunit and allow EMT of PCa cells (22). HIF-1α can also be viewed as a mandatory crossroad that drives cancer cells toward malignancy (6,10,13,35,54) in which miR-205 and oxidative stress play a pleiotropic role. The role of oxidative stress, a common feature of aggressive cancers, in regulating miRNAs that control EMT is still unclear. Indeed, although oxidative stress has been widely reported as positively correlated with EMT activation, expression of miR-200a leads to an increase in oxidative stress in ovarian tumors (36).
Interestingly, activation of COX-2 and HIF-1α is just a transient phenomenon during CAF-induced EMT. For this reason, targeting such factors may be not promising for therapeutic purposes. We actually observed that once PCa cells have experienced EMT, silencing of either COX-2 or HIF-1α is ineffective. In contrast, ectopic overexpression of miR-205, which represents the final mediator of CAF-induced EMT by directly repressing ZEB1/2 (19,25), can both prevent and revert the acquisition of mesenchymal features induced by stromal signals. The effects of miR-205 overexpression spread to several key features of metastatic cells, including the ability to cross the ECM barrier and invade tissues, the resistance to anoikis allowing survival in the bloodstream, and the achievement of stem-like traits. To the best of our knowledge, miR-205 is the first example of a miRNA that, once exogenously modulated in tumor cells, can impede activation of and by surrounding fibroblasts. In keeping with this, forced expression of miR-205 leads to interruption of the “efferent” pathway engaged by cancer cells and leading to stromal reactivity. Such evidence further justifies the opportunity to utilize miR-205 as a tool to prevent or counteract PCa metastasis. In vivo data generated in the present study support the capability of miR-205 to reduce tumor growth, presumably by inhibiting local invasion, and lung colonization. The latter effect may depend on either increased anoikis of miR-205-expressing PCa cells, which therefore would not survive in circulation, or reduced extravasation (or both), thus ultimately leading to impaired metastasis formation. The results also highlight the relevance of MET during tumor dissemination. EMT in tumor cells needs indeed to be transient. During early steps of metastasis, tumor cells undergo EMT in response to external cues at the invasive front to gain a more motile and invasive mesenchymal phenotype. However, once a metastatic cell has invaded a new tissue, it needs to revert to a more epithelial phenotype to efficiently settle down and grow as a metastasis. The need to maintain a certain degree of plasticity justifies the reversibility of CAF-induced EMT, as observed in the present study. From our standpoint, MET has to occur after cells exit from the bloodstream, likely due to stromal signals from the host secondary organ. Cells undergoing MET when still in the circulation, such as miR-205-expressing cells in our metastasis assay, are actually unable to metastasize. This is in contrast with data of Korpal et al. (32) showing that miR-200s replacement in 4TO7 mouse mammary tumor cells can even favor colonization when cells are intravenously injected. Presumably, the roles of miR-205 and miR-200s in EMT/MET are not completely overlapping.
Overall, the evidence collected so far will be instrumental to properly shape miR-205-based therapeutic approaches. Overexpression of miRNA in the primary tumor by local delivery could be specifically envisaged to prevent local invasion and intravasation, thus blocking initial dissemination. However, effective anti-metastatic therapy should impair the survival and colonization capabilities of already disseminated tumor cells and not simply prevent detachment from the primary tumor. Notably, circulating tumor cells can be detected in patients with very early neoplastic lesions (42). For this purpose, systemic therapeutic approaches should be preferable. Obviously, this can be accomplished only when systems to selectively deliver therapeutic miRNAs to the cells of interest become available, thus avoiding their overexpression in unwanted tissues and limiting potential side effects.
Materials and Methods
See Supplementary Data for a detailed description of miRNA expression profiling, miRNA and gene expression analysis, ChIP, western blot analysis, and Boyden invasion assay.
Experimental models
Human PCa cells (PC-3, DU145, LNCaP, and 22Rv-1) were obtained from the European Collection of Cell Cultures and maintained at 37°C/5% carbon dioxide in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. For hypoxia experiments, PC-3 cells were maintained at a constant gas mixture of 1% oxygen, 94% nitrogen, and 5% carbon dioxide in a specially designed hypoxia incubator (Galaxy® 48 R Eppendorf). Control cells were maintained in 20% oxygen. CoCl2 was administered at the final concentration of 150 μM.
Human prostate fibroblasts (HPFs and CAFs) were isolated from surgical explantation after patient informed consent in accord with the Ethics Committee of the Azienda Ospedaliera Universitaria Careggi. Briefly, HPFs and CAFs were extracted from healthy and intratumoral regions of the prostate of PCa-bearing patients (average Gleason=4+4, all pT3aN0). Tissue samples were aseptically obtained from patients undergoing radical prostatectomy. Tissues were digested overnight in 1 mg/ml collagenase I, and cells were plated in DMEM containing 10% fetal bovine serum. CM from HPFs and CAFs was obtained by 48-h serum-starved cells, clarified by centrifugation, and used freshly.
Fibroblast activation
HPFs were grown to subconfluence and treated for 24 h with 10 ng/ml rTGF-β1, 50 ng/ml IL-6, or CM from PC-3 cells. Fresh serum-free medium was added for an additional 24 h before collection of CM.
Transfection
miR-205 precursor and negative control (miR-Neg) were purchased as Pre-miR™ miRNA precursor molecules (Life Technologies, Carlsbad, CA). miRCURY LNA Inhibitor specific for miR-205 (LNA-205) and the negative control (LNA-Neg) were purchased from EXIQON (Woburn, MA). Cells seeded at the appropriate density were transfected for 4 h at 37°C with 20 nM miRNA precursors or 100 nM miRNA inhibitors using Lipofectamine-2000 (Life Technologies), according to the manufacturer's instruction, and processed at different time intervals.
For silencing experiments, siCOX-2 (sc-29279; Santa Cruz Biotechnology, Santa Cruz, CA), siHIF1-α (5′-AAAGGACAAGUCACCACAGGA-3′; Qiagen, Hilden, Germany), control siRNA (16), or siPRKCE (19) were administered at a final concentration of 20 nM, using Lipofectamine-2000.
miRNA expression profiling
Total RNA was isolated from PC-3 cells using TRIzol (Life Technologies) reagent, then miRNAs were profiled on the Illumina Human v2 MiRNA expression beadchip (Illumina, San Diego, CA), according to the manufacturer's instructions. All array data have been deposited in National Center for Biotechnology Information's Gene Expression Omnibus (GEO,
Analysis of publicly available datasets
Processed gene and miRNA expression data used in Taylor et al. (52) were downloaded from GEO (GSE21032). miR-205 expression levels in normal prostate tissue, primary prostate carcinoma, and metastatic lesions were extracted. Primary tumors from patients who underwent chemo-, radio-, or androgen ablation therapy prior to prostatectomy were excluded from the analysis. The Cox model and Kaplan–Meier method were used to test the association between miR-205 expression and biochemical relapse. Expression values were dichotomized defining as “miR-low” 17% of primary tumors with the lowest expression levels.
Using the 98 samples (normal prostate tissue, primary prostate carcinoma, metastatic lesions, and cell lines) with available gene and miRNA data, miR-205 expression levels were correlated with expression levels of all 19,839 genes (Spearman correlation). Hence, genes were ordered according to their correlation with miR-205, and a GSEA was performed (GSEA 2.0, Preranked analysis), testing the “C2” gene sets collection of the Molecular Signatures Database v3.0 (
miRNA and gene expression analysis
miR-205, miR-200b, pri-miR-205, E-cadherin, Drosha, Dicer, ZEB1, ZEB2, SNAI1, SNAI2, TWIST1, PRKCE, and IL6 mRNA expression was assessed by qRT-PCR as detailed in Supplementary Data.
Chromatin immunoprecipitation
The presence and position of putative HIF-1α binding sites within the miR-205 promoter region was predicted using the Jaspar algorithm (
Prostasphere formation and clonogenicity assay
PC-3 cells, modulated for miR-205 expression, were incubated for 72 h with CM from CAFs and then detached using Accutase (Sigma-Aldrich, St. Louis, MO). For prostasphere formation, single cells were plated at 150 cells/cm2 on low-attachment 100-mm plates (Corning Inc., Corning, NY) in DMEM/F12 (Life Technologies) supplemented with B27 and N2 (Life Technologies), 5 μg/ml insulin, 20 ng/ml basic fibroblast growth factor, and 20 ng/ml epidermal growth factor. Cells were grown under these conditions for 15–20 days and formed non-adherent P0 spheres termed prostaspheres. For the evaluation of self-renewal, a single prostasphere was dissociated in single cells with Accutase, and a dilution of one cell per well into 96-well low-attachment plates was performed to isolate individual P1 prostaspheres.
Flow cytometry
PC-3 cells (1×106), modulated for miR-205 expression, were incubated for 72 h with CM from CAFs and then were labeled with FITC-anti-CD44 (clone G44-26) and PE-anti-CD24 (clone ML5) antibodies for 1 h at 4°C in the dark. Cells were washed, and flow cytometry was performed using a FACSscan (BD Biosciences, Franklin Lakes, NJ).
IL-6 quantification by ELISA
Determination of IL-6 concentration in cell culture media was performed by Chemiluminescence ELISA Kit according to the manufacturer's instructions (Life Technologies, Cat. No. KHC0069). The IL-6 ELISA was linear between 5 and 4000 pg/ml. Cell culture supernatant samples were diluted fourfold in Standard Diluent Buffer and 100 μl of the diluted samples were used to run the assay.
Xenograft experiments
In vivo experiments were carried out in accord with national guidelines and approved by the Ethics Committee of the Animal Welfare Office of the Italian Work Ministry and conformed to the legal mandates and Italian guidelines for the care and maintenance of laboratory animals. Male SCID-bg/bg mice (6–8 weeks old; Charles River Laboratories International Inc., Wilmington, MA) were injected subcutaneously, both in the right and left lateral flanks, with 1×106 PC-3 cells (modulated in miR-205 expression) plus 0.5×106 CAFs, in a total volume of 0.1 ml phosphate-buffered saline. Animals (eight per group) were monitored daily, and tumor size was measured every 2–3 days by a caliper. Tumor volumes were determined by the length (L) and the width (W): V=(LW 2)/2. Mice were sacrificed before the tumor masses exceeded a size to produce evident physical discomfort, such as a rough looking hair coat, lack of grooming activity, or abnormal posture (huddling, hunching, or being stiff). Excised tumors were fixed overnight at 4°C in formalin (5% in phosphate buffered saline) for histologic analyses. Formalin-fixed, paraffin-embedded tissue blocks were cut into 5 μm consecutive sections and mounted on positively charged slides. Tissue sections were deparaffinazed and rehydrated before staining with hematoxylin and eosin.
Lung colonization assays
Male SCID-bg/bg mice (6–8 weeks old) were injected with PC-3 previously transfected with miR-205 precursor or miR-Neg and, after 48 h, treated ex vivo with CM from CAFs for an additional 72 h. Mice (eight animals per group) were injected in the lateral tail veins with the differently treated PC-3 cells (1×106 in 0.1 ml of phosphate-buffered saline). Animals were monitored every 3 days and sacrificed after 8 weeks. Lungs were inspected for micrometastases by histological analyses.
Statistical analysis
Data are presented as mean values±standard deviation from at least three independent experiments. Statistical analysis of the data was performed by two-tailed Student's t test. P-values<0.05 were considered statistically significant. Statistics applied to microarray analyses is described in the relative sections.
Footnotes
Acknowledgments
We thank Dr. Valentina Profumo for useful suggestions; Dr. Valentina Doldi and Dr. Luigi Carlessi for technical advice and institutional Functional Genomics Core Facility for microarray analysis. We also thank Mrs. Betty Johnston for editing the article.
The work in the authors' laboratory is supported by the Italian Association for Cancer Research (AIRC), Special Program “Innovative Tools for Cancer Risk Assessment and Early Diagnosis,” 5×1000, (project No. 12162), (N.Z., R.V., M.A.P.); AIRC grants (No. 11542, 8797), (P.G., P.C.); Istituto Toscano Tumori (No. 0203607), (P.C.); and MIUR-PRIN 2008, (P.C.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.