
Abstract
Introduction
A
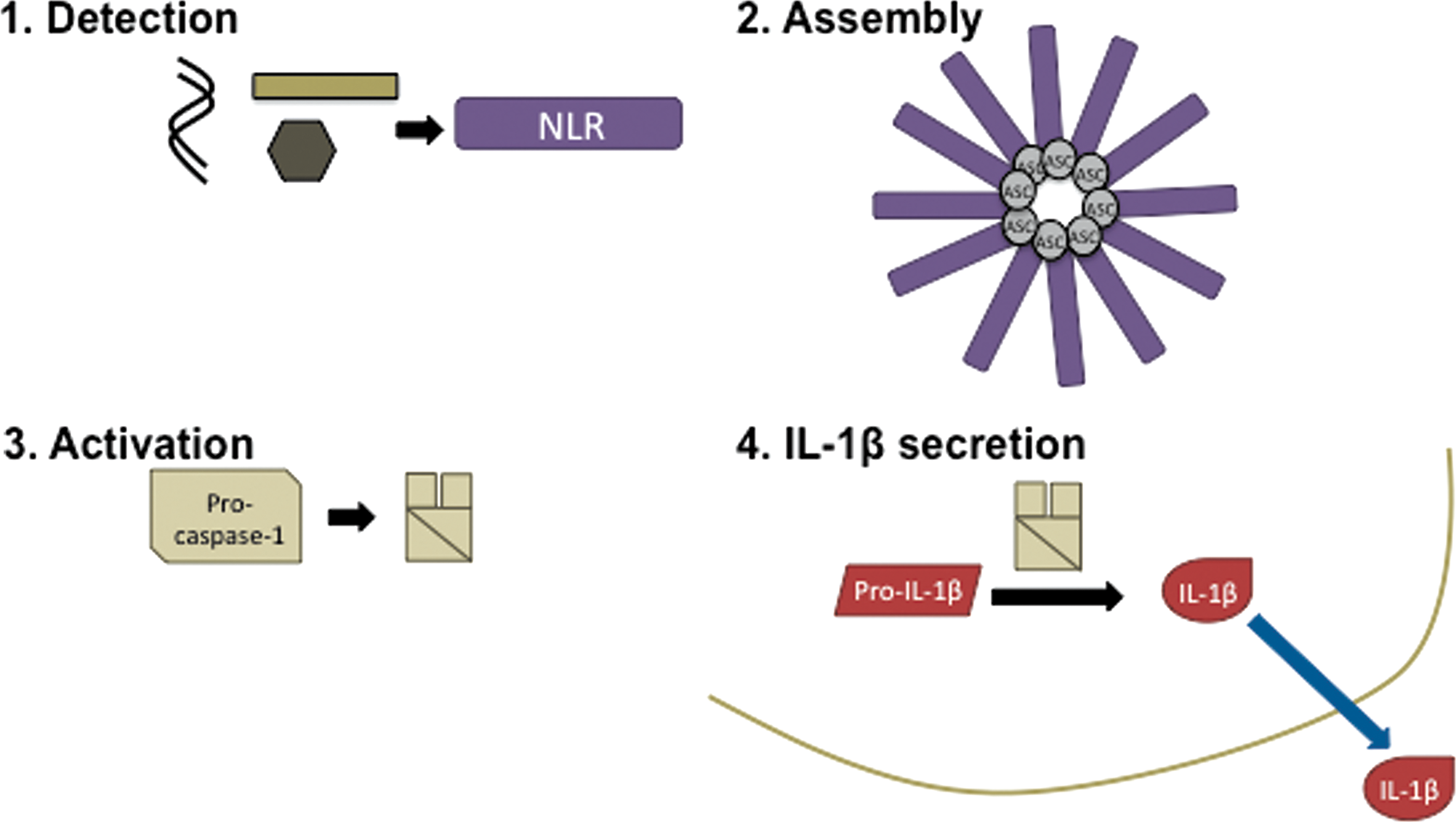
Inflammasome complexes induce the cleavage of pro-caspase-1 to the p10 and p20 subunits of active caspase-1 enzyme, which in turn cleaves the presynthesized pro-IL-1β cytokine into its active 17 kDa form that is then secreted from the cell (Fig. 1). While caspase-1 is able to cleave itself in an inflammasome complex in vitro (24), the mechanism and the possible roles of additional proteases in vivo remain unknown. IL-1β release requires two PRR activating signals: signal one activates the NFκB-dependent expression of pro-IL-1β, usually through toll-like receptor (TLR) activation, and signal two activates caspase-1 to cleave pro-IL-1β through NLR-mediated inflammasome formation. The mechanism of IL-1β release after cleavage is unclear, although several noncanonical models have been proposed, as previously reviewed (23). IL-18 and cell death by pyroptosis also follow inflammasome activation and have also previously been reviewed (64). Very recently, it was shown that the caspase-1 knockout mouse previously used to determine the role of caspase-1 in inflammasome activation also had a truncation mutation in the caspase-11 gene, which was subsequently shown to regulate noncanonical inflammasome activation leading to pyroptosis (4, 8, 34, 47, 76). Although caspase-11 is not required for inflammasome responses to some sterile stimulants, such as K+ efflux, it is required for responses to gram-negative bacterial infections (4, 8, 34, 76). Early work has hinted at potential molecular mechanisms of caspase-11-mediated regulation of inflammasome activation, but additional work is required to determine what the targets of caspase-11 are and how they might mediate pyroptosis.
Phagocytes are the only cell type known to contain inflammasomes and these same cells rely upon autophagy to monitor for the presence of pathogens. Autophagy is a “self-eating” process by which a cell collects protein aggregates, bacteria, viruses, dying cells, and damaged organelles into a double-membrane compartment called an autophagosome, which fuses with the lysosome to degrade the contents for recycling and antigen presentation. Of the three types of autophagy, macroautophagy is the most relevant to inflammasome studies and will be referred to as “autophagy” in this review. Autophagy ensures an energetic homeostasis in the cell by providing nutrients during metabolic stress and also serves a protective role by removing damaged and damaging components of the cytosol. Given these important roles, it is not surprising that autophagy is a tightly regulated process that is linked to other major pathways, such as phagocytosis, biosynthesis, and innate immune signaling. Autophagy is normally kept to a minimum in unstressed cells, but it is quickly enhanced during nutrient deprivation, infection, and cellular damage.
Autophagy occurs in four phases: induction, nucleation, elongation, and fusion, with points for regulation at each step (Fig. 2) (49). During the induction step, repression of autophagy is relieved by one of many possible initiation signaling cascades involving class I phosphatidylinositol 3 kinase/Akt/molecular target of rapamycin (PI3K/Akt/mTOR), 5′-adenosine monophosphate activated protein kinase (AMPK), elongation initiation factor 2α (eIF2α), tumor protein 53 (p53), c-Jun N-terminal kinase 1 (JNK1), inositol-phosphate requiring enzyme 1 (IRE-1), inositol-triphosphate receptor (IP3R), or intracellular calcium. Vesicle nucleation occurs during the second step, which results in the assembly of a phagophore, the double membrane vesicle that will engulf cytosolic cargo. The phagophore begins to form upon the assembly of a protein complex consisting of the lipid kinase Vps34 and the regulatory proteins Vps15 and Beclin1, which initiates Vps34 enzymatic activity to phosphorylate phosphatidylinositols to generate phosphatidylinositol 3-phosphate (PI3P). The presence of PI3Ps recruits FYVE and PX domain containing proteins to the membrane, labeling it as an autophagophore. The autophagophore is elongated during the third step of autophagy via a ubiquitin E3 ligase complex consisting of autophagy proteins (Atg) Atg5, Atg12, and Atg16L1, which conjugates phosphatidylethanolamine (PE) to microtubule-associated protein 1A/1B, light chain 3 (LC3). LC3-PE remains associated with the inner and outer membranes of the phagophore as it extends in length until it has completely engulfed its cargo to become an autophagosome. During the final fusion step of autophagy, the autophagosome fuses with the lysosome to create an autolysosome, which degrades its contents via hydrolases and exports the remaining materials to the cytoplasm for reuse.

Both autophagy and inflammsome proteins have been genetically linked to human autoinflammatory diseases, suggesting that these processes might be functionally related. Indeed, recent work has uncovered exciting new regulatory connections between autophagy and inflammasome activation, which is the focus of this review. Autophagy is required for inflammasome activation and inflammasome complexes are turned off upon targeted degradation in autophagolysosomes. Consequently, NLR proteins bind and regulate autophagy proteins to initiate autophagophore formation. These topics and the unanswered questions they contain will be discussed in detail in the following sections of this review.
The Inflammasomes
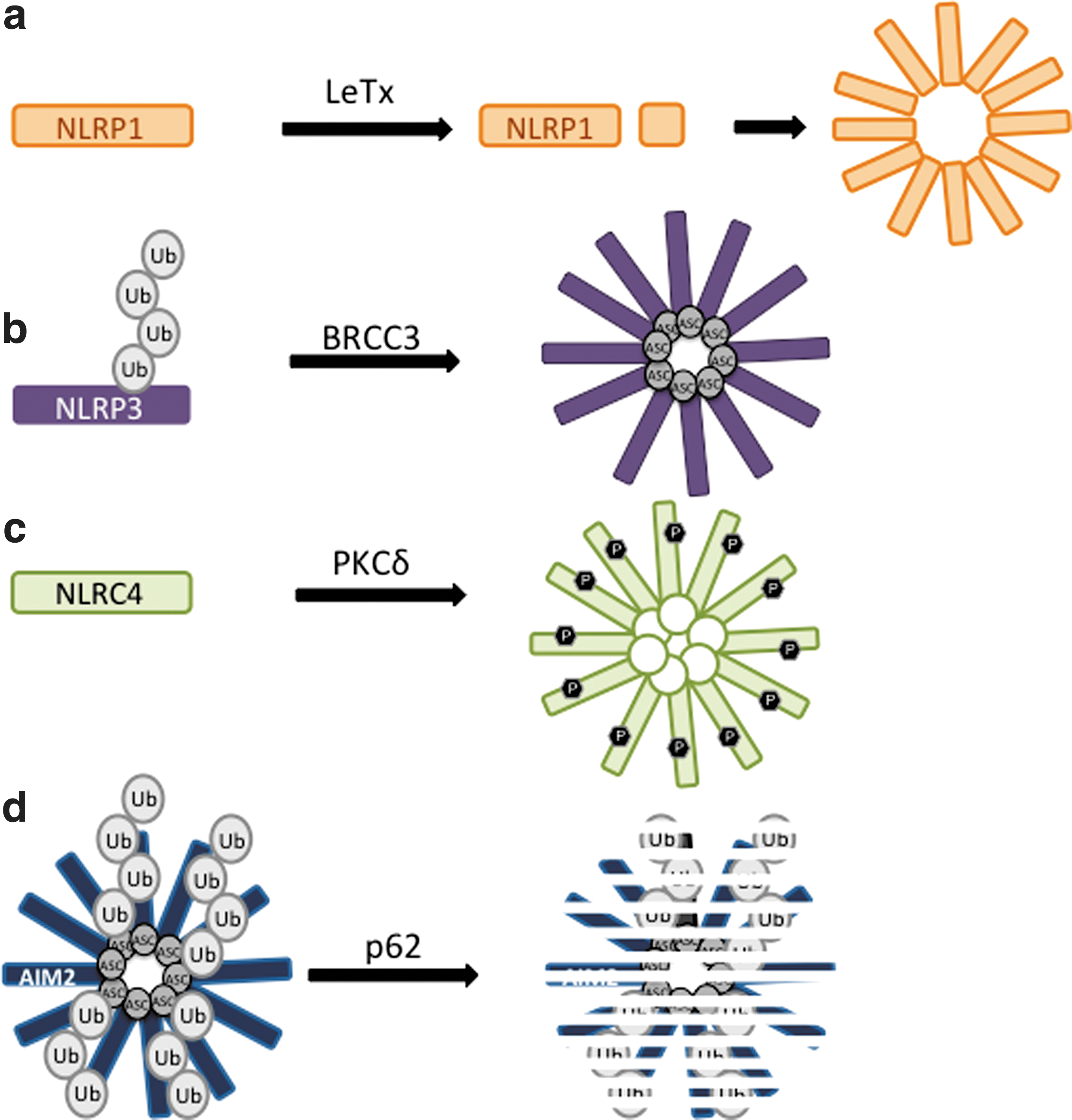
The NLRs which have been shown to form inflammasomes in response to PAMPs or DAMPs in macrophage cells are NLRP1 (Nalp1, Nlrp1b), NLRP3 (Cias1, Nalp3), NLRC4 (Ipaf), NLRP7 (NOD12), NLRP12 (monarch-1), and absent in melanoma 2 (AIM2) (PYHIN). Although absent in melanoma 2 (AIM2) is structurally unrelated to the other NLRs, it is functionally related and will be discussed below. NLRP1 has been shown to respond to anthrax lethal toxin (LeTx) and the muramyl dipeptide (MDP) moiety of peptidoglycans (7, 18, 25, 31, 56), although experiments with bone-marrow derived macrophage (BMDM) from the first NLRP1 knockout mouse have recently shown that NLRP1 is only required for the LeTx inflammasome response and not the MDP response, which instead requires NLRP3 (53). Mechanistically, LeTx directly cleaves NLRP1, which activates NLRP1 to form inflammasomes, activate caspase-1, and cleave IL-1β (18, 31). Both N-terminal and FIIND domain cleavage sites have been reported for NLRP1, although only cleavage at the N-terminal site has been shown to be LeTx-dependent (18, 31, 56) (Fig. 3). NLRP1 has recently been reported to respond to reduced ATP levels in the cytoplasm in response to nutrient deprivation, but NLRP1 cleavage was not detectable under these stimulation conditions (57). The NLRP1 inflammasome does not include ASC (25, 67) possibly because NLRP1 has a pyrin domain that could bind to the pyrin domain of Caspase-1 independently of ASC. Interestingly, ASC is required for the response to MDP (69), further supporting a role for NLRP3 and not NLRP1 in MDP stimulation.
NLRP3, the most well-characterized NLR family member, is activated by a wide range of pathogens and sterile DAMP signals, including K+ efflux, MDP, monosodium urate (MSU), cholesterol crystals, mitochondrial reactive oxygen species (mtROS), Listeria monocytogenes, Candida albicans, Salmonella typhimurium, and influenza virus, among others (29). The role of NLRP3 in caspase-1 activation and IL-1β secretion in response to these signals has been evaluated with the NLRP3 and ASC knockout mice, which both had defects for all tested stimuli (29, 41, 44, 69), indicating that NLRP3 utilizes the ASC adapter. Moreover, NLRP3 has also been shown to bind ASC (3, 21), and ASC foci are visible by immunofluorescence microscopy upon NLRP3 stimulation (71). Given the structurally diverse range of NLRP3 stimulants, it is not surprising that the molecular mechanism of NLRP3 activation remains unclear and will be discussed further in the following sections. While much effort has focused on defining the potential activating factors of NLRP3, relatively less is known about what signals regulate turning off active NLRP3. Recent evidence suggests that intracellular Ca2+, cyclic AMP (cAMP), and nitric oxide (NO) negatively regulate NLRP3 activation through binding or modification of NLRP3 (39, 51, 54). However, Ca2+, cAMP, and NO levels are controlled by several pathways and thus, the negative regulation of NLRP3 is likely a complex balance of converging signals.
NLRC4 is stimulated by intracellular flagellin and type III secretion systems from bacteria, including S. typhimurium, Shigella flexneri, and Escherichia coli (28, 62, 63). NLRC4 utilizes ASC (59, 71), Naip5, and Naip2 adapter proteins depending on the bacterial stimulus (29). While NLRC4 can directly detect the N-terminus of flagellin, Naip5 association is required to respond to the C-terminus of flagellin (52, 99). The Naip2 adapter protein detects a rod component of type III secretion systems and binds NLRC4 to activate inflammasome formation (52, 99). The ASC adapter protein is important but not essential for all types of NLRC4 inflammasomes, as ASC deficient BMDMs have a partial defect in caspase-1 activity and IL-1β secretion upon flagellin stimulation, suggesting that ASC enhances the activity of the NLRC4 inflammasome (9, 62, 99). However, ASC binds NLRC4 (33) and ASC deficient BMDMs have a complete defect in caspase-1 cleavage and NLRC4 inflammasome foci formation upon infection with S. typhimurium or Legionella pneumophila (12, 59, 71). As a further complication, caspase-1 dependent cell death, or pyroptosis, is intact in ASC deficient BMDMs upon infection with Pseudomonas aeruginosa, L. monocytogenes, or S. flexneri (30, 81, 90). Thus, the role of ASC in NLRC4 activation might depend on the context of inflammasome stimulation. Activation of NLRC4 during S. typhimurium infection also requires phosphorylation at serine 533 (S533) between the NACHT and LRR domains, which is likely targeted for phosphorylation by the PKCδ kinase (73) (Fig. 3). This is the first evidence of the phosphorylation-mediated regulation of inflammasomes and it will be of interest to determine whether additional NLRs are regulated by phosphorylation and dephosphorylation.
Recent work has identified two new NLRs as pathogen sensors; NLRP12 detects Yersinia pestis and NLRP7 detects acylated lipopeptides from bacteria (48, 95). NLRP12 binds ASC (96) and is required for IL-1β secretion and caspase-1 activity in response to Y. pestis infection in vivo and in cultured phagocytes, although NLRP3 was also required in these experiments (95). The ligand for the NLRP12 inflammasome activation is unknown, but the Y. pestis type III secretion system is required (95). NLRP7 also detects specific PAMPs from intracellular bacterial pathogens. The acylated lipopeptides that activate NLRP7 in human macrophage THP-1 cells are from Mycoplasma spp., which are common contaminants in cell cultures. ASC foci are also colocalized with NLRP7 upon stimulation, suggesting an inflammasome forms (48). Notably, since lipopeptides were recognized by TLR2, no additional priming of the cells was required for IL-1β release, making mycoplasma contamination a threat to inflammasome studies performed in cell culture (48). Significantly less is known about the NLRP12 and NLRP7 inflammasomes in comparison to NLRP3 and subsequent studies will provide more insight into the mechanisms and roles of their activation.
AIM2 is structurally unique from other NLRs because it has a HIN-200 domain instead of an LRR, and similarly to NLRP1, AIM2 has a pyrin domain at its N-terminus (82). AIM2 recognizes cytosolic double-stranded DNA (dsDNA) from bacteria and viruses, and utilizes the ASC adapter protein as shown by ASC knockout mice and ASC foci formation upon AIM2 stimulation (26, 43, 75). Although it is unclear what the molecular determinants are for distinguishing bacterial DNA molecules in the cytosol, genetic evidence clearly shows that AIM2 is required for IL-1β secretion upon infection of BMDMs with Mycobacterium tuberculosis and Francisella tularensis in addition to the viral pathogens, such as vaccinia virus and cytomegalovirus (50, 75, 79). AIM2 is also partially responsible for IL-1β secretion in response to L. monocytogenes, which is also detected by NLRP3 (50, 75). Notably, in vivo infections of AIM2 knockout mice with mouse cytomegalovirus revealed that early control of virus replication is AIM2-dependent (75), highlighting the critical role of inflammasomes in innate immunity in vivo.
Autophagy Studies Uncover Regulation of Inflammasomes by ROS
Deletion or depletion of autophagy by genetic or pharmacological means has a clear enhancing effect on IL-1β secretion upon inflammasome stimulation or LPS treatment conditions. Specifically, IL-1β secretion was apparently increased upon inhibition of autophagy by 3-methyladenine (3MA) treatment, expression of the Atg4B dominant negative mutant, deletion of LC3B, deletion of Atg16L1, or depletion of Beclin1 (Fig. 1) (38, 66, 80, 85, 100). A study utilizing the Atg16L1 knockout mouse model system has found that the TIR-domain-containing adapter-inducing interferon-β (TRIF) TLR adapter protein, K+ efflux, and ROS are required for inflammasome activation (80). ROS is produced by NADPH oxidases or mitochondria in response to phagocytosis and TLR stimulation (68, 97). Although initial reports suggested that NADPH oxidases might be important for ROS production during inflammasome stimulation in the human THP-1 cell line (20, 86), studies examining PBMCs from human patients lacking the p22phox, p47phox, or NOX2 subunits of the NADPH oxidases conclusively showed that NADPH oxidases were not required for IL-1β secretion (93, 94). These conflicting results might be due to differences between primary and transformed cells, since primary BMDM from the gp91 NADPH oxidase subunit knockout mouse were also competent for IL-1β secretion in a subsequent study (86). Although NADPH oxidase-derived ROS is not required for inflammasome activation, ROS from an alternative source is required since treatment with a general ROS scavenger reduced IL-1β production in NADPH oxidase deficient human PBMCs and mouse bone-marrow derived dendritic cells (38, 93, 94). This left mitochondria as the likely source of ROS required for inflammasome activation, but it was not clear how autophagy was able to regulate its production in response to inflammasome stimulants.
An important clue came when blockage of autophagy by treatment of macrophage cells with 3MA (PI3K inhibitor) resulted in the production of mitochondrial ROS (mtROS) and NLRP3-dependent IL-1β secretion in the absence of traditional inflammasome stimulants (Fig. 4) (38, 100). This suggested that when damaged mitochondria were not cleared by autophagy, they released mtROS, which stimulated NLRP3 inflammasomes. If all traditional NLRP3 stimulants induce mitochondrial damage, then the mtROS model would present a unified mechanism for the activation of NLRP3 by a structurally diverse set of stimulants. The mtROS model of NLRP3 inflammasome activation was further supported by the colocalization of NLRP3 with mitochondrial markers, suggesting that the inflammasome activation site is physically located near the source of mtROS (100). Mitochondria function was specifically important for mtROS production and IL-1β secretion because shRNA-mediated depletion of voltage dependent anion channel 1 (VDAC1), which is important for mitochondrial function and mtROS production (88), reduced caspase-1 cleavage and IL-1β secretion in response to sterile NLRP3 stimuli (100). Notably, this effect was not observed for NLCR4 or AIM2 stimuli, making mtROS a specific requirement of NLRP3 inflammasome activation, although the role of mtROS in additional inflammasomes must also be examined further (87, 100). High levels of Bcl-2, which partially blocks VDAC, reduces mtROS production, and blocks apoptosis, also reduced NLRP3-inflammasome activation in BMDMs and an immortalized human macrophage cell line (87, 100). In contrast, it has previously been shown that a Bcl-2 overexpression in the human THP-1 macrophage cell line has lower levels of caspase-1 activity and IL-1β secretion in response to MDP and ATP stimulation (10, 100), making it likely that the role of Bcl-2 may differ between cell lines or that MDP and ATP stimulation is unique from other NLRP3 stimulants. Although the role of Bcl-2 in THP-1 cells was initially investigated in the context of NLRP1 stimulation, the response to MDP and ATP treatment used in this study has since been shown to be NLRP3-dependent and not NLRP1-dependent in knockout mice (53), limiting the known requirement of mtROS to NLRP3-inflammasomes.
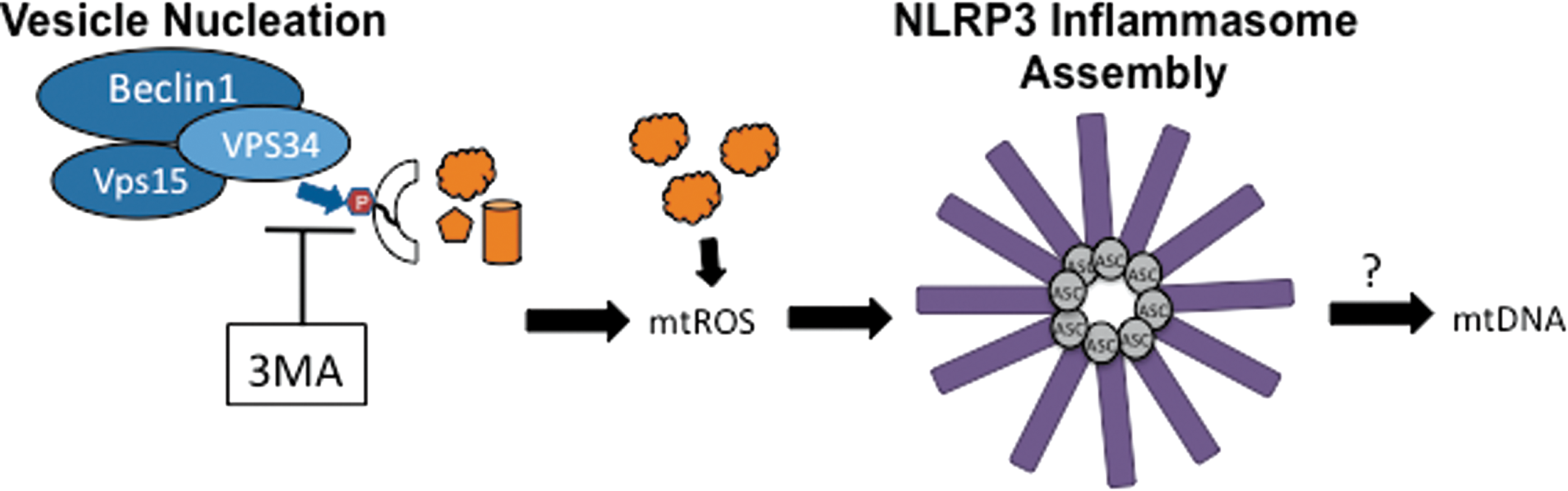
A major advance in understanding the molecular mechanism of the mtROS-mediated activation of NLRP3 was achieved in experiments using mice deficient for autophagophore formation and elongation due to the knockout of LC3 (LC3 −/−) or the depletion of Beclin1 (beclin1+/− ) (66). BMDMs from these mice had elevated caspase-1 activation and IL-1β secretion upon stimulation of the NLRP3 inflammasome with ATP, which causes a K+ efflux in the cell through the P2X7 channel (46). An increase in swollen and damaged mitochondria was visible by EM, confirming that mitochondrial damage resulted from K+ efflux due to ATP treatment (2, 32, 40, 66). Remarkably, BMDMs lacking mitochondrial DNA (mtDNA), called ρ0 cells, were unable to secrete IL-1β in response to NLRP3 stimuli and DNAseI treatment reduced caspase-1 activation and IL-1β secretion in normal BMDM, suggesting that mtDNA is involved in NLRP3 inflammasome activation (66, 87). In a series of elegantly designed experiments, it was revealed that mtDNA is released to the cytosol upon stimulation with ATP in an mtROS and NLRP3-dependent manner (66), indicating that caspase-1 activation is downstream from NLRP3-mediated translocation of mtDNA to the cytosol (Fig. 4). Since mtDNA release to the cytosol in response to ATP was impaired in BMDMs from either ASC or NLRP3 knockout mice, the inflammasome itself is likely involved in mtDNA release (66). This is in contrast to a more recent model proposed by Shimada et al., in which NLRP3 is activated after it directly binds to mtDNA released upon mitochondrial damage due to apoptosis triggered by NLRP3 stimulants (87). Shimada et al. argue that mtDNA was not detected in NLRP3 KO BMDM upon ATP treatment because the mtDNA was degraded in the absence of NLRP3-binding, although this possibility has not been tested. Intriguingly, oxidized nucleoside 8-hydroxy-guanosine (8-OH-dG), a marker for oxidized mtDNA, was detectable in endogenous NLRP3 immunoprecipitations and was even capable of blocking IL-1β production when it was added in excess to BMDM by competitively binding to endogenous NLRP3 (87). This strongly supports a role for oxidized mtDNA in NLRP3 inflammasome activation, although it is not clear whether NLRP3 may also facilitate mtDNA release before binding (Fig. 4).
The roles of other PRRs have also been examined in mtROS and mtDNA production. First, while the AIM2-inflammasome is also activated by exogenous mtDNA, endogenous mtDNA did not directly bind endogenous AIM2 in BMDM and ATP-dependent IL-1β production was AIM2 independent (66, 87). Furthermore, AIM2 activation by dsDNA was not affected by an inhibitor of mtROS production, AIM2 stimulation did not change mitochondrial membrane potential, and Bcl-2 did not affect AIM2 stimulation (66, 87). Thus, cytosolic mtDNA activates NLRP3 specifically. Secondly, TLRs have also been shown to induce mtROS production by a separate mechanism, which is important for clearance of intracellular bacterial pathogens (97). Upon stimulation of TLR1, 2, or 4, the TRAF6 signaling adapter protein translocates to the mitochondria and interacts with evolutionarily conserved signaling intermediate in Toll pathways (ECSIT), resulting in the ubiquitination of ECSIT and the increased ROS and mtROS (97). Cells must be primed by TLR activation to induce pro-IL-1β expression before NLR stimulation in inflammasome studies, making it likely that TLR signalling contributes to some mtROS production. Indeed, both overnight and 6 h stimulation of TLR4 with LPS lead to the production of ROS and mtROS (97), which are commonly used priming conditions, for inflammasome activation. Since TLR signaling induces mtROS production (84, 97), and ROS induces autophagy (17), it is possible to hypothesize that TLR signaling initiates and limits inflammasome activation through the mtROS-mediated activation of NLRP3, the NFκB-mediated expression of pro-IL-1β, and the autophagy-mediated degradation of pro-IL1β.
Regulation of Inflammasomes by Autophagosomal Degradation
In addition to its role as a regulator of NLRP3-inflammasome activation, autophagy also negatively regulates inflammasomes through newly discovered autophagy-dependent degradation of inflammasome proteins and IL-1β (38, 85). Studying the negative regulation of inflammasome activation is important for understanding how this potent signal is “turned off” to avoid acute tissue damage. Not surprisingly, many NLRs and autophagy factors are linked to autoimmune and autoinflammatory diseases (74, 83), which are characterized by high levels of inflammatory cytokines. As a negative regulatory mechanism of inflammasome activation, pro-IL1β is targeted to autophagosomes for degradation in response to TLR stimulation (38). Specifically, it has been shown that IL-1β is sequestered in the LC3-positive autophagosomes upon TLR stimulation and pro-IL-1β protein levels decreased when autophagy was induced by rapamycin (Fig. 5) (38). This suggests that TLR stimulation induces both pro-IL-1β expression and degradation by autophagy, thereby limiting the amount of available pro-IL-1β protein in the absence of NLR stimulation. A recent report suggests that mature IL-1β uses the autophagy machinery for secretion in a noncanonical secretory pathway (22). However, more evidence is required to vigorously evaluate this hypothesis.
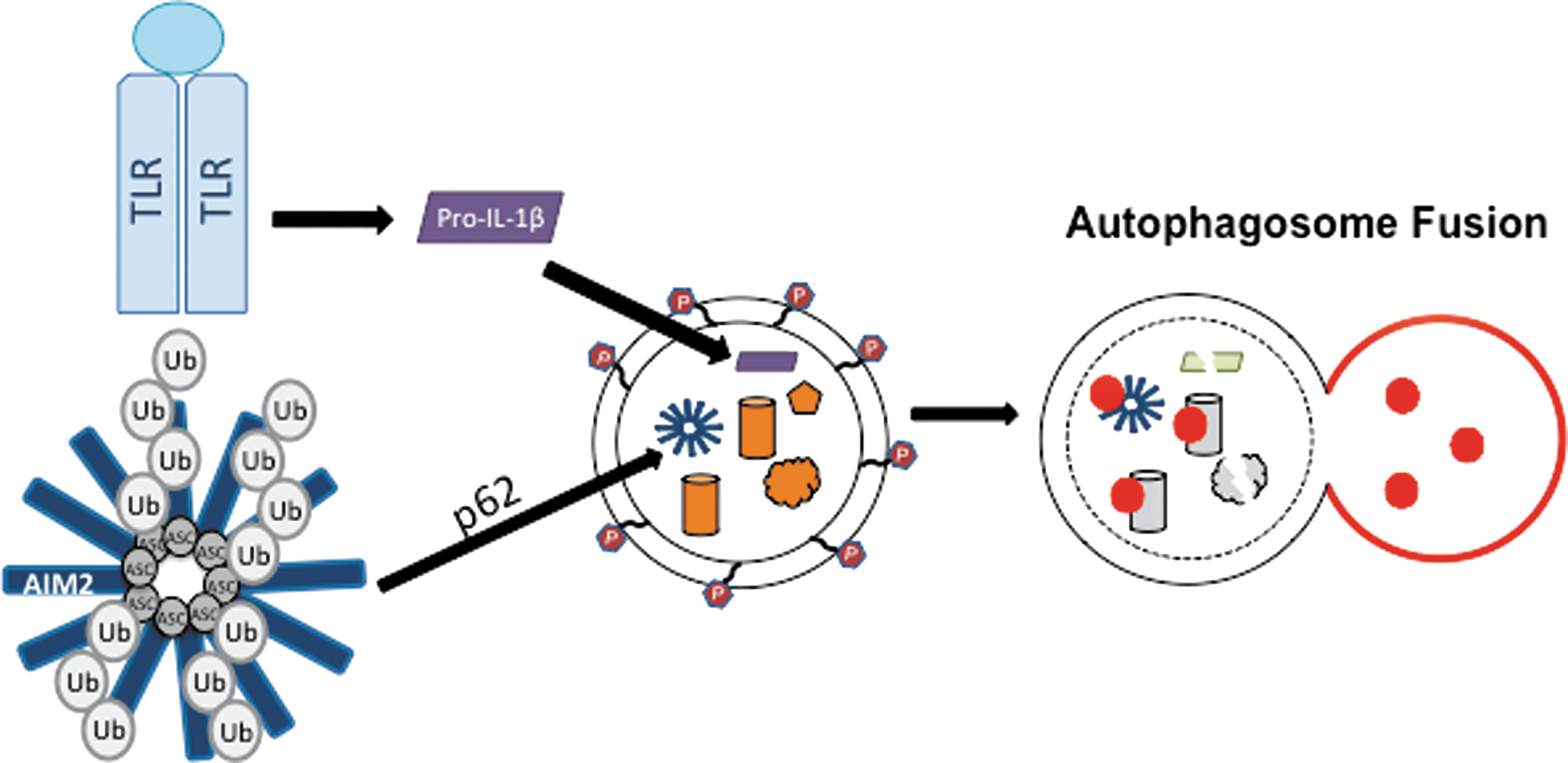
Inflammasomes are also negatively regulated by autophagy upstream from IL-1β secretion. It has been shown that a portion of ASC-containing inflammasomes is redirected towards autophagosomes and autophagolysosomes upon NLRP3 or AIM2 stimulation in THP-1 and primary human macrophage cells (85). Mechanistically, ASC localization to autophagosomes was dependent on both Beclin-1 and p62, a protein that specifically recruits ubiquitinated proteins to autophagosomes for degradation (Fig. 5) (85). ASC and ASC-containing inflammasome complexes could be recruited to autophagosomes by p62 since the K63-ubiquitination of ASC was detectable upon AIM2 stimulation (Fig. 3) (85). However, many new questions will need to be answered to elucidate the molecular mechanism of this potential negative regulation of inflammasomes. The ubiquitin ligase that modifies ASC and the trigger for ubiquitination are unknown. However, ASC protein levels did not change upon AIM2 stimulation and AIM2 levels actually increased at the timepoints in this study, making the significance of ASC localization to the autophagosome unclear. Perhaps the degradation of ASC-containing inflammasomes is balanced by increased protein expression, or proportionally very few inflammasomes are recruited to autophagosomes under stimulation conditions. Since experimental evidence is lacking, further investigation into the molecular mechanisms, regulation, and purpose of targeting inflammasomes to autophagosomes is imperative for determining how autophagy may work as a potential ‘off switch’ for activated inflammasomes.
In addition to ASC, NLRP3 is also ubiquitinated (45, 58, 72), although binding to p62 has not been reported so it is not known whether NLRP3 can be independently recruited to autophagosomes. NLRP3 is ubiquitinated in the LRR domain with a mix of lysine 63 (K63) and lysine 48 (K48) linked chains by unknown ubiquitin ligases (Fig. 3) (72). Ubiquitinated NLRP3 is inactive because its deubiquitination is required for inflammasome activation and is triggered by TLR activation, mtROS, and ATP (45, 58, 72). The deubiquitinase responsible for NLRP3 activation, BRCA1-BRCA2-containing complex 3 (BRCC3), is a member of the BRCC36-containing isopeptidase (BRISC) deubiquitination complex, which has been suggested to specifically cleave K63-linked ubiquitin chains and not K48-linked chains (15, 16, 27), making the mechanism of K48-linked ubiquitin removal unclear. Although K48-linked ubiquitinated proteins are often targeted for proteasomal degradation (27), MG132 proteasomal inhibitor treatment did not affect ubiquitinated NLRP3 protein levels in the presence of a BCC3 inhibitor (72), suggesting that ubiquitination of NLRP3 does not lead to proteasomal degradation of NLRP3. Thus, the mechanism of inhibition of NLRP3 by ubiquitination remains unknown.
NLRs Upregulate Autophagy
The positive and negative regulation of inflammasomes by autophagy is complemented by NLR-mediated control of autophagy (Fig. 6). NLRX1, a resident mitochondria protein (65), enhances autophagy during viral infection through an interaction with mitochondrial Tu translation elongation factor (TUFM) (55). TUFM likely increases autophagy through its interaction with the Atg5-Atg12 complex, which is an essential component of the vesicle elongation step of autophagy and is unable to bind NLRX1 on its own (Fig. 6) (55). Although it is not clear how the NLRX1/TUFM interaction with Atg5-Atg12 elevates autophagy, the resulting decrease in vesicular stomatitis virus (VSV) production in NLRX1 and Atg5 knockout mouse embryonic fibroblast (MEF) cells suggests that NLRX1 and autophagy are proviral factors during VSV infection (55). NLRX1 knockout MEFs had a larger decrease in VSV replication than Atg5−/− MEFS, which might be due to the proposed negative regulation of RIG-I-like helicase (RLH) signaling through the mitochondrial antiviral signaling (MAVS) adapter protein by NLRX1 (55, 98). This function has been called into question because different methods of knocking out NLRX1 in mice have given conflicting results. While two strains of NLRX1−/− mice have intact RLH/MAVS signaling and virus replication, a third strain has a defect in RLH/MAVS signaling and enhanced virus replication (5, 77, 89, 98). The latter strain was used to study the role of NLRX1 in TUFM-mediated autophagy enhancement, so it will be important to evaluate whether this effect is consistent in the other two NLRX1−/− strains as well.
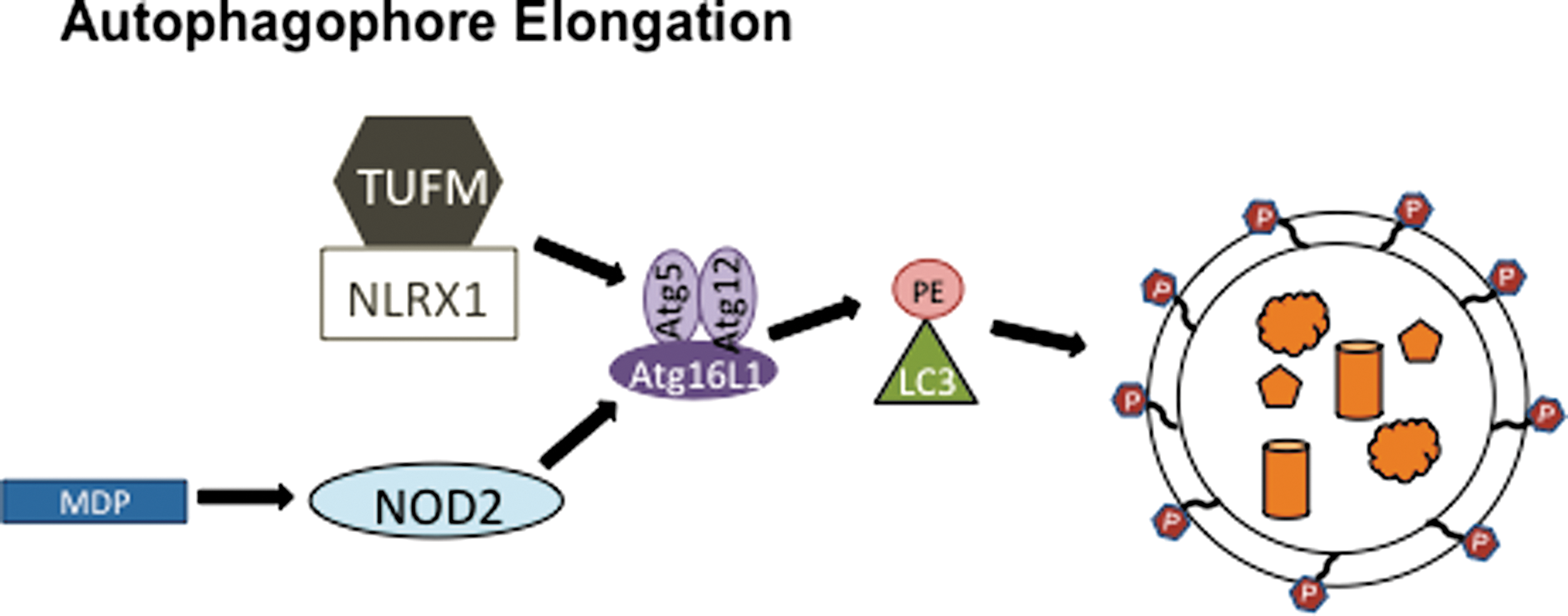
In addition to NLRX1, NOD2 also positively regulates autophagy. NOD2 is an NLR family member that utilizes the receptor interacting protein 2 (Rip2) adapter protein kinase to form a protein complex resembling the inflammasome, called a nodosome, which forms upon stimulation with MDP. Instead of activating caspase-1, the NOD2 nodosome activates NFκB, resulting in inflammatory cytokine production (6). Both NOD2 and Atg16L1 have been identified by several groups as the genes with risk alleles for Crohn's disease, a chronic inflammatory bowel disease characterized by high levels of inflammatory cytokines (1, 13, 37, 61, 78). A T300A mutation in the N-terminus of the WD repeats of Atg16L1 is associated with Crohn's disease in the presence or absence of NOD2 LRR domain point mutations R702W, G908R, or L1007C(frameshift) (6, 74). Recently, NOD2 and Atg16L1 have been functionally linked in a mechanism of bacterial pathogen clearance, which may explain why they are both genetically linked to Crohn's disease (14, 42, 70, 92). NOD2 induces autophagophore formation upon MDP stimulation by binding and recruiting Atg16L1 to the bacterial entry site of the plasma membrane to initiate autophagic destruction of the bacterial pathogen (Fig. 6) (42). Strikingly, dendritic cells from Crohn's patients with NOD2 or Atg16L1 variant alleles have wild type levels of autophagy in response to a TLR1/2 ligand, but have significantly reduced autophagy levels in response to MDP, a NOD2 stimulant (14). Consequently, cells from Crohn's patients have a defect in lysosomal destruction of bacterial pathogens (14, 92). The resulting higher load of bacteria in cells from Crohn's patients may contribute to higher levels of inflammatory cytokines, including IL-6 and IL-1β, which are detected in Crohn's patients' tissues. The increase of IL-1β cytokine production in Crohn's patients could be due to either increased pro-IL-1β protein levels or increased caspase-1 activity. Consistent with a role for NOD2 in Crohn's disease, the enhanced IL-1β secretion in Crohn's patients' cells in response to MDP is due to increased pro-IL1β mRNA levels and not due to caspase-1 activity, which remains unchanged when compared to healthy patients (70). NOD2 is constitutively expressed in immune cells and inducibly expressed in intestinal epithelial cells upon pro-inflammatory stimulation (6, 35). Thus, a potential mechanism for the trigger that controls NOD2-mediated autophagy could be the induction of NOD2 expression. Since RIP2 is ubiquitinated and phosphorylated to regulate NOD2 activation, additional post-transcriptional mechanisms may also regulate NOD2 protein levels (19, 91). As a potential mechanism for disease pathogenesis, NOD2-mediated autophagy may serve as a model for future studies since many additional autophagy and NLR proteins are also linked to chronic inflammatory diseases with unknown mechanisms.
Conclusions
The intersection of inflammasomes and autophagy is an exciting area of research with many unanswered questions that should be addressed by future investigation. Of course, for every host defense there is an opposing pathogen offense, making it likely that bacterial or viral proteins may exist that target or exploit the newly discovered links between autophagy and inflammasome regulation to avoid detection or destruction. Thus, as we uncover new mechanisms of regulation between the ancient innate immune pathways of inflammasomes and autophagy, we may also find novel host-pathogen interactions that may be targeted therapeutically. Moreover, treatments for chronic inflammatory diseases in which NLRs and autophagy proteins are implicated will rely on these findings as well.
Footnotes
Acknowledgments
This work was partly supported by 1F32AI096698 (M.R.); CA082057, CA31363, CA115284, DE019085, AI073099, AI083025, HL110609, GRL, the Hastings Foundation, and the Fletcher Jones Foundation (J.U.J.).