
Abstract
Introduction
H
High mobility group box-1 (HMGB1) in its oxidized (disulfide) form, most likely determined by the redox state of the extracellular milieu, potentiates N-methyl-
HMGB1 is a redox-sensitive protein containing three cysteines, namely C23, C45, and C106. C23 and C45, which are in the first HMG-box domain of HMGB1 (BoxA), can readily form a disulfide bond (48). C106, within the second HMG-box domain of HMGB1 (BoxB), is unpaired and required for HMGB1 binding to toll-like receptor 4 (TLR4) (68). Intracellular HMGB1, either nuclear or cytosolic, is in a reduced state (22), while on its extracellular release, it can form the disulfide bond (62, 69). HMGB1 containing the C23–C45 disulfide bond and free C106 have proinflammatory activity that is mediated by TLR4, thus inducing cytokine production and release from immunocompetent cells (68, 69). In contrast, HMGB1 containing all reduced cysteine residues (all-thiol HMGB1) has chemotactic properties that are mediated by the formation of a heterocomplex with the chemokine CXCL12, which activates the CXCR4 receptor (61, 62). Both HMGB1 activities can be irreversibly inactivated by sulfonation of cysteine residues, which probably represents a physiological mechanism to terminate the signaling of extracellular HMGB1 (69).
In addition to its immune-related functions, HMGB1 acts on brain cells by promoting neuronal differentiation, neurite outgrowth, and glia activation (29, 42
–44). Recently, we described a key role of HMGB1 in the generation and recurrence of seizures in animal models of epilepsy, thus unveiling its neuromodulatory effects in vivo (26, 37). HMGB1-induced neuronal hyperexcitability in seizure models was chiefly mediated by TLR4 and involved N-methyl-
In order to elucidate the mechanism by which HMGB1 contributes to NMDAR-mediated hyperexcitability and excitotoxicity (26, 37, 72), we asked whether HMGB1 enhances NMDAR-induced Ca2+ increase in neuronal somata. We also investigated the impact of the redox state of HMGB1, the identity of its receptors, and the mode of intracellular signaling in neurons.
Results
NMDAR-mediated neuronal Ca2+ influx
Figure 1A depicts the dose-dependent increase in intracellular Ca2+ evoked by 2.15 min exposure to NMDA in hippocampal neuronal cultures preloaded with fura-2 acetoxymethyl-ester (fura-2AM). While 1 μM NMDA was ineffective, 10 and 20 μM NMDA induced an average 50% and 80% increase in the peak value of intracellular Ca2+ (+39% and +64% for area under the curve [AUC], respectively) (p<0.01 vs. control Krebs buffer by one-way analysis of variance [ANOVA]). The maximal effect was reached at 100–500 μM NMDA (p<0.01 vs. control). NMDA-induced intracellular Ca2+ increase reflected ion influx through the Ca2+-permeable channel, as it was prevented by 5 min preincubation and co-incubation with 10 μM MK801, an open-channel and use-dependent NMDAR antagonist (67) (Fig. 1B). NMDA half maximal effective concentration was ∼11 and ∼15 μM for peak effect and AUC, respectively. In subsequent experiments, we used 10 μM NMDA, unless otherwise indicated, which corresponds to the concentration previously used to study the functional interactions of NMDA and interleukin-1 beta (IL-1β) or HMGB1 in vitro (25, 31, 64).

Effect of HMGB1 redox state on NMDA-induced Ca2+ increase
Preexposure of neurons to disulfide HMGB1 (0.004–400 nM) for 5 min did not affect Ca2+ levels per se but potentiated the NMDA-induced Ca2+ response dose dependently (Fig. 2A, B). Disulfide HMGB1 at 0.004 nM was ineffective, while a maximal increase in peak value and AUC of ∼40% and 83%, respectively, was reached between 0.4 and 400 nM (p<0.01 by two-way ANOVA). Disulfide HMGB1 (40 nM) was unable to promote Ca2+ influx when incubated with an ineffective concentration of NMDA (1 μM, Fig. 2A). When neurons were exposed to all-thiol HMGB1 (reduced form) in the presence of 10 μM NMDA, no effect was observed in the maximal increase in peak value while the AUC value was enhanced (data not shown). This suggested that all-thiol HMGB1 might be ineffective, but would start oxidizing in the incubation medium during the course of the experiment to induce its effect. To elucidate, therefore, the impact of the redox state of HMGB1 on NMDA-induced Ca2+ influx, we used a mutant form of HMGB1, 3S-HMGB1 (61), in which cysteines have been replaced by serines and cannot be oxidized. Incubation with 3S-HMGB1 (0.004–40 nM) failed to potentiate the NMDA-induced intracellular Ca2+ increase (Fig. 2C).
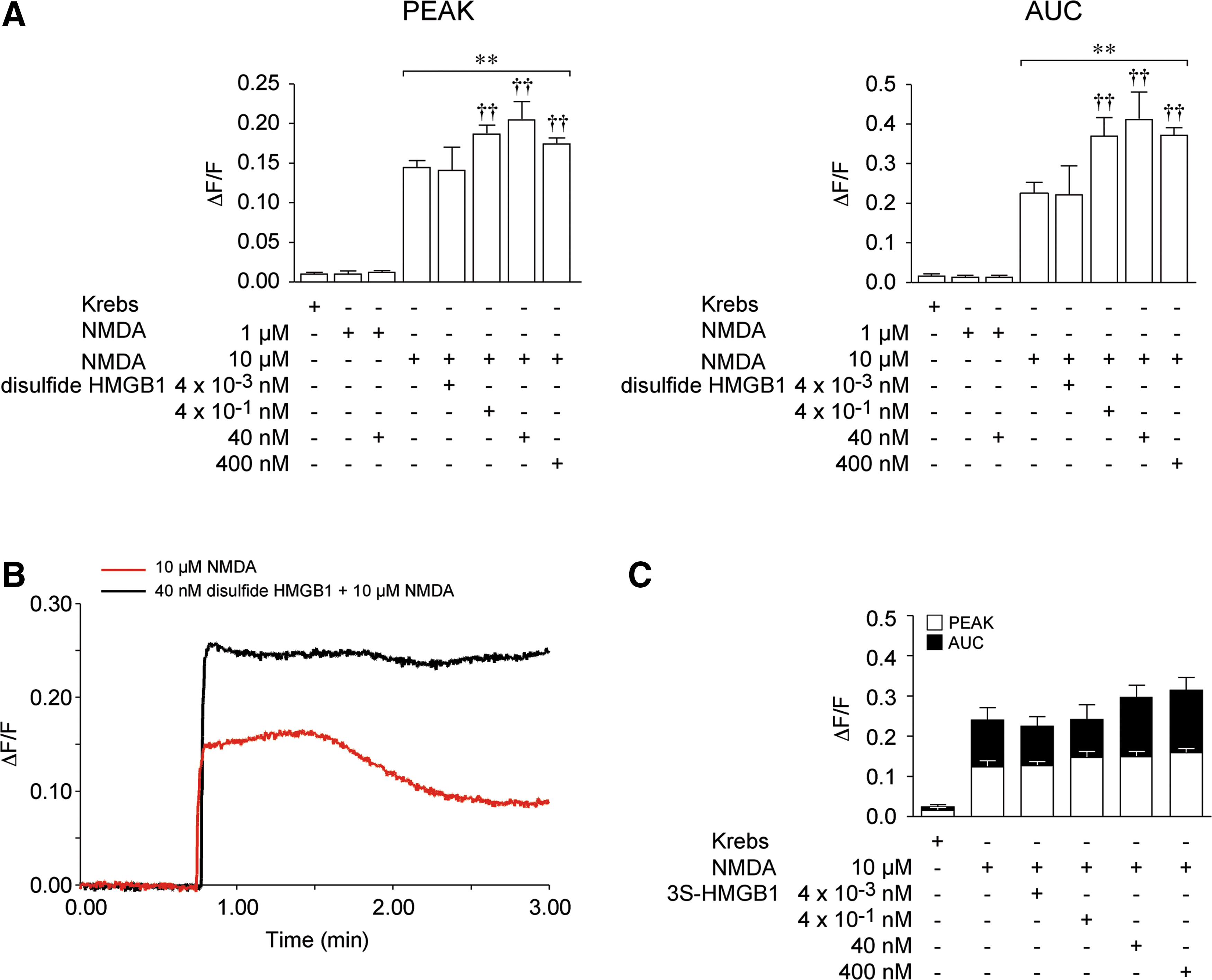
Receptor-mediated effect of disulfide HMGB1
The effect of 40 nM disulfide HMGB1 on the NMDA-mediated Ca2+ response of hippocampal neurons was abolished by 100 nM BoxA, an HMGB1 competitive inhibitor (70) (Fig. 3A; p<0.05 by two-way ANOVA), and by 100 μg/ml Rhodobacter sphaeroides lipopolysaccharide (LPS-RS), a selective TLR4 antagonist (Fig. 3B; p<0.01 by two-way ANOVA). These two antagonists per se neither evoked Ca2+ responses (not shown) nor affected Ca2+ fluxes induced by NMDA alone (Fig. 3A, B).
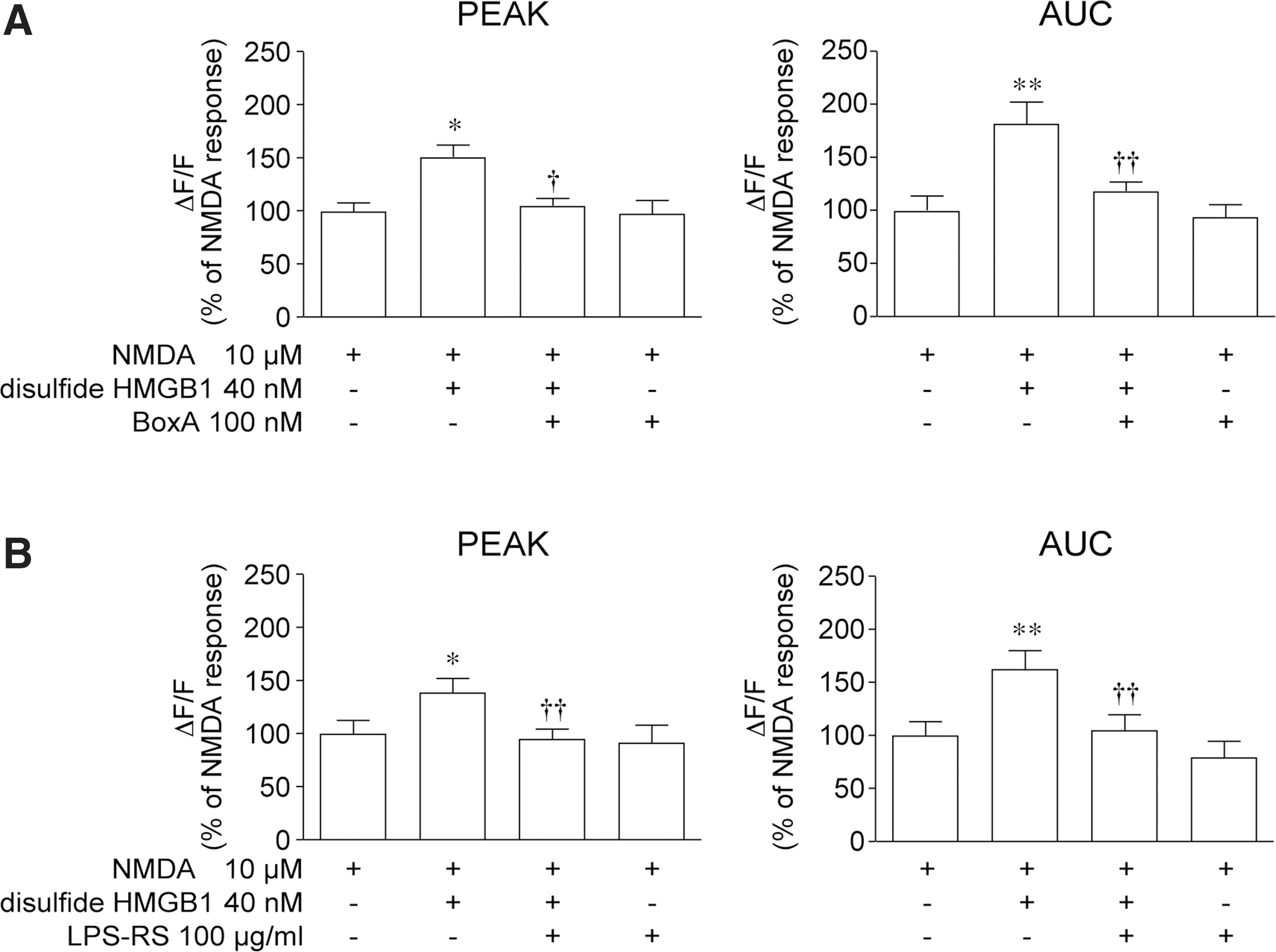
To test the involvement of RAGE and TLR4 in mediating disulfide HMGB1 interaction with NMDAR, we used neuronal cultures from Rage−/− (Fig. 4A) or Tlr4−/− mice (Fig. 4B). Ca2+ increase induced by 10 μM NMDA was similar in Rage−/− and wild-type neurons (wt, peak 0.17±0.02; AUC 0.22±0.03; Rage −/− peak, 0.15±0.01; AUC, 0.22±0.02). Disulfide HMGB1 enhanced the NMDA-induced Ca2+ response similarly in Rage −/− and wild-type hippocampal neurons (wt, peak +39%, AUC +82%; Rage −/− peak +35%, AUC +77%, Fig. 4A). The potentiation of NMDA response induced by disulfide HMGB1 in Rage −/− neurons was abolished by 100 μg/ml LPS-RS (Fig. 4A), as in wild-type neurons (Fig. 3B).
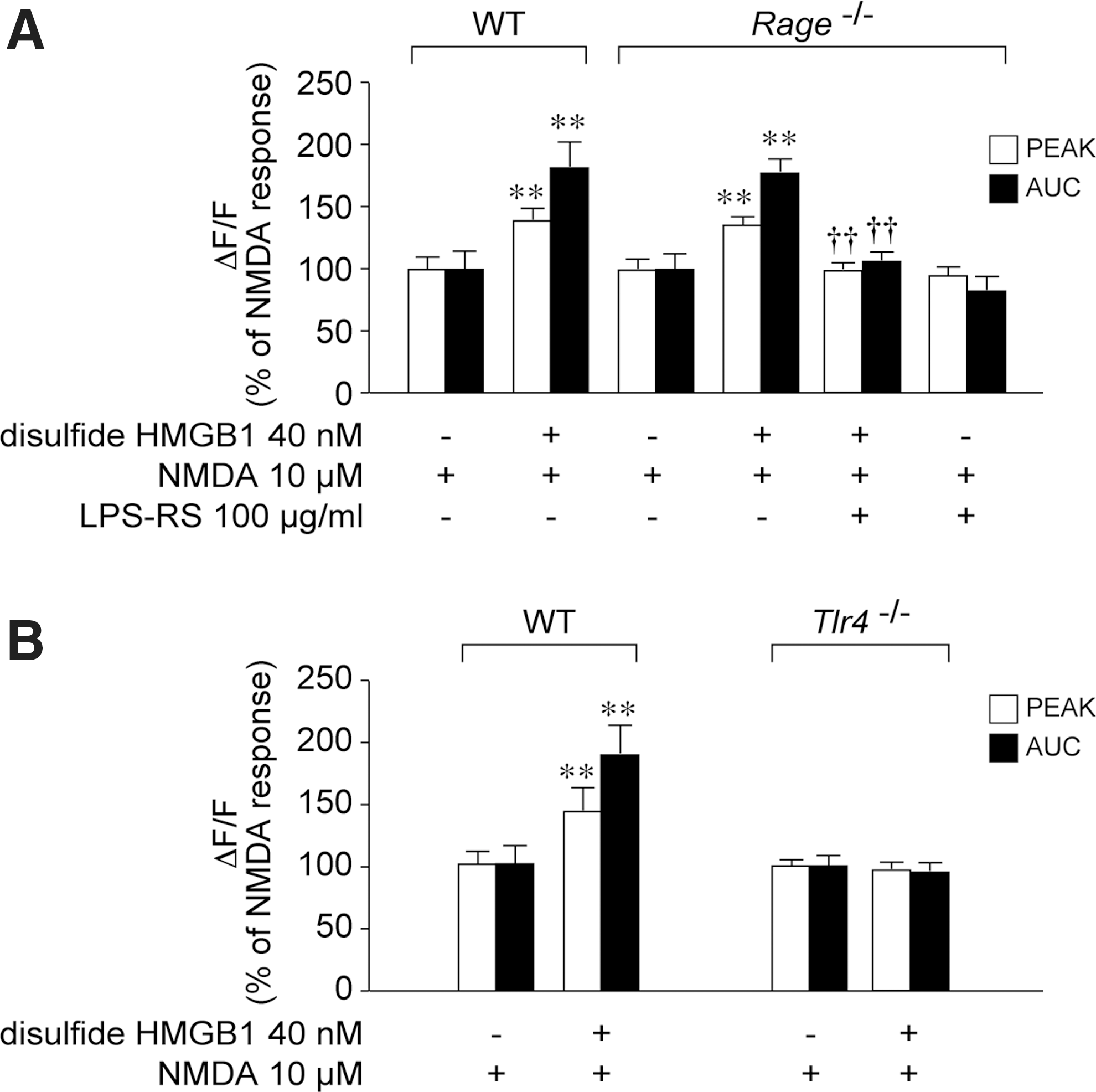
NMDA-mediated Ca2+ influx in neuronal cultures from Tlr4−/− was similar to wild-type cultures (wt, peak 0.16±0.01; AUC 0.23±0.04; Tlr4−/− peak, 0.14±0.01; AUC, 0.23±0.02). Disulfide HMGB1 failed to potentiate NMDA-induced Ca2+ response in Tlr4−/− derived neurons (Fig. 4B).
Molecular pathway underlying disulfide HMGB1 potentiation of the NMDA response
Previous evidence showed that activation of the IL-1β/IL-1R1 axis in neurons potentiates NMDA-induced Ca2+ influx via neutral sphingomyelinase (nSmase) and Src-family kinases, leading to the phosphorylation of the NR2B subunit (64). This pathway also mediates the proconvulsive activity of IL-1β (4). Since IL-1R1 and TLR4 have a similar cytosolic Toll/IL-1 receptor (TIR) domain and interact with the adaptor protein myeloid differentiation primary response protein (MyD88) (40), we investigated whether disulfide HMGB1 exerts its action on NMDA response by activating the same post-translational pathway as IL-1β.
Preincubation of hippocampal neurons with either 25 μM 3-O-methylsphingomyelin (3-O-MS), a selective nSmase inhibitor (57), or 10 μM 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo [3,4-d] pyrimidine (PP2), a selective antagonist of Src-family kinases (19), prevented the enhancement of the NMDA-induced Ca2+ response caused by 40 nM disulfide HMGB1 (p<0.05 by two-way ANOVA, Fig. 5A, B). Both inhibitors given alone were ineffective on NMDA-induced Ca2+ influx. PP3, the inactive PP2 analog (23), was ineffective per se, and did not affect disulfide HMGB1 action (Fig. 5B).
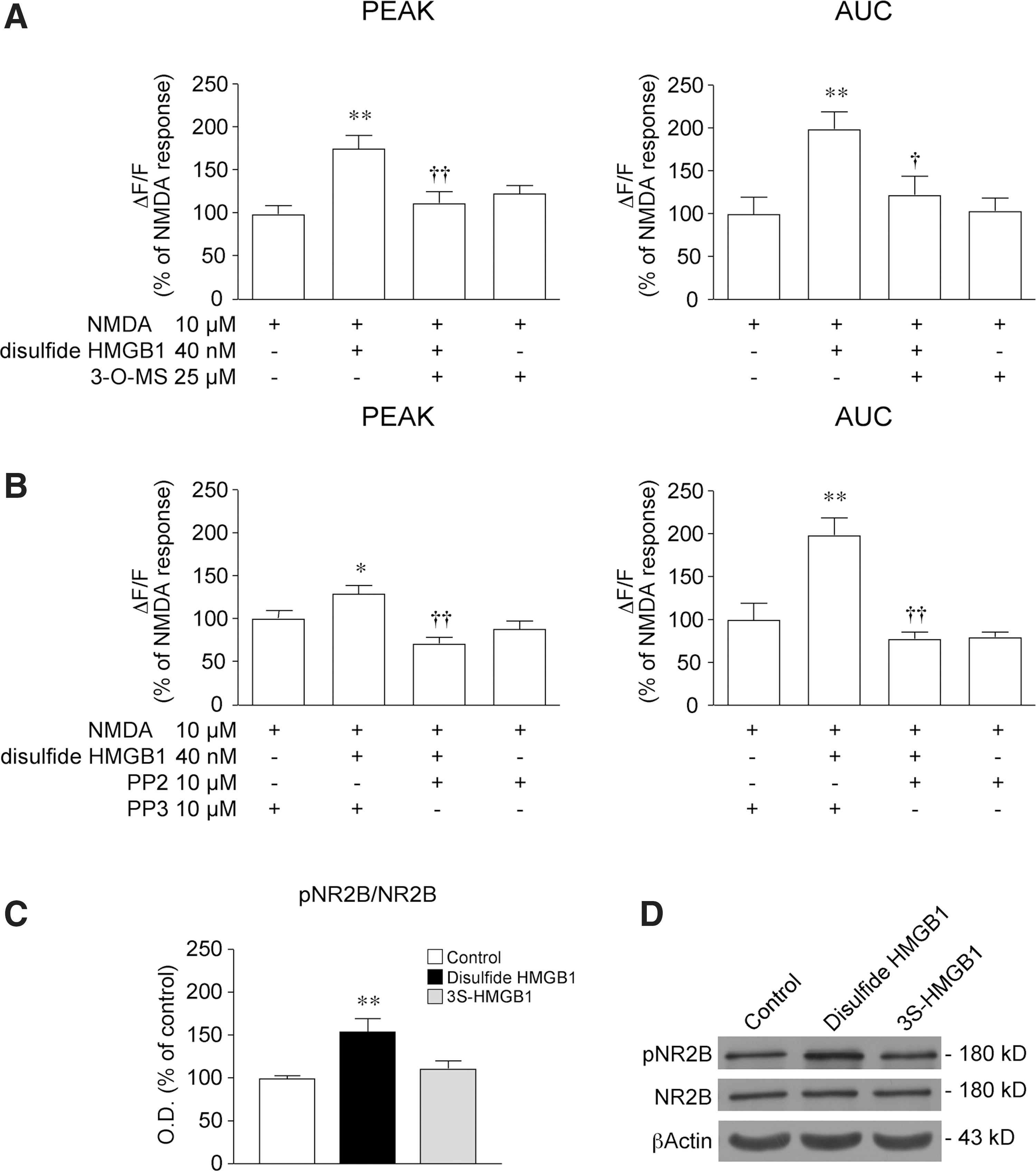
Since Src-family kinases increase NMDAR channel-gating properties by phosphorylating the NR2B subunit (49), we assessed by Western blot the level of Tyr1472 NR2B phosphorylation in neurons after 5 min of incubation with either disulfide or 3S-HMGB1. NR2B Tyr1472 phosphorylation was increased by about 55% by disulfide HMGB1 (Fig. 5C, p<0.01 by one-way ANOVA), while 3S-HMGB1 was ineffective. The total level of NR2B was not changed by the treatments (Fig. 5D).
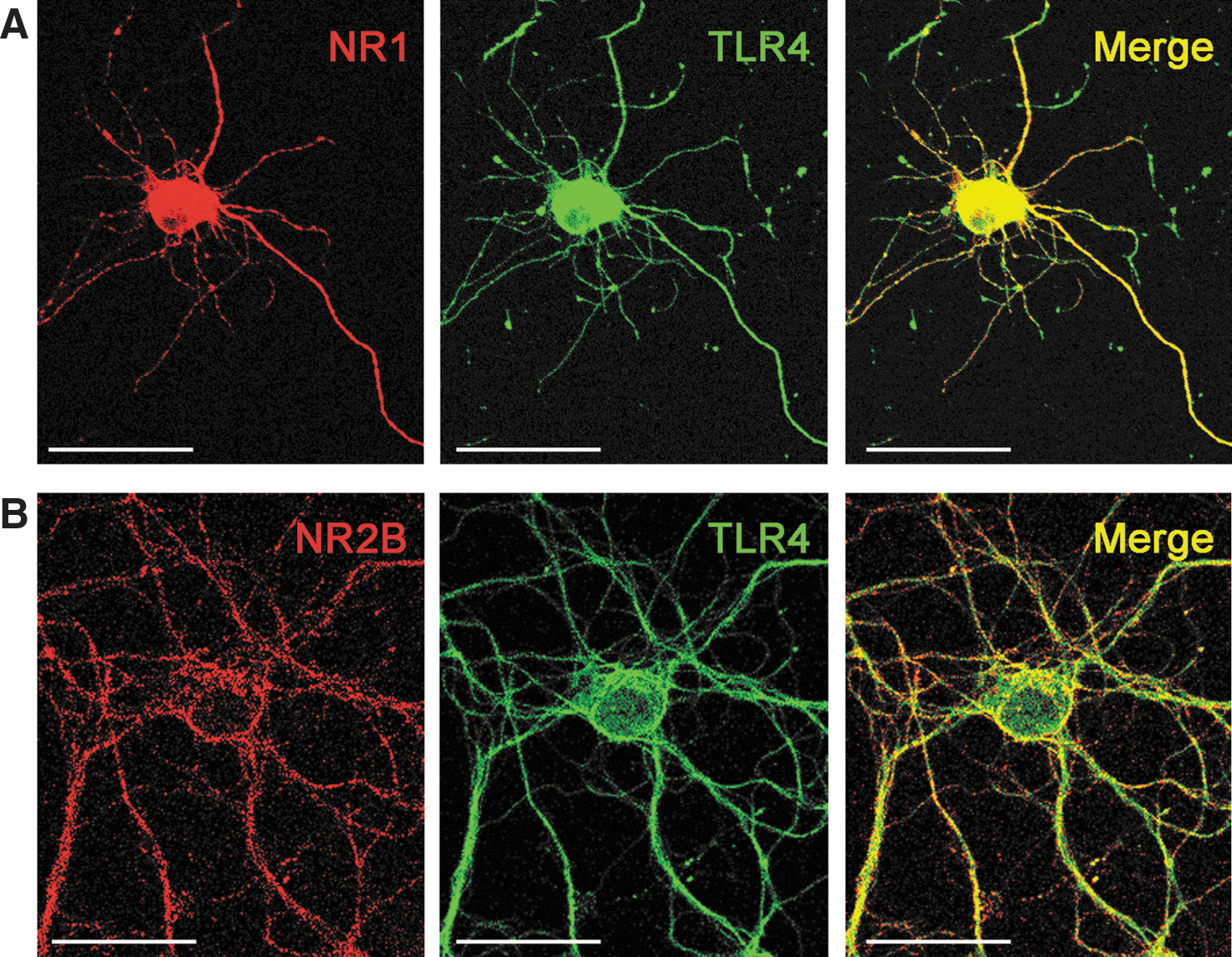
The mechanism we describe involves short-range interactions between TLR4, its interactors and downstream effectors, and the NMDAR as a target. We expected, therefore, that TLR4 and the target NMDAR would colocalize in the same neuronal structures. Double immunostaining showed a co-localization of TLR4 with N-methyl-
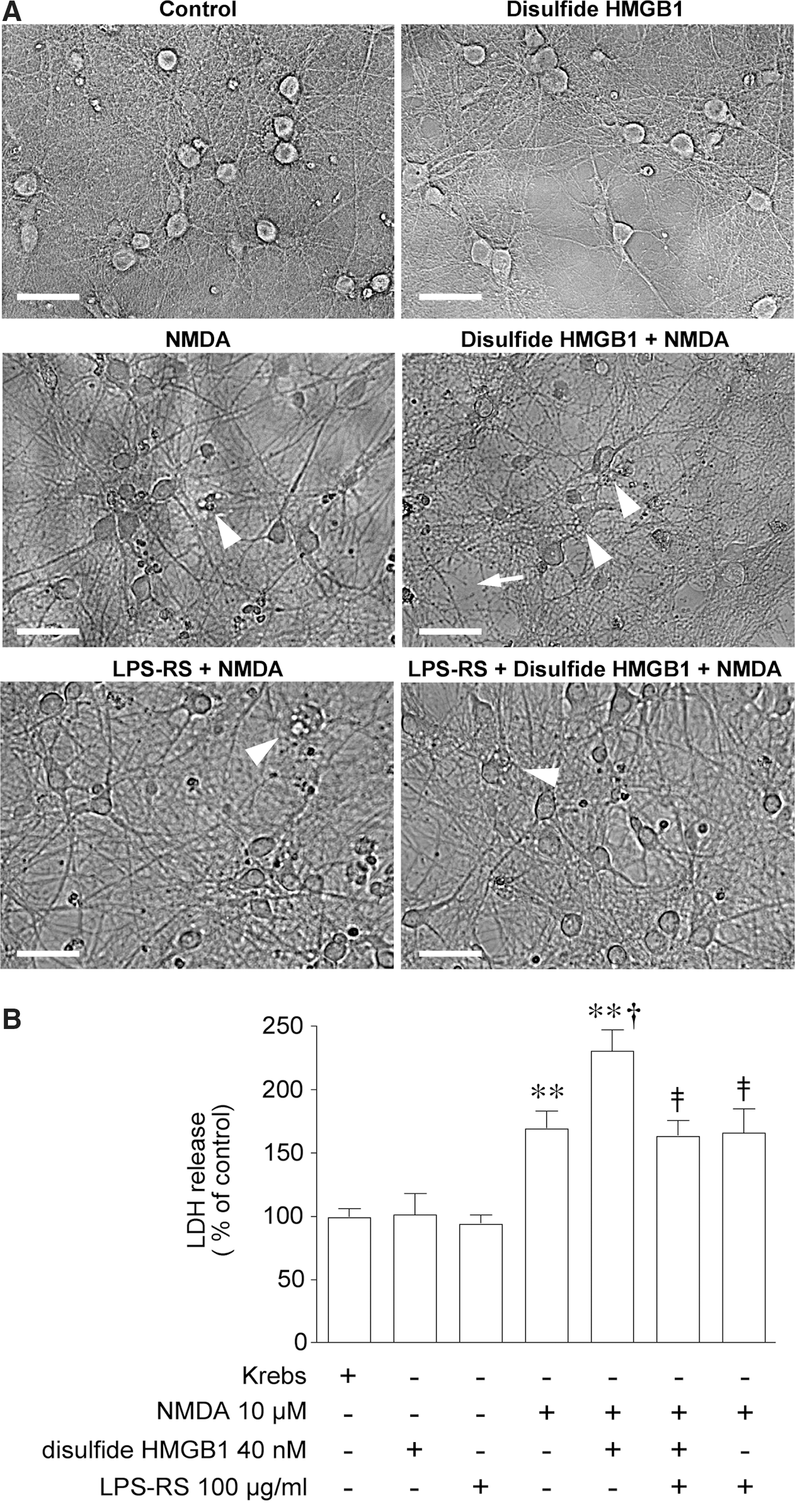
Effect of disulfide HMGB1 on NMDA-induced excitotoxicity
To evaluate the consequences of the disulfide HMGB1-TLR4 signaling on NMDAR function, we measured neuronal death. Phase-contrast microscopy of control and disulfide HMGB1-treated neurons showed viable cells characterized by bright compact somata of round-oval shape, with intact processes (Fig. 7A). Neurons exposed to 10 μM NMDA showed signs of neurodegeneration such as loss of brightness, somatic swelling, and vacuolation (highlighted in Fig. 7A). These features were exarcerbated in cultures pretreated with disulfide HMGB1, showing also loss of membrane integrity and neurite disintegration (highlighted in Fig. 7A). Morphological assessment of cell degeneration was confirmed by quantification of lactate dehydrogenase (LDH) release in the medium (Fig. 7B): NMDA increased LDH release by 60% (p<0.01 by two-way ANOVA), and this effect was doubled by co-incubation with disulfide HMGB1 (p<0.05 by two-way ANOVA). Preincubation of hippocampal neurons with 100 μg/ml LPS-RS, a selective TLR4 antagonist, prevented disulfide HMGB1 enhancement of NMDA-induced cell death (Fig. 7A, B; p<0.01 by two-way ANOVA). LPS-RS given alone was ineffective on NMDA-induced cell death (Fig. 7A, B).
Proictogenic effect of disulfide HMGB1
When we first demonstrated that HMGB1 increased chemoconvulsant-induced seizures in mice (37), the distinction between all-thiol and disulfide HMGB1 was not appreciated, nor was the possibility that all-thiol HMGB1 could be readily oxidized in vivo. To elucidate the impact of the redox state of HMGB1 on seizures (Fig. 8A), we used 3S-HMGB1 (61), which cannot be oxidized. The intrahippocampal injection in mice of 5.5 μg all-thiol HMGB1 increased the frequency of kainic acid (KA)-induced seizures (Fig. 8B; p<0.05 by one-way ANOVA) and the total time spent in seizures by about twofold (Fig. 8C; p<0.01). An intrahippocampal injection of 5.5 μg disulfide HMGB1, 15 min before KA, induced similar proictogenic effects by increasing seizure number and duration (Fig. 8B, C; p<0.05 by one-way ANOVA), whereas an injection of 5.5 μg 3S-HMGB1 was ineffective on these parameters (Fig. 8B, C). The sample of 3S-HMGB1 we used was fully bioactive as a chemoattractant for mouse 3T3 fibroblasts (data not shown).
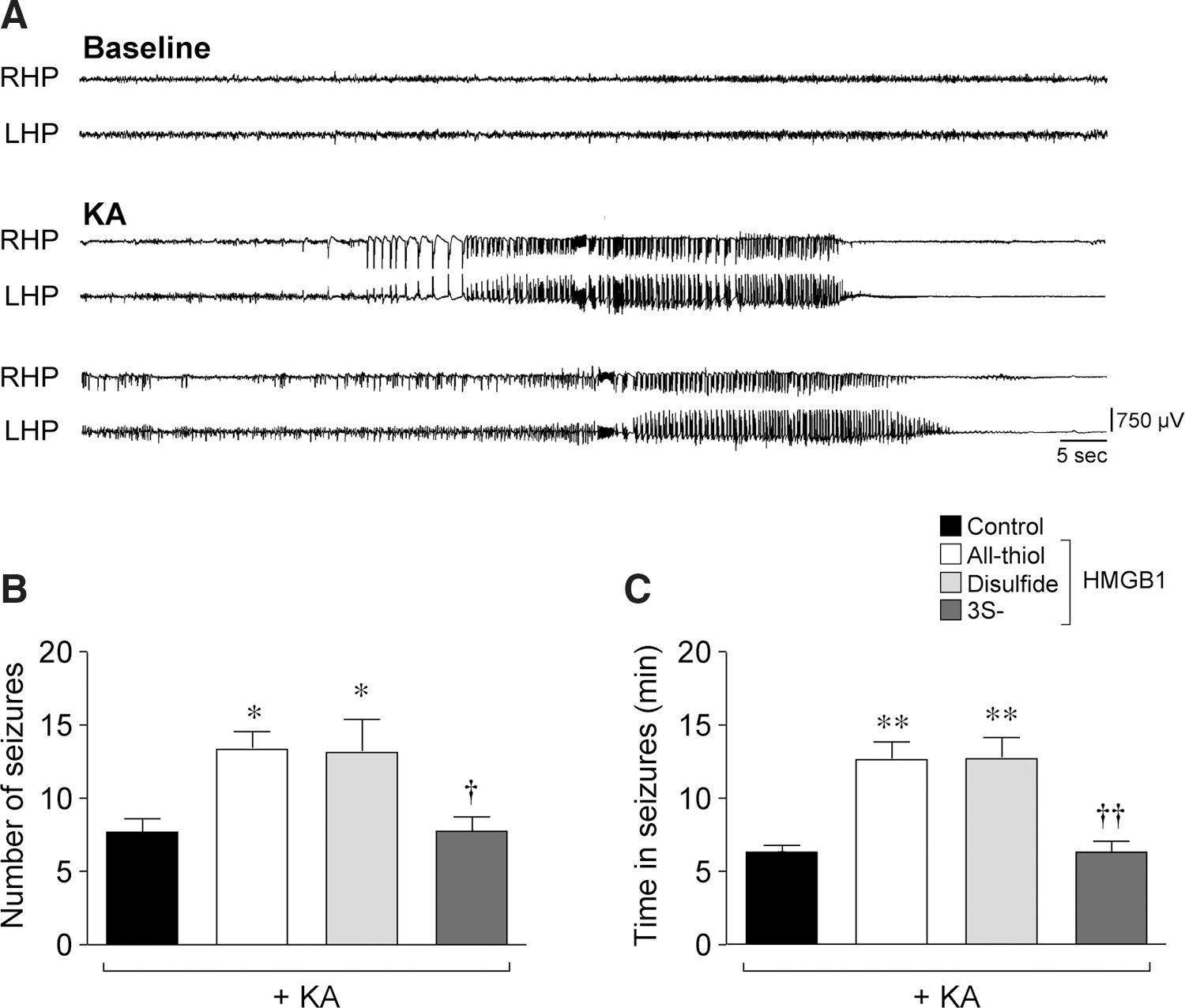
We suggest, therefore, that all-thiol HMGB1 is oxidized on intracerebral injection, thus acquiring the same proictogenic activity as disulfide HMGB1.
Discussion
Our study reports the novel finding that extracellular HMGB1 in its disulfide form enhances the Ca2+ influx evoked in neuronal cell body by NMDAR stimulation. The lack of this effect when HMGB1 is in nonoxidizable form underscores the key role of the redox state of this molecule for determining its neuronal action. The functional interaction between HMGB1 and NMDAR is mediated by the activation of TLR4, while RAGE signaling is not implicated. The TLR4-dependent molecular signaling activated by HMGB1 involves nSmase and Src kinases. Notably, the same signaling is induced in neurons by IL-1β activating IL-1R type 1 (IL-1R1) and similarly, underlies the cytokine potentiation of NMDA-mediated Ca2+ response (64). The common activation by both IL-1β and HMGB1 of a fast post-translational pathway in neurons which affects NMDAR function is compatible with the evidence that both TLR4 and IL-1R1 (64) are colocalized with NMDAR containing the NR2B subunit in neuronal cell body and dendrites. Moreover, IL-1R1 and TLR4 are a part of the same receptor superfamily sharing a common cytosolic TIR domain and the adapter protein MyD88, both of which are instrumental for intracellular signaling activation (40).
In analogy with IL-1β, we propose that the phosphorylation of the NR2B subunit is the final molecular target of the activation of the HMGB1-TLR4 signaling, which leads to the potentiation of NMDA-evoked neuronal Ca2+ influx. The following evidence supports this conclusion: (i) the disulfide, but not 3S-HMGB1, promotes both NR2B phosphorylation and NMDA-induced Ca2+ influx; (ii) the disulfide HMGB1 form activates Src, which is known to be involved in Tyr phosphorylation of NR2B protein and the consequent increase in magnitude and duration of NMDAR channel opening (1, 73); (iii) the disulfide HMGB1 form activates nSmase, which via ceramide production (33) alters Ca2+ currents in neurons through Src (4, 50, 64); and (iv) TLR4 and NR2B subunit are colocalized in the same neuronal compartments where HMGB1 potentiation of NMDA-mediated Ca2+ influx is measured.
Our data suggest that the activation of this rapid onset, post-translational pathway represents the key mechanism by which HMGB1 promotes neuronal excitability and seizure activity in vivo and potentiates NMDA-induced cell death (our study; 26,31,37). Notably, this was indeed the case for the pro-convulsive and pro-neurotoxic effects of IL-1β (4, 64). Concerted physiopathological actions of IL-1β and HMGB1 can be envisaged, as both these molecules are released by glia and neurons during brain injury (11, 53) or seizures (4, 37), and they cooperate to induce proinflammatory cytokines, chemokines and activate metalloproteinases (24, 52). Our evidence suggests that HMGB1 and IL-1β may also act in synergy by promoting the activation of neuronal NMDAR by endogenous ligands (e.g., glutamate,
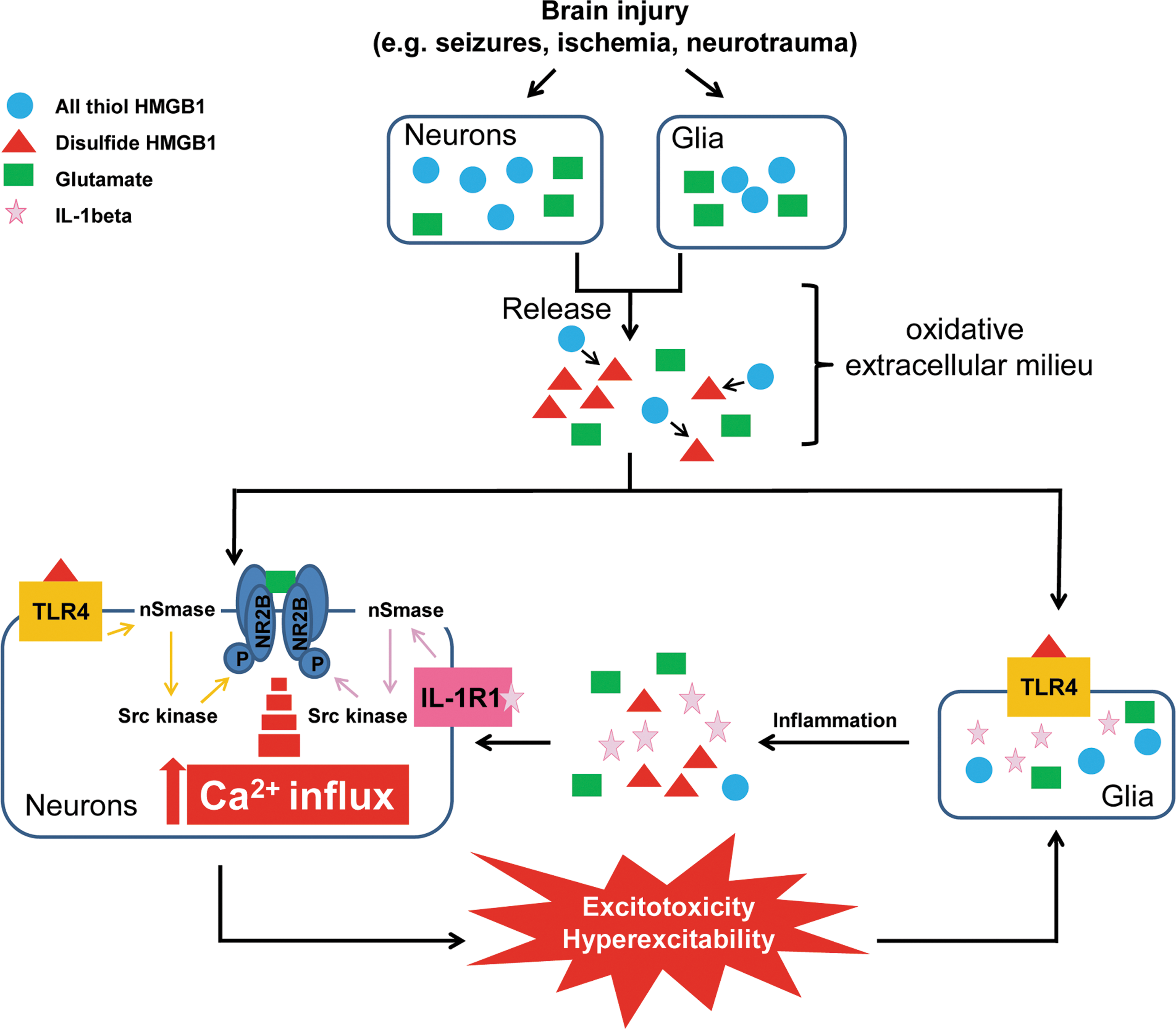
The redox state of HMGB1 determines its pleiotropic effects (61, 62, 69). We previously showed that convulsive stimuli cause HMGB1 nuclear-to-cytoplasmatic translocation in neurons and glia, and its cellular release (37). This phenomenon has been also described during brain ischemia (14, 44) or traumatic brain injury (18, 41). In these studies, released HMGB1 has pro-excitatory (37) and pro-neurotoxic effects (7, 14, 21, 28, 31, 41, 53, 72) as shown by pharmacological and genetic interventions in the animal models. Notably, brain injury, seizures, and the associated tissue inflammation induce the rapid production of free radicals and ROS (56, 65), thus favoring the oxidation of released HMGB1 and the consequent TLR4-dependent augmentation of neuronal NMDAR function. Disulfide HMGB1 has, indeed, been measured in experimental models of hepatic inflammation or muscle injury, showing that HMGB1 released in a permissive environment can change its redox state from a reduced to an oxidized form (62, 69). We suggest, therefore, that all-thiol HMGB1 is converted to disulfide HMGB1 in the hippocampus of mice undergoing seizures. Unfortunately, this hypothesis is difficult to prove rigorously, because the ex vivo manipulation of hippocampi in order to measure extracellular/released HMGB1 requires constant oxygenation to maintain cell viability, which would convert all-thiol HMGB1 to disulfide HMGB1 in any case.
We provide evidence that HMGB1-TLR4 functional interaction with NMDAR promotes cell death and seizures; therefore, it could critically contribute to the excitatory and neurotoxic role of HMGB1 described in brain injury models (30, 31, 44). The contribution of HMGB1 to CNS pathology may be amplified by the induction of cytokines and ROS release from immunocompetent cells and glia (17, 44, 68, 69).
There is evidence of increased neuronal HMGB1 release after incubation with NMDA at doses 2–10 higher than the one we used, and for prolonged incubation times ranging from 30 min to 24 h. These experiments were done in mixed cell cultures containing both astrocyes and neurons (31). HMGB1 levels in our neuronal cell culture medium ranged between 5 and 20 ng/ml, as assessed after 15 min and 24 h incubation in Mg2+-free Krebs solution. These concentrations were not significantly modified by 2.15 min exposure to NMDA followed by drug washout (data not shown). In accordance with our in vitro evidence, we demonstrated that HMGB1 in vivo is massively released by glia during seizures associated with cell loss (37), whereas glial cells are virtually absent in our culture conditions. We can, therefore, expect that glia contributes to the substantial concentrations of HMGB1 reached in vivo in the extracellular milieu in pathologic conditions. In this regard, HMGB1 brain levels have been reported to reach 2.5 μg/mg tissue after in vivo brain ischemia (66). These concentrations are in the range of those we used in our study.
HMGB1 potentiation of postsynaptic NMDAR function may also have a role in the cognitive deficits described in animals after systemic (8) or intracerebral HMGB1 administration in mice (39). Thus, a relatively high dose of HMGB1 induces loss of dendritic spines in pyramidal neurons that are associated with memory dysfunction (8). Notably, NR2B phosphorylation has been shown to mediate dendritic spine loss induced by amyloid-β-oligomers in hippocampal cultures (58).
A recent study indicated that HMGB1 can interact directly with presynaptic NMDAR without the concomitant activation of TLR4 or RAGE (42). The mechanism we describe here is different: (i) We monitored Ca2+ influx induced by NMDA in cell somata, therefore our mechanism involves postsynaptic NMDAR. Presynaptic NMDAR-mediated effects can be excluded, as our experiments were run in the presence of tetrodotoxin (TTX), which blocks synaptic transmission; (ii) the molecular interaction between HMGB1 and presynaptic NMDAR facilitates receptor activation at sub-stimulatory amounts of agonist (42); in our studies, HMGB1 potentiates the postsynaptic NMDA response only when NMDAR is activated by an effective agonist concentration. This likely depends on the ability of HMGB1 to amplify the magnitude and duration of the NMDA-activated Ca2+ permeable channel (1, 73) by inducing NR2B phosphorylation; (iii) the physical interaction between HMGB1 and NMDAR was observed at picomolar HMGB1 concentrations and promoted cell motility, neurite outgrowth, cell differentiation, and glutamate release from presynaptic terminals, which are phenomena with putative physiological effects during brain development, cell repair/regeneration, and long-term potentiation (13, 45, 46, 60). We instead showed the activation of a pathological chain of events triggered by nanomolar HMGB1 concentrations, which are similar to those measured in rodent brain after damage or seizures (37, 66). The combination of our data and those already published, therefore, indicates that the functional consequences of extracellular HMGB1 on neurons depend on its redox state, extracellular concentration, and the neuronal compartment where HMGB1–NMDAR interactions take place (i.e., cell somata vs. presynaptic terminals).
In conclusion, our findings provide novel evidence of a TLR4-mediated interaction between extracellular HMGB1 and neuronal NR2B containing NMDAR, leading to pathologic outcomes. Agents interfering with this HMGB1 action, such as BoxA or TLR4 antagonists, may be, therefore, considered for developing pharmacological interventions (20, 71) to prevent the excessive NMDAR stimulation leading to hyperexcitability and cell damage in various acute and chronic CNS disorders (15).
Materials and Methods
Experimental animals
We used C57BL6N wild-type (Charles River), Rage−
/−, and Tlr4−
/− 18 day-old mouse fetuses for neuronal cultures and male C57BL6N mice (60 day-old, 25 g) for EEG recording. The generation of the Rage
−/− mouse line has been previously described in detail (3, 35). Tlr4
−/− mice were obtained from The Jackson Laboratories (Strain Name: C57BL/10ScNJ). They have a deletion of the Tlr4 gene, resulting in the absence of both mRNA and protein; thus, these mice do not respond to lipopolysaccharide (LPS) stimulation. The phenotypic characterization of these mice is reported in detail in the vendor Web site (C57BL/10ScNJ, Stock Number 003752;
Procedures involving animals and their care were conducted in accordance with the ethically approved institutional guidelines that are in compliance with national and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1, Dec.12, 1987; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996).
Primary hippocampal neuronal cultures
Neuronal cultures were prepared from C57BL6N wild-type Rage−
/− and Tlr4−
/− 18 day-old mouse fetuses as previously described (9). Hippocampi were isolated in cold Hank's Buffered Salt Solution (HBSS; Life Technologies Invitrogen), then incubated 20 min at 37°C in HBSS containing 0.1% trypsin (Life Technologies Invitrogen). The digestion was then blocked with 10% heat-inactivated fetal bovine serum (Life Technologies Invitrogen) and 0.05% DNAse (Sigma) in Neurobasal media (Life Technologies Invitrogen). The hippocampi were mechanically dissociated by repeated passage through a constricted pasteur pipette. The cells were resuspended in Dulbecco's Modified Eagle Medium (Life Technologies Invitrogen) containing 10% horse serum, and they were plated on eight-well microslides (Ibidi) coated with 25 μg/ml poly-
Single-cell Ca2+ imaging
Cultured hippocampal neurons plated into eight-well microslides were loaded for 30 min at 37°C with 10 μM fura-2AM form (Molecular Probes) in Krebs–Ringer–Hepes (KRH) buffer (NaCl 125.0 mM, KCl 5.0 mM, MgSO4 1.2 mM, CaCl2 2.0 mM, glucose 10.0 mM Hepes 25.0 mM, and pH 7.4) supplemented with 1% bovine serum albumin (BSA) (63). After washing out the dye-containing medium, cells were imaged on the stage of an inverted Olympus IX81 microscope equipped with a Ca2+ imaging unit (CellR Olympus; 51). Image sequences were acquired for 3 min by alternate excitation at 340 and 380 nm, and measurement of emission at 510 nm every 235 ms (9 ms exposure time, 2×2 binning) with 40×fluorescence objective and a digital CCD camera. Fura-2AM fluorescence intensity, which reflects the concentration of intracellular Ca2+, was measured in all single neuronal somata present in the recorded field and was expressed as DeltaF/F (ΔF/F), which corresponds to (Ft−F0/F0), where F0 is the initial fluorescence value and Ft is the fluorescence value at the time t of measurement. All imaging experiments were carried out at 37°C in 5% CO2 Mg2+-free KRH containing 0.5 μM TTX to prevent synaptic activity.
The response to drug application was calculated as the peak amplitude defined as the maximum Ca2+ rise and the AUC obtained from the integral of the area of the response for each cell (Prism Graphpad Software) as an index of the total Ca2+ entry (34).
Drug application
NMDA (Tocris) was dissolved in Mg2+-free KRH and applied at 1–500 μM to neuronal hippocampal cultures for 2.15 min as a bolus application after 45 s of baseline fluorescence recording (64). MK-801 (10 μM; Tocris), a use-dependent open channel NMDAR antagonist (67), was incubated for 5 min before 10 μM NMDA, and along with NMDA for 2.15 min (64).
Disulfide HMGB1 (HMGBiotech) was expressed in Escherichia coli and purified using a continuous descending gradient of ammonium sulfate (HiLoad 26/10 Phenyl Sepharose High Performance column connected to an Akta Purifier FPLC system; GE Healthcare) followed by ionic-strength chromatography (Hi-trap Q column; GE Healthcare). The purity and integrity of purified HMGB1 was verified by Coomassie blue staining after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Contaminating LPS was removed from protein preparations by Triton X-114 extraction. Disulfide HMGB1 activity was measured by testing its ability to stimulate cytokine production in human macrophages.
Nonoxidizable HMGB1 (3S-HMGB1) was generated using the QuikChange XL Site-Directed Mutagenesis kit according to the manufacturer's instructions (Stratagene; Agilent Technologies), checked by sequencing, expressed, and purified as Cytokine-HMGB1. The activity of 3S-HMGB1 was measured by its ability to induce migration of mouse 3T3 fibroblasts in migration assay.
Full-length, LPS-free recombinant HMGB1 (all-thiol) is the unmodified protein, with no tags or additional amino acids. It is produced in E. coli from an expression plasmid coding for the rat protein, and it was concentrated for an injection in the brain using Microcon 10000 MWCO filters (Millipore). In in vitro experiments, disulfide, all-thiol, or 3S-HMGB1 (0.004–400 nM) was added 5 min before, and during, 10 μM NMDA stimulation.
In in vivo experiments, disulfide, all-thiol or 3S-HMGB1 (5.5 μg/μl) was unilaterally injected 15 min before KA into the septal pole of the hippocampus.
BoxA (HMGBiotech) corresponds to one of the highly conserved DNA-binding domains of HMGB1 protein, aa 1–89. It functions as endogenous HMGB1 competitor (70). BoxA is extensively tested by HMGBiotech to exclude LPS contamination for HMGB1. BoxA (100 nM) was incubated 5 min before HMGB1, and for the following incubation times as reported in the respective figures. This was the dose effective in suppressing cortical neuronal cell death induced by conditioned media from NMDA-treated cultures (31).
LPS-RS (100 μg/ml; InvivoGen), a selective TLR4 antagonist (10), was added 5 min before HMGB1. One-hundred-fold excess relative to HMGB1 was previously used to inhibit LPS-induced intracellular Ca2+ increase in trigeminal sensory neurons (12).
3-O-MS (25 μM; BioMol Research Laboratories, Inc.), a selective nSmase inhibitor (57, 74), was added 30 min before HMGB1. This dose was shown to inhibit the IL-1β-induced IL-6 synthesis in primary neurons (57).
Ten micromolar PP2 (Calbiochem), an Src-family of tyrosine kinase inhibitor (19), was added 30 min before HMGB1 and for the following incubation times as reported in the respective figures. This dose was shown to block IL-1β-mediated hyperpolarization of warm-sensitive hypothalamic neurons (50). Control cultures were treated with 10 μM PP3, the ineffective analog of PP2.
Immunofluorescence labeling and confocal imaging
Hippocampal neurons were fixed in 4% paraformaldehyde for 30 min, then washed thrice with phosphate-buffered saline (PBS). Cells were permeabilized at 4°C in PBS containing 0.1% Triton X-100 (Sigma) for 3 min followed by 15 min incubation in 10% BSA in PBS to inhibit nonspecific antibody binding sites. Then, neurons were incubated overnight with primary antibodies against NR1 subunit (1:200; Merck Millipore), NR2B subunit (1:50; Santa Cruz Biotechnology, Inc.), and TLR4 (1:200; Abcam, Inc.) at 4°C in 0.2% BSA in PBS. After three washing steps, sections were incubated in PBS containing anti-mouse (for NR1), anti-goat (for NR2B), or anti-rabbit (for TLR4) secondary antibody conjugated to Alexa488 or Alexa546 (1:500; Molecular Probes). Double immunostaining was examined with an Olympus Fluorview laser scanning confocal microscope (microscope BX61 and confocal system FV500) using dual excitation of 488 nm (Laser Ar) and 546 nm (Laser He–Ne green) for Alexa488 and Alexa546, respectively. The emission of fluorescent probes was collected on separate detectors. To eliminate the possibility of bleed-through between channels, the sections were scanned in a sequential mode. The specificity of the TLR4 antibody was double checked using neurons from Tlr4 −/− mice. In these neurons lacking Tlr4, no staining was observed. As additional control condition, omission of the primary antibody against NR1, NR2B, or Tlr4 abrogated the immunohistochemical signal.
Western blots
Hippocampal neuronal cultures were treated for 5 min with 40 nM disulfide or 3S-HMGB1. After two washes with ice-cold PBS, the cells were harvested into ice-cold solubilization buffer containing 20 mM Tris acetate, 270 mM sucrose, 0.1% deoxycholate, 1 mM ethylenediaminetetraaceticacid, 1 mM ethylene glycol tetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na ortovanadate, 50 mM NaF, 5 mM Na pirophosphate, 10 mM Na-β-glicerophosphate, 1 mM dl-dithio-threitol, 1% Triton X-100 in the presence of a complete set of protease (Complete; Boehringer Mannheim), and phosphatase inhibitors (Sigma). Total proteins (30 μg per lane; Bio-Rad Protein Assay) were separated using 8% acrylamide SDS-PAGE, and each sample was run in duplicate. Proteins were transferred to Hybond nitrocellulose membranes by electroblotting. For immunoblotting, we used anti-pTyr1472-NR2B (1:1000; Thermo Scientific) rabbit polyclonal antibodies. Blots used to measure pTyr1472-NR2B were stripped, then re-probed to measure total levels of NR2B as previously described (16). Total NR2B levels were assessed using rabbit polyclonal anti-NR2B antibody (1:500; Invitrogen). Immunoreactivity was visualized with enhanced chemiluminescence (ECL) using peroxidase-conjugated goat anti-rabbit (1:1000; Sigma) immunoglobulin as secondary antibodies. Densitometric analysis of immunoblots was done to quantify the changes in protein levels (Quantity One Software; Bio-Rad Laboratories) using film exposures with maximal signals below the photographic saturation point. Optical density values in each sample were normalized using the corresponding amount of β-actin (1:15,000; Santa Cruz Biotechnology).
Cellular viability assay
Hippocampal neurons were exposed to vehicle (Mg2+-free KRH), 40 nM disulfide HMGB1 alone for 5 min, 10 μM NMDA alone for 2.15 min, or LPS-RS (100 μg/ml)±disulfide HMGB1 for 5 min followed by co-incubation with 10 μM NMDA. At the end of incubation, all drugs were removed and neurons were incubated in fresh culture medium for 24 h. Neuronal cell damage was assessed by measuring LDH released in the extracellular medium (LDH detection kit; Roche). LDH release is a well-established index of cell injury (32, 36). Fifty microliter of culture medium were combined with 50 μl of NAD+ and tetrazolium salt solution for 30 min; then, the absorbance was detected at 492 nm with a spectrophotometer (ELISA reader; Infinite M200). The results were expressed as % of LDH values in control culture medium. At the end of the experiments, cell cultures were fixed in 4% paraformaldehyde for 30 min to assess cell viability morphologically by phase-contrast microscopy.
Mouse model of seizures
Male C57BL6N mice (60 day-old, 25 g) mice were surgically implanted with an injection guide cannula and recording electrodes under general gas anesthesia (1.4% isoflurane in a mixture of 70% N2O–30% O2) and stereotaxic guidance (37). Two nichrome-insulated bipolar depth electrodes (60 μm OD) were implanted bilaterally into the dorsal hippocampus (from bregma [mm]: nose bar 0; anteroposterior—1.7, lateral 1.5 and 1.9 below dura mater). A 23-gauge cannula was unilaterally positioned on top of the dura mater and glued to one of the depth electrodes for the intrahippocampal infusion of KA. The electrodes were connected to a multipin socket and, along with the cannula of injection, secured to the skull by acrylic dental cement. The correct position of the electrodes and injection needle was evaluated by histological analysis of brain tissue at the end of the experiments.
An intrahippocampal injection of KA in freely moving mice was adiministered 7 days after surgery as previously described (37). KA (7 ng in 0.5 μl; Sigma) was dissolved in 0.1 M PBS, pH 7.4 and injected unilaterally in the dorsal hippocampus by using a needle protruding 1.9 mm from the bottom of the guide cannula. This dose of KA was proved to induce EEG ictal episodes in the hippocampus in 100% of mice without mortality (37).
Acute seizures assessment and quantification
EEG seizures induced by an intrahippocampal injection of KA in mice have been extensively described earlier (26, 37). Briefly, a 30 min baseline EEG recording was done before KA injection to assess the spontaneous activity pattern, and for 180 min after kainate injection. Kainate was injected over 1 min (CMA/100 pump) using a 30-gauge injection needle connected to a 10.0 μl Hamilton microsyringe via PE20 tubing; the needle was left in place for one additional min to avoid backflow through the cannula. Ictal episodes are characterized by high frequency (7–10 Hz) and/or multispike complexes and/or high-voltage (700 μV–1.0 mV) synchronized spikes simultaneously occurring in the injected and contralateral hippocampi (Fig. 8A). EEG activity was monitored using the GRASS 79D EEG Recording System. The signal was digitalized with a PowerLab 16/S data acquisition system and analyzed with LabChart 7 software (ADInstruments Pty Ltd.). The EEG recording of each animal was analyzed visually to detect any activity different from baseline. Seizures were quantified by their total number and duration (by summing up the duration of every ictal episode during the EEG recording period). Seizures occurred with an average latency of about 10 min from KA injection, then recurred for about 90 min from their onset, and were associated with motor arrest of the mice. EEG analysis was done by two independent investigators who were blinded to the treatment, who visually reviewed all the EEG tracings.
Statistical analysis
Data represented the means±standard error of the mean (n=number of individual wells). The effects of treatments were analyzed by one-way ANOVA followed by Tukey's test (NMDA dose–response, western blot analysis, and EEG seizures) or by two-way ANOVA followed by Tukey's test or Student t test. A significance level of 95% (p<0.05) was accepted. Statistical analysis was done using absolute values.
Footnotes
Acknowledgments
This work was supported by Cariplo Foundation grant 2009-2426 (A.V. and M.E.B.), Fondazione Monzino (A.V.), and Ministero della salute (RF-2009-1506142 to A.V. and M.E.B.). The authors are grateful to Pietro Veglianese, Assunta Senatore (Mario Negri Institute), and Alessandro Preti (HMGBiotech) for their contribution to a part of these experiments.
Author Disclosure Statement
The authors declare no direct financial interest. However, M.E.B. is founder and part-owner of HMGBiotech