
Abstract
Introduction
I
The treatment strategies for retinopathy of prematurity remain unsatisfactory. The main approach is the use of laser photocoagulation to decrease areas of damaged and hypoxic retina (31, 45). However, this treatment only provides benefits in the end stage of the condition, and unfortunately, neither prevents disease progression nor completely prevents vision loss. Therefore, new strategies that target the underlying mechanisms involved in the development of retinopathy of prematurity are urgently required (10, 12). A likely critical pathomechanism is an excessive generation of reactive oxygen species (ROS). The neonatal retina maintains a high rate of oxidative metabolism, which is exacerbated in retinopathy of prematurity by an inadequate ability to scavenge ROS due to reduced levels of antioxidants (35). Clinical studies have evaluated the use of antioxidants, such as vitamins C and E. However, the results have been inconsistent (35, 42), which is likely to be due, at least in part, to these approaches having suboptimal effects in reducing ROS accumulation.
Our study provides novel insight into the role of particular NADPH oxidase (NOX) isoforms in the development of vascular injury, inflammation, and oxidative stress in ischemic retinopathy. Furthermore, the presence of NOX isoforms in retinal vascular and nonvascular cells suggests that particular cell populations drive reactive oxygen species (ROS)/NOX-mediated damage. We speculate that the inhibition of pathological NOX isoforms may provide a new avenue for the treatment of ischemic retinopathies, such as retinopathy of prematurity, where preventative mechanism-based therapies are urgently required.
There is considerable interest in the role of the NADPH oxidase (NOX) family in a wide range of ischemic and ROS-mediated vascular pathologies (3, 5, 17, 59). This is due to the uniqueness of this enzyme family as they are the only enzymes with their sole function being the generation of ROS. Seven isoforms of the NADPH oxidase catalytic unit, NOX (NOX1–5, DUOX1, DUOX2), have been identified, of which NOX1, NOX2, and NOX4 are considered to be relevant to vasculopathy (3, 5, 17, 56, 59). Strategies to reduce NOX activity and the generation of ROS, such as naturally occurring flavonoids, have recently been shown to have benefits in retinal pathology (7, 25). However, the relative contribution of individual NOX isoforms to retinal neovascularization is not known. Identifying the pathologically relevant NOX isoform(s) has the potential to provide a new and effective preventative treatment approach for retinopathy of prematurity. Furthermore, the advent of more specific pharmacological approaches to inhibit certain NOX isoforms, such as GKT137831, represents a new treatment strategy. Indeed, this compound is an orally active NOX inhibitor with a relative specificity for NOX1 and NOX4 (4, 19).
To determine the contribution of NOX isoforms to retinopathy of prematurity, we used a robust in vivo model known as oxygen-induced retinopathy (OIR). We induced OIR in mice with deletions of the NOX1, NOX2, or NOX4 isoforms and also evaluated the effects of GKT137831 in mice and rats with OIR. Vascular injury and inflammation as well as the levels of proangiogenic and proinflammatory markers and ROS were examined in retina. To determine the cellular source of NOX in retina, we studied primary cultures of retinal vascular and neuro-glial elements and examined the effects of GKT137831 on hypoxic-induced ROS production in retinal microglia. We have clearly identified NOX1 as the first molecular treatment target for vasculopathy and the excessive production of ROS in retinopathy of prematurity. Our findings therefore have implications for the treatment of various ischemic retinopathies.
Results
Body weight gain
As reported previously, the body weight gain of animals with OIR was reduced compared to room air controls (14). There was no effect of NOX1 and NOX4 isoform deletion on body weight gain. However, NOX2 knockout (KO) mice with OIR had reduced body weight gain compared to NOX2 WT OIR mice. GKT137831 had no effect on body weight gain compared to untreated OIR rats (Supplementary Table S1; Supplementary Data are available online at
NOX1 KO mice with OIR exhibit reduced retinal neovascularization, vaso-obliteration, and vascular leakage
To determine if NOX1, NOX2, and NOX4 are present in retina, we qualitatively evaluated gene expression in C57BL/6 mice. Given that all the three isoforms were expressed (Fig. 1A), we next determined which NOX isoform contributes to vasculopathy in OIR. Therefore, we studied NOX1 KO, NOX2 KO, and NOX4 KO mice. We first determined that retina from NOX KO mice exhibited a deficiency in the relevant NOX isoform (Fig. 1A) and KO mice did not have compensatory changes in NOX isoform expression (Supplementary Fig. S1A–C). In room air controls, vascularization of the retina was normal. As expected, in all WT mice with OIR, retinal neovascularization and an avascular vaso-obliterated central retina were present (Fig. 1B–D). In NOX1 KO mice with OIR, retinal neovascularization and the avascular central retina were clearly markedly reduced compared to NOX1 WT mice with OIR. However, in NOX2 KO mice and NOX4 KO mice with OIR, neovascularization and the avascular central retina were not reduced compared to respective WT OIR (Fig. 1B–D). The findings for neovascularization were confirmed in paraffin sections of retina (Supplementary Fig. S2A, B). Breakdown of the blood–retinal barrier and subsequent vascular leakage is a major contributor to vision loss (16, 39). Indeed, retinal vascular leakage occurred in WT mice with OIR compared to room air controls (Fig. 1E). In NOX1 KO mice with OIR, retinal vascular leakage was reduced compared to NOX1 WT controls with OIR. However, in NOX2 KO mice and NOX4 KO mice with OIR, retinal vascular leakage was not affected compared to respective WT mice with OIR (Fig. 1E).

NOX1 KO mice with OIR exhibit reduced leukostasis
As inflammation is known to contribute to the development of vasculopathy in OIR (23, 29, 61), we evaluated the extent of leukocyte adherence to the retinal vasculature. In room air controls, few adherent leukocytes were detected in retinal blood vessels (Fig. 2A, B). As expected, in WT mice with OIR, retinal leukostasis was increased compared to room air controls (Fig. 2A, B). Importantly, in NOX1 KO mice with OIR, retinal leukostasis was reduced compared to NOX1 WT mice with OIR to the level seen in room air controls. However, in NOX2 KO and NOX4 KO mice with OIR, retinal leukostasis was not reduced compared to respective WT OIR mice (Fig. 2A, B).

NOX1 KO mice with OIR exhibit reduced labeling for the microglial marker, ionizing binding adaptor molecule-1
Microglia are resident inflammatory cells of the retina, which contribute to vascular remodeling in OIR (43) and are known source of ROS (55). Using the microglial marker, ionizing binding adaptor molecule-1 (Iba1), we investigated the degree of immunolabeling in the region of the retina associated with pre-retinal neovascularization. As previously reported (14), the degree of Iba1 immunolabeling was increased in WT mice with OIR compared to room air controls. Immunolabeling for Iba1 was often associated with blood vessels (Fig. 3A, B). In NOX1 KO mice with OIR, Iba1 immunolabeling did not increase as seen in NOX1 WT mice with OIR (Fig. 3A, B). In NOX2 KO mice with OIR, the extent of Iba1 immunolabeling was similar to NOX2 WT mice with OIR, with this labeling associated with pre-retinal blood vessels. In NOX4 KO mice with OIR (Fig. 3A, B), Iba1 immunolabeling was not elevated to the same extent as NOX4 WT mice with OIR, although still present in pre-retinal blood vessels.
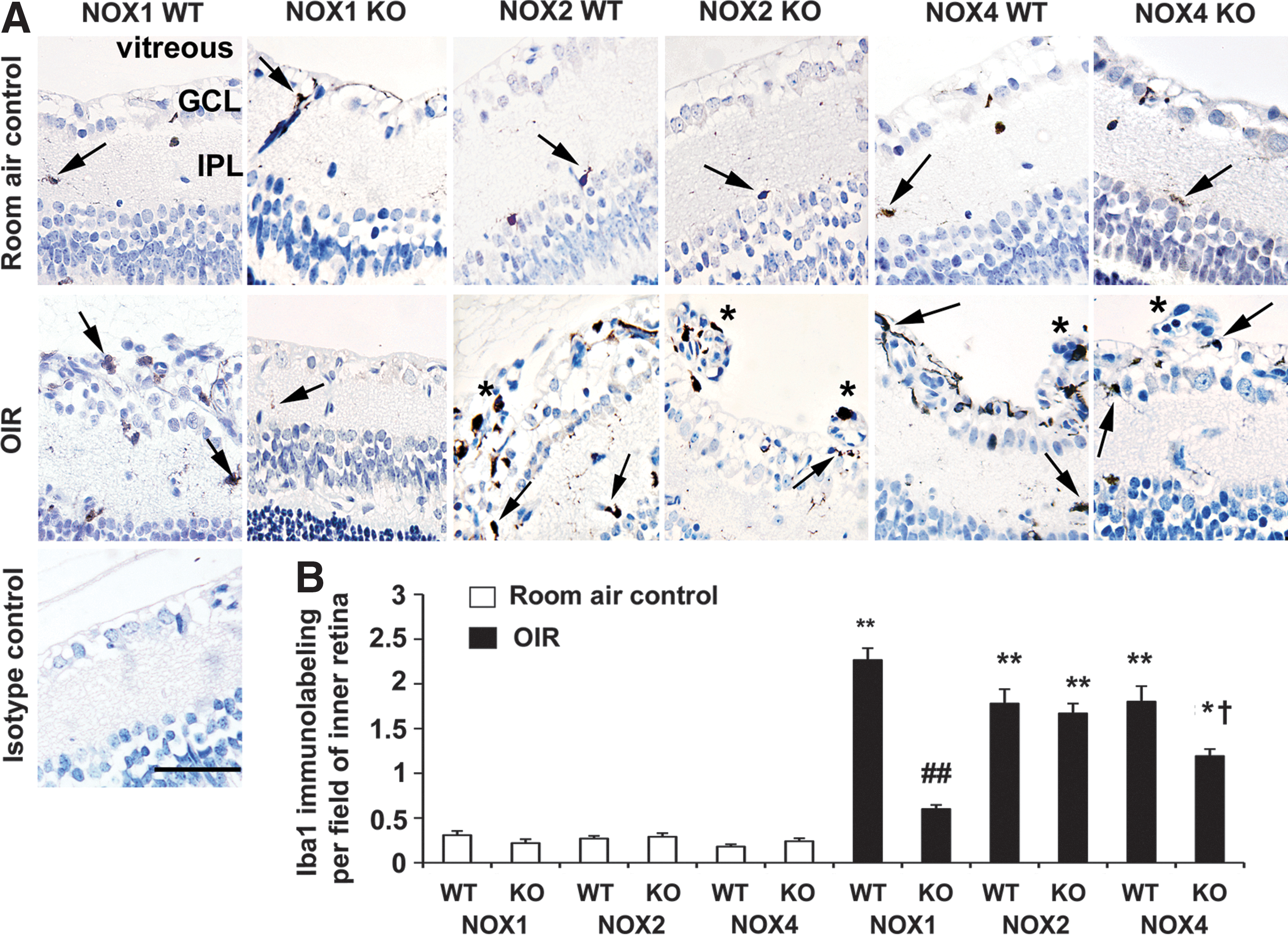
NOX1 KO mice with OIR exhibit reduced expression of angiogenic and inflammatory factors
To determine if angiogenic and inflammatory factors, which are known to be involved in the development of OIR (6, 8, 37, 40), were reduced in the KO mice, we studied the gene expression of VEGF, erythropoietin, angiopoietin-2, and intercellular adhesion molecule-1 (ICAM-1) in retina by quantitative polymerase chain reaction (PCR). As expected, in WT mice with OIR, retinal mRNA levels of all factors were increased compared to room air controls (Fig. 4A–D). Consistent with the decrease in retinal vasculopathy and leukostasis, in NOX1 KO mice with OIR, retinal mRNA levels of these particular molecules implicated in angiogenesis and inflammation, were reduced compared to NOX1 WT mice with OIR (Fig. 4A–D). By contrast, in NOX2 KO and NOX4 KO mice with OIR, the mRNA levels of all these factors were unaltered compared to respective WT mice with OIR (Fig. 4A–D).
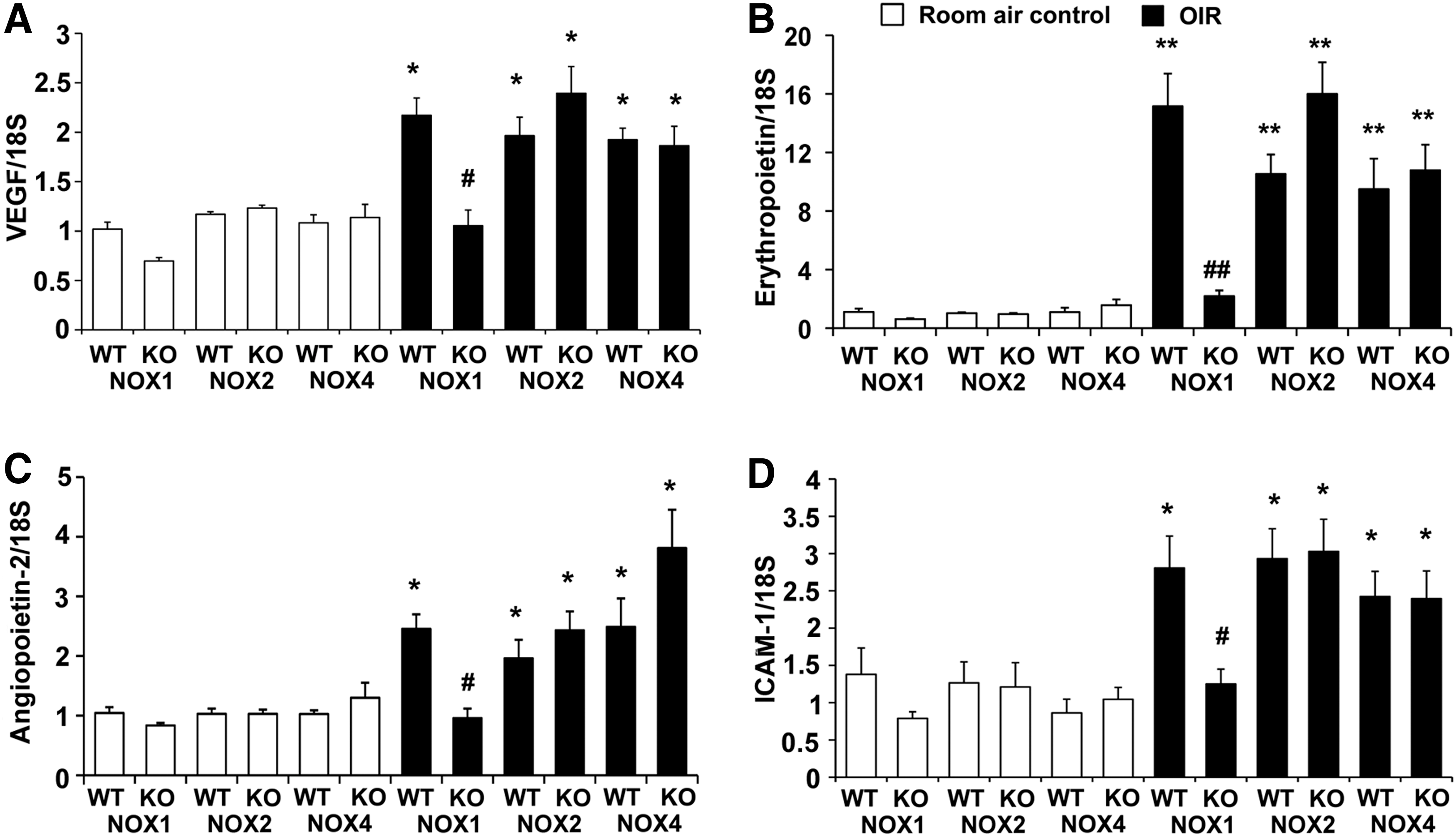
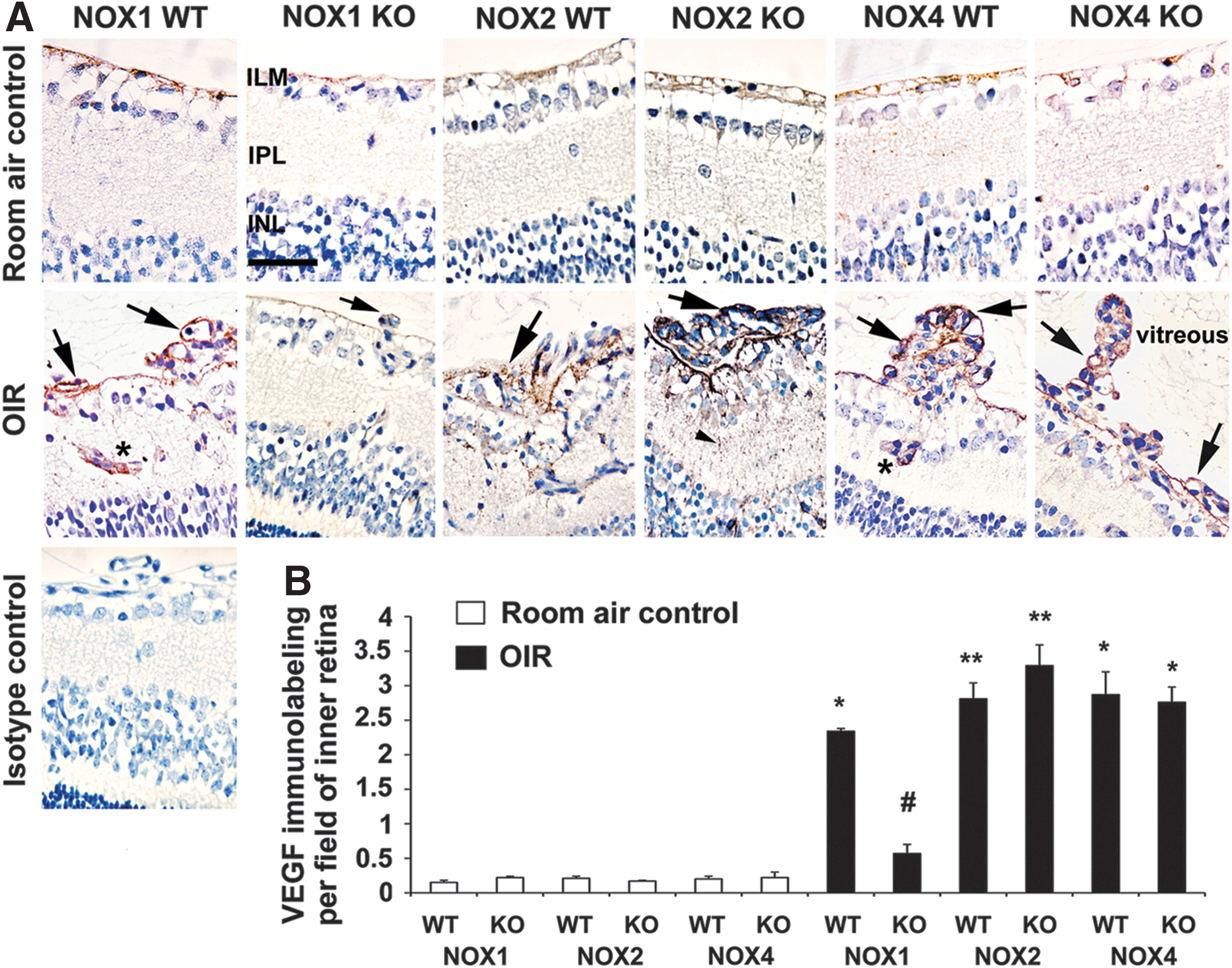
NOX1 KO mice with OIR exhibit reduced retinal VEGF protein
Given VEGF's predominant role in retinal vasculopathy and inflammation in OIR (40), we performed VEGF immunolabeling. In room air controls, VEGF immunolabeling was barely detectable and mainly observed on the inner limiting membrane and occasionally in ganglion cells (Fig. 5A). In WT mice with OIR, intense VEGF immunolabeling was observed in pre-retinal neovascular vessels protruding into the vitreous cavity, in some intraretinal blood vessels, ganglion cells, and processes of macroglial Müller cells. In NOX1 KO mice with OIR, but not in NOX2 KO and NOX4 KO mice with OIR, VEGF labeling in all regions was markedly reduced compared to respective WT mice with OIR (Fig. 5A, B).
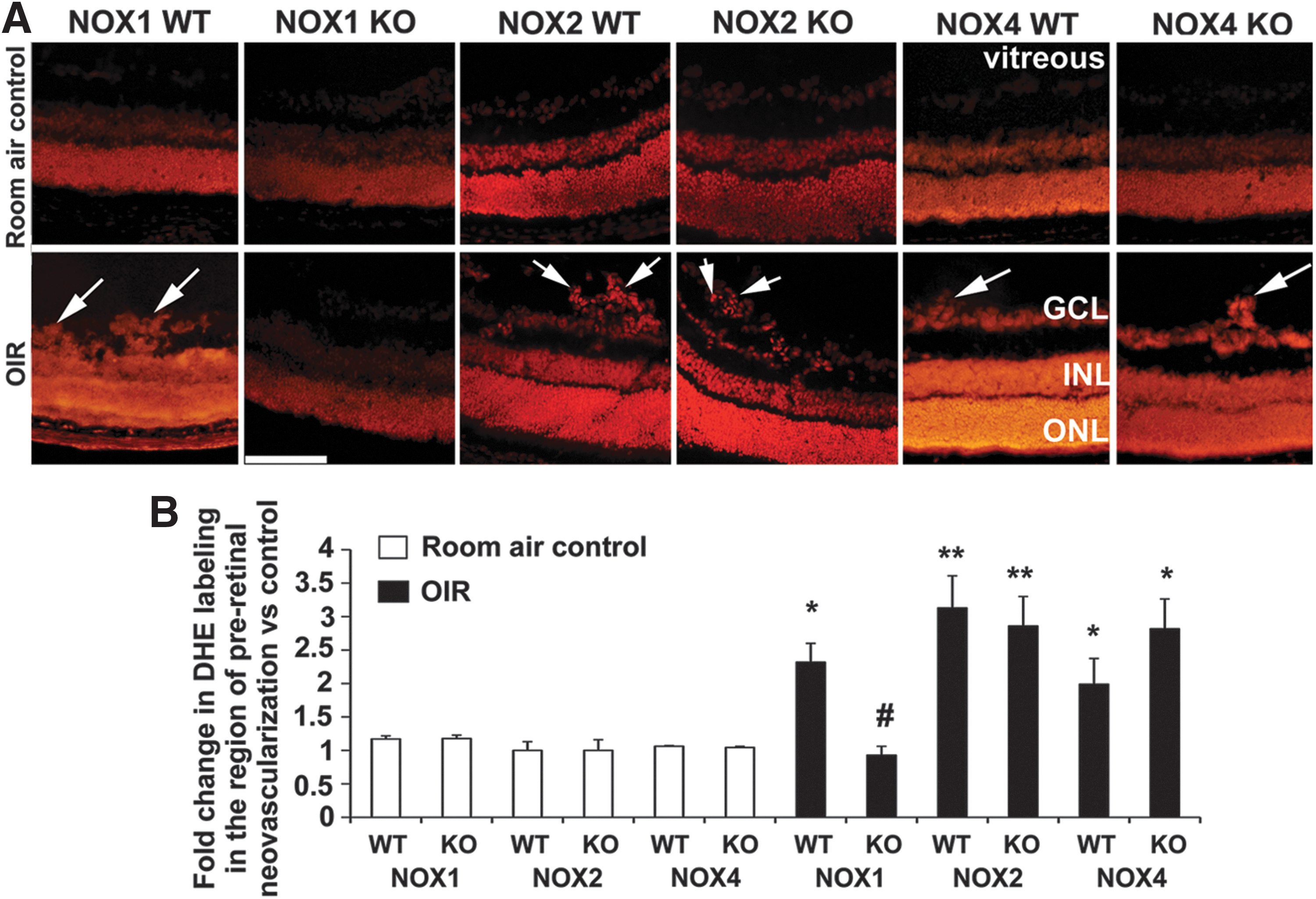
NOX1 KO mice with OIR exhibit reduced levels of ROS in retina
We next evaluated whether the improvements in OIR that occurred in NOX1 KO mice were accompanied by a reduction in ROS levels in the retina. As expected in all groups, dihydroethidium (DHE) labeling for ROS was present in regions of high oxygen consumption, such as the outer nuclear layer (photoreceptor nuclei) and the inner nuclear layer (mainly macroglial Müller cell nuclei) (Fig. 6A). However, in WT mice with OIR, DHE labeling was also detected in the area of pre-retinal neovascularization at the retinal surface (inner limiting membrane and ganglion cell layer) (Fig. 6A). Consistent with our findings of a reduction in neovascularization in NOX1 KO mice with OIR, retina from these mice exhibited reduced DHE labeling in the region of pre-retinal neovascularization compared to WT mice with OIR (Fig. 6A, B). However, in NOX2 KO and NOX4 KO mice with OIR and pronounced retinal neovascularization, DHE labeling persisted in the region of neovascularization (Fig. 6A, B). Similar to findings for DHE labeling, we found that labeling for nitrotyrosine, a marker of peroxynitrite-induced damage, was localized to pre-retinal blood vessels in WT mice with OIR and was virtually absent in retina from room air controls (Fig. 7A, B). This pattern of labeling is consistent with previous reports in the OIR model (53). In NOX1 KO mice with OIR, but not in NOX2 KO and NOX4 KO mice with OIR, nitrotyrosine labeling was reduced compared to respective WT mice with OIR (Fig. 7A, B).

GKT137831 inhibited hypoxia-induced ROS generation by microglia and reduced retinal vasculopathy in OIR
As it is not clear which retinal cell populations express particular NOX isoforms, we evaluated NOX expression profiles in primary retinal cultures. A qualitative rather than quantitative assessment was required as retina and cultures were obtained from different species (rats and cows). Studies were not performed in mouse retinal cells as the small size of the neonatal mouse retina hinders the acquisition of a sufficient number of cells for culture. In cultures of retinal rat microglia, ganglion cells and pericytes, NOX1, NOX2, and NOX4, were expressed, whereas cultured retinal glia expressed NOX1 and NOX4 and cultured retinal bovine endothelial cells expressed NOX2 and NOX4 (Fig. 8A–C). Previously, the role of NOX5 has not been evaluated in retina due to its absence from rats and mice, although it is present in other mammals, including humans and cows (59). As NOX5 may be relevant to vascular pathology (21), we evaluated its expression in human retina and in bovine retinal endothelial cells and bovine retinal pericytes. NOX1, 2, 4, and 5 were present in human retina (Supplementary Fig. S3A), and NOX5 was present in both bovine retinal endothelial cells and pericytes (Supplementary Fig. S3B).

Based on our findings that retinal microglia express NOX1, NOX2, and NOX4, and that NOX1 contributes to vasculopathy in OIR, we examined whether microglia are a source of ROS. We exposed primary cultures of microglia to hypoxia to mimic the in vivo environment of OIR. Hypoxia for 4 h resulted in increased levels of ROS in cell lysates compared to normoxia controls. GKT137831 reduced the elevated ROS levels to that of normoxia controls (Fig. 8D). We next determined if GKT137831 reduced vasculopathy in OIR. In OIR rats, and NOX4 WT and KO mice, GKT137831 reduced neovascularization and the avascular retina compared to untreated OIR animals (Fig. 8E–J). Both early intervention and late intervention were equally effective in rats with OIR (Fig. 8H–J). GKT137831 also reduced the levels of DHE labeling in C57BL/6 mice with OIR (Supplementary Fig. S4).
Discussion
The present study provides the first evidence that NOX1, but not NOX2 or NOX4, has an important role in the development of OIR. Using mice with genetic deficiencies in NOX isoforms, we demonstrated that deletion of NOX1, but not NOX2 or NOX4, protected against OIR-induced retinal neovascularization, capillary vaso-obliteration, vascular leakage, and vascular adherence of leukocytes. Furthermore, NOX1 KO mice exhibited in retina reduced the expression of proangiogenic and proinflammatory factors as well as reduced microglial density and ROS levels. The novel dual NOX1 and NOX4 inhibitor, GKT137831, reduced retinal vasculopathy in mice and rats with OIR and also in NOX4 KO mice with OIR. We determined that the vascular and neuro-glial elements involved in the development of retinal vascular injury in retinopathy of prematurity are sources of NOX1, 2, and 4 and that NOX5 is present in human retina. Furthermore, we identified microglia as a source of hypoxia-induced ROS generation, which is responsive to GKT137831. This is the first report to provide direct evidence that NOX1 has a pathological role in OIR, a finding that has major implications for the medical treatment of retinopathy of prematurity and other neovascular ischemic retinopathies.
Vision loss and blindness caused by retinopathy of prematurity is largely a consequence of hypoxia-induced neovascularization and vascular leakage (10). VEGF is arguably the most important mediator of this pathology (30), with contributions from other factors, such as erythropoietin (9) and angiopoietin-2 (37). Our data revealed a predominant role for NOX1 in promoting the expression of these putative mediators of OIR. Few studies have compared the role of NOX isoforms in angiogenesis in vivo (17, 56), and to our knowledge, none have been performed in retinopathy. Our findings are in agreement with other studies investigating the role of NOX isoforms in angiogenesis, although these experiments were performed in other organs. For example, basic fibroblast growth factor-stimulated angiogenesis in matrigel plugs was reduced in NOX1 KO mice but not in NOX2 KO or NOX4 KO mice (17). A key role for NOX1 in angiogenesis has also been reported in tumors where NOX1 was suggested to be the angiogenic switch for VEGF-related vascular growth (5). These results for NOX1 contrast those studies that showed angiogenic functions for NOX2 and NOX4. In NOX2 KO mice with VEGF-loaded sponge implants, angiogenesis was reduced (54). Angiogenesis was blunted in NOX4 KO mice with femoral artery ligation (47), whereas in cardiomyocyte-specific NOX4 overexpressing mice, myocardial capillary density was increased (62). The reasons for the divergent angiogenic roles of the NOX isoforms are not clear; however, it is possible that the influence of particular NOX isoforms on angiogenesis depends on the tissue environment. In this regard, it can be speculated that the drivers for neovascularization in OIR, namely intense hypoxia and contributions from inflammatory cells (23, 43), favor NOX1 over other NOX isoforms. Of interest are the mechanisms by which hypoxia influences NOX1 expression and activity. Furthermore, it will be important to determine how NOX1 expression and activity influence the production of angiogenic and inflammatory genes during the development of OIR, and particularly the contribution of hypoxic-inducible factor-1α, which is upregulated in the acute stage of OIR (34).
The early stage of OIR in mice features hyperoxia-induced capillary vaso-obliteration resulting in an avascular central retina. The extent of damage is viewed to predict the severity of neovascularization in OIR (28). Elevated levels of ROS are thought to be involved in this capillary degeneration as intravitreal administration of liposome-encapsulated superoxide dismutase (36) and systemically administered Trolox, a water-soluble analogue of vitamin E (39), reduce the avascular area in OIR. Our data extend these findings by highlighting that NOX1, but not NOX2 or NOX4, is likely to be a source of ROS, which mediates capillary degeneration in OIR.
Leukocytes contribute to OIR by remodeling the retinal vasculature and/or delivering inflammatory mediators, cytokines and possibly ROS to influence neovascularization (23, 43). In OIR, leukocytes adhere to the retinal vasculature (58). We demonstrated that a deficiency in NOX1, but not in NOX2 or NOX4, reduces retinal leukostasis in OIR. Furthermore, there was a reduction in ICAM-1 levels in NOX1 KO mouse retina consistent with the previously reported major role of ICAM-1 in retinal leukostasis (32). To our knowledge, little is known about the contribution of NOX2 and NOX4 to retinal inflammation in OIR. It has been reported that lipopolysaccharide-induced as well as diabetes-induced leukostasis and ICAM-1 expression were reduced in NOX2 KO mice (2). The reasons for the differences between these findings and our results may, as previously mentioned, relate to the particular retinal disease being evaluated. However, it is possible that given NOX2's role in inflammation (41, 48), it may influence inflammatory processes in the early stages of OIR. Nevertheless, strategies to inhibit NOX2 in patients are not likely to be appropriate since deficiencies in NOX2 are associated with compromised phagocytic defense resulting in life-threatening bacterial and fungal infections (48), a situation that could have severe consequences for children with retinopathy of prematurity who are already prone to infections (29).
Several reports have shown that ROS levels are increased in the retina after OIR (1, 6, 44, 53) and that untargeted scavenging of ROS with agents such as vitamin C derivatives and the nonspecific NOX inhibitor, apocynin (49), reduce retinal neovascularization and vaso-obliteration (1, 44). Consistent with previous studies (1, 53), we found increased ROS levels in WT mice with OIR in the region of pre-retinal neovascularization. Importantly, we also demonstrated that a deficiency in NOX1, but not in NOX2 and NOX4, reduced this increase in ROS levels. This finding was substantiated in WT mice with OIR, where ROS levels in the inner retina were reduced with GKT137831. A previous study in OIR reported that ROS levels were reduced when retinal sections were incubated with NOX2-tat (a peptide that inhibits NOX assembly by binding to p47phox and blocking its interaction with NOX2) (1). However, despite these reported results, we did not find a reduction in retinal vasculopathy in NOX2 KO mice with OIR, suggesting that any ROS generated from NOX2 is likely to have little impact on retinal neovascularization. Indeed, it remains controversial as to whether NOX2-tat is specific for NOX2 (59), and thus, its effects may also be mediated by inhibition of NOX1.
To determine whether the inhibition of NOX isoforms is a potential treatment of retinopathy of prematurity, we chose a complementary pharmacological approach by evaluating the NOX inhibitor, GKT137831, which inhibits both NOX1 and NOX4. Studies were performed in NOX4 KO mice with OIR and also in rats with OIR as the retinopathy, which develops in rats, more closely mimics that of children with retinopathy of prematurity (22). GKT137831 had a similar effect to NOX1 deletion in suppressing retinal neovascularization and reducing the extent of loss of retinal avascularity in OIR. These findings demonstrate the potential of specific NOX isoform inhibition as a treatment of retinopathy of prematurity in children.
Neovascularization in OIR is largely driven by the hypoxia-induced upregulation of VEGF and other angiogenic factors produced in neurons and glia (52). Therefore, the next logical step would be to determine the cellular location of NOX isoforms in retina. However, this is difficult due to the paucity of reliable and specific NOX antibodies. Furthermore, antibody approaches are unlikely to provide information about the activities of individual NOX isoforms. We therefore performed a qualitative assessment of NOX isoform mRNA levels in primary cultures of retinal cells involved in neovascularization. Under basal conditions, only microglia, ganglion cells, and pericytes expressed NOX1, NOX2, and NOX4. Given that microglia contribute to OIR (43) and are important producer of ROS and cytokines (50, 55), further evaluation of this particular cell type was performed. In OIR, immunolabeling for retinal microglia in the inner retina was reduced in NOX1 KO mice but remained elevated in NOX2 KO and NOX4 KO mice, although there was a small reduction in NOX4 KO mice. However, the presence of microglia in the neovascular tufts of retina from NOX4 KO mice with OIR may indicate why retinal neovascularization was not reduced in these animals. We complemented these in vivo studies by demonstrating that hypoxia induces the increased production of ROS in cultured microglia. The ability of GKT137831 to suppress this increase in ROS confirms the role of NOX in excess ROS formation in microglia. However, these findings do not preclude a role for retinal cells other than microglia as a source of ROS derived from NOXs that contribute to retinal neovascularization in OIR. For instance, a recent study has shown that retinal ganglion cells exhibit a preferential increase in NOX1 mRNA rather than NOX2 and NOX4 mRNA after ischemia (15).
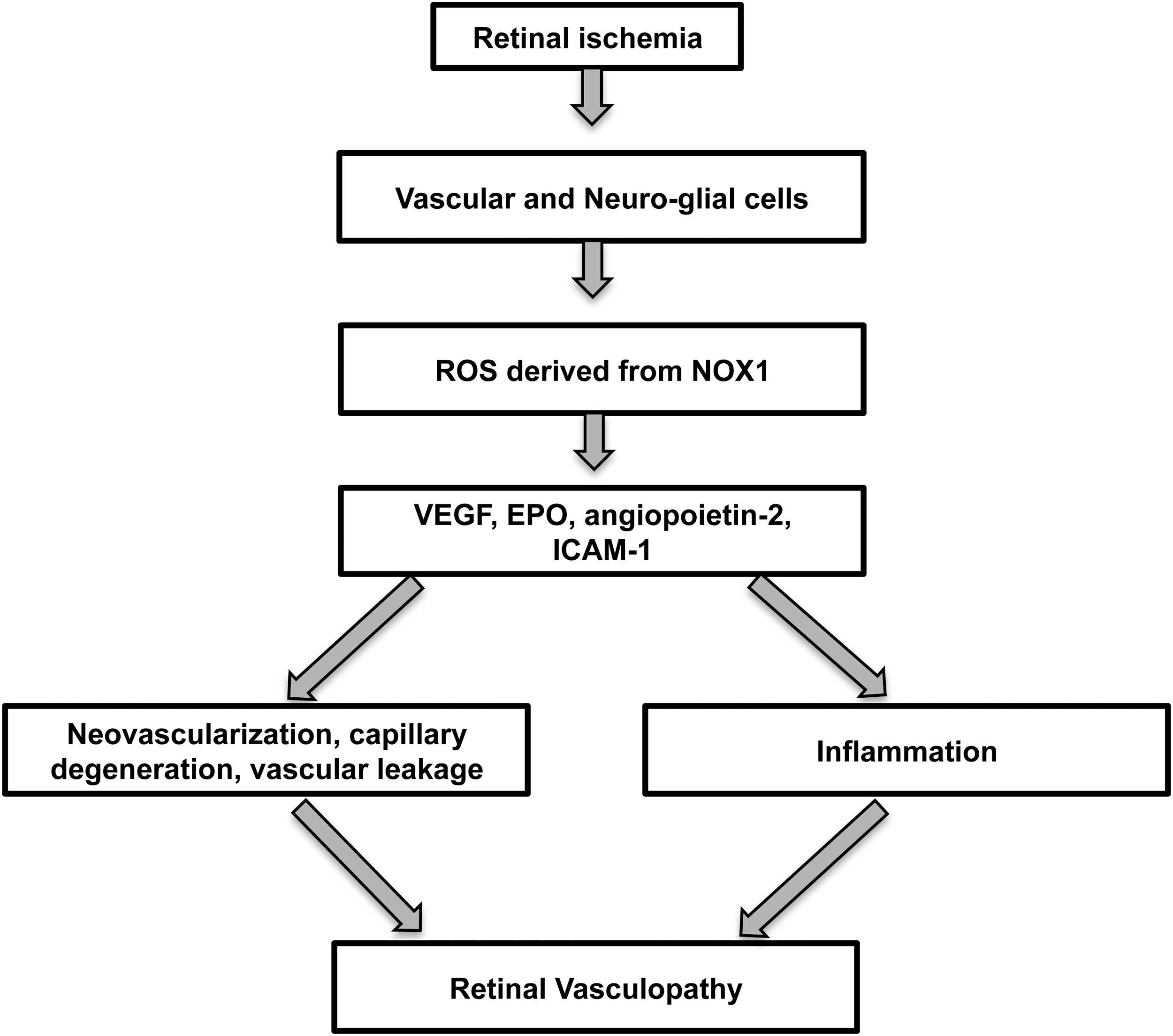
In summary, retinopathy of prematurity is the major ocular condition causing blindness of the neonate, and its prevalence has escalated due to the increased survival of younger and smaller babies. Currently, there are no effective strategies that prevent or attenuate the vasculopathy of this vision-threatening disorder. Although anti-VEGF therapies have been suggested as a treatment option, there are concerns about detrimental effects of such a treatment approach on retinal and organ development (13). Our data provide the first evidence that NOX1 influences retinal vasculopathy in OIR by the increased production of ROS and subsequent generation of angiogenic and inflammatory factors (Fig. 9). NOX1 inhibition has the potential to become an effective mechanism-based treatment strategy by alleviating the increases in pathologically relevant ROS. Certainly, further studies evaluating the effects of NOX1 inhibition and GKT137831 on other aspects of retinal pathology including neuronal and glial dysfunction are necessary before these findings can be translated to patients. Furthermore, the ocular safety profiles of NOX isoform inhibitors such as GKT137831 in patients with retinopathy will need to be established. Finally, a role for NOX5 in ischemic retinopathy cannot be entirely excluded given that it is expressed in human retina and bovine retinal vascular cells, but unfortunately at present cannot be studied in relevant rodent models. Overall, our findings are not only relevant for retinopathy of prematurity but also for other retinopathies and organ pathologies that feature neovascularization associated with amplified ROS.
Materials and Methods
Animals
All procedures were approved by the Alfred Medical Research and Education Precinct (AMREP) animal ethics committee and performed according to the National Health and Medical Research Council (NHMRC) of Australia's Guidelines for the Care of Animals in Scientific Research. In rodents, OIR develops over two phases comprising vaso-obliteration (Phase I) and neovascularization (Phase II) (40). In mice, Phase I was induced using a previously published protocol with a minor modification (57). Mice were exposed between postnatal day (P)7 and P11 to 75% O2 cycled with 20% O2 for 3 h/day. In rats, Phase I was induced by exposure between P0 and P11 to 80% O2 cycled with 20% O2 for 3 h/day (14, 33, 58). Phase II of OIR occurred when mice and rats were placed in room air from P12 to P18. Controls were in room air for the entire study (P0–P18). NOX1 KO, NOX2 KO (Jackson Laboratories, Bar Harbour, ME), and NOX4 KO (26) mice were bred on a C57BL/6 background. WT for NOX1 KO and NOX4 KO were littermates and for NOX2 KO were C57BL/6 mice. NOX1 KO mice were a gift from Professor Karl-Heinze Krause, Geneva, Switzerland (18). Sprague Dawley rats were purchased from the Australia Research Centre (Perth, Western Australia). Rats were treated with GKT137831 at a dose of 60 mg/kg/day between P0 to P18 (early intervention) and P12 to P18 (late intervention). GKT137831 was dissolved in dimethyl sulfoxide and administered by subcutaneous injection (100 μl). Controls received dimethyl sulfoxide by subcutaneous injection (100 μl). A separate set of NOX4 WT and NOX4 KO mice with OIR mice were treated with GKT137831 (60 mg/kg/day) from P7 to P18 by subcutaneous injection (100 μl). GKT137831 was provided by Genkyotex SA (Geneva, Switzerland). Mice and rats were killed at P18 with sodium pentobarbitone (170 mg/ml; Virbac, Peakhurst, New South Wales, Australia).
NOX inhibition
GKT137831, a member of the pyrazolopyridine dione family is an efficient inhibitor of both NOX1 and NOX4 isoforms (Ki in the range of 100–150 nM in cell-free assays of ROS production using membranes prepared from cells heterologously overexpressing specific NOX enzyme isoforms) (4). GKT137831 is also a weak inhibitor of the NOX2 isoform in cell-free assays but does not significantly inhibit neutrophil oxidative burst at concentrations up to 100 μM and does not inhibit innate microbial bacterial killing in vitro or in vivo (when used at a concentration of up to 20 μM or administered at 100 mg/kg orally, respectively). Furthermore, GKT137831 has no scavenging or antioxidant activity when tested at 10 μM and has an excellent specificity for NOX1 and NOX4 enzymes as shown in an extensive in vitro off-target pharmacological profiling on 170 different proteins, including ROS producing and redox-sensitive enzymes (24, 27).
Retinal neovascularization and vaso-obliteration in retinal wholemounts
Retinal wholemounts were prepared as described previously (14, 57). More information can be found in Supplementary Data. Seven to nine mice per group were evaluated from three litters of animals, and six to seven rats per group were examined. Neovascularization (neovascular tufts) was defined as clumps of blood vessels that were positively labeled for BS-I lectin. Neovascular tufts over the whole retina were psuedocolorized in red, and Image J software (v3.1; National Institutes of Health, Bethesda, WA) was used to set a threshold for red fluorescent labeling, which was then applied to all sections. Neovascular tufts were quantitated over the whole retina and expressed as a % of neovascular tufts per retina. The extent of vaso-obliteration (avascular area) in the central retina (for mice) and mid-peripheral retina (for rats) was quantitated using the freehand line tool in Image J. The avascular area was expressed as a percentage of the total retinal area. Retinal neovascularization was confirmed in 3-μm paraffin sections from a separate set of NOX1, NOX2, and NOX4 WT and KO mice, as previously described (14, 57).
Retinal vascular leakage and leukostasis
Elevated levels of albumin in retina are an indicator of vascular leakage and blood–retinal barrier breakdown (38). Albumin levels in freshly frozen single retina were measured according to the manufacturer's instructions using a commercially available Mouse Albumin Enzyme-linked immuno sorbent assay quantification set (Bethyl Laboratories, Montgomery, TX). Albumin values were normalized to dry retinal weight. Six to eight mice per group were evaluated. To measure the extent of adherence of leukocytes to the retinal vasculature, leukostasis was quantitated as described previously (57). More information is found in Supplementary Data.
Immunohistochemistry for microglia and VEGF
Immunohistochemistry for the microglial marker, Iba1, and for VEGF was performed as described previously (14). Four 3-μm paraffin sections at least 60 μm apart were randomly selected from each eye, dewaxed in histolene, and hydrated in graded ethanols. Antigen retrieval was achieved by microwaving in 0.1 M citrate buffer for 10 min. After washing with 0.1 M phosphate-buffered saline (PBS), pH 7.4, sections were incubated with 1% normal goat serum in PBS for 1 h for Iba1 labeling or 10% donkey serum in PBS for 1 h for VEGF labeling. The sections were then incubated overnight at 4°C with either an anti-Iba1 antibody (1:1000; Wako, Tokyo, Japan) or a goat anti mouse VEGF164 antibody (R&D systems, Minneapolis, MN). The sections were washed three times with PBS and incubated for 30 min with either a biotin-conjugated goat anti-rabbit IgG (1:200; DakoCytomation, Glostrup, Denmark) for Iba1 labeling or a biotinylated donkey anti-goat IgG antibody (1:500; Jackson Immunoresearch, West Grove, PA) for VEGF labeling. After washing with PBS, the sections were then incubated with the Vectastain ABC standard kit (Vector Laboratories, Burlingame, CA) for 30–45 min and liquid DAB+substrate chromagen system (Dakocytomation) for 15 s. The sections were then counterstained with Harris' Haematoxylin for 7 min, rinsed in tap water, dehydrated through graded ethanols, cleared in histolene, and coverslipped with DPX (VWR International Ltd., Poole, England). A negative control without the primary antibody and an isotype IgG antibody control were included to detect nonspecific labeling. In each section, four nonoverlapping fields at ×400 magnification were captured using a Spot digital camera (SciTech, Preston, Victoria, Australia). Image J software was used to set a threshold for brown Iba1 immunolabeling and brown VEGF immunolabeling, which was then applied to all captured fields. Immunolabeling was quantitated using the threshold tool in the inner retina, which included pre-retinal blood vessels attached to the retinal surface and protruding into the vitreous cavity. The inner retina comprised the inner limiting membrane, ganglion cell layer, and inner plexiform layer. Results are expressed as Iba1 or VEGF immunolabeling per field of inner retina. Five to six mice per group were evaluated.
Real-time PCR and qualitative assessment of NOX isoform expression
In mice, this was performed for NOX1, NOX2, NOX4, VEGF, erythropoietin, angiopoietin-2, and ICAM-1, as described previously (14, 57). More information is found in Supplementary Data. Total RNA was isolated from single retinas of 6–10 mice per group. To qualitatively determine the expression profile of the NOX1, NOX2, NOX4, and NOX5 isoforms in distinct retinal cell types, RNA was isolated from cultured rat ganglion cells, glia, and microglia, and bovine retinal endothelial cells and pericytes. The method for the primary culture of rat ganglion cells, glia and bovine retinal endothelial cells and pericytes is described previously (14, 58). NOX isoform expression was qualitatively assessed in a human retinal cDNA preparation (BioChain, Newark, CA). Confirmation of the absence of NOX1 mRNA in NOX1 KO retinas, NOX2 mRNA in NOX2 KO retinas, and NOX4 mRNA in NOX4 KO retinas was achieved from amplifications using primers located within the exonic regions disrupted in each NOX KO mouse strain. The primer sequences used in the study are found in Supplementary Table S2.
Reactive oxygen species
The DHE method was used to measure ROS levels in single retina. Eyes were enucleated and immediately frozen in OCT compound (Sakura Finetechnical Co., Ltd., Tokyo, Japan). Unfixed cryosections (20 μm) were incubated with sterile PBS containing 0.1%
Primary cultures of rat retinal microglia under hypoxic conditions
The methods for culturing primary rat retinal microglia are as described previously (14). Characterization of cultured retinal microglia was performed with immunohistochemistry for Iba1 (14). Dishes containing purified retinal microglia were placed in a Modular Incubator Chamber (MIC101; QNA International Pty Ltd, Glenferrie South, Victoria, Australia). The hypoxic conditions were established by flushing the chamber for 4 min with 0.5% O2, 94.5% N2, and 5% CO2. The same gas mixture was then pumped into the chamber and immediately the chamber was sealed. Microglia were exposed to GKT137831 at concentrations of 0.1, 5, and 10 μM. The chamber was then placed in a 37°C incubator for 4 h. Comparisons were made to microglia exposed to normoxic conditions.
ROS levels in rat retinal microglial lysates
Primary cultured rat retinal microglia were washed with Hank's balanced salt solution (HBSS) for 2 min followed by a 30-min incubation with 5 μM DHE at 37°C in 5% CO2, in the dark. After incubation, cells were washed with HBSS and then imaged immediately using an Eclipse TE2000 inverted fluorescence microscope (Nikon Instruments, Inc.) and image-processing computer software (NIS-Elements version 2.20; Nikon Instruments, Inc.). Fluorescence intensity of cells was used as a measure of intracellular ROS levels using Image J analytical software (version 1.44). For quantitation, a minimum of 15 randomly selected cells from three different fields from each dish was evaluated. The intensity of fluorescence in a blank space between two cells was measured as background intensity. Three independent experiments were performed in each group with a minimum of three replicates.
Statistics
All data were analyzed using the GraphPad Prism software (v.5; GraphPad Software, Inc., LA Jolla, CA). Both animal and cell culture data sets were assessed for normality, and in the case of more than two treatments, a one-way ANOVA (parametric) was followed by a Bonferroni post hoc analysis or Kruskal–Wallis or by Mann–Whitney U tests (nonparametric). Either an unpaired t-test (parametric) or a Mann–Whitney U test (nonparametric) was conducted when there were only two treatments. Investigators were masked to the groups. A value of p<0.05 was considered significant.
Footnotes
Acknowledgments
The authors thank Mariam Sofi and David R. Berka for technical assistance. The authors thank Monash Micro Imaging for assistance with confocal microscopy. JDRF and the NHMRC of Australia generously supported this research. Both J.L.W.-B. and K.A.J.-D. are NHMRC Senior Research Fellows. M.E.C. is an Australian Fellow of the NHMRC and JDRF Scholar. H.H.H.W.-S. is supported by the EU (Marie Curie International Reintegration Grant and ERC Advanced Grant) and Dutch Kidney Foundation. D.D. was awarded a Postgraduate Publication Award from the Monash University.
Author Disclosure Statement
C.S. is an employee of Genkyotex, Geneva, Switzerland. The other authors have nothing to disclose.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.