
Abstract
Introduction
T
Knockout of antioxidant enzymes glutathione peroxidase 1 (GPX1) and superoxide dismutase 1 (SOD1) alone or together impaired glucose-stimulated insulin secretion (GSIS), but the molecular mechanism and signaling pathway remain unclear. Using islets isolated from the GPX1 and (or) SOD1 knockout mice, we have demonstrated that the GPX mimic ebselen rescued impaired GSIS in the GPX1 knockout islets and mice via regulating glucokinase, glucose transporter type 2, pancreatic and duodenal homeobox 1, and uncoupling protein 2 by activating peroxisome proliferator-activated receptor gamma coactivator 1 alpha-mediated antioxidant response element/glucocorticoid receptor signaling pathways. In contrast, the SOD mimic copper diisopropylsalicylates showed different roles and mechanisms in regulating GSIS. Our results revealed a novel metabolic effect of ebselen in promoting GSIS, and provided a new strategy to treat disorders related to insulin secretion.
Se-dependent glutathione peroxidase-1 (GPX1) and Cu,Zn-superoxide dismutase (SOD1) represent two major intracellular antioxidant enzymes that can modulate intracellular H2O2 status. Despite low GPX1 and SOD1 activities in islets (only 2% and 29% of that in liver, respectively) (39), we have found that knockout of GPX1 (GPX1 knockout [GKO]) and SOD1 (SOD1 knockout [SKO]) alone or in combination (double knockout of GPX1 and SOD1 [dKO]) caused substantial impairment of GSIS (68). Consistently, a Gpx1 gene variant (C198T) lowering the enzyme activity was identified in the South Indian population, which resulted in increased incidences of type 2 diabetes (C/T, 1.4-fold and T/T, 1.8-fold) (54). Intriguingly, the overt phenotypes of GSIS impairments in the GKO and SKO mice were similar (68), although GPX1 catalyzes H2O2 breakdown whereas SOD1 catalyzes H2O2 formation. It is puzzling that the presumed opposite effects of these two knockouts on islet intracellular H2O2 production might have induced seemingly similar biochemical and signaling regulation of GSIS. The biochemical regulation of GSIS depends on four key proteins: glucose transporter type 2 (GLUT2), glucokinase (GK), pancreatic and duodenal homeobox 1 (PDX1), and uncoupling protein 2 (UCP2) (30). However, it remains unclear whether the impacts of GKO and SKO on GSIS were mediated by altering functional expressions of these proteins in a coordinated fashion. Although we previously observed changes of PDX1 and UCP2 (68) in the whole pancreas of these knockout mice, systematic responses of GK, GLUT2, PDX1, and UCP2 in their islets have not been studied. More importantly, there is no information on the signaling cascade and molecular mechanism to link these knockout—initiated islet intracellular ROS changes to the responses of these four proteins and the observed GSIS phenotypes.
The gene promoter regions of GK, GLUT2, PDX1, and UCP2 may share common domains that bind transcriptional factors involved in signaling pathways related to the question described earlier (45). The first is the peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α)-mediated antioxidant response element (ARE) signaling pathway. While PGC-1α is involved in responses of various genes to redox regulation (62) and regulates GSIS in human islets (40), two of its gene variants are associated with increased risks of type 2 diabetes in the Indian population (6, 73). The second is the glucocorticoid receptor (GR) pathway that is negatively regulated by ROS or H2O2 in inflammation and immune responses (31). Polymorphisms of gr are associated with type 2 diabetes with low insulin levels and attenuated first phase of GSIS in women (65). The third is the wingless-type MMTV integration site (Wnt) pathway that is positively regulated by ROS or H2O2 (70). Elevated oxidation status by selenium (Se) deficiency (33) or GPX3 knockout (4) activated the Wnt pathway in mice. This pathway activated gk gene transcription and insulin secretion in isolated islets of C57BL/6 mice (56), and has at least four variants of TCF7L2 that are associated with increased risks of type 2 diabetes (63). The fourth is the nuclear factor of activated T-cells (NFAT) pathway that stimulates gk, glut2, and pdx1 expression and insulin secretion in β-cell (24). This pathway in mouse C141 cells is activated by superoxide, but inhibited by H2O2 (32). Since many members in these four signaling pathways are ROS responsive and involved in GSIS, and can bind to domains in the gene promoter regions of GLUT2, GK, PDX1, and UCP2, it is fascinating for us to explore whether GKO, SKO, and dKO initiated their impacts on GSIS via these pathways.
The GPX mimic ebselen has been shown to protect against oxidative injuries (15), suppress (14) the diazinon-induced hyperglycemia in rats, and decrease islet UCP2 (69). The SOD mimic copper diisopropylsalicylate (CuDIPs) attenuated the streptozotocin (STZ)-induced diabetes in rats (20) and restored the suppressed foxa2 expression in the SKO islets (68). As an essential micronutrient, Se is a component of 25 human selenoproteins (36) that are involved in antioxidant defense (10, 11) and regulation of β-cells (61). Either overexpression or deficiency of selenoproteins disturbed glucose homeostasis in mice (37). It has been reported that Se functioned as an insulin mimetic in isolated adipocytes (17) and STZ-induced diabetic rodents (5). Despite all these well-documented features, little is known on the roles and mechanisms of ebselen, CuDIPs, and Se in regulating GSIS. Due to the specific inactivation of the target GPX1 and(or) SOD1, islets isolated from the GKO, SKO, and dKO mice are ideal experimental models for such studies. Therefore, our objectives were (i) to determine whether ebselen, CuDIPs, and sodium selenite could rescue the impaired GSIS in GKO, SKO, and dKO islets and(or) mice; (ii) to reveal whether these yet-illustrated GSIS rescues were mediated by regulating islet GK, GLUT2, PDX1, and UCP2 expressions and functions; and (iii) to elucidate whether the ultimate control of this regulation was initiated by the mimics-restored ROS-responsive signaling via the ARE, GR, Wnt, and NFAT pathways.
Results
Effects of ebselen, CuDIPs, and Se on GSIS of islets
Ebselen, CuDIPs, and Se were used as the GPX mimic, SOD mimic, and compound incorporated as selenocysteine into the active center of GPX, respectively, and incubated with islets isolated from the GKO, SKO, and dKO mice, along with the wild type (WT), to rescue their impaired GSIS. Ebselen enhanced (p<0.01) GSIS of islets of all four genotypes treated with 16.7 mM glucose (Fig. 1A–D), compared with their respective controls. Notably, such enhancement was so strong in the GKO islets that their GSIS even exceeded the WT level. The ebselen treatment also elevated (p<0.01) in the baseline insulin secretions of WT and GKO islets (at 2.8 mM glucose). In contrast, CuDIPs promoted GSIS and the baseline insulin secretion only in the SKO islets (p<0.01). Meanwhile, Na2SeO3 elevated islet GSIS of SKO (p<0.05) and dKO (p<0.01).

Responses of GK, GLUT2, PDX1, UCP2, lactate dehydrogenase, glutathione reductase, and ROS
To explore biochemical mechanisms for the GSIS rescues observed earlier by ebselen, CuDIPs, and Se, we determined their effects on the activity or protein levels of four key regulators (GK, GLUT2, PDX1, and UCP2) of GSIS. Ebselen elevated (p<0.05) GK activity by 74%, 20%, 86%, and 58% and GLUT2 by 4.9-fold, 2.1-fold, 6.4-fold, and 85%, respectively, in the WT, GKO, SKO, and dKO islets (Fig. 2A–D and Supplementary Fig. S1; Supplementary Data are available online at
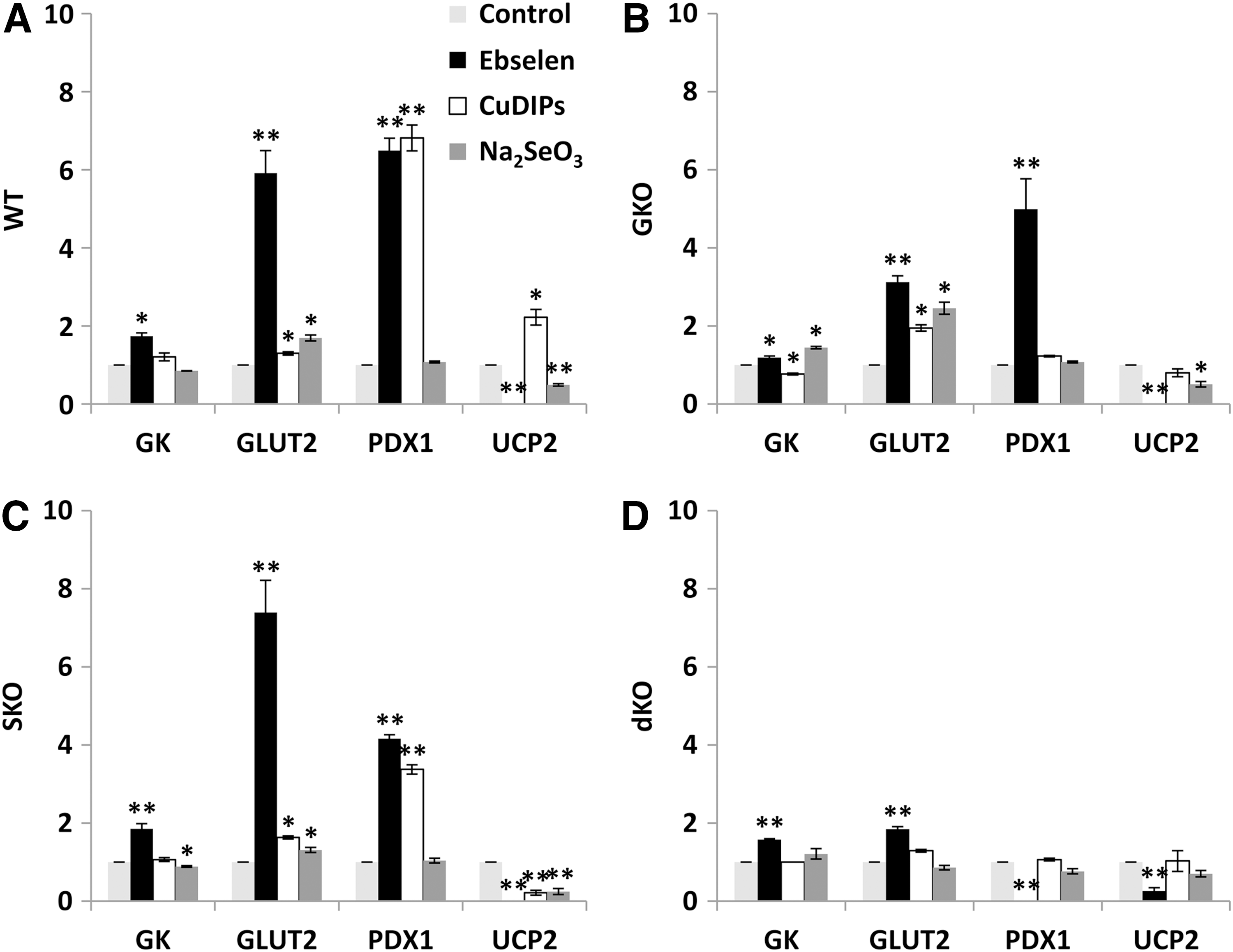
Impacts of the GKO and ebselen on islet intracellular H2O2 status were indirectly assessed by a non-specific ROS probe (2′,7′-dichlorofluorescin diacetate [DCFH-DA], Supplementary Fig. S2A). Ebselen decreased the intracellular ROS by 74% in the GKO islets and removed their initial genotype difference from the WT islets. However, the ebselen treatment showed no effect on the activities of two thiol-containing enzymes: lactate dehydrogenase (LDH) and glutathione reductase (GSR) in the WT and GKO islets (Supplementary Fig. S2B, C).
Signaling mapping for GSIS regulation by ebselen
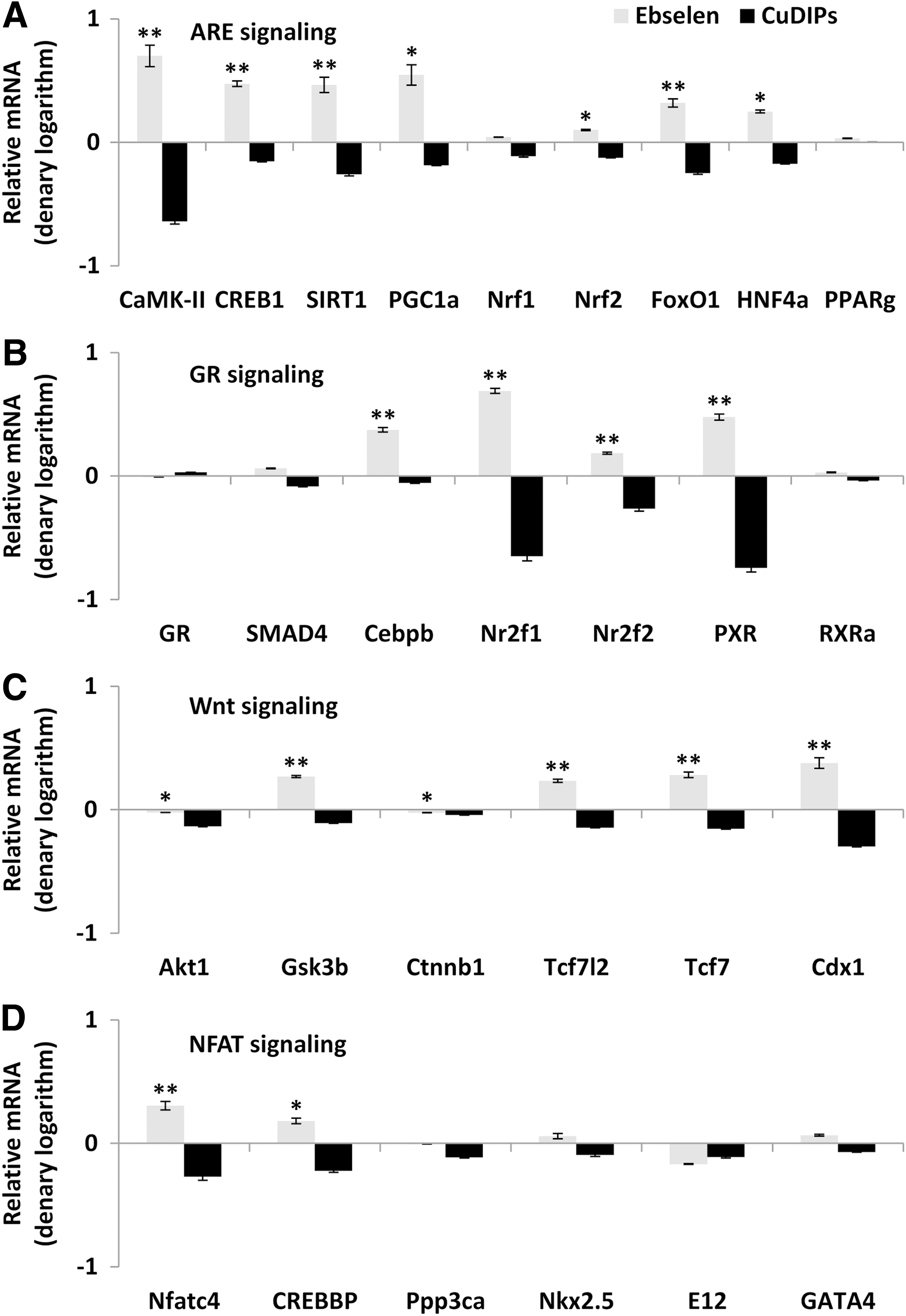
To reveal whether the illustrated effects of ebselen on islet GK, GLUT2, PDX1, and UCP2 were mediated by transcriptional regulation, we determined their mRNA responses to ebselen in the GKO islets by quantitative real-time polymerase chain reaction (Q-PCR). Since their mRNA changes resembled those of their protein responses (Fig. 3A), we proposed a transcriptional regulation for this cascade and analyzed their gene promoter regions to search for shared binding domains that might mediate the regulation. After 1000 bp upstream genomic sequences of each gene had been retrieved from EMBL database (

PGC-1α as a main mediator for the upstream signaling regulation
The overall positive responses of the ARE and GR pathway genes to the ebselen treatment led us to project PGC-1α (the central effector of the ARE pathway) and CCAAT/enhancer binding protein (C/EBP), beta (C/EBPβ, the main effector of the GR pathway) as the primary mediators for the upstream signaling regulation of GSIS by ebselen. Subsequently, we applied small interfering RNA (siRNA) to knock down these two genes in islets to assess their relative importance in the event. The pgc-1α siRNA suppressed the gene expression of both pgc-1α and c/ebpβ by more than 70% (p<0.01), whereas the c/ebpβ siRNA decreased (p<0.01) only its own expression (Fig. 5A). The double siRNA did not produce suppression of either gene further than the single treatment. Most striking, knockdown of pgc-1α blocked the ebselen-induced up-regulation of gk, glut2, and pdx1 mRNA levels; down-regulation of ucp2 mRNA level (Fig. 5B); and GSIS rescue (Fig. 5C) in the GKO islets.

Rescue of GSIS in the GKO mice by ebselen
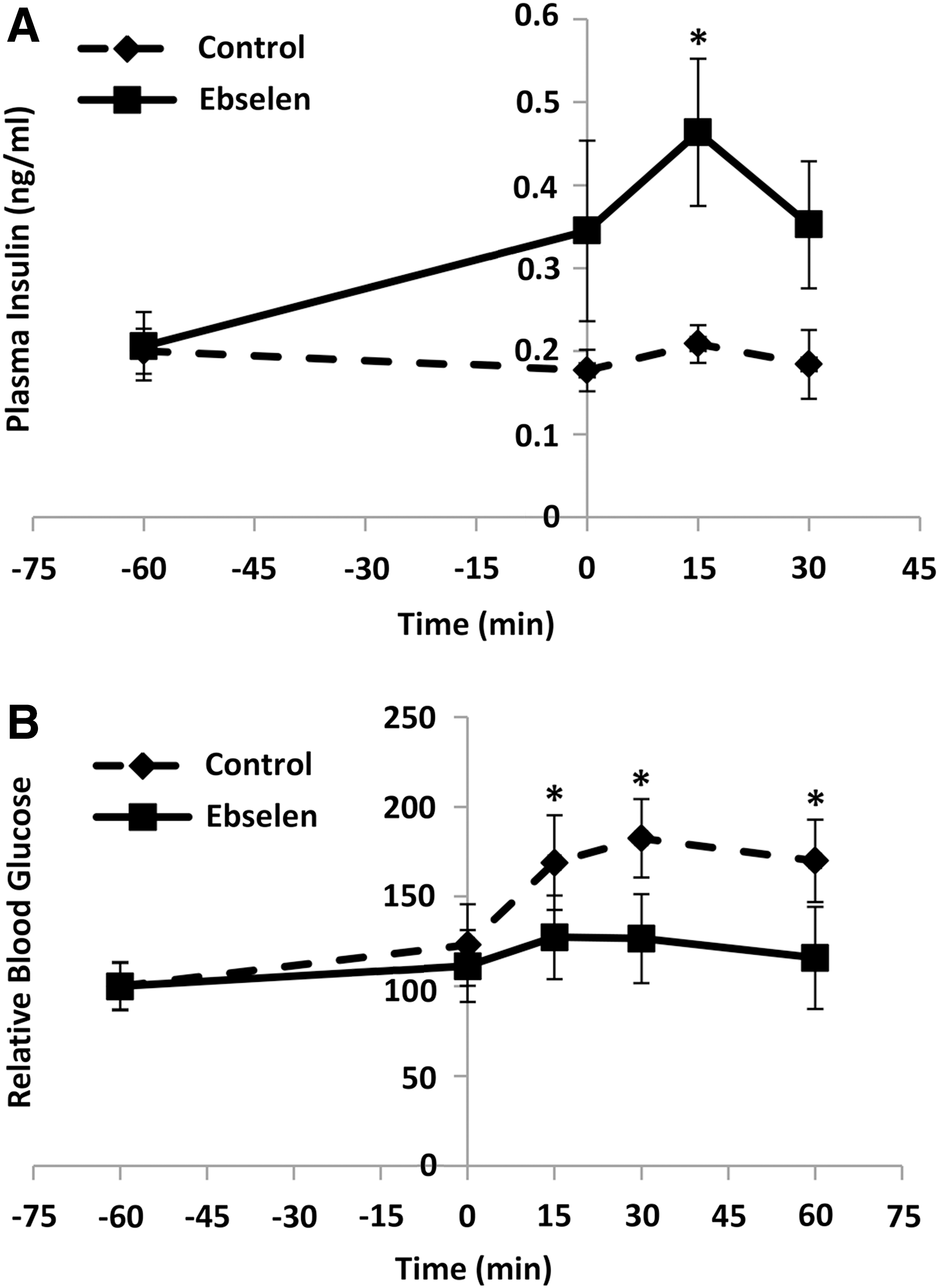
To determine whether the observed rescue of GSIS in GKO islets by ebselen was reproducible at physiological conditions and whether ebselen caused any “off-target” toxicity, we gave the fasted GKO mice an intraperitoneal (i.p.) injection of ebselen at 1 h before the GSIS test. The injection elevated their plasma insulin concentrations at 0 min (baseline) by 95%, 15 min by 1.2-fold (p<0.05), and 30 min by 91% after the glucose challenge (Fig. 6A). Consequently, glucose clearance was improved by 17%, 18%, and 21% (p<0.05) at 15, 30, and 60 min, respectively, by the ebselen injection (Fig. 6B). The injection produced no differences from the control in activities of plasma alanine aminotransferase (ALT) and alkaline phosphatase (AKP) or hepatic LDH and GSR (Supplementary Fig. S4).
Discussion
The most exciting, novel finding of the present study was that the GPX mimic ebselen (57), at a relatively low dose (14), rescued GSIS in the GKO islets and mice. Despite its recognition as the GPX mimic in 1984 (47) and subsequent extensive research on its protections against ROS, ischemic damage, and inflammation (15), only a couple of studies have explored its involvement in insulin synthesis (16) or hypoglycemic effect (14). Unprecedentedly, our study provides the direct evidence for a novel metabolic effect of ebselen in promoting GSIS. Due to the recently discovered association between the GPX1 mutation and increased risk of type 2 diabetes (54), our findings offer a potentially new therapy of insulin secretion defects that are associated with diabetes and hyperglycemia (41). Despite its lipophilic property, ebselen was prepared in 4% w/v hydroxypropyl-β-cyclodextrin for an i.p. injection to C57BL/6 mice (58) or was directly suspended in water for oral administration to humans (72). The ebselen that was delivered in both ways was absorbed to blood and even crossed the blood–brain barrier (58, 72). Since ebselen is included in the National Institutes of Health Clinical Collection (3), it has a history of use in human clinical trials and known safety profiles (
Ebselen rescued GSIS in the GKO islets by up-regulating GK, GLUT2, and PDX1, and down-regulating UCP2. Two strong evidences supported that this rescue was executed by ebselen via ROS scavenging instead of simply being an Se carrier. The first evidence was the 74% decrease of intracellular ROS level in the ebselen-treated GKO islets compared with the control. The second evidence was the different effects of sodium selenite and ebselen on the four GSIS regulators or GSIS itself in the GKO islets. In fact, the potential of ebselen as an Se carrier or transporter was questioned by a recent study due to the lack of stimulation of GPX activity or selenoprotein P expression in HepG2 cells (27). By taking four consecutive steps of genomics and bioinformatics analyses into consideration, we have revealed that the regulation of ebselen on the four key regulators of GSIS took place at transcription and was mediated by PGC-1α via the ARE and(or) GR pathways. In the first step, the Q-PCR analysis depicted parallel responses of mRNA and protein levels of GK, GLUT2, PDX1, and UCP2 in the GKO islets to ebselen, and suggested transcriptional regulation as the action site of ebselen. In the second step, four signaling pathways (ARE, GR, Wnt, and NFAT) were identified as the potential mediator for the initial action of ebselen by analyzing gene promoters of the four key regulators of GSIS. Indeed, these pathways are highly responsive to redox regulation (31, 32, 62, 70) and involved in transcriptional regulation of the key regulators of GSIS (8, 24, 59, 65). In the third step, the candidate pathways (ARE and GR) and mediators (PGC-1α and C/EBPβ) were chosen based on their gene expression responsiveness to ebselen. In the final step, we applied siRNA to prove that PGC-1α, indeed, served as the main mediator to link the ebselen-initiated intracellular ROS decrease to the downstream gene expression of gk, glut2, pdx1, and ucp2 for the GSIS rescue in the GKO islets.
There are both scientific and clinical significances to elucidate the novel role of PGC-1α in initiating the positive effect of the GPX mimic ebselen on GSIS. This reveals not only a new signaling pathway to explain how GPX1 and(or) ebselen regulate insulin secretion but also a new potential therapeutic target to treat insulin secretion disorders (26). Expression and function of PGC-1α is highly responsive to ROS (62). Down-regulation of PGC-1α decreased GSIS and(or) insulin production in both human and rat islets (40). Two of its gene variants were associated with increased risks of type 2 diabetes in the Indian population (6, 73). As a transcriptional co-activator for peroxisome proliferator activated receptors and LXR (53), it may play a dual role in GSIS on the metabolic conditions (22, 25, 64). It is also interesting to note that PGC-1α activates gene expression of GPX1 and several other antioxidant enzymes (60). Thus, the ebselen-mediated activation of PGC-1α (71) may constitute a positive feedback between GPX1 and PGC-1α. However, potential roles of other redox-sensitive or ARE-related factors such as nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (and nuclear respiratory factor 1 [Nrf1]) (62) in the ebselen-induced cascade events should also be considered. In fact, Nrf2 was a downstream target molecule of PGC-1α in the pathway scheme derived from the present study, and its mRNA was up-regulated by ebselen in the GKO islets. Thus, the roles of these two proteins were cooperative or coordinated in activating the cascade. In the future, plasmid reporter assays and/or chromatin immune-precipitation analyses should be conducted to discern the true initiation of transcription of GK, GLUT2, PDX1, and UCP2 by PGC-1α, Nrf2, and other factors in the ebselen-mediated GSIS rescue.
Another significant finding of the present study was that the GPX and SOD mimics demonstrated distinctly different regulation of signal transduction and gene expression related to GSIS. For many years, many antioxidants such as these mimics have been perceived to be the same “pathway” or “family” members for ROS scavenging (7). Using the GKO, SKO, and dKO islets, we were able to metabolically control the intracellular H2O2 and superoxide status (68) without confounding factors as encountered in conventional models (38, 46, 66), for comparing the functions and mechanisms of the GPX and SOD mimics in regulating islet GSIS. Overall, these two mimics depicted at least three distinctly different features. First, ebselen promoted GSIS in all four genotypes, whereas CuDIPs helped only SKO islets. Second, ebselen caused consistent up-regulation of GK and GLUT2 and down-regulation of UCP2 in islets of the four genotypes; while CuDIPs produced an opposite effect on UCP2 between the WT and SKO islets. As an uncoupler of respiration and oxidative phosphorylation, UCP2 is activated by endogenously generated superoxide (35) and is a negative regulator of insulin secretion in β-cells (74). Thus, down-regulation of UCP2 in all genotypes by ebselen and in SKO by CuDIPs was consistent with their effects on GSIS. However, these two mimics exhibited an opposite effect on UCP2 in the WT islets (CuDIPs showed no effect on GSIS in the WT islets). Lastly, gene expression of the four identified pathways related to GSIS was largely suppressed by CuDIPs, but promoted by ebselen in the dKO islets. This helps explain why ebselen but not CuDIPs promoted GSIS in the dKO islets. Comparatively, these impacts of ebselen and CuDIPs on islet GSIS and related pathways were in line with those of GKO and SKO on hepatotoxicity of acetaminophen (38), femoral mechanical characteristics (67), islet β-cell mass and insulin synthesis (68), and hepatic energy metabolism (66). Although antioxidants are often perceived to be beneficial to islet β-cell function and survival, clinical applications remain controversial or restricted to a narrow window of therapeutic dosage (7). Our finding highlights the importance of discretionary use of antioxidants in clinical treatment of GSIS to avoid harmful effects based on reciprocal results of seemingly “similar” antioxidant compounds. Furthermore, altering GPX1 and SOD1 activities might be applied to manipulate or restore intracellular H2O2 and superoxide balance in islets as a new or more effective strategy to treat insulin secretion disorders, in comparison with the insulin-centric therapy of insulin stimulators or analogues (55).
However, it seems puzzling that the increased H2O2 by GKO impaired GSIS and the removal of H2O2 by ebselen increased GSIS, whereas the blocking of enzymatic H2O2 production from superoxide by SKO also impaired GSIS and the recovery of H2O2 production by CuDIPs in the SKO islets rescued GSIS. This again underscores the complexity of the ROS regulation on GSIS, and may be explained by the concentration-dependent dual effect of H2O2 (29). Although excessive H2O2 could suppress GSIS (50), a minimal amount of H2O2 from the glucose metabolism is an essential signaling molecule for triggering GSIS (49). Thus, the SKO islets might lack sufficient H2O2 or appropriate ratios of H2O2 to superoxide to initiate GSIS, and the CuDIPs treatment rescued this function by restoring H2O2 generation. However, this notion could not explain the positive effect of ebselen on GSIS in the SKO islets. Likewise, sodium selenite improved GSIS in the SKO and dKO islets, but not in the WT or GKO islets. This might be due to a low SOD1-like catalytic activity of selenite in transforming superoxide to H2O2 (18) in the SKO and dKO islets. However, selenite did not affect GSIS of the WT and GKO islets, which was different from that reported in rat islets and min6 cells (13). Since GPX1 and ebselen were supposed to reduce both H2O2 and organic hydroperoxides (43), the latter were likely involved in the impaired GSIS in GKO mice and the rescue by ebselen. Although lipid profiles (total cholesterol, total triglyceride, and nonesterified fatty acid) were not different between the GKO and WT mice (66), the role of organic hydroperoxides in the ebselen-rescued GSIS needs further research. In addition, ebselen stimulated insulin secretion in the WT and GKO islets at 2.8 mM glucose, and CuDIPs did that in the SKO islets. Although low glucose level at 2.8 mM often inhibits insulin secretion by other stimuli, for example, glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide (23), certain therapeutic insulin secretagogues such as glibenclamide elevated islet insulin secretion at both basal (1 mM) and high (15 mM) glucose levels (26). Likewise, GK activators also increased beta cell cytosolic calcium and insulin secretion at 1 mM glucose (44). Despite this, stimulation of insulin secretion by ebselen and CuDIPs in the respective genotypes was still stronger at 16.7 than 2.8 mM glucose in the present study.
In summary, we have demonstrated a novel metabolic role and therapeutic potential of the GPX mimic ebselen (57) in rescuing GSIS in the GKO islets and mice. This rescue constituted coordinated regulation of GK, GLUT2, PDX1, and UCP2 by activating the PGC-1α-mediated ARE and(or) GR signaling. In comparison, the SOD mimic CuDIPs exerted different impacts on GSIS and the related gene expression. Thus, our findings have clarified that GPX1 and SOD1, as two important intracellular antioxidant enzymes, function distinguishably in regulating insulin secretion. Most likely, evolution has selected unique signaling pathways for different antioxidant enzymes or compounds to precisely control insulin secretion in response to complicated metabolic conditions. Clinically, these unique features of different antioxidant enzymes or mimics may be used to fine-tune islet ROS status and the related signaling for effective treatments of insulin secretion and diabetic disorders.
Materials and Methods
Mouse models, animal care, and in vivo study
Our mouse experiments were approved by the Institutional Animal Care and Use Committee at Cornell University and were conducted in accordance with National Institutes of Health guidelines for animal care. The GKO, SKO, and their dKO mice were derived from the C57BL/6 line. Deletions of gpx1 and sod1 genes in respective genotypes were verified by PCR analysis using tail DNA as templates and enzyme activity assays of various tissues (38). All experimental mice were 3–6 month-old males, weaned at 3 weeks of age, reared in plastic cages in an animal room at a constant temperature (22°C) with a 12 h light–dark cycle, and were given free access to a Torula yeast and sucrose-based diet added with 0.3 mg Se/kg (as sodium selenite) (68) and distilled water.
To examine the in vivo effect of ebselen on insulin secretion, fasting (overnight for 8 h) GKO mice (3 month-old male, n=6 per group) were given an i.p. injection of ebselen (at 50 mg/kg body weight) at 1 h before the glucose tolerance test (1 g of glucose/kg) and GSIS test (46). Ebselen was dissolved in dimethyl sulfoxide (DMSO) and injected with saline (1:1 by volume) together. The same volume of DMSO and saline was used as the solvent control. Blood glucose concentrations were measured by clipping tails and using the Glucometer Elite system (Bayer, Elkhart, IN). Plasma insulin concentrations were determined using a rat/mouse Insulin enzyme-linked immunosorbent assay kit with mouse insulin as standards (Crystal Chem, Downers Grove, IL).
In vitro experiments
All chemicals were purchased from Sigma (Saint Louis, MO) unless indicated otherwise. Detailed protocols, reagents, and instruments for islet isolation, culture, and insulin secretion were the same as previously described (69). Briefly, Langerhans' islets were isolated from the WT, GKO, SKO, and dKO mice using a standard procedure (21) with minor modifications. Isolated islets (50 per sample, n=3 per group) were recovered in RPMI 1640 (Gibco, Grand Island, NY) with 5.5 mM glucose and 10% fetal bovine serum for 2 h before treatment. For the islet experiments, ebselen, CuDIPs, and Se (sodium selenite) were used at 50, 10, and 0.1 μM and prepared in DMSO, ethanol, and saline, respectively. The same amounts of vehicle were used as the respective controls. The chemicals were removed after 5 h of incubation, and the islets were transferred to Krebs–Henseleit buffer for 30 min and then incubated for 60 min with 2.8 or 16.7 mM glucose in the same buffer. The supernatants were collected for insulin analysis, and the islets were used for mRNA, protein, and enzyme activity analyses.
Real-time Q-PCR and Western blot analyses
Total RNA was extracted from islets using Trizol reagent (Invitrogen, Carlsbad, CA). Reverse transcription was performed using Super Script III reverse transcriptase, RNaseOUT Ribonuclease Inhibitor, and Oligo(dT)12–18 (Invitrogen). The cDNA obtained from 1 μg of total RNA was used as a template for PCR amplification. Oligonucleotide primers were designed based on Genebank entries and IDT PrimerQuest. Primer sequences for the GSIS-related genes are described in Supplementary Table S1. Relative mRNA levels were determined by real-time Q-PCR (7900HT; Applied Biosystems, Foster City, CA) as previously described (48).
Islet samples used for Western blot analysis were homogenized in phosphate buffer (50 mM, pH 7.4) containing 0.1% Triton X-100 and protease inhibitor mixture (AEBSF, aprotinin, bestatin hydrochloride, E-64-[N-(transepoxysuccinyl)-
Enzyme activity measures
Islet GK activity was assayed at 28°C by measuring absorbance increases of NADPH at 340 nm in a coupled enzyme system. One unit of GK activity was defined as the amount that catalyzes the formation of 1 μmol of glucose 6-phosphate per minute. Islet and liver LDH activities were measured using a kit from Sigma according to the manufacturer's instructions. Islet and liver GSR activities were measured as previously described (75), and one unit of activity was defined as 1 nmol of glutathione disulfide reduced per minute. Plasma ALT activity was assayed using a kit (Thermo Scientific, Waltham, MA) according to the manufacturer's instructions. Plasma AKP activity was measured by the hydrolysis of p-nitrophenol phosphate to p-nitrophenol, and the enzyme activity unit was defined as the amount of activity that releases 1 μmol of p-nitrophenol per minute at 30°C (9).
Intracellular ROS
Islet ROS levels were determined using the fluorescent probe, 2′,7′-dichlorofluorescin diacetate. Briefly, the treated islets were washed and incubated with DCFH-DA (20 μM) for 10 min at 37°C. After removal of the probe, cells were washed with pre-warmed phosphate-buffered saline. Fluorescence was monitored at 488 nm excitation and 525 nm emission wavelengths.
RNA interference
Silencer® Selected Pre-Designed siRNAs (Invitrogen) duplex sequences that specifically target pgc-1α [NM_008904.2] and c/ebpβ [NM_009883.3] were employed. The Silencer Selected Negative Control from Invitrogen was used as non-targeting scramble siRNA. The transfection was performed according to the manufacturer's recommendations with minor modifications. Briefly, islets were dispersed into single cells by mechanical shaking at 37°C for 3 min in 0.05% trypsin with 0.5 mM ethylenediaminetetraaceticacid (28). The enzymatic reaction was stopped by adding 10% bovine serum albumin. Cells were washed and recovered in full culture medium at 37°C for 2 h. Thereafter, islet cells were divided equally into 24-well plates in culture medium without antibiotics or fetal bovine serum. The siRNAs (20 nM) was transfected twice using Lipofectamine™ RNAiMAX (Invitrogen). The second transfection was performed 24 h after the first. The efficiency of siRNA was verified by real-time Q-PCR analysis of the target genes. After the second transfection, the islets were recovered in RPMI 1640 culture medium with 5.5 mM glucose and 10% fetal bovine serum for 2 h. Subsequently, the ebselen treatment and the GSIS test were performed as described earlier. The medium supernatants and the islets were collected for insulin and mRNA analyses, respectively.
Bioinformatics analyses of gene promoter sequences
Upstream (1000 bp) genomic sequences of gk, glut2, pdx1, and ucp2 were retrieved from EMBL database (
Statistical analysis
Data were analyzed using SAS (release 6.11; SAS Institute, Cary, NC). Treatment effects were determined by one-way or two-way analysis of variance. Results are presented as mean±standard error of the mean, and significance level was set at p<0.05.
Footnotes
Acknowledgment
This research was supported in part by NIH grant (DK53018).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.