
Abstract
Introduction
T
It is no accident that oxidative stress is common to most forms of CNS disease. Onset of planetary photosynthesis, increased oxygen in the atmosphere, and evolution of oxidative phosphorylation required defenses against the propensity of oxygen to gain an additional electron in its outer shell, which enables reactivity with cellular constituents to amplify oxidative events. Indeed, most primitive life forms that failed to evolve enzymatic defenses against this process failed to survive (134). Superoxide dismutase (SOD) is the most ubiquitous enzymatic defense among life forms. First recognized in bacteria and later identified in all kingdoms of life (128, 134), SOD was also identified early in the brain (21). Its unique importance in the normal brain was demonstrated in a study of sustained dietary transitional metal depletion in rats. While all other tissues showed a marked decrease in SOD activity, there was no change in the brain (151), indicating preferential brain SOD synthesis.
Interest in pharmacologic intervention for oxidative stress associated with acute CNS injury was invigorated in 1978 with the report that feline brain ascorbate concentrations became depleted in a time-dependent fashion after the onset of middle cerebral artery occlusion (MCAO) (61). This finding implicated depletion of endogenous antioxidant defenses in the pathogenesis of CNS injury and fostered a discussion of replenishing these defenses with exogenous antioxidants. In vitro work showed that oxidation of brain Na+,K+-ATPase could be prevented by the addition of exogenous SOD (76). Relevance in in vivo systems was soon reported. Wei et al. (213) observed that topical SOD preserved vasoreactivity and decreased endothelial lesion density in feline traumatic brain injury (TBI). The therapeutic potential of exogenous SOD was subsequently pursued in experimental models, including intraventricular hemorrhage (132), cardiac arrest (32), TBI (33), and ischemic stroke (119) where efficacy was typically, but not always (64, 217), observed.
From a clinical therapeutic perspective, exogenous SOD posed numerous limitations, including immunogenicity, a plasma half-life of 6 min, and poor blood–brain barrier (BBB) penetration. Conjugation of SOD with polyethylene glycol (PEG-SOD) dramatically increased its plasma half-life to 40 h, but BBB penetration remained poor (208, 221). However, acute CNS injury often disrupts the BBB. Hence, PEG-SOD was investigated in both preclinical models and a clinical TBI trial. Although preclinical results were mixed (74), a phase II human TBI study predicted efficacy (137). A large population of severe TBI patients was subsequently given a single dose of PEG-SOD or placebo within 8 h of injury (222). While PEG-SOD provided a slight numerical improvement in outcome scores, a statistically significant effect was absent. This failure led to general abandonment of PEG-SOD as a therapeutic, although ongoing efforts to identify novel conjugates may raise further investigation of the native enzyme in CNS disorders (200). At the same time, the failed TBI trial caused a question as to whether supplemental SOD activity is a valid therapeutic strategy.
This concern was short lived. The advent of mice genetically modified to either overexpress SOD (56) or with targeted deletions causing SOD deficiency (105) provided an opportunity to specifically confirm the critical role of this enzymatic system in the responses of the brain and spinal cord to oxidative stress (34, 100, 178, 212), which confirmed expectations that manipulation of SOD activity can be exploited to alter the course of disease.
Concurrent with efforts to exploit endogenous/exogenous native SOD, the development of small molecules was initiated with the intent to derive pharmaceutical profiles superior to the native enzyme, that is, SOD mimics. Metalloporphyrins constitute one class of SOD mimetics and are characterized by use of porphyrin as the ligand, typically incorporating either Fe or Mn transitional metals, thus offering the required redox activity. The rational design, through various substitutions in the core porphyrin ligand structure, afforded optimized redox potential matching that of the native enzyme. Structure-activity relationships have been reviewed extensively elsewhere (17, 18, 20, 135, 156).
This article focuses on metalloporphyrins (Fig. 1) and provids a detailed review of the experience with these compounds in a diverse array of CNS disorders (Fig. 2), all of which present a commonality of oxidative/nitrosative stress being central in disease onset and progression.

While originally synthesized as SOD mimics, the body of evidence clearly demonstrates that metalloporphyrins offer a substantially broader mechanism of action than endogenous SOD. Metalloporphyrins have been reported to interact with superoxide, peroxynitrite, nitric oxide, and nuclear transcription factors, as oxidoreductants. However, mechanisms of action have been insufficiently investigated for individual molecules to explain purported therapeutic effects. Most metalloporphyrins also have limited CNS bioavailability due to their inherent charge and poor BBB penetration. Structural modifications have been employed to overcome this limitation. However, until the mechanism of action is fully detailed, it is unlikely that appropriately designed clinical trials will be empowered to provide clinical confirmation of the ubiquitous preclinical reports of improved functional outcome by metalloporphyrins in CNS disorders (Table 1).
Charges are indicated in all complexes. While in solid state, FeTM-4-PyP carries 5+ charges; once dissolved at pH 7.8, the Fe center will have axially bound OH− ligand and total charge of the biologically relevant molecule will, therefore, be 4+(11, 201).
See Abbreviations Used section for chemical names.
ALS, amyotrophic lateral sclerosis; SAH, subarachnoid hemorrhage; SCI, spinal cord injury; TBI, traumatic brain injury; IV, intravenous; IP, intraperitoneal; ICV, intracerebroventricular; IT, intrathecal; SC, subcutaneous; PO, per os.
Amyotrophic Lateral Sclerosis
The study of metalloporphyrins in amyotrophic lateral sclerosis (ALS) was triggered by early reports that oxidative stress is important in ALS pathogenesis. Mutant copper, zinc superoxide dismutase (Cu,Zn SOD) is present in a subset of familial ALS patients (48), and this mutation has been associated with progressive motor neuron death. This mutation can be mimicked in the laboratory by the use of transgenic mice and rats overexpressing a Cu,Zn SODG93A mutation (69, 77). Concentrations of spinal cord SODG93A in the rat mutants are in the range of 16–26 μM (161), consistent with supra-therapeutic and toxic concentrations of the Mn porphyrin Mn(III) meso-tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin (MnTDE-2-ImP) studied in primary mixed neuronal/glial cell cultures exposed to oxygen–glucose deprivation (179). The SODG93A mutants develop progressive motor neuron death with neurologic deficits manifesting at 3–6 months of age depending on the exact mutant. The primary metabolic defect of the mutation remains controversial (75). However, numerous investigations indicate that increased peroxynitrite formation is central in ALS pathogenesis (22, 57, 166). Nitration-induced motor neuron and astrocyte cell death has been extensively documented in vitro (152, 219).
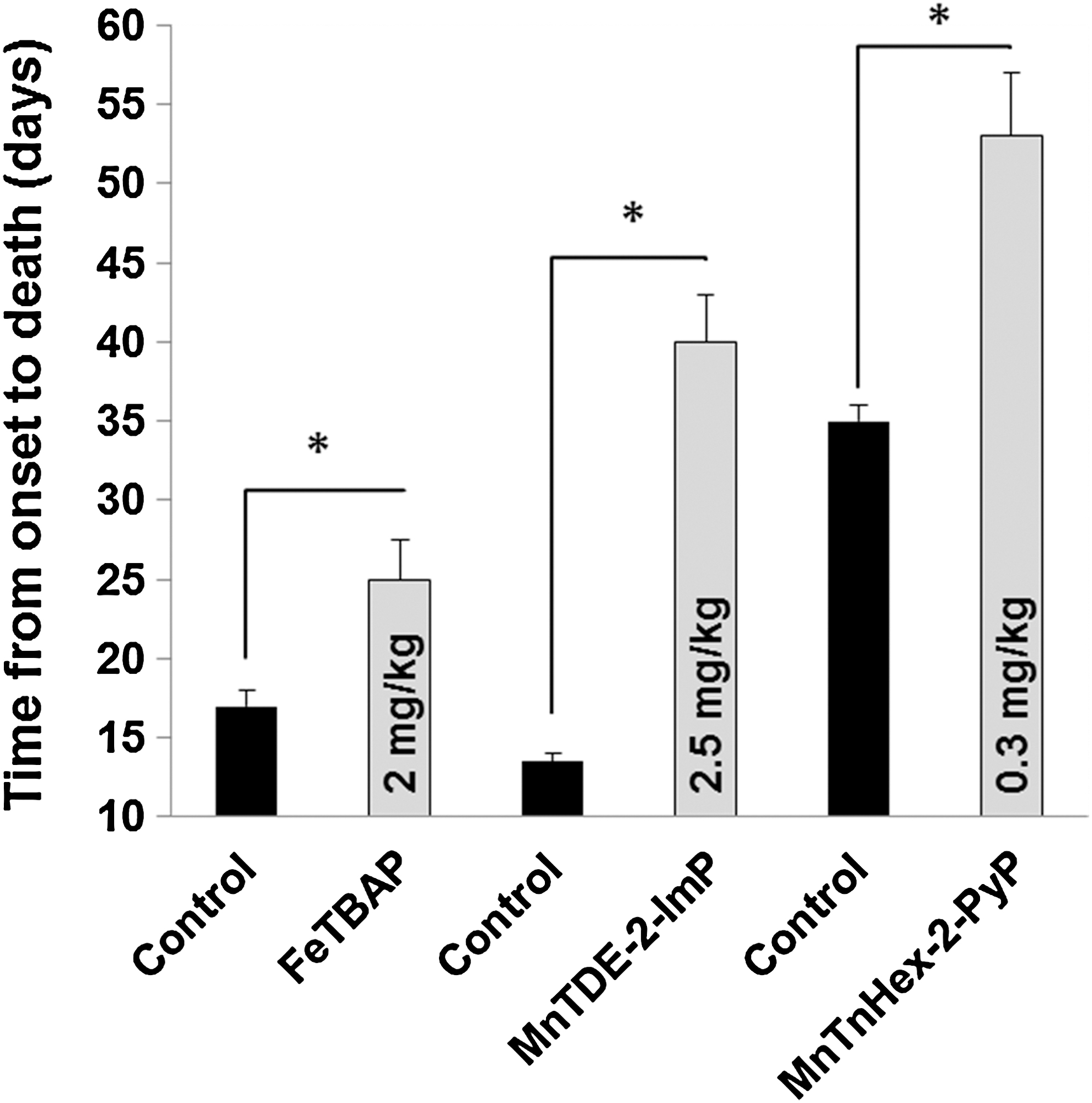
Although Mn porphyrins, including Mn(III) meso-tetrakis(4-carboxylatophenyl)porphyrin (MnTBAP, MnTCPP), were the first to be shown effective against oxidative stress (58), the Fe analog Fe (III) meso-tetrakis(4-carboxylatophenyl)porphyrin (FeTBAP, FeTCPP) was the first metalloporphyrin investigated in SODG93A mice, because its reactivity toward peroxynitrite was reported greater than that of MnTBAP (42). Wu et al. (218) chronically treated SODG93A mice with FeTBAP (2 mg/kg/day intraperitoneal) and measured the age of terminal motor deficit onset. FeTBAP poorly penetrated the BBB with concentrations in the brain and spinal cord being ∼2%–4% of those in the kidney and liver. Survival was increased by 7–9 days depending on the timing of treatment onset. Concentrations of malondialdehyde and protein-bound carbonyl groups were used as markers of oxidative stress. FeTBAP decreased both. Thus, while FeTBAP did not reverse lethality associated with the SODG93A mutation, disease progression was slowed. Although bioavailability was confirmed, brain and spinal cord concentrations were low, suggesting that greater therapeutic effects might be achievable with larger tissue concentrations. This might now be tested with recently synthesized Mn porphyrins offering markedly greater lipophilicity (16, 103).
This experiment was followed by work using the Mn porphyrin MnTDE-2-ImP, also known as AEOL10150. Crow et al. (44) studied SODG93A mutant mice that typically develop signs of motor dysfunction at 90 days of age. Experiments using high-dose MnTDE-2-ImP (2.5 mg/kg/day) were performed with and without creatine as a dietary supplement and rofecoxib, a cyclooxygenase-2 (COX-2) inhibitor. Treatment was begun at onset of signs of motor dysfunction. Survival was increased from 102 days in vehicle-treated controls to 129 days in MnTDE-2-ImP-treated mice. This was held in contrast to an increase in survival of 6 days from the combination of creatine and rofecoxib. The addition of creatine and rofecoxib did not potentiate MnTDE-2-ImP efficacy. MnTDE-2-ImP efficacy was similar whether given intraperitoneal or subcutaneous. Tissue MnTDE-2-ImP concentrations were not measured to define bioavailability. However, at 10 days after symptom onset, a greater number of spinal motor neurons were present in the MnTDE-2-ImP group.
MnTDE-2-ImP has also been evaluated as an adjunct to the histone deacetylase inhibitor phenylbutyrate (PBA). Earlier work had shown transcriptional dysregulation in both SODG93A mice and human sporadic ALS (84, 145), potentially attributable to histone hypoacetylation and diminished access of transcription factors to target genes. PBA inhibits hypoacetylation and decreases apoptotic cell death in SODG93A mice (164). This was associated with increases in survival time, nuclear factor–κB (NF-κB) p50 nuclear translocation, and beta cell lymphoma 2 (bcl-2) expression. Petri et al. (153) studied PBA (400 mg/kg/day) with or without MnTDE-2-ImP (2.5 mg/kg/day) begun at disease onset in SODG93A mice (153). Survival was increased by 11% with MnTDE-2-ImP, 13% with PBA, and 19% by the two drugs in combination. This combined treatment benefit corresponded to a greater number of intact motor neurons observed at a standardized preterminal assessment interval. However, it is not obvious how this interaction biochemically improved survival. While increased histone acetylation is associated with increased NF-κB activity, Mn porphyrins, including MnTDE-2-ImP, have been consistently shown to inhibit NF-κB-induced gene transcription (162, 180, 182, 225), most likely due to oxidation (205) and/or glutathionylation (87, 89) of the promoter binding elements. This competition is consistent with the observed increase in NF-κB-dependent manganese superoxide dismutase (MnSOD) and bcl-2 expression caused by PBA alone, while these increases were markedly muted by combination with MnTDE-2-ImP. This cumulative preclinical data indicating consistent efficacy from metalloporphyrins in ALS models (Fig. 3) allowed initiation of clinical investigation.
In unpublished Phase I clinical trials in ALS patients (23, 146), MnTDE-2-ImP was given subcutaneously in a single dose-escalation study of up to 75 mg/kg or twice daily (40 mg/kg) for approximately 6.5 days. These doses markedly exceeded predicted therapeutic doses extrapolated from mouse SODG93A mutant studies. No adverse effects were observed other than irritation at the injection site, which was mild to moderate and of limited duration. There was no effect on QTc interval, neurologic examination, or vital signs. Due to the short-term treatment, efficacy analysis was not performed. This study constitutes the first and only use of metalloporphyrins in humans to date.
Stroke
Stroke represents loss of brain function due to disturbance in its blood supply. Stroke, as a generic term, broadly encompasses ischemic stroke induced by a temporary or permanent occlusive vascular event, subarachnoid hemorrhage (SAH) from a cerebral aneurysm or arteriovenous malformation and consequent delayed arteriopathy (also known as cerebral vasospasm), or rupture of small vessels leading to intraparenchymal hemorrhage. While many responses of brain to these unique events overlap, it is important to independently consider therapeutic efficacy in each stroke subtype due to different etiologies, mechanisms of pathogenesis, and temporal course of disease progression. Stroke, a regional brain disorder, does not pertain to cardiac arrest or other anoxic brain insults, which present homogeneous and global deprivation of oxygen to the brain.
Ischemic stroke
Ischemic stroke has been intensively investigated with regard to Mn porphyrin therapeutic efficacy. Although preclinical efficacy has been known for more than a decade, development of pentacationic Mn porphyrins as clinical therapeutics has been hampered by limited bioavailability attributable to the very structure of the molecule which offers its physicochemical characteristics, that being the 5+ charge designed to attract anionic reactive oxygen species (ROS)/reactive nitrogen species (RNS). The 5+ charge is associated with hydrophilicity, which simplifies drug formulation, as Mn porphyrins are highly soluble in water or saline. However, the net 5+ charge limits BBB penetration due to poor lipophilicity. Despite this, work with early-generation molecules compounds indicated major therapeutic potential.
The first metalloporphyrin report was derived from a study of Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin (MnTE-2-PyP) (11, 14) in rats and mice subjected to temporary MCAO (125). Pilot studies identified a critical concern regarding Mn porphyrins. Arterial blood pressure was decreased by intravenous MnTE-2-PyP in a dose-dependent manner. Blood pressure can be a major determinant of focal ischemic outcome due to modulation of cerebral blood flow through collateral circulation (41, 93). This hypotensive effect was formally investigated and found to be true of Mn porphyrins as a compound class, most prominent in the rat. Other species, including mouse, guinea pig, dog, baboon, humans (146, 163), and macaque monkeys (unpublished data), have a substantially less hemodynamic response when clinically relevant doses are studied. Rats have very low endogenous extracellular superoxide dismutase (EC-SOD) relative to the other species. It was postulated that interactions between superoxide and nitric oxide regulation of vascular tone by Mn porphyrins could explain the large blood pressure decrease in the rat (163). However, neither intravenous Cu,Zn SOD nor PEG-SOD altered blood pressure in the rat arguing against modulation of vascular tone due to scavenging ROS/RNS by Mn porphyrin. In contrast, co-administration of the selective histamine H1 antagonist chlorpheniramine fully blocked the Mn porphyrin hypotensive effect in rats, plausibly by interaction with redox-regulated histamine release from mast cells (70). This matter has not been investigated in further detail. Blood pressure is still of concern, particularly when large loading doses are given to rapidly penetrate the BBB to rescue ischemic neurons. Since rats constitute the primary species for preclinical evaluation of stroke drugs, hypotensive effects of Mn porphyrins in this species could confound definition of efficacy and make conclusions unreliable.
Consequently, early work with Mn porphyrins in rat stroke models deferred to acute and chronic intracerebroventricular administration. Given this route, both MnTE-2-PyP and MnTDE-2-ImP were found to be highly efficacious in experimental ischemic stroke (125, 179, 182). Of major importance, efficacy was retained when treatment onset was delayed for 7.5 h after onset of the ischemic insult. This wide therapeutic window is of considerable relevance to clinical stroke where patient presentation to medical care is often delayed. MnTE-2-PyP and MnTDE-2-ImP were equivalent in efficacy (Fig. 4), as defined by cerebral infarct volume and neurologic scores. Evidence of attenuation of oxidative stress was confirmed by measurement of aconitase activity, a marker of superoxide production (65), which was preserved by Mn porphyrin, and corresponded to a decrease in ischemia-induced 8-hydroxy-2′-doexyguanosine (8-OHdG) formation. Therapeutic brain concentrations were found to be similar to concentrations required to obtain protection against oxygen–glucose deprivation in primary mixed neuronal/glial cell cultures.

Short-term outcome studies conducted in mice also showed efficacy from both MnTE-2-PyP and MnTDE-2-ImP when given intravenously (125, 179). Kinetic studies found that while brain MnTDE-2-ImP uptake was negligible in nonischemic tissue, MnTDE-2-ImP uptake was accelerated in postischemic tissue (179). These results indicate acute parenchymal bioavailability of hydrophilic Mn porphyrins after BBB disruption has occurred.
Hydrophilic Mn porphyrin bioavailability through a disrupted BBB may explain other reports of hydrophilic Mn porphyrin efficacy in ischemic stroke, some of which have provided additional insights into the mechanism of action. Huang et al. (78) reported that 1.5 mg/kg Mn(III) meso-tetrakis(N-methylpyridinium-4-yl)porphyrin (MnTM-4-PyP) (58) decreased cerebral infarct size and neurologic deficit measured 24 h post-MCAO when intravenous treatment was begun 30 min before ischemia onset. MnTM-4-PyP decreased mitochondrial cytochrome c release, caspase 3 activation, and apoptosis. Others have reported Mn porphyrin-mediated (MnTBAP) inhibition of postischemic apoptosis and attenuation of caspase-independent nuclear translocation of apoptosis-inducing factor (AIF) (106). Inhibition of apoptosis by Mn porphyrins is further supported by work in cultured cerebellar granule neurons. Tauskela et al. (196) examined multiple Mn porphyrins and found consistent and major inhibition of staurosporine-induced caspase 3 activation and improved cell survival. Curiously, efficacy was independent of Mn porphyrin SOD activity, indicating a major mechanism of action different from simple ROS/RNS scavenging. The same group reported neuroprotection from Mn porphyrins in the context of N-methyl-D-aspartate (NMDA) excitotoxicity (196, 197). A broad spectrum of metalloporphyrins was effective in inhibiting propidium idodide uptake and increased intracellular calcium levels, again largely independent of SOD activity. Others have reported that Mn porphyrins inhibit oxygen–glucose deprivation Ca2+ release from the endoplasmic reticulum in primary cortical neuronal cultures (78). Thus, the neuroprotective effect of metalloporphyrins, at least in part, can be dissociated from their antioxidant potential as a mechanism for the repeatedly demonstrated efficacy. These findings are likely relevant across the spectrum of CNS disorders. For example, Mn porphyrins with low SOD activity have been reported efficacious in Parkinson's disease and epilepsy models (111, 112).
However, some of these studies included critical determinations made from commercially available MnTBAP. Commercial MnTBAP has major impurities (159), which can confound interpretation of results. Pure MnTBAP does not possess SOD activity (11, 15), while impurities may (159). In contrast, both commercial and pure MnTBAP are weak peroxynitrite scavengers (13). This has created considerable confusion in understanding mechanisms of metalloporphyrin action, as have claims that a specific metalloporphyrin is either a selective SOD mimic or peroxynitrite decomposition catalyst.
A linear relationship has been established between log kcat(O2 ·−) and log kred(ONOO−) for the Mn porphyrin series (60). The more potent SOD mimics are also the more potent peroxynitrite scavengers. Peroxynitrite is one of the strongest oxidants. Consequently, it can oxidize even those metalloporphyrins (e.g., MnTBAP) that have poor redox properties and are electron rich. Electron-rich Mn porphyrins would not favor binding of electron-rich peroxynitrite to the metal center in a first step of its reduction. Further, electrostatics does not favor the approach of anionic ONOO− to anionic Mn porphyrins. Indeed, the log kred(ONOO−) for MnTBAP is 5.02, while it is 7.53 for MnTE-2-PyP (202). For details, see Batinic-Haberle et al. (20a)
The mechanistic picture for pentacationic Fe porphyrins also has been confused. Early work demonstrated Fe porphyrins to be potent peroxynitrite reductants (42, 190), leading to claims that these molecules offered selective peroxynitrite scavenging. This is incorrect. Since SOD-like activity parallels the ability to reduce ONOO−, Fe porphyrins are also potent SOD mimics (19, 148), and similar to Mn porphyrins (187), they react with nitric oxide (104). The magnitude of such reactivities is dependent on numerous features of their molecular design. Thus, while Mn porphyrins and Fe porphyrins are often categorized as SOD mimics and peroxynitrite reductants, the redox chemistry of both is complex, thus limiting the interpretation of in vivo mechanisms of action under conditions of oxidative/nitrosative stress (201).
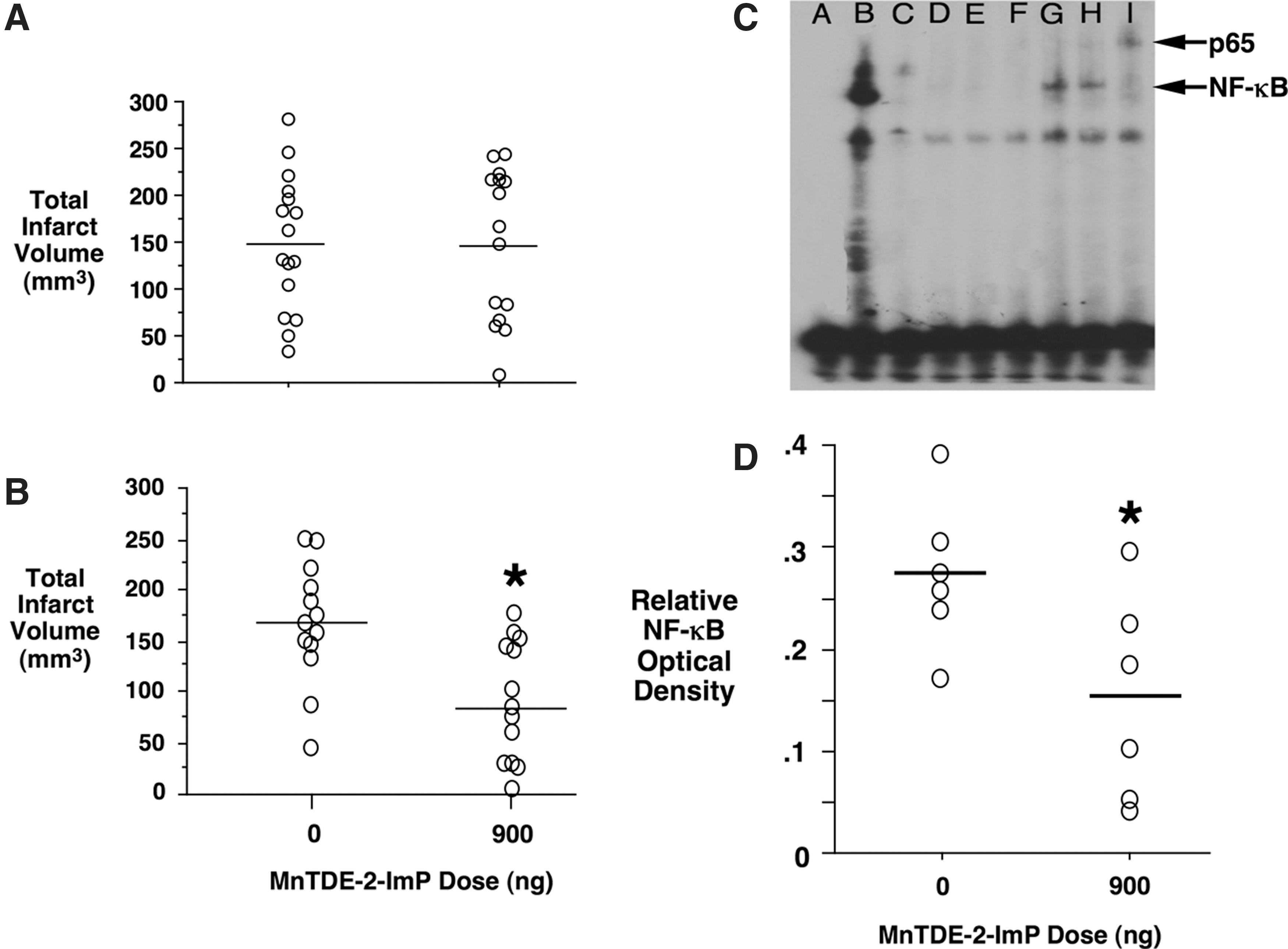
While preclinical efficacy results reported for ischemic stroke are highly encouraging, from a clinical perspective, an efficacious stroke drug should produce sustained outcome improvement, which cannot be defined in a recovery observation interval of one to several days. Of potential clinical importance, Mn porphyrins have been shown to provide sustained outcome improvement when functional and histologic analysis was assessed 8 weeks after the ischemic insult (182). However, this was highly dependent on dosing strategy. While a single intracerebroventricular MnTDE-2-ImP dose was associated with decreased neurologic deficit and cerebral infarct size at 1 week postischemia, there was no benefit when rats were allowed to survive 8 weeks. In contrast, when intracerebroventricular treatment was sustained for 1 week, benefit persisted for the full 8-week observation interval (Fig. 5). This implicates redox-regulated events that persist into at least the sub-acute recovery interval presenting implications for dosing strategies should clinical efficacy trials be proposed.
The studies described earlier are of importance with regard to proof of principle, that is, hydrophilic Mn porphyrins present therapeutic properties of major relevance to stroke treatment (e.g., wide therapeutic window and sustained efficacy). However, use of hydrophilic Mn porphyrins in clinical stroke is highly unlikely if an intracerebroventricular route is required to deliver drug to brain parenchyma. Intracerebroventricular administration, while used for more chronic disorders such as lymphoma and dystonia, is an impractical route of administration for the larger stroke population, and the parenchymal distribution would be expected to be inconsistent (170). Thus, poor bioavailability halted the development of Mn porphyrins for ischemic stroke.
Synthetic efforts have recently provided novel Mn porphyrin analogs designed for improved BBB penetration (19, 214). In some compounds, SOD activity remains preserved, while lipophilicity is increased >4 orders of magnitude (20a). Improved lipophilicity should allow intravenous treatment with preserved therapeutic benefit in acute treatment paradigms. The prototype Mn(III) meso-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTnHex-2-PyP) was found to provide 9-fold greater brain concentration than MnTE-2-PyP after a single intraperitoneal dose in normal mice, and is also orally bioavailable (214). In focal ischemic stroke, parenteral (intraperitoneal or subcutaneous) MnTnHex-2-PyP efficacy in rats was similar to that previously reported for intracerebroventricular administration of more the hydrophilic analogs MnTE-2-PyP and MnTDE-2-ImP and offered sustained efficacy and the same 6-h therapeutic window (180) (Fig. 6). Due to better BBB penetration, markedly smaller doses of MnTnHex-2-PyP are sufficient to provide similar efficacy. This finding was also reported for MnTnHex-2-PyP in the SODG93A mouse ALS model (43) (Fig. 3). The requirement of low doses also allowed extension of studies to the rat, as arterial blood pressure effects were less pronounced at lower blood concentrations.
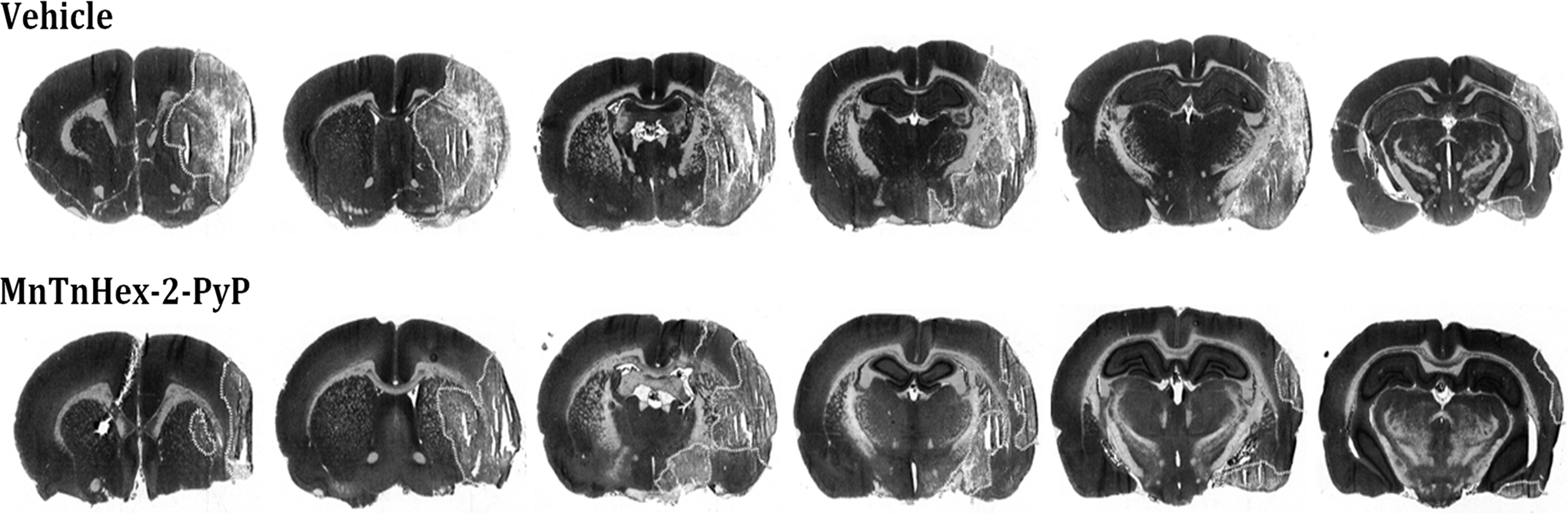
Despite this advance, there is neurotoxicity associated with all Mn porphyrins evaluated to date. The mechanism has not been defined. In both rats and mice, supra-therapeutic Mn porphyrin doses produce a constellation of signs that is consistent with the “Straub phenomenon,” first recognized with an injection of morphine into mice (24). Specifically, catatonic tail rigidity, hyperexcitability, and proptosis are observed. This response has not been observed in the very limited human exposure to Mn porphyrins to date (146). However, additional toxicity was observed with MnTnHex-2-PyP that included malaise and death. This could be attributable to the potential for saponification due to the combination of high lipophilicity and the polar MnTnHex-2-PyP structure. Hence, while MnTnHex-2-PyP again advanced proof of principle regarding potential value of Mn porphyrins in stroke by demonstration of clinically relevant efficacy, further synthetic efforts were required to decrease toxicity so as to enable advances to the clinical domain.
Mn(III) meso-tetrakis(N-n-butoxyethylpyridinium-2-yl)porphyrin (MnTnBuOE-2-PyP) appears to offer an opportunity for a greater therapeutic index (156). This molecule is structurally similar to MnTnHex-2-PyP but includes insertion of a single oxygen atom into each of the lipophilic side chains. This serves to preserve lipophilicity and SOD activity, but toxicity is markedly decreased. For example, a single 5-mg/kg MnTnHex-2-PyP intraperitoneal injection is lethal in the mouse. In contrast, the same dose of MnTnBuOE-2-PyP produced no observable adverse effects. Mice treated with approximately 9 mg/kg/day of MnTnBuOE-2-PyP for 7 days retain body weight. Rotarod performance, as a measure of neurologic function was unchanged. SOD-deficient Saccharomyces cerevisiae yeast fail to thrive in aerobic environments. Addition of MnTnHex-2-PyP dose dependently increases SOD-deficient yeast growth to a concentration of approximately 10 μM, while greater concentrations are toxic. In contrast, MnTnBuOE-2-PyP supports SOD-deficient yeast growth at the highest concentration tested (30 μM). Formal in vivo efficacy analysis in ischemic stroke and other forms of CNS pathology have not been reported for MnTnBuOE-2-PyP.
Fe porphyrins also have been examined in experimental ischemic stroke. Thiyagarajan et al. (198) studied temporary MCAO (tMCAO) in rats. Rats were given either vehicle or ascending intravenous doses of either (Fe (III) meso-tetrakis(N-methylpyridinium-4-yl)porphyrin (FeTM-4-PyP) or Fe (III) meso-tetrakis(4-sulfonatophenyl)porphyrin (FeTSPP). The therapeutic window was also evaluated with treatment onset beginning at 2, 6, 9, or 12 h postischemia. Outcome was measured at 70 h after reperfusion. Both compounds substantially decreased cerebral infarct and edema volumes when given as late as 6 h postischemia. However, neurologic function was improved only when FeTM-4-PyP was given at 2 h postischemia. There was no effect of FeTSPP on neurologic outcome at any dose or postischemic treatment onset interval. Blood pressure effects of neither compound were measured. Data were presented implicating attenuation of peroxynitrite formation and apoptosis. While limited by a very short observation recovery interval, this study provided some evidence of potential Fe porphyrin efficacy in stroke, which is consistent with previously reported mechanisms of action, albeit neurologic function was not improved with a clinically relevant therapeutic window.
In a nearly contemporaneous report, the same group explored the combination of a Fe porphyrin and a poly(adenosine diphosphate[ADP]-ribose) polymerase (PARP) inhibitor (177). FeTM-4-PyP was administered 5 min before reperfusion alone (1 or 2 mg/kg) or in combination with 1,5-isoquinolinediol administered 60 min after reperfusion in a tMCAO rat model. Outcome was measured 22-h post-MCAO. At that interval, either compound alone provided a substantial decrease in cerebral infarct size. Combination treatment provided a greater benefit. However, while both compounds improved neurologic score compared with vehicle-treated cohorts, combination treatment failed to provide superior functional outcome to either drug alone. Notably, FeTM-4-PyP alone was found to produce a dose-dependent decrease in PARP activity at 2 h after reperfusion onset, indicating convergence of mechanisms of action. While promising, clinical implications of this study are limited by preischemic FeTM-4-PyP treatment and the very short recovery observation interval.
Suofu et al. (191) extended the therapeutic potential of Fe porphyrins in ischemic stroke. Rats were subjected to 3.5 h tMCAO and treated with intravenous FeTM-4-PyP or vehicle just before reperfusion onset. FeTM-4-PyP had no effect on cerebral blood flow (laser Doppler) or blood pressure. FeTM-4-PyP inhibited activation of matrix metalloproteinase (MMP-9 and MMP-2) at 8 h after ischemia onset. Collagen type IV and microvascular basal lamina were better preserved by FeTM-4-PyP, implicating amelioration of endovascular reperfusion injury. Palomares et al. (147) performed a similar experiment with FeTM-4-PyP given 10 min before reperfusion onset from tMCAO. This experiment segregated MCAO severity with regard to magnitude of CBF decrease (laser Doppler) during MCAO. When ischemia was mild, but not severe, FeTM-4-PyP decreased cerebral infarct size, as measured at 2 h after reperfusion. Severe ischemia was associated with poor reperfusion suggesting that failure of FeTM-4-PyP to offer benefit may be attributable to failure of the compound to reach its target. Neurologic function was not assessed. While neither study provided evidence for long-term neurologic efficacy, this body of evidence indicates that further work is warranted to investigate therapeutic potential of Fe (or Mn) porphyrins, given before therapeutic recanalization with tissue plasminogen activator so as to diminish acute reperfusion injury.
Spontaneous intracerebral hemorrhage
Approximately 10%–15% of clinical strokes constitute nonaneurysmal intra-parenchymal hemorrhage, with etiologies including arterial hypertension, use of anti-coagulants and thrombolytics, cerebral amyloidopathy, and hemorrhage into ischemic infarcts. Although hematoma expansion is typically completed within 3–12 h postictus (95), lesion volume continues to expand and persist due to perihematomal edema formation for approximately 14 days (83).
Perihematomal edema is associated with intracranial hypertension, global oligemia, and mitochondrial dysfunction. There are numerous factors that contribute to edema, including thrombin-induced BBB breakdown, MMP activation, oxidative stress induced by iron products, and NF-κB activation and cytokine expression (113, 209). Thus, there are numerous targets by which Mn porphyrins could have therapeutic benefit in this disorder, but to date, neither Mn nor Fe porphyrins have been evaluated. The spin trap nitrone, disodium 2,4-disulfophenyl-N-tert-butylnitrone (NXY-059) has undergone a human trial in intracerebral hemorrhage (ICH). No benefit was observed (121). This is consistent with its SOD-like activity being more than two orders of magnitude less than MnTE-2-PyP. Further, NXY-059 was ineffective in an SOD-deficient Escherichia coli aerobic growth assay (203), while Mn porphyrins have proved consistently effective (156). Further, NXY-059 has negligible BBB and cell membrane penetration (47). This is most likely due to its anionic charges, which would disfavor BBB penetration. Hence, NXY-059 potency and bioavailability to interact with cellular responses to ICH would be predicted to offer little or no benefit. Despite this, the failure of NXY-059 to offer benefit in clinical ICH has cast a pall over the use of redox-active therapeutics for this and other stroke disorders (94). Hence, a direct comparison of metalloporphyrins to NXY-059 would be of considerable interest in experimental ICH.
SAH and delayed arteriopathy
Rupture of a cerebral aneurysm causes extravasation of blood into the subarachnoid space. An immediate vasoconstrictive response occurs, the mechanism of which is undefined, but this appears to be unrelated to blood components because the magnitude of constriction is independent of the volume of blood extravasated (55, 216). This progresses to a delayed arteriopathy that is characterized by focal occlusions and microvascular flow defects culminating in delayed neurologic deficits. Although intensively investigated (30), the pathogenesis of this arteriopathy is incompletely defined, and, as a result, therapeutic options remain crude and often ineffective. Long-term neurologic and cognitive deficits are common (5, 139, 171).
Oxidative stress clearly contributes to this morbidity. This arises from both the initial ictal event, where increased intracranial pressure may present an incomplete global ischemic insult, and biomolecular responses to hemorrhage that propagate an inflammatory response in both parenchyma and the vasculature. NF-κB activation, cytokine expression (226), uncoupling of endothelial nitric oxide synthase (NOS) (165), and free iron central to the Fenton and Haber–Weiss reactions (107) have been implicated. Thus, there are numerous mechanisms by which metalloporphyrins could play therapeutic roles in SAH.
There have only been two reported experimental studies of Mn porphyrins in SAH and no studies of Fe porphyrins. Both Mn porphyrin studies identified efficacy. Aladag et al. (2) employed a rat model in which autologous intracisternal blood injections are given at 0 and 48 h. Rats were treated with intraperitoneal vehicle or 5 mg/kg MnTBAP (commercial grade) twice daily beginning 6 h after the first injection with treatment continued for 5 days. Blood injection alone decreased basilar artery diameter to 26% of that in normal controls. MnTBAP treatment preserved basilar artery diameter at 88% of control. This was impressive, but neurologic function was not evaluated. In the second study, Sheng et al. (180) subjected mice to endovascular perforation of the anterior cerebral artery. The Mn porphyrin MnTnHex-2-PyP (225 μg/kg intraperitoneal) or vehicle was given twice daily for 3 days. MnTnHex-2-PyP had no effect on blood pressure. SAH alone caused 30%–40% decrease in diameters of the middle and anterior cerebral and internal carotid arteries. MnTnHex-2-PyP-treated mice had vessel diameters similar to shams. Although neurologic deficits remained in MnTnHex-2-PyP-treated mice, deficit severity was markedly less than that in vehicle-treated mice. The efficacious MnTnHex-2-PyP dose was ∼1/20th of that for MnTBAP. This most likely represents dependence of efficacy on Mn porphyrin lipophilicity, charge, and size of the molecule. A direct comparison of the two Mn porphyrins has not been made, but would be valuable to increase our insight into the impact of different physicochemical factors on the therapeutic potential.
These data indicate efficacy of Mn porphyrins in experimental SAH. Mechanisms of efficacy have not been defined in either of the models evaluated. Mn porphyrins decrease tissue oxidation and nitration, NF-κB activation, and cytokine expression in other models of CNS injury, but this should be specifically examined in the context of SAH. Furthermore, both outcome studies employed very short observation intervals (i.e., 3–5 days postictus). Since clinical neurological and cognitive deficits progress in presentation over 10–14 days postictus and may persist at least 5–7 years (154), it is imperative that any clinical trial of Mn porphyrin in this disorder be based on preclinical data demonstrating benefit from Mn porphyrin intervention over long-term recovery intervals and that dosing regimens be defined to ensure sufficient dosing duration is employed to sustain long-term efficacy, as has been noted in ischemic stroke (182).
Global Cerebral Ischemia
Global ischemia is distinguished from focal ischemia in that there is homogeneous reduction of flow throughout the brain (or in some models, homogeneous ischemia isolated to the forebrain). Thus, there is no opportunity for collateral perfusion to establish an ischemic penumbra and tissue that could be salvaged from a sustained hypoperfusion state. Global ischemia causes rapid onset of irreversible delayed ischemic neuronal injury, with ischemic intervals of 5–7 min being sufficient to cause substantial cell death in selectively vulnerable neuronal populations.
There have been two studies of Mn porphyrins in global ischemia insults. The first utilized a severe forebrain ischemia model in mature rats (125). A single dose of MnTE-2-PyP (150 or 300 ng) was given intracerebroventricularly 60 min before 10 min ischemia onset. At 5 days postischemia, neither neurologic function nor histologic damage was improved. The dose used was identical to that shown to provide short-term improved outcome from focal cerebral ischemia (125). Thus, responses of global and focal ischemia to Mn porphyrins are clearly different.
A different result was found in the immature brain. Drobyshevsky et al. (54) subjected pregnant rabbits to aortic occlusion sufficient to cause severe fetal brain ischemia so as to mimic the pathogenesis of cerebral palsy. Surviving kits are characterized by a high incidence of hypertonia. Treatment of the dam with MnTnHex-2-PyP 30 min before ischemia and then repeated 30 min after reperfusion decreased the incidence of hypertonia from 80% to 20%. However, MnTnHex-2-PyP given only at 30 min after reperfusion onset had no benefit. This was held in contrast to posttreatment with a combination of ascorbate and Trolox, which provided benefit. Kinetic analysis provided a probable explanation. Fetal tissue MnTnHex-2-PyP uptake was insufficiently rapid to ameliorate injury, while ascorbate and Trolox rapidly increased fetal brain antioxidant activity. Importantly, pretreatment with MnTE-2-PyP failed to protect under conditions where MnTnHex-2-Pyp was efficacious. In this model, none of the membranes are damaged, which would have otherwise provided an alternative pharmacokinetic route for the hydrophilic MnTE-2-PyP.
Together, these studies indicate important limitations of Mn porphyrins in global ischemic insults. Although MnTnHex-2-PyP was effective in the cerebral palsy model, this was only true for a pretreatment dosing paradigm (54), which limits clinical importance. MnTE-2-PyP pretreatment provided no benefit in adult global ischemia (125). This could represent lack of sufficient time for parenchymal distribution when given by the intracerebroventricular route. Perhaps more important, both studies only utilized a single acute dose, yet measured outcome 7 days later. Further studies with sustained treatment regimens are required before Mn porphyrins can be fully dismissed as not having therapeutic value for global cerebral ischemia.
Traumatic Brain Injury
Despite the prevalence and magnitude of individual and societal implications from TBI, there is no pharmacologic therapy with proven efficacy for this disorder (127). TBI pathogenesis invokes mechanisms of action similar to those identified as targets for metalloporphyrins in other CNS disorders (e.g., oxidative stress, transcription factor activation, mitochondrial dysfunction, and apoptosis) (73). To date, an investigation of metalloporphyrin efficacy has been limited. Leinenweber et al. (108) exposed mice to closed cranial impact. Pilot studies were performed to determine the maximal intravenous MnTE-2-PyP dose tolerated. The toxic end point selected was the “Straub phenomenon,” identical to that observed with direct intracerebroventricular MnTE-2-PyP injection (125). A 6 mg/kg dose was found to produce “just noticeable” effects. Mice were treated with a single dose of vehicle or 3 or 6 mg/kg MnTE-2-PyP at 15 min postimpact. TBI caused a marked decrease in aconitase activity, a marker of superoxide production (66), which was partially and transiently abated by MnTE-2-PyP. However, MnTE-2-PyP had no effect on 21-day functional recovery, Fluoro–Jade staining, or glial fibrillary acidic protein immunoreactivity. It is unlikely that bioavailability can explain lack of effect, because behavioral responses to intravenous MnTE-2-PyP treatment were similar to those observed with intracerebroventricular injection, and closed cranial impact is associated with major BBB disruption (127). Once again, it should be questioned whether the duration of action of a single Mn porphyrin dose is sufficient to abate long-term oxidative and nitrosative responses to an acute brain insult. An examination of sustained treatment efficacy and confirmation of enhanced bioavailability with recently synthesized lipophilic Mn porphyrins would be required to rule out the therapeutic potential for metalloporphyrins in TBI.
Spinal Cord Injury
Numerous studies of redox-active metalloporphyrins have been reported in experimental spinal cord injury (SCI). This is because numerous targets for metalloporphyrin efficacy (MMP-9 and NF-κB activation, apoptosis, PARP activation, etc.) are present in acutely injured spinal cord. Most studies have used MnTBAP. None have defined MnTBAP purity in their preparation. While this was later defined as a potential confound in the definition of molecular mechanisms of action (159), early studies employed MnTBAP as tools to examine the role of oxidative/nitrosative stress in SCI, most likely due to ready commercial-grade availability.
Nitric oxide has long been recognized as participating in spinal cord nociceptive systems (72) and responses to injury (40). Use of metalloporphyrins to define interactions between nitric oxide and superoxide as mediators of secondary injury in SCI first appeared in 2000 (117). Rats were subjected to spinal cord impact. The time courses of nitric oxide, superoxide, and peroxynitrite formation were characterized over the first 2–3 h postimpact. Concentrations of all three ROS/RNS increased, and these increases could be attenuated by perfusion of the spinal cord with SOD and the NOS inhibitor nitro-L-arginine. It was concluded that superoxide and nitric oxide reacted to form peroxynitrite, leading to further tissue damage. This was supported by immunohistochemical measurement of nitrotyrosine positive cells over 72 h, which showed peak nitrotyrosine staining at 12 h postimpact. Rats treated with a combination of intraperitoneal nitro-L-arginine (1 mg/kg) and MnTBAP (10 mg/kg) exhibited a 63% decrease in nitrotyrosine-positive spinal cord cells in both gray and white matter. Similarly, Luo et al. (120) used flow cytometry with hydroethidium as a superoxide indicator in spinal cord tissue explanted from guinea pigs subjected to spinal cord compression. Exposure of tissue to high-dose (100 μM) MnTBAP immediately after compression partially inhibited superoxide formation. Leski et al. (109) observed increased DNA and protein oxidation after spinal cord impact in rats. This was linked to degradation of neurofilament protein, a marker of axonal injury. Pretreatment with the combination of nitro-L-arginine and MnTBAP markedly decreased the number of injured axons. Although neurologic function was not assessed, inhibition of axonal injury by the combination of nitro-L-arginine and MnTBAP was taken as evidence that ROS/RNS was the causal mechanism of axonal loss.
Direct evidence that MnTBAP decreases peroxynitrite-induced SCI was provided when the peroxynitrite donor, 3-morpholinosydnonimine was microdialyzed into rat spinal cord (8). This caused a marked increase in protein nitration. Microdialysis of an extremely high concentration of MnTBAP (2.5 mM) into tissue concurrent with 3-morpholinosydnonimine completely blocked protein nitration, which was consistent with scavenging of peroxynitrite. Liu et al. (116) used the same system and observed decreased hydroxynonenal (4-HNE) and myeloperoxidase by MnTBAP. Bao et al. (9) complimented this work by microdialyzing Fenton's reagents (H2O2 and Fe2+) into rat spinal cord. Formation of hydroxyl radical and neuron loss was tracked. Fenton's reagents increased both parameters by two to four-fold compared with the control. These increases were nearly completely inhibited when 500 μM MnTBAP was included in the dialysate. Collectively, these studies provided initial evidence of the broad-spectrum scavenging properties of Mn porphyrins in SCI. Further, Ling et al. (114) suggested a link between MnTBAP efficacy and inhibition of apoptosis by measuring a decreased number of terminal deoxynucleotidyl transferase-mediated dUTP nick-end-labeling (TUNEL)-positive cells after spinal cord impact, but specific markers of apoptosis (e.g., caspase-3) were not measured. Despite providing important insights into the mechanisms of action for Mn porphyrins in SCI, the therapeutic significance of these collective observations was not defined, as neurologic function was not assessed in any of these studies.
Extensive clinical research has explored the use of methylprednisolone with the intent of attenuating oxidative and inflammatory responses to injury. While there was initial optimism for methylprednisolone, this has failed to translate to routine practice, and adverse effects may outweigh any benefits (27). Thus, clinical use of methylprednisolone is not currently recommended (79). However, methylprednisolone has been the standard against which other pharmacologic interventions are commonly compared in experimental investigation of new drug efficacy for SCI.
Three laboratory studies have directly compared Mn porphyrins with methylprednisolone. Sheng et al. (181) examined mice subjected to spinal cord compression injury. Treatment with intravenous MnTDE-2-ImP (0.5 mg/kg bolus followed by 1 mg/kg/h), methylprednisolone (30 mg/kg bolus followed by 5.4 mg/kg/h), or vehicle was begun 5 min after injury and continued for 24 h. Neurologic and histologic outcomes were measured over a 21-day recovery interval. Neither drug provided benefit. Since other work had shown poor BBB penetration of MnTDE-2-ImP (179), a second experiment was performed with a single MnTDE-2-ImP intrathecal injection (2.5 or 5.0 μg/kg) or vehicle. Neurologic and histologic outcomes at 21 days were dose dependently improved, demonstrating Mn porphyrin efficacy superior to methylprednisolone, but dependence of Mn porphyrin efficacy on the route of administration was consistent with the low lipophilicity of MnTDE-2-ImP. Hachmeister et al. (71) provided mechanistic evidence of superiority of Mn porphyrins compared with methylprednisolone. Rats were subjected to spinal cord impact and treated with intrathecal MnTBAP before (1 mg/kg) or 4 h postimpact (2.5 mg/kg) or intravenous methylprednisolone 4 h after impact (30 mg/kg followed by 5.4 mg/kg intravenously for 5.5 h). Both pre- and post-treatment with MnTBAP decreased impact-induced membrane lipid peroxidation, formation of 4-HNE, and neuron loss. Methylprednisolone offered no benefit. Most recently, Liu et al. (118) examined rats subjected to spinal cord impact injury. In the biochemical experiments, rats were nonrandomly assigned to intrathecal MnTBAP (4 mg/kg), or methylprednisolone (9.2 mg/kg) infusion before impact. The source and purity of MnTBAP was not stated. Both drugs prevented increased SCI-induced H2O2 tissue concentrations. In contrast, only MnTBAP inhibited SCI-induced superoxide tissue concentrations. MnTBAP was also found to inhibit protein nitration. An outcome study was then performed. Rats were assigned to receive intravenous vehicle, MnTBAP (10 mg/kg IP), or methylprednisolone (30 mg/kg IV) beginning 4 h postimpact. The drug doses (50% of first dose) were repeated 2 h later. Rats underwent neurologic assessment (BBB score and inclined plane) over a 10-week recovery interval. Both methylprednisolone and MnTBAP improved function, although the efficacy of MnTBAP exceeded that of methylprednisolone. Cumulatively, these studies indicate consistent efficacy of Mn porphyrins in improving neurologic function that exceeds that of methylprednisolone. However, Mn porphyrin efficacy was highly dependent on bioavailability, and none of the lipophilic Mn porphyrins have been studied in the context of SCI.
Yu et al. (223) studied MnTBAP in the context of promoting neural stem cell grafts. It was reasoned that most donor stem cells fail to survive because of oxidative stress associated with inflammation. Formation of nitric oxide and peroxynitrite was independently stimulated in human fetal brain progenitor cell cultures. Peroxynitrite, but not nitric oxide, induced apoptosis. Co-treatment with MnTBAP inhibited both peroxynitrite formation and apoptotic cell death. The next experiment employed a scaffold seeded with the stem cells. This system was stressed in vitro to determine whether MnTBAP could sustain cell viability in the presence of a peroxynitrite donor. Efficacy was confirmed. Rats were then subjected to spinal cord hemisection, and the scaffold was implanted containing (or not containing) MnTBAP. Without MnTBAP, the majority of stem cells succumbed to caspase-3 activation and apoptosis over 24 h. MnTBAP impeded caspase-3 activation and mitigated cell death by 40%–70%. Neurologic function was not assessed.
Fe porphyrins have also been evaluated in SCI. Studied as a peroxynitrite decomposition catalyst (91, 136), intraperitoneal FeTSPP (10–100 mg/kg) was given to mice at 1 and 6 h after spinal cord compression injury and compared with dexamethasone (1 mg/kg) (68). FeTSPP produced a dose-dependent improvement in 10-day functional outcome. This was associated with decreased neutrophil infiltration, nitrotyrosine formation, lipid peroxidation, nuclear NF-κB translocation, and cytokine expression. However, dexamethasone was as effective as the most efficacious FeTSPP dose. Thus, in this experiment, while a mechanistic pathway for FeTSPP-induced improvement in outcome was delineated, FeTSPP failed to provide benefit superior to readily available corticosteroid therapy. One limitation in concluding that Fe porphyrins may have no advantage compared with corticosteroids was the use of only two FeTSPP doses (at 1 and 6 h postinjury). Other work with Mn porphyrins in stroke has shown that sustained treatment is necessary to produce enduring functional benefit (182). Whether sustained treatment with FeTSPP over the 10-day recovery interval would have led to a different conclusion remains to be determined. This may, in part, have been answered by subsequent experiments from the same group (67), in which the purported peroxynitrite decomposition catalyst WW-85 (30–300 μg/kg intraperitoneal) was given to mice for 10 days post-SCI. A dose-dependent improvement in neurologic function was observed in WW-85-treated mice. This was associated with depressed markers of oxidative stress and apoptosis. However, the WW-85 structure has not been provided to confirm it to be a metalloporphyrin (175).
Cumulatively, extensive data support a role for metalloporphyrins both as experimental tools to investigate mechanisms of secondary injury associated with SCI and as potential therapeutic agents to either abate this secondary injury or improve success in stem cell transplants intended to restore spinal cord function. The mechanisms of action associated with metalloporphyrins in SCI are similar to those identified in other forms of CNS injury. Limited bioavailability of evaluated compounds presented a major hurdle to future development, although this plausibly could be overcome with recent developments in enhancing metalloporphyrin lipophilicity. Three out of four studies demonstrated superiority of metalloporphyrins compared with corticosteroids in improving functional outcome. Further study of the robustness of sustained metalloporphyrin dosing regimens in the context of long-term functional outcome analyses is necessary to justify consideration for clinical development.
Parkinson's Disease
Although multifactorial in etiology, numerous experimental models of Parkinson's disease have identified a role for oxidative stress in its pathogenesis (10, 26, 142, 184). Post mortem examinations of Parkinson's disease patients have confirmed these findings (3, 6, 49, 224). Most models induce oxidative stress to generate selective dopaminergic neurodegeneration (25). In 1996, Patel et al. (150) provided the first evidence that metalloporphyrins could have relevance to Parkinson's disease. Cultured neurons were exposed to paraquat, a superoxide generator. MnTBAP dose dependently inhibited paraquat-induced aconitase inactivation and lactate dehydrogenase release. The structural analog Zn(II) meso-tetrakis(4-carboxylatophenyl)porphyrin [ZnTBAP (46)] was markedly less potent than MnTBAP. Neither MnTBAP nor ZnTBAP are SOD mimics, and ZnTBAP is not redox active. The fact that both exhibit some efficacy suggests that mechanisms of action other than superoxide scavenging are involved. Indeed, Mn porphyrin (MnTDE-2-ImP) protection against paraquat toxicity was later linked to inhibition of apoptotic pathways in a dopaminergic neuronal cell line (36).
Kalivendi et al. (92) also examined cultured neurons. Superoxide production, capase-3 activation, TUNEL staining, and lactate dehydrogenase release were stimulated by 1-methyl-4-phenylpyridinium (MPP
In vivo Mn porphyrin efficacy has been reported in at least two laboratory studies. Liang et al. (111) subjected mice to MPTP neurotoxicity. A novel lipophilic Mn porphyrin Mn(III) 5,15-bis(methoxycarbonyl)-10,20-bis(trifluoromethyl)porphyrin (AEOL11207) was synthesized. Proof of bioavailability and BBB penetration was provided by formal pharmacokinetic analysis. However, a price was paid for the increased lipophilicity. Based on units of SOD activity per mg, the early-generation MnTDE-2-PyP activity was approximately threefold greater than native SOD; while AEOL11207 SOD activity was only 0.8% that of native SOD. AEOL11207 has electron-withdrawing trifluoromethyl groups in meso positions, but bears no charge for electrostatic facilitation of O2 ·− dismutation, accounting for the decrease in SOD-like activity (158). Despite the profound decrease in SOD activity, a clinically relevant oral route of administration for AEOL11207 markedly decreased MPTP-induced lipid peroxidation, nitrotyrosine formation, and depletion of glutathione and striatal dopamine. While this study provides encouraging evidence of in vivo Mn porphyrin efficacy in a clinically relevant Parkinson's disease model, therapeutic implications are limited, because behavior was not assessed. However, Chen et al. (35), using a different Mn porphyrin (MnTDE-2-ImP) in a mouse paraquat model, reported improved function in two different neurologic tests, portending potential therapeutic value from this class of molecules.
The AEOL11207 study raises an important question in that biochemical efficacy was achieved despite a low SOD activity. Although lipophilicity for AEOL11207 was not reported, it may be presumed that this accounted for its documented BBB penetration (204). This, again, suggests that SOD activity may not be the only important characteristic for predicting Mn porphyrin efficacy in CNS disorders. This is consistent with the lack of dependence on SOD activity levels reported for the potency of other Mn porphyrins as inhibitors of staurosporine-induced apoptosis in neuronal cultures (196, 197). Thus, mechanisms of action for Mn porphyrins, while closely associated with inhibition of oxidative stress, may depend on properties not yet appreciated, which may contribute to the consistently reported neuroprotective properties of this class of drugs across a range of CNS disorders.
Finally, Fe porphyrins have been examined in methamphetamine-induced dopaminergic neurotoxicity. Imam et al. (82) subjected mice to intraperitoneal methamphetamine with or without FeTM-4-PyP. Methamphetamines caused increased body temperature, formation of striatal 3-nitrotyrosine, and depletion of dopamine and its metabolites. FeTM-4-PyP largely inhibited these responses, but functional responses were not assessed.
Collectively, and to the extent that paraquat, MPP
Epilepsy
Primary oxidative stress may contribute to epileptogenesis, and seizure-induced oxidative stress may lead to further tissue damage (1, 37, 90, 110, 210). Treatment with anti-epiletic drugs may not be sufficient to arrest oxidative stress (131). To the extent that metalloporphyrin efficacy in other CNS disorders may be attributed to attenuation of oxidative stress, it can be hypothesized that metalloporphyrins could also be of benefit in epilepsy (112, 183). No clinical studies have been conducted, and preclinical work is limited. Two studies appeared nearly simultaneously reporting Mn porphyrin efficacy in experimental epilepsy models (63, 112). Folbergrova et al. (63) infused 12-day-old rats intracerebroventricularly with DL-homocysteic acid to induce seizures with characteristics of status epilepticus. Anatomically generalized superoxide production, detected by hydroethidine, was increased by 50%–60% at 60 min after seizure onset. Diffuse neuronal necrosis, absent at 60 min, was evident several hours later. Treatment with compounds previously proved anticonvulsant in immature brain [e.g., the NMDA-receptor antagonist AP7 (62)] partially blocked increased superoxide production and histologic damage. Both intraperitoneal Tempol and MnTM-4-PyP inhibited superoxide production and partially protected tissue from histologic damage. The effect of MnTM-4-PyP, at the doses studied, was greater than Tempol. This work provides evidence of seizure-generated superoxide production preceding cell death and offers a window of opportunity for Mn porphyrins to limit irreversible injury. Further work examining prolonged recovery intervals and functional outcome would be important in defining potential clinical relevance.
Liang et al. (112) studied mitochondrial oxidative stress using homozygous MnSOD-deficient mice. This mutation is uniformly fatal shortly after birth with anecdotally reported tonic–clonic seizures beginning in the 2nd to 3rd week of life (122). Using video-electroencephalographic monitoring, Liang et al. (112) confirmed presence of frequent spontaneous motor seizures. Subcutaneous AEOL11207 (5 mg/kg/day), beginning on day 5, decreased seizure frequency and duration, but not severity. AEOL11207 increased lifespan by ∼5 days. This improved survival was associated with decreased inactivation of mitochondrial aconitase and 3-nitrotyrosine formation, and preserved ATP.
As mechanisms of epileptogenesis become better defined, the role of oxidative stress as a pathomechanism has become more prominent. The aforementioned studies indicate a potential therapeutic role of metalloporphyrins in protecting tissue against seizure-induced damage and potentially epileptogenesis, with current evidence implicating efficacy derived from decreased oxidative stress. This is fertile ground for advanced inquiry.
Alzheimer's Disease
Oxidative stress (172), microglial activation (173), β-amyloid (Aβ) aggregation (195), and tau phosphorylation (133) have been implicated in Alzheimer's disease (AD) pathogenesis. Hence, metalloporphyrins have also been studied in AD models, albeit most often as tools to define AD mechanisms.
Mitochondrial dysfunction and superoxide production are of central relevance to the development of oxidative stress in AD (7, 123). Early work with homozygous MnSOD-deficient mice implicated brain mitochondrial MnSOD in protection against neurodegenerative disorders. Treatment of neonatal MnSOD−/− mice with MnTBAP (5 mg/kg/day intraperitoneal) served to double survival from this otherwise lethal mutation (e.g., MnTBAP inhibited development of dilated cardiomyopathy) (130). However, since MnTBAP has poor BBB penetration, brain pathogenesis progressed unabated. MnTBAP-treated mice developed progressive gait abnormalities, tremor, and loss of righting reflex. Histologic examination revealed spongiform abnormalities in the cortex and brainstem.
Development of lipophilic Mn porphyrins now allows a study of metalloporphyrins in MnSOD−/− mouse brain (112). Mice treated with AEOL11207 (5 mg/kg/day) provided evidence of brain mitochondrial penetration by AEOL11207, decreased mitochondrial oxidative stress, and a marked decrease in seizure activity. While lifespan remained only double that of untreated controls, similar to the benefit reported for MnTBAP (130), a histologic examination showed the familiar spongiform changes in untreated animals. AEOL11207 decreased the severity of these changes. It is not clear, however, why lifespan was not appreciably increased by the lipophilic Mn porphyrin. It could be postulated that the partial arrest of neurodegeneration was not sufficient to enable viability. Whether this indicates the limit of metalloporphyrins in this model and potential relevance to AD is not known. AEOL11207 has low SOD activity, but may offer fair ability to reduce ONOO−. Whether greater efficacy could be obtained from a lipophilic metalloporphyrin with greater SOD activity can only be speculated, but remains interesting, as the effect of AEOL11207 was dose dependent and a ceiling effect was not reported. This is consistent with the report of Melov et al. (129), who reported doubling of the effect of anionic MnTBAP on MnSOD−/− murine lifespan when mice were treated instead with a neutral salen manganese complex offering higher lipophilicity, more appropriate electrostatics, and fair SOD activity. Most ROS and RNS are anionic electron-rich species, and cationic electron-deficient Mn porphyrins would unambiguously enable better scavenging of ROS/RNS (20).
Others have used MnTBAP to study abnormalities in cerebrovascular reactivity associated with AD. In addition to its direct neurotoxic effects, Aβ impairs cerebral circulation. Overexpression of Cu,Zn SOD or topical SOD application can inhibit this impairment (80), thereby invoking superoxide regulation as a mechanism for Aβ-induced dysfunction. Consistent with this, Niwa et al. (143) demonstrated in vivo that Aβ−related cerebral vasoconstriction can also be inhibited by MnTBAP. These results, however, are difficult to interpret, because pure MnTBAP does not offer SOD activity, but instead may act as a peroxynitrite decomposition catalyst (11, 15). The MnTBAP purity and source were not stated.
Finally, Sompol et al. (186) studied primary neuronal cultures derived from wild-type, amyloid precursor protein (APP)/presenilin 1 (PS1) knock-in mice. The knock-in genotype was associated with oxidative damage (e.g., 4-HNE, and 3-nitrotyrosine), particularly as the neurons matured in vitro. Mature neurons had decreased MnSOD activity and mitochondrial respiratory activity and increased vulnerability to Aβ. Overexpression of MnSOD or MnTE-2-PyP (100–1000 pg/ml) abated these responses. This work implicates a critical role of MnSOD in responses of vulnerable neurons to Aβ and a potential therapeutic role for metalloporphyrins in AD pathogenesis.
Investigation of the potential therapeutic effects of Mn porphyrins in AD has otherwise been limited. To the extent that AD is associated with mitochondrial oxidative stress, data from MnSOD−/− mice predict value from further investigation, as does evidence for a potential beneficial interaction between Mn porphyrin and Aβ-induced vasoconstriction and vulnerability to Aβ.
Pain
Metalloporphyrins have been extensively used in pain research as purported selective peroxynitrite decomposition catalysts. Although most metalloporphyrins have since been shown to be less selective than originally thought, also having the capacity to react with numerous other ROS/RNS species, and transcription factors, insights gained from the use of metalloporphyrins have been valuable in pain research.
Central sensitization-induced hyperexcitability to acute pain may confer sustained hyperalgesia and allodynia. The etiology of chronic pain is multi-factorial (85), but ROS/RNS contributions to the modulation of molecular elements of nociception are becoming better understood and believed important (28, 115, 192, 194). The implication of superoxide and peroxynitrite in neuropathic pain is not novel to the discussion of metalloporphyrins (4, 211). Other redox-active compounds have been found effective in ameliorating hyperalgesia (97). Metalloporphyrins have proved efficacious in a variety of preclinical models, and most work has focused on Fe porphyrins because of their purported selective decomposition of ONOO−.
In diabetic neuropathy, Fe(III) meso-tetrakis(N-(1–2(-2(-2-methoxyethoxy)ethoxy)ethyl)pyridinium-2-yl)porphyrin (FP-15) (155, 193) partially decreased hyperalgesia in mice, and this was associated with decreased nitrotyrosine and PARP immunofluorescence in the sciatic nerve (53, 144). Subsequent work compared structurally diverse Fe porphyrins (FP-15 and FeTMSP (Fe(III) meso-tetrakis(2,4,6-trimethyl-3,5-disulfonatophenyl)porphyrin)) and found persistence of improved function and decreased nitro-oxidative stress independent of the Fe porphyrin employed (207). Similar effects of vastly different redox-active and charged (pentacationic vs. heptaanionic) Fe porphyrins, if reliable, suggest that the effects may be due to released Fe, as suggested by Tovmasyan et al. (201).
Negi et al. (141) similarly demonstrated attenuation of streptozotocin-diabetic neuropathy in rats observed over an 8-week interval when treatment with FeTM-4-PyP, or the PARP inhibitor 4-amino-1,8-napthalimide (4-ANI) was begun 6 weeks after a streptozotocin injection. Both interventions decreased mechanical allodynia, malondyaldehyde and peroxynitrite formation, and increased nicotinamide adenine dinucleotide concentrations. 4-ANI also decreased thermal hyperalgesia. However, combined treatment with FeTM-4-PyP and 4-ANI had a larger therapeutic effect than either treatment alone. While this remains supportive in defining peroxynitrite as a causative agent in diabetic neuropathy and the contention that this could be optimally abated by treatment with a combination of peroxynitrite decomposition catalysts and PARP inhibition, neither FeTM-4-PyP nor 4-ANI was subjected to a dose-escalation examination which would enable confirmation that a peak effect of either compound was employed, bringing into question the necessarily greater efficacy of combination treatment. This study offers strength, because in contrast to most or all studies examining the efficacy of metalloporphyrins in ameliorating the evolution of chronic pain, the observation interval of 8 weeks greatly exceeded the more typical interval of several days used by other investigators.
Carrageenan is used in a variety of inflammatory and injury-induced pain models, because it induces activation of protein kinase B (Akt) and its upstream activator phosphatidylinositol 3-kinase in dorsal horn neurons (38, 39). Carrageenan also induces peroxynitrite formation at the peripheral injection site (168). In rats subjected to paw carrageenan injection, Fe porphyrins (FeTMSP or FeTM-4-PyP) decreased paw edema and markers of peroxynitrite formation (169). Yeo et al. (220) used an intradermal carrageenan injection to study the role of peroxynitrite in orofacial pain. Intracerebroventricular FeTSPP had no effect on responses to von Frey fiber stimulation in control rats, indicating that the Fe porphyrin has no intrinsic analgesic effect. However, intracerebroventricular FeTSPP dose dependently decreased allodynic responses in carrageenan-injected rats. This was speculated to be associated with inhibition of microglial activation, but that was not measured.
Ndengele et al. (140) provided further insights into Fe porphyrin mechanism of action in pain therapeutics. Intraplantar injection of O2 ·− (donor not stated) produced an intense local inflammatory response that was dose dependently blocked by co-treatment with either L-NAME or Fe-TM-4-PyP, implicating nitric oxide as both a source of local peroxynitrite production and an initiator of the inflammatory response. Intraplantar ONOO− injection (donor not stated) dose dependently produced the same inflammatory response, which was associated with activated NF-κB, inducible COX-2 expression, and prostaglandin E2 (PGE2) formation. Hyperalgesia was inhibited with selective and nonselective COX-2 inhibitors or anti-PGE2 antibody. To link this to previous observations made with carrageenan injection, rats were treated with FeTM-4-PyP, indomethacin (a nonselective cyclooxygenase inhibitor), the selective COX-2 inhibitor NS398, or combinations of these compounds. While FeTM-4-PyP alone was effective in blocking carrageenan-induced hyperalgesia, paw edema, and PGE2 release, it also synergized with indomethacin and NS398; that is, doses of both the COX-2 inhibitors and Fe porphyrin could be decreased 10-fold below the doses required for either intervention alone to achieve maximal benefit. From a clinical perspective, this interaction could allow low doses of combined drugs to avoid adverse effects while still allowing maximal efficacy to be achieved.
Doyle et al. (52) asked similar questions regarding the respective roles of nitric oxide, superoxide, and peroxynitrite in the development of thermal hyperalgesia. It was postulated that hyperalgesia is mediated, at least in part, through induced sphingosine-1-phosphate (S1P) synthesis. S1P, acting through G-protein-coupled S1P receptors present in neurons and glia, is a known mediator of inflammation and pain perception (189, 215). S1P receptor stimulation activates both nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and NOS, thereby generating both superoxide and nitric oxide. Rats were subjected to a paw injection of S1P receptor agonists or antagonists. S1P agonists induced a dose- and time-dependent thermal hyperalgesia onset. This was inhibited by co-treatment with an S1P antagonist. Apocynin (an NADPH oxidase inhibitor) and L-NAME independently caused partial inhibition of S1P-induced hyperalgesia. FeTM-4-PyP and MnTE-2-PyP also caused a dose-dependent inhibition of S1P-induced hyperalgesia. Although this study provides strong evidence for a redox-active mechanism in S1P signaling, it does not fully isolate the pathway to peroxynitrite. MnTE-2-PyP is known to catalytically reduce superoxide, peroxynitrite, and nitric oxide when assayed in isolated in vitro systems (187, 188). Although reactivity of FeTM-4-PyP toward nitric oxide has not yet been assessed, it is likely similar to the reactivity of analogous Mn porphyrins, as FeTM-4-PyP has similar SOD activity (157). There is some evidence that superoxide pain signaling is independent from that of nitric oxide (99).
While metalloporphyrins have provided insights into a redox-regulated mechanism for the development of hyperalgesia, such studies will continue to be limited by the pleiotropic redox activity of both Fe and Mn porphyrins (176). However, the consistent amelioration of objective measures of neuropathic pain in diverse preclinical animal models offers major promise of potential clinical application and a mechanism of action that could either be sufficient alone, or in combination with other therapeutics to treat neuropathic pain.
One limitation to the development of most metalloporphyrins for clinical application is bioavailability. The inherent 4+ charges of four electron withdrawing N-alkylpyridinium groups in the meso or ortho positions of the porphyrin ring not only render these molecules their high potency, but may also limit BBB penetration and any potential central effect. This has to be assessed, as cationic charges may attract Mn porphyrins toward phosphates of lipid membranes. The proof is in the high ability of cationic Mn porphyrins to enter mitochondria and nucleus (20a). Further, this does not necessarily abrogate opportunity for such molecules to function in peripheral tissue and chronic conditions; BBB penetration over time may be sufficient to deliver efficacy. Moreover, recent efforts to optimize “drugability” of metalloporphyrins through side-chain substitutions for CNS application has offered an improved opportunity for use in chronic pain. Rausaria et al. (157) have synthesized a series of novel lipophilic Mn porphyrins, Mn(III) hexahydro-29H,31H tetrabenzo[b-,g-,l-,q]porphine (MnTCHP). At least one of these molecules has high oral absorption. Perhaps, of greatest scientific value, these substitutions have entirely depleted any SOD-like activity, while peroxynitrite decomposition activity is retained. Hence, specific investigation of peroxynitrite activity in isolation of superoxide signaling may be performed. Similar to AEOL11207 discussed earlier in the context of Parkinson's disease and epilepsy (111, 112), MnTCHP retains therapeutic activity in the absence of SOD activity. In both carrageenan-induced hyperalgesia and sciatic-nerve constriction-induced neuropathic pain, MnTCHP provided dose-dependent efficacy (157).
Thus, we are presented with substantive data that peroxynitrite formation plays a prominent role in the development of delayed diabetic neuropathy and also in acute models of neuropathic pain. This may be attributable to activation of upstream signaling cascades, including those governed by NF-κB and S1P. Metalloporphyrins with documented activity as peroxynitrite decomposition catalysts abrogate these hyperalgesia states. Early-generation metalloporphyrins were nonselective peroxynitrite decomposition catalysts, also having high affinity for superoxide. However, metalloporphyrins synthesized to increase lipophilicity and BBB penetration to treat neuropathic pain sacrificed SOD activity, yet retained efficacy. It will be of interest to determine whether this loss of SOD activity has compromised efficacy, which might be determined by direct comparison with other lipophilic metalloporphyrins with retained SOD activity [e.g., MnTnBuOE-2-PyP (156)]. However, a direct comparison would be encumbered by multiple differences among compounds; no two molecules differ with regard to only one parameter.
Opioid Tolerance
Although multi-modal approaches to chronic pain are employed (101), opioids remain central to the management of this disorder. While opioids are initially effective in most patients, tolerance, addiction, and adverse effects limit sustained efficacy and safety. A molecular investigation of mechanisms responsible for opioid tolerance has identified desensitization of μ-opioid G-protein receptor signaling by β-arrestin-2 (29, 45). Despite this, little breakthrough in clinical therapeutics has been appreciated.
Peroxynitrite also has been implicated in opioid tolerance (50, 51, 138, 149, 167). Although no link has been established between peroxynitrite and β-arrestin-2-mediated μ-opioid G-protein receptor desensitization, interactions between β-arrestin-2 and nitric oxide, including S-nitrosylation, have been suggested in other disorders (206).
Morphine induces increased superoxide production by NADPH oxidase 1 (51, 81). This superoxide may serve to accelerate guanosine triphosphate hydrolysis in the G-protein-coupled receptor Gα subunit, thereby terminating morphine receptor signaling. Morphine also increases nitric oxide production (124, 199). A simultaneous increase in both superoxide and nitric oxide, therefore, can be predicted to result in increased peroxynitrite and an inflammatory response, which, in turn, contributes to evolution of morphine tolerance (167).
Morphine induces peroxynitrite formation in both the dorsal lumbar spinal cord and brain (50, 138). This can be inhibited by either L-NAME or a spectrum of metalloporphyrins, and this has been associated with inhibition of morphine tolerance in mice (50, 138). However, opioid tolerance is more complex than modulation of G-protein-coupled receptor signaling, because chronic morphine exposure is also associated with PARP activation and loss of MnSOD enzymatic activity (50).
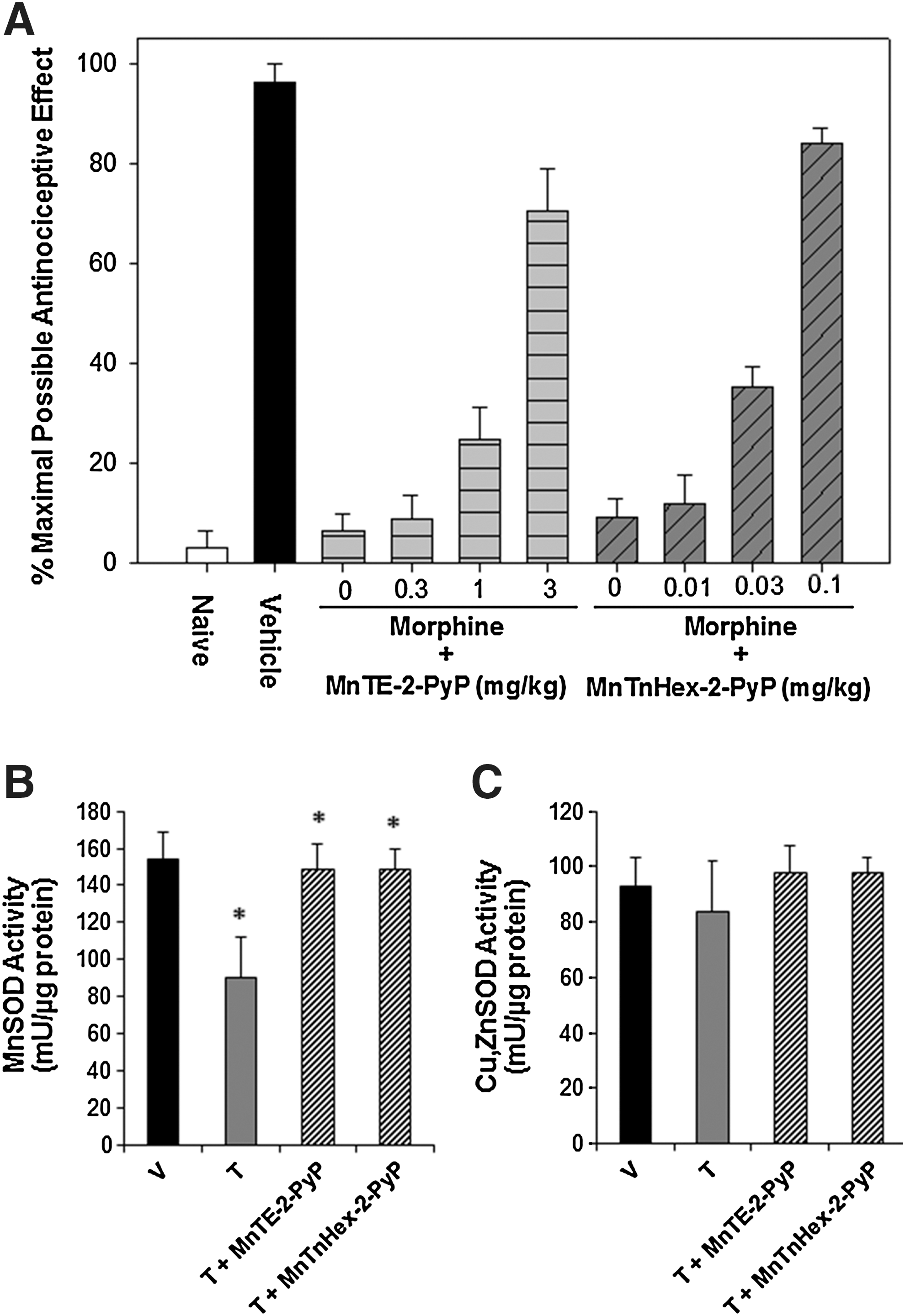
With regard to therapeutic potential from metalloporphyrins, there is limited data of substantive clinical relevance to date. Most studies evaluated morphine exposure intervals of 5–7 days, while clinical exposure can extend to months or years. Thus, the robustness of metalloporphyrin inhibition of morphine tolerance over time is unknown, particularly in the context of the various clinical conditions for which opioid therapy is indicated. What is known is that metalloporphyrin lipophilicity plays a critical role in efficacy (16). Metalloporphyrins with greater lipophilicity and presumably greater BBB penetration offer greatest efficacy in experimental opioid tolerance and at significantly lower doses than are required for hydrophilic metalloporphyrins (Fig. 7).
Mechanisms and Limitations
From research in CNS disorders and other disease states, the pharmacologic properties of an abundant portfolio of metalloporphyrins have emerged, and a current synopsis of mechanisms of action is provided (Fig. 8). However, it is likely that mechanisms are far more complex than are currently appreciated.

Although the etiology and pathogenesis differ among the CNS disorders outlined earlier, metalloporphyrins as a class have a highly consistent record of efficacy in preclinical models emulating these diseases. These diseases were chosen as potential candidate systems for the study of metalloporphyrins, because they share, at least in part, a component of oxidative/nitrosative stress that contributes to disease progression which is typically attributed to reactions of superoxide, nitric oxide, and peroxynitrite. Hence, the study of metalloporphyrins appeared to be rational. Decreased peroxynitrite formation, either through superoxide and nitric oxide scavenging or through increased peroxynitrite decomposition, has been reported for ALS (44), ischemic stroke (198), acute SCI (223), Parkinson's disease (36), epilepsy (63), AD (186), diabetic neuropathy (207), and opioid tolerance (138).
Although metalloporphyrins were originally synthesized as SOD mimics (58), we now know that the magnitude of SOD activity varies markedly among the various efficacious Mn porphyrins. Thus, SOD activity alone cannot account for their full range of biochemical effects in vivo. It is likely that mechanisms accounting for metalloporphyrin efficacy are far more complex than simple superoxide scavenging, based on several observations. The wide range of activities of different metalloporphyrins may also be a consequence of various degrees of their cellular distribution.
It is not certain whether it is stoichiometrically reasonable to expect nanomolar or even micromolar tissue metalloporphyrin concentrations to add appreciably to the already abundant enzymatic and scavenging anti-oxidant defenses endogenously present, unless SOD enzymes are down-regulated, as is the case with some types of cancer. This is consistent with reports that metalloporphyrin efficacy can be achieved with molecules having negligible SOD activity (111, 112, 196, 197). At the same time, SOD activity cannot be completely dismissed as an important feature, because rate constants in some metalloporphyrins are >107 M −1 s−1, thus very nearly approaching the activity of SOD itself [kca(O2 ·−) ∼109 M −1 s−1].
It has been shown that the therapeutic potential of Mn porphyrins parallels their SOD activity [for details, see Batinic-Haberle et al. (20a)]. The explanation has been offered on thermodynamic and kinetic grounds. The reason lies in the fact that electron-deficient cationic porphyrin-based SOD mimics, which are electrophiles, favor reactions with electron-rich anionic nucleophiles. Such negatively charged nucleophiles are essentially all reactive species: O2 ·−, ONOO−, CO3 ·−, ClO−, protein-S−, GS−, HO2 ·−, and so on. Thus, a more potent SOD mimic is more likely to be a more efficient drug. The reader is encouraged to consult for details other reviews in this Forum on “SOD Therapeutics” and original contributions. Peroxynitrite is a major candidate species that is removed by metalloporphyrins (15), resulting in the prevention of protein nitration and subsequent inactivation [such as is the case with MnSOD (15)]. It is important to note that the peroxynitrite reactivity of metalloporphyrins parallels their superoxide reactivity. Thus, any potent SOD mimic is a potent peroxynitrite scavenger [for details on diverse reactivities of metalloporphyrins, see Batinic-Haberle et al. (20a)]. Presently, there is essentially no compound that is specific for any of the reactive species. At the same time, there is evidence that under certain conditions, MnTBAP can promote nitration of tyrosines of phenyl rings by peroxynitrite, as shown by Ferrer–Sueta et al. (59). MnTBAP is a special case of an “antioxidant,” as it has no SOD activity but has frequently been shown by different authors as possessing therapeutic potential. Other longer-lived species, especially hydrogen peroxide, may also be an important target through mechanisms that are independent of SOD activity. Mn porphyrins with weak SOD activity have been documented to decrease hydrogen peroxide production in mitochondria (31).
Further, it has been reported that metalloporphyrins utilize hydrogen peroxide for glutathionization of thiols in proteins such as NF-κB, thereby inhibiting expression of downstream gene products (87, 88 and [Jaramillo, et al., unpublished data]). This action involves mimicking glutathione peroxidase or cysteine oxidase activities and may be conferred through a specific pro-oxidant property of catalytic Mn porphyrins enabled by local cellular chemistry, which is consistent with earlier reports of inhibited DNA binding of NF-κB by Mn porphyrins, originally speculated to reflect an oxidation of the p50 subunit (205). Inhibition of other redox-regulated transcription factors by Mn porphyrins has been reported (e.g., hypoxia inducible factor-1α [HIF-1α], activating factor-1 [AP-1], and specificity protein-1 [SP-1]), although the mechanisms for this have not been explored (17), particularly in the CNS. In other words, Mn porphyrins may act as either oxidants or reductants depending on their immediate milieu. This is consistent with their enzymatic structure.
Another candidate for metalloporphyrin redox chemistry is nitric oxide. Mn(II) porphyrins bind nitric oxide rapidly in reduced form, which may affect peroxynitrite formation or specific nitric oxide signaling (187). While Mn porphyrins will not, the other potent cyclic polyamine-based SOD mimics (such as frequently explored M40403) may affect signaling by reducing NO to nitroxyl, nitric oxide, and subsequently release its protonated form, HNO which seems to be involved in signaling (133a). This could be either beneficial or detrimental, depending on the stage of disease progression. In the CNS, nitric oxide is closely associated with inflammation in the acute phase, but may promote neurogenesis during recovery (96).
Distribution of metalloporphyrins is thought to, at least in part, partition selectively to the nucleus and mitochondria, and this may be influenced by lipophilicity (20). If so, this could offer higher local concentrations to interact directly with ROS/RNS and disproportionately abate oxidative/nitrosative damage to critical elements of cell survival.
Thus, while a specific metalloporphyrin may exhibit a specific property in an in vitro analysis, it is not necessarily the case that that property accounts for the efficacy demonstrated in preclinical models, even if the same mechanism of action is assessed in vivo. Thus, for example, when a metalloporphyrin is declared to be a peroxynitrite decomposition catalyst, this may or may not represent its critical mechanism of action. This reductionism has served to confuse the delineation of metalloporphyrin mechanisms of action, which has relevance to defining appropriate molecular targets for intervention, creating appropriate dosing paradigms, and confirmation of the scientific basis for the widely demonstrated in vivo efficacy.
Confusion has also been introduced by ubiquitous use of impure compounds. Commercially available MnTBAP preparations suffer from vast impurities (15, 159, 160), and, thus, its mode of action and efficacy need thorough re-investigation with a pure compound. The same holds true for other commercially available metalloporphyrins. Minimally, investigators are encouraged to explicitly define the source and purity of the metalloporphyrin used in their investigations. Finally, confusion is also introduced with different, and sometimes incorrect, abbreviations for the same compound, such as MnTBAP and MnTCPP, and FeTBAP and FeTCCP.
Finally, it has to be noted that in addition to the redox activity of metalloporphyrins, the accumulation within cell and differential distribution within cellular compartments will markedly affect the therapeutic potential of these compounds. For example, the 10-fold increase in lipophilicity of meta MnTE-3-PyP has compensated fully for its 10-fold lower SOD activity (when compared with ortho MnTE-2-PyP) in protecting SOD-deficient E. coli while growing aerobically (102).
Summary
It is curious that no metalloporphyrin has progressed to clinical trials, with the exception of AEOL 10150 used in a single Phase I ALS study (146). No adverse effects were observed that would discourage further development. With regard to the CNS, a major barrier has been the very property which renders optimal potency to most metalloporphyrins, that being the pentacationic electrostatic charge that facilitates approach of anionic reactive species toward the transition metal. This limits BBB penetration and rapid onset of action that are critical in some CNS disorders (e.g., ischemia), but novel lipophilic metalloporphyrins have surmounted this challenge by incorporating side-chain substitutions. This came, at some cost in that SOD activity, was often, but not always decreased. Nevertheless, efficacy persisted, raising further questions regarding precise mechanisms of action.
Most or all CNS disorders have experienced innumerable futile efforts to prove efficacy of purported pharmacologic therapeutics in clinical trials. In particular, antioxidants, most being stoichiometric rather than catalytic and few having proven BBB penetration, have developed poor track records in clinical efforts to treat CNS disorders. Hence, a stigma, by association, has emerged that plagues the development of metalloporphyrins as a class of molecules which offer distinctly different properties as catalytic oxidoreductants. Few studies have directly compared metalloporphyrins with other purported CNS antioxidant therapeutics, and this will be required to convince skeptics that metalloporphyrins constitute a novel class of therapeutics. Novel molecules typically arise from university laboratories and small biotech companies that do not have resources to sufficiently investigate preclinical efficacy to enable rational clinical trial design. At the same time, those making development decisions for therapeutics should be encouraged by the robust nature and remarkable stability of this class of compounds across a diverse array of diseases and models, all of which have clearly documented oxidative/nitrosative stress as common and pivotal contributions to the evolution of CNS disease. Collective efforts will be required to enable metalloporphyrins, as catalytic oxidoreductants, to be subjected to the series of development steps that are necessary to enable a valid analysis of their potential as therapeutics in CNS injury and neurodegeneration.
Footnotes
Acknowledgment
The authors would like to thank Ines Batinic-Haberle, PhD, for critical reading of the manuscript.
Author Disclosure Statement
Duke University is the assignee of patents associated with Mn Porphyrin composition of matter and use. Some patents have been licensed to Biomimetix Pharmaceutical. David S. Warner, MD, is an inventor on a use patent for Mn Porphyrins and holds minor equity in Biomimetix Pharmaceutical. Huaxin Sheng, MD, is an inventor on a use patent for Mn Porphyrins. There are no conflicts of interest for Drs. Chaparro, Sasaki, Izutsu, Pearstein, and Tovmasyan. Neither Biomimetix Pharmaceutical nor Duke University officials participated in the preparation of this article.
Abbreviations Used
Charges on the abbreviated names of metalloporphyrins are omitted throughout text for simplicity, but are shown here and in Figure 2 and ![]() .
.