
Abstract
Introduction
I
Among all these PTMs, redox-mediated PTM is an important category of PTMs that targets the thiol group of cysteine residues. Recently, redox-mediated PTMs are receiving increasing attention, as they are found in both physiological and pathological conditions including oxidative stress. S-glutathionylation is a major redox-mediated thiol modulation mechanism, involving the addition of a glutathione (GSH) moiety to the protein. Oxidative stress, or reactive oxygen species (ROS), facilitates S-glutathionylation. Protein S-glutathionylation has been extensively discussed in many excellent reviews with a variety of different emphases (31, 44, 45, 75, 76, 88, 93, 100, 107). Over the past few years, S-glutathionylation has been increasingly observed in many ion channels, such as voltage-gated calcium channels, ryanodine receptor (RyR), and ATP-sensitive potassium channels (KATP channels), all of which contribute to critical cellular functions (4, 123, 137, 138). In this review, we introduce redox-mediated thiol modulation first and then focus on S-glutathionylation of ion channels. Criteria for testing S-glutathionylation, methods and reagents used in designing experiments are also discussed. Besides S-glutathionylation, other important thiol modifications that contribute to ion channel modulation (e.g., S-nitrosylation, S-palmitoylation, and S-sulfhydration) are briefly discussed, mainly to elaborate on the possible interplay and crosstalk among these thiol modifications. Finally, we consider how the regulation of mechanosensing would be affected by thiol modifications of the mechanosensitive channels, specifically the KATP channel that is functionally inhibited upon S-glutathionylation.
Thiol Groups (−SH) as a Reactive Center in PTM
The thiol groups of protein cysteine residues are susceptible to oxidative modification, through which PTMs may occur. Thiol groups could be modified in response to exogenous stimuli or changes in local redox environment, thus affecting the function of the proteins with such accessible thiol groups (80). In the oxidative stress condition, where the redox balance is shifted toward oxidation, the thiol groups could be modified differently depending upon the accessibility of oxidants or other small molecules. For example, (i) If glutathione (GSH) is accessible to the thiol group, oxidation may lead to S-glutathionylation. (ii) In the presence of abundant nitric oxide (NO), S-nitrosylation may occur. (iii) If two cysteine residues are close to each other, oxidation may cause the formation of a disulfide bond between them; either within the protein or between two separate proteins. (iv) Oxidation of cysteine residues may also result in the sequential formation of cysteine radicals (P-S•), followed by sulfenic (PS-OH), sulfinic (PS-O2H), and eventually sulfonic (PS-O3H) acids. In addition, S-palmitoylation and S-sulfhydration can also modify the thiol groups and attach lipid or hydrogen sulfide (H2S) to the cysteine residues, respectively (Fig. 1).
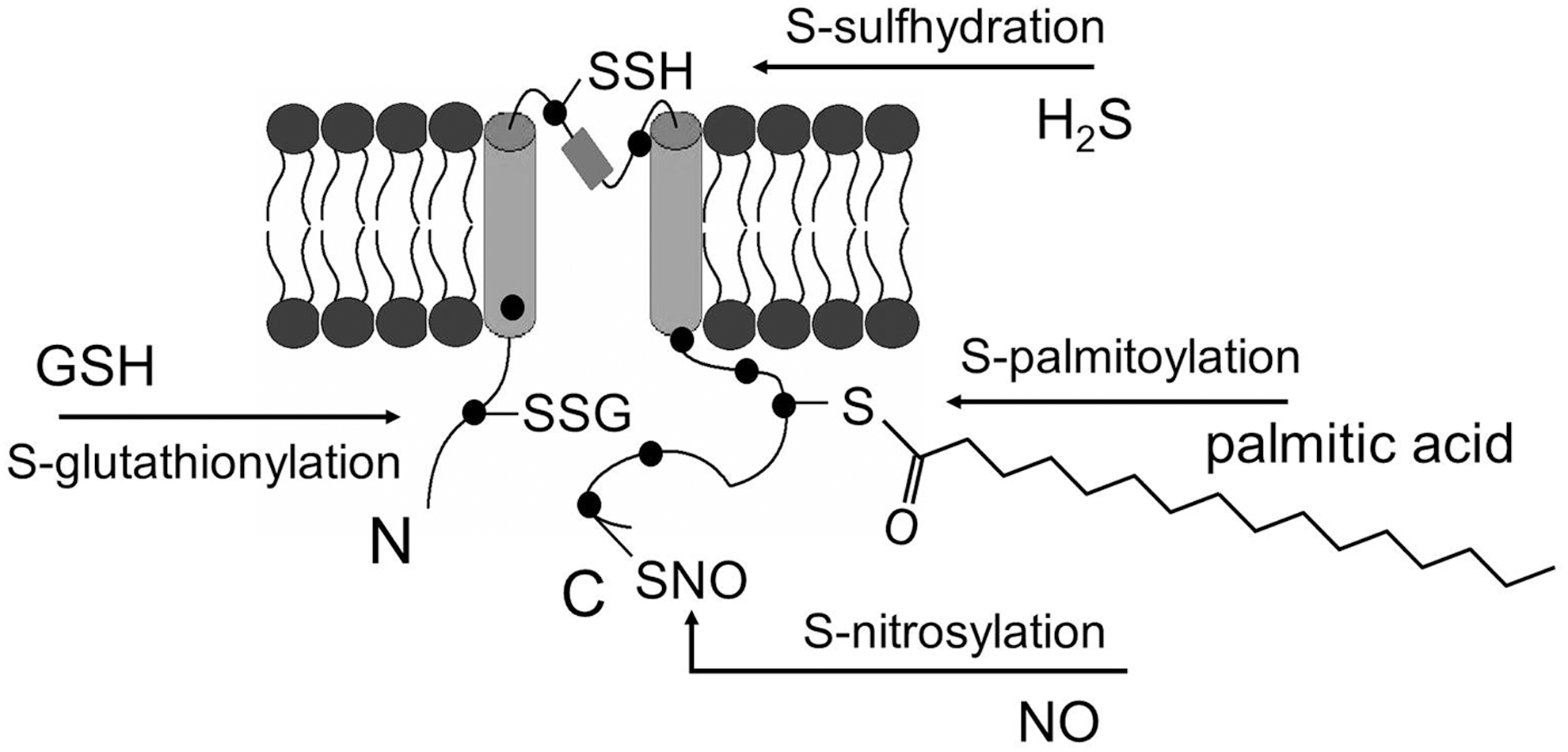
S-Glutathionylation Overview
S-glutathionylation is a PTM of proteins at the cysteine residues by adding a GSH moiety (31). Protein S-glutathionylation may occur in physiological conditions but is generally facilitated by oxidative stress when excessive ROS are present and GSH is locally available to the target protein (33). The ratio of reduced and oxidized glutathione (GSH/GSSG) and deglutathionylation enzymes also contribute to the protein S-glutathionylation. S-glutathionylation has been found in a large number of proteins, affecting a variety of cellular processes (32, 51). Many different thiol modification mechanisms have been associated with oxidative stress but as GSH is naturally abundant within the cells, GSH is likely to serve as a major electron donor, making S-glutathionylation a preferred mechanism for protein modification during oxidative stress (33, 44).
Glutathione
Glutathione (GSH) exists in virtually all cells in the millimolar concentration range. It is the major non-protein thiol compound that acts as an inherent antioxidant, and works together with oxidized glutathione (or alternatively called glutathione disulfide, GSSG) as an intracellular redox buffer (31). GSH is a tri-peptide, containing a cysteine, a glycine, and a glutamate. Cysteine is attached to glycine via a normal peptide bond, whereas the carboxyl group of the glutamate side-chain is bound to the amine group of cysteine via an unusual peptide bond (80). In mammalian cells, the concentration of GSH ranges from ∼1 to 10 mM depending on the cell type.
Deglutathionylation enzymes
Protein deglutathionylation is catalyzed by specific enzymes. Glutaredoxin (Grx) is suggested to be a major deglutathionylation enzyme in mammalian cells (74, 76). The mammalian cytosolic form of Grx (Grx1) is very selective and effective for protein-SSG compared with other forms of disulfides (e.g., S-S disulfide bond, S-nitrosylation, etc.), and thus is considered as a specific deglutathionylating enzyme (44). Moreover, in Grx1 knockout mice, no deglutathionylating activity is detected (74), further supporting its critical role. Besides Grx1, other enzymes have been implicated in deglutathionylation, for example, sulfiredoxin (39) and many other enzymes are known to contribute to antioxidant defense, including thioredoxins, peroxiredoxins (Prx3/5) (41), thioredoxin reductases, glutathione reductase, and so on. (3).
Specificity of protein S-glutathionylation
The specificity of protein S-glutathionylation is still under wide debate (31, 44, 75). In general, it is believed that not all cysteine residues are equally susceptible to S-glutathionylation upon stimuli. However, no consensus motif has been found that is related to S-glutathionylation site (32). Although enzymes have been suggested to promote glutathionylation (44, 51, 124), this process is still considered as a reaction mainly mediated by oxidants. Several contributing factors conferring specificity have been pointed out (32, 51), including the accessibility of the thiol group, the reactivity of the cysteine residues, and the like.
Criteria for Testing S-Glutathionylation
As various thiol modifications arise from cysteine oxidation, multiple approaches should be employed to test S-glutathionylation specifically and to avoid identifying S-glutathionylation that is artificial or has no functional consequences. Several criteria should be taken into consideration: (i) Physiological or pathological relevance of the experimental conditions. Experimental conditions that closely mimic the in vivo physiological or pathophysiological situation such as micromolar concentrations of hydrogen peroxide (H2O2) are better than millimolar concentrations of H2O2. Using enzyme systems such as the xanthine oxidase (XO) or NADPH oxidase (NOX) to produce endogenous ROS are also preferred choices. (ii) Using specific reactive species. Peroxynitrite (ONOO−) is one of the commonly used reactive species that has significant physiological relevance, however, the application of exogenous peroxynitrite could result in both S-nitrosylation and S-glutathionylation, which need to be further distinguished by other methods. (iii) The combination of biochemical and functional assays. Biochemical evidence for protein S-glutathionylation is critical, but to rule out non-functional modifications, assays that assess the functional consequence of S-glutathionylation: for example, channel activities measured by patch-clamp electrophysiology or Ca++ level measured by Ca++ imaging, need to be employed. (iv) Identification of specific cysteine residues responsible for S-glutathionylation. It has been previously shown that for a single protein, some of the cysteine residues are more likely to be S-nitrosylated and others are more likely to be S-glutathionylated (72). Besides, in some proteins only one cysteine residue is modified while for other proteins, the thiol modulation could occur on multiple sites. Moreover, S-glutathionylation may occur on a different protein associated with the target protein being studied. Without identifying the exact residues, it is hard to distinguish all these possibilities and pinpoint the exact modulation mechanism. (v) Reversibility. S-glutathionylation is considered to be a reversible reaction while some other modifications (e.g., the formation of PS-O2H and PS-O3H) are considered irreversible (32). Using a general reducing agent (e.g., dithiothreitol [DTT]) or specific deglutathionylation enzyme (Grx) to demonstrate the reversibility would aid in distinguishing S-glutathionylation from other kinds of modifications (45).
Oxidative Stress and ROS
Oxidative stress is the condition in which excessive ROS production overwhelm the cellular antioxidant system, leading to an imbalance of the cellular redox state (71). S-glutathionylation is a modification that could be facilitated by both exogenous ROS and ROS producing enzymes.
Reactive oxygen species
ROS that are of importance in biological systems include superoxide (O2 •−), hydroxyl radicals (•OH), H2O2, singlet oxygen (1O2), and so on. (94).
H2O2 is well established to participate both in oxidative modifications-mediated damage and in activation of signaling molecules. H2O2 has a relatively long lifespan and can travel in the cytosol or across the membrane to execute its effect (40). H2O2 is a relatively weak oxidizing agent but its cytotoxic effect is potentiated by interaction with a free metal, for example, iron. Because of its stability and relatively long lifespan, H2O2 is often chosen as the primary exogenous source of ROS to perform experiments.
Superoxide is another major endogenous ROS that participates in a limited range of signaling functions (48). Superoxide rapidly reacts with NO or iron-sulfur-containing proteins forming peroxynitrite or hydroxyl radicals. Dismutation of superoxide spontaneously or via catalysis by superoxide dismutase (SOD) is a major source of endogenous H2O2.
Hydroxyl radicals are the most reactive ROS and the strongest oxidizing agent, considered as the most damaging ROS that exist endogenously. However, they have a very short half-life and are in general membrane impermeable.
When ROS and NO are present together, the formation of peroxynitrite (ONOO−) is the dominant reaction as NO and ROS react at an extremely fast rate (8, 116). Therefore, in the presence of ROS, the bioavailability of NO will drastically decrease (20). Moreover, ONOO− is a strong and relatively stable reactive species (86), which could facilitate the formation of both S-nitrosylation and S-glutathionylation.
ROS-producing enzymes
ROS can also be produced by enzymes including NOX and XO. The NOX family generates ROS (mainly O2 •−) through electron transfer from NADPH to molecular oxygen (9). NOX activity is regulated by Ca++, which is in turn affected by the membrane excitability of the cells (117). NOX has been found to regulate ion channel function including TWIK-related acid-sensitive potassium channel (63), T-type Ca++ channel and RyR (115). Interestingly, the activation of endothelial NADPH oxidase is also ion channel (KATP channel)-dependent (143). The closure of the KATP channel leads to membrane depolarization, which triggers NOX activation and further generation of ROS (27). These studies suggest a potentially important interplay between ROS signaling and ion channel regulation.
XO was initially found to be involved in metabolizing hypoxanthine, purine degradation, and xanthine degradation to uric acid with the generation of superoxide but has since been widely used in experiments to generate ROS (11).
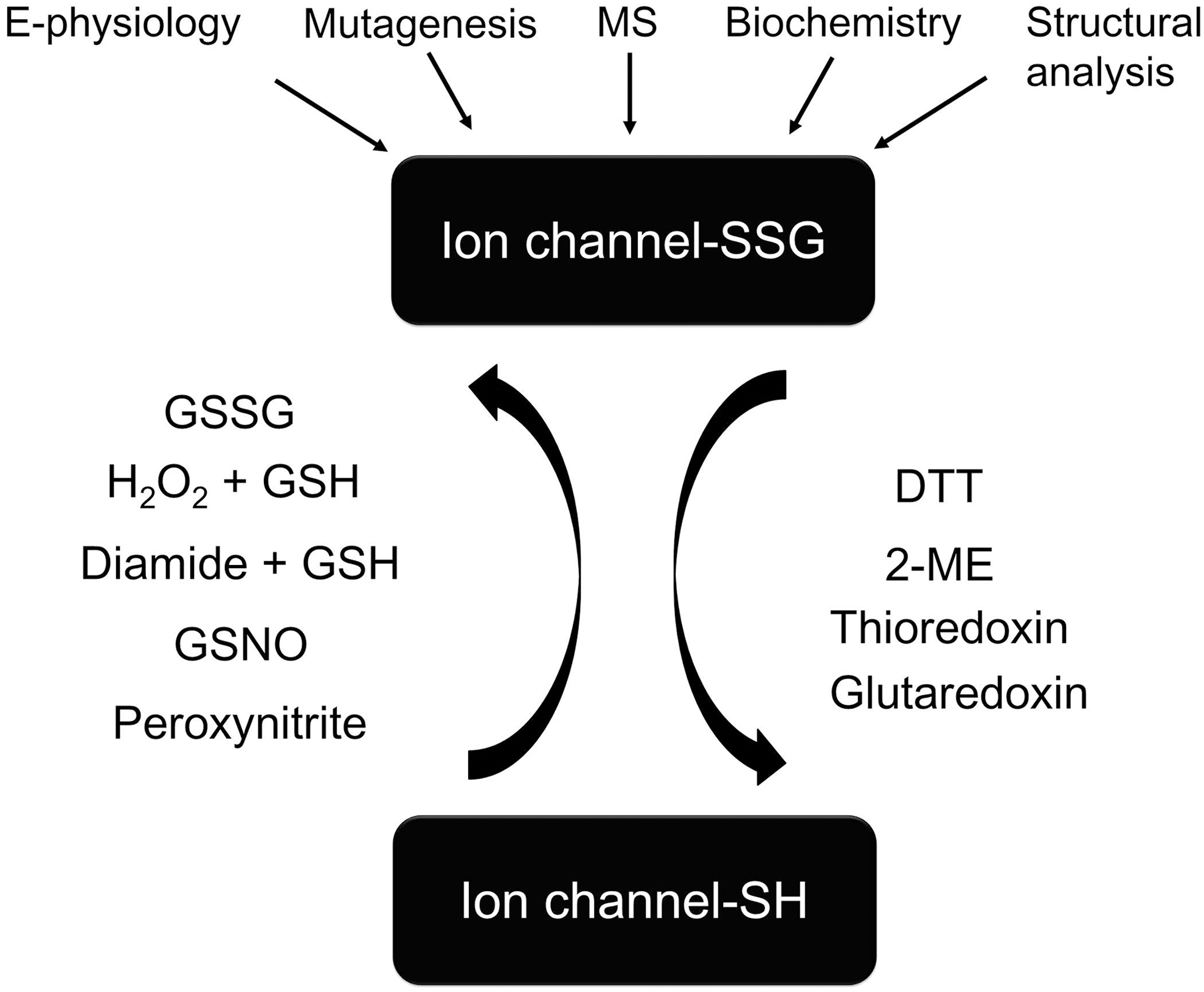
Methods to Test S-Glutathionylation
The methods and reagents for testing ion channel S-glutathionylation are illustrated in Figure 2.
Induction of S-glutathionylation
H2O2, discussed earlier, is a classic reagent to induce S-glutathionylation, provided the system has sufficient GSH. H2O2 concentrations ranging from 10 μM to 10 mM have been used to induce oxidative stress in different experimental conditions (65, 71, 141). However, it is worth keeping in mind that low concentration of H2O2 applied in an experimental condition is more relevant to physiological or pathophysiological conditions.
Using enzymatic systems (e.g., XO system) to manipulate the endogenous redox system may also be a good choice (13, 34, 56). Xanthine (50 μM) plus XO (50 mU ml−1) are the common ways to generate superoxide. Literature shows that XO (0.75 mU ml−1) and hypoxanthine (125 μM) can not only raise superoxide levels but also increase H2O2 (59). Since SOD decreases superoxide and increases H2O2 while catalase decreases H2O2, using SOD and catalase together with the XO system can provide more specific information to further distinguish the involvement of different ROS.
GSSG (1–10 mM) is often used to induce S-glutathionylation as well (28, 129, 138). The combined treatments of GSH (100 μM–5 mM) with oxidants (e.g., H2O2, 10 μM–1 mM; diamide 100 μM–1 mM) are also commonly used (138, 145). Both of these treatments are relatively specific to S-glutathionylation.
In addition, the application of peroxynitrite (ONOO−) or S-nitrosoglutathione (GSNO, 10–500 μM) to induce S-glutathionylation is also found in the literature (12, 38, 125, 129). The application of peroxynitrite is of high physiological relevance but as peroxynitrite can also induce other modification including S-nitrosylation, additional experiments should be performed to further dissect the exact modification mechanism. Peroxynitrite is generally made with the combination of NO and ROS (e.g., superoxide) (8, 19, 34, 66). NO can be generated using sodium nitroprusside (10 nM) or 3-morpholinosydnonimine (SIN-1, 1 mM) and superoxide can be made using the X/XO system (xanthine, X, 0.1 mM and XO, 0.01 U ml−1). The combination of these compounds thus would lead to the formation of ONOO−.
N-ethylmaleimide is often used to block the S-glutathionylation (129), and reactive disulfides (pyridine disulfides), for example, 2,2′-dithiodipyridine and 2,2-dithiobis-5-nitropyridine are sometimes used to mimic the addition of the GSH moiety to the cysteine residues (137, 138). These reagents, however, are also used for the study of S-nitrosylation (140). Thus, the effects of reactive disulfides should not be directly interpreted as the exclusive effect of S-glutathionylation. Rather, the results of the reactive disulfide should be viewed as supportive evidence in addition to other S-glutathionylation detection and induction methods.
Functional assays
S-glutathionylation often renders the protein active/inactive, thus the protein activity change upon S-glutathionylation should be monitored. For instance, in the case of enzymes such as glyceraldehyde phosphate dehydrogenase, cAMP-dependent protein kinase, creatine kinase, and protein kinase C, the altered enzyme activities by S-glutathionylation could be detected using biochemical assays (16, 35, 53, 60, 79, 96, 130). For ion channels, patch-clamp electrophysiology is the gold standard to test ion channel function and Ca++ imaging is also commonly used to study a variety of Ca++-permeable channels.
Biochemical tests
The most popular method to test S-glutathionylation biochemically is to use BioGEE (a membrane permeable biotinylated glutathione ethyl ester) in western blot experiments (87, 138). In principle, the cells expressing proteins of interest are loaded with BioGEE (100 μM–500 μM) first. The cells are then challenged with oxidants, for example, H2O2 (10 μM–1 mM) or diamide (10 μM–1 mM) for a time period (5 min–a few hours). After that, the cells are lysed and streptavidin-conjugated agarose beads (or magnetic beads) are used to pull down the BioGEE-conjugated protein complex, and the protein of interest is detected by the protein-specific antibodies (145). Alternatively, the BioGEE-protein complex could be immunoprecipitated (IP) first with specific antibodies against the protein of interest and then immunoblotted (IB) with streptavidin or anti-biotin antibody (125). A variation of this experiment is to avoid using BioGEE. Instead, the cells are directly challenged with oxidants and then IP with anti-GSH followed by IB with antibodies against the protein of interest (2) (or vice versa, IP with specific antibodies and IB with anti-GSH) (38). Additionally, the use of Biotin-GSSG (2 mM) is reported (10).
Mass spectrometry
Mass spectrometry (MS) could be employed to detect S-glutathionylation specifically. GSH has a molecular weight (MW) of 307 Da. If the protein of interest were glutathionylated, the MW of the protein would change accordingly and could be detected by MS. If one GSH is attached, then the protein MW would be shifted by 305 Da. If two were attached, then the protein MW would shift by 610 Da (2*305) and so on. By using MS, additional information regarding the number of GSH moieties attached to the target protein could be obtained. Moreover, if the proteins are digested with enzymes (e.g., trypsin or LysC) and followed by tandem mass spectroscopy (MS/MS) analysis, the specific cysteine sites that are S-glutathionylated can also be revealed (10, 28). It is worth noting that obtaining enough ion channel protein in acceptable purity for MS analysis is relatively challenging and this method has been mainly used for soluble protein so far.
Mutagenesis and chimeras
Systemic site-directed mutagenesis is one of the gold standards to test any kind of PTMs including S-glutathionylation. Single, double, triple, or even cysteine-free constructs could be generated with this method. Alanine and serine are the two major substitutions for cysteine. Mutation of cysteine to serine has an advantage over alanine when one considers the structural similarity. However, the substitution of serine may introduce potential unexpected phosphorylation site.
If the proteins of interest have a homology that responds to S-glutathionylation differently, chimeras could also be created using these homologies to determine which domains or motifs are responsible for S-glutathionylation (137).
Structural analysis
The incorporated GSH moiety is likely to interact with the surrounding amino acids, structurally affect the protein conformation, and thus change the protein function. The GSH moiety can affect the protein structure in at least two ways: First, GSH is a tripeptide so its relatively large size may have a steric hindrance effect on proteins that are normally tightly packed. Second, GSH has charged groups that may interact with the charged groups in the protein, either attracting or repelling certain amino acids to cause protein conformational change.
Structural modeling and molecular dynamics are useful tools to study ion channel structure and function (54, 134, 135) including S-glutathionylation-mediated structural changes of ion channels (55, 137). Homologous ion channel crystal structures determined by X-ray are the most common templates for structural modeling. If suitable crystal structure templates are not available, the structural information obtained by other methods, for example, cryoelectron microscopy and nuclear magnetic resonance (NMR) spectroscopy, may also be valuable for modeling.
Reversibility
The S-glutathionylation of protein is reversible. Reducing agents, for example, DTT (1–20 mM), 2-mercaptoethanol (2-ME, 1–10 mM), or specific enzymes like Grx1 (1 μM–10 μM), could be used to test the reversibility of S-glutathionylation. Grx1 is considered as the only specific deglutathionylation enzyme so far (45), so Grx1-mediated recovery is often essential to demonstrate the functional S-glutathionylation reversibility of the protein of interest (129, 138).
S-glutathionylation of Ion channels
Here, we discuss the S-glutathionylation of a variety of ion channels in detail.
S-glutathionylation of KATP channels
KATP channels are modulated by neurotransmitters and changes in the metabolic state thus contribute to the regulation of membrane excitability, which in turn is involved in insulin secretion, vascular tone regulation, shear-sensing, cardioprotection, and so on. (7, 27, 29, 77, 84, 110, 111, 136, 138). KATP channels are made of four pore-forming Kir (inward rectifier K+ channel) subunits and four regulatory sulphonylurea receptors of the ATP-binding cassette (SUR) subunits (102). Different combinations of Kir and SUR have been found in distinct tissues including Kir6.2/SUR1 in β-cells, Kir6.2/SUR2A in cardiac muscle, Kir6.1/SUR2B in smooth muscles, and others. Earlier studies have found that native KATP channel can be modulated by H2O2 and other oxidants but the molecular mechanisms of these modulations are not clear. Recently, Yang et al., reported that the vascular KATP (Kir6.1/SUR2B) channel is inhibited in the oxidative stress condition via S-glutathionylation of the Kir6.1 subunit in a physiologically relevant context (137, 138). Functional studies show that the treatments with a variety of S-glutathionylation inducers including GSH/H2O2, (GSH: GSSG)/H2O2, GSSG, or GSH/diamide lead to functional channel inhibition. The formation of a disulfide bond between the channel protein and GSH was further tested in biochemical experiments with BioGEE. Using site-directed mutagenesis, Cys176 was identified as the primary S-glutathionylation site and Cys43 as a contributing site.
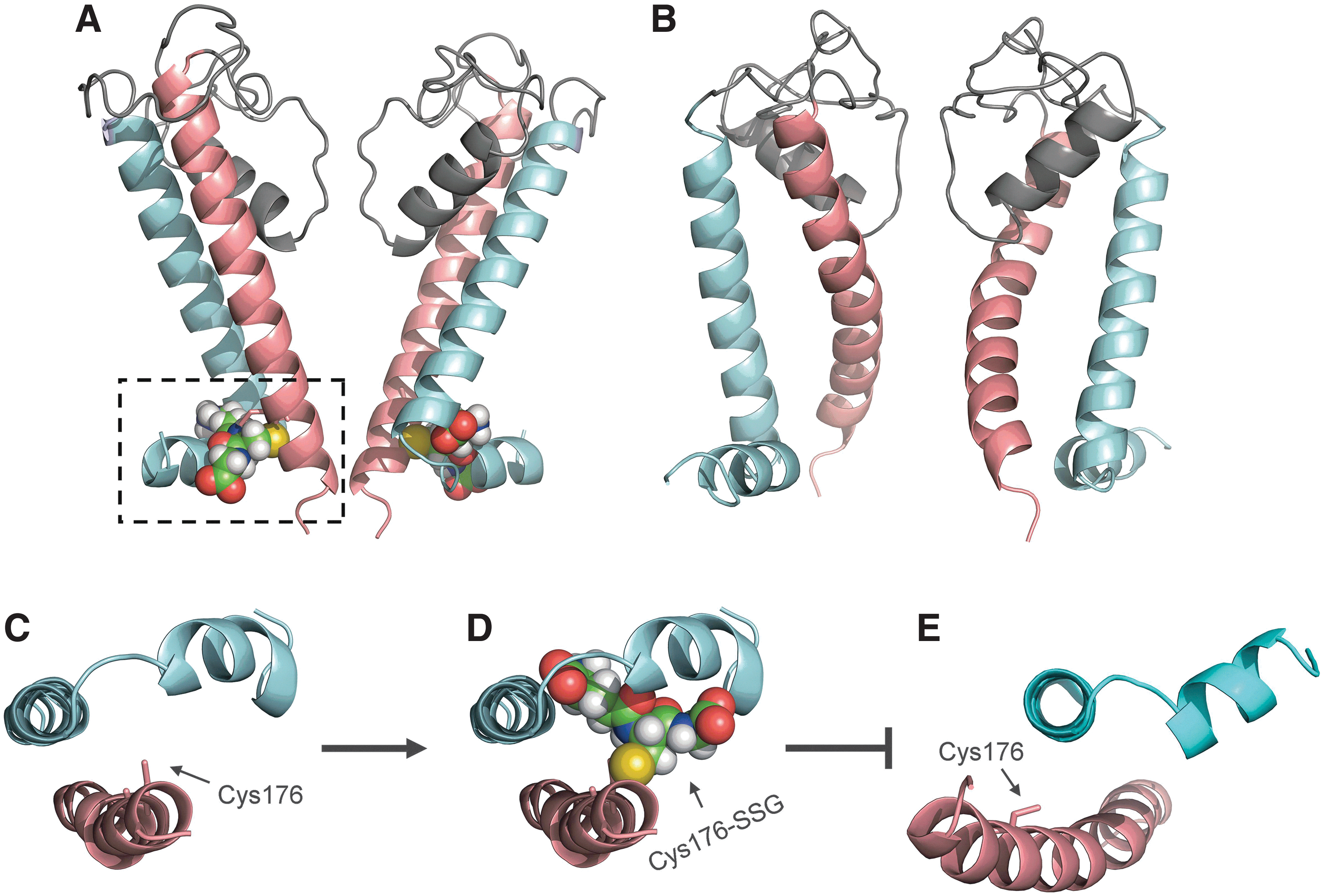
To understand the structural consequence of KATP channel S-glutathionylation, structural modeling of KATP channel has been performed. In particular, the Kir6.1 pore-forming subunit in its closed and open states were modeled, based on the recently crystalized chicken Kir2.2 and the bacterial Kir channel as templates, to study how S-glutathionylation affects the KATP channel gating (137) (Fig. 3A, B). From the model of Kir6.1 channel in its closed state, it was found that Cys176, the primary cysteine site of Kir6.1 for S-glutathionylation, is located in the critical region of the inner helix, close to the activation gate residue Phe178 and the hinge residue Gly175 (residue numbers are based on rat Kir6.1 protein). Phe178 is suggested to form the activation gate and block the ion-conducting pathway when the channel is in its closed state. As the gating hinge, Gly175 is the site where the channel bends to open (61). Cys176 locates between Phe178 and Gly175 and its side chain faces a pocket formed by inner helix, outer helix, and slide helix (Fig. 3C). When the GSH moiety is modeled onto Cys176, it is found that GSH moiety can fit into this pocket nicely after energy minimization (Fig. 3D). The Kir6.1 channel structure in its open state is also modeled and shows that the inner helix undergoes a significant leftward movement (137), resulting in the disruption of the binding pocket for the GSH moiety (Fig. 3E). Therefore, based on these models, it is proposed that incorporation of the GSH moiety into the Kir6.1 subunit prevents the channel from entering its open state, thus retaining the channel in its closed state.
S-glutathionylation of Kir4-Kir5 channel
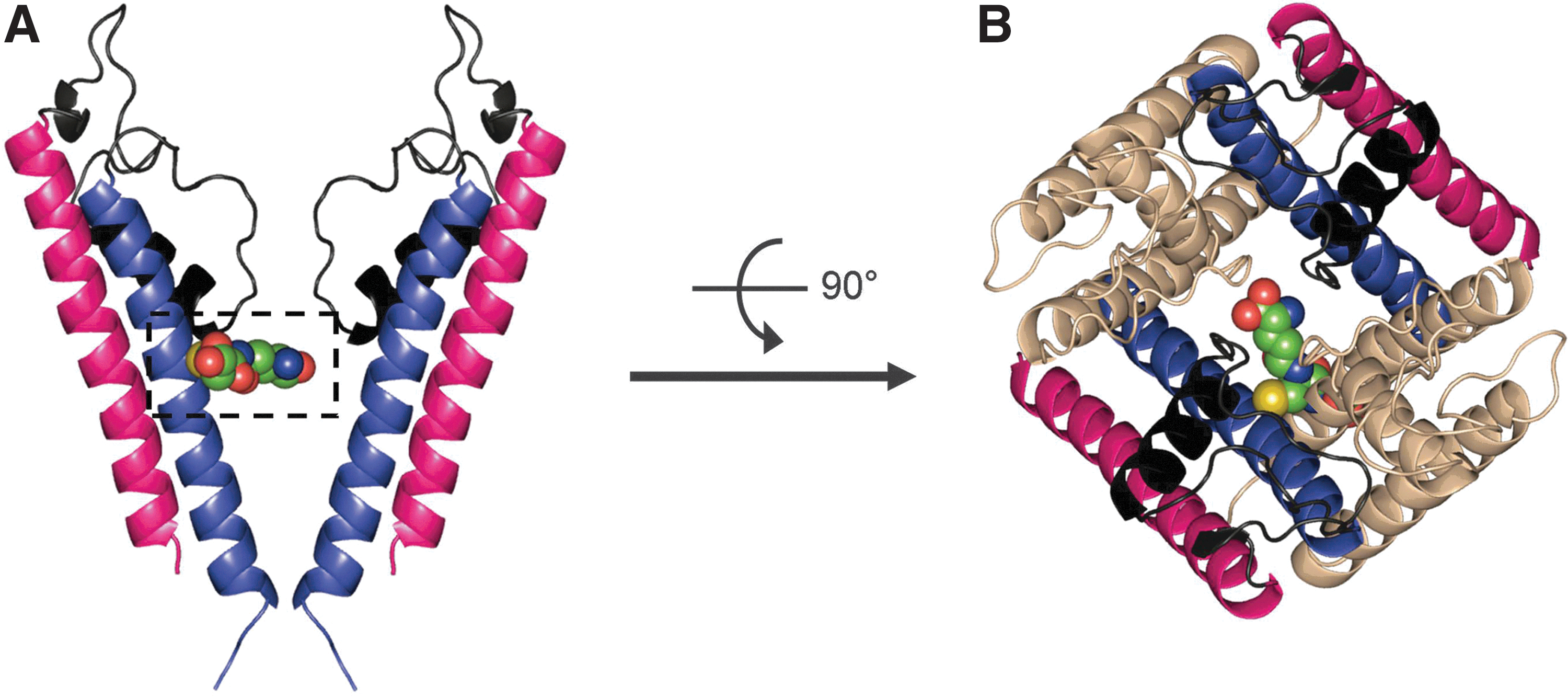
Kir4.1 channel is found in the kidney, retina, and central nervous system as a homodimeric or heterodimeric (together with the Kir5.1) channel, acting as a K+ transporter or a pH-dependent membrane potential regulator (50, 62). Jin et al. has found that the presence of Kir5.1 in the Kir4-Kir5 heterodimeric channel enables channel sensitivity to S-glutathionylation. Using diamide or H2O2 together with GSH, it was found that the Kir4-Kir5 channel could be inhibited via S-glutathionylation while the homodimeric Kir4.1 channel is not sensitive to this modification (55). Further study showed that Cys158 in the S6 helix of Kir5.1 is the major residue for S-glutathionylation (Fig. 4A, B). In addition, it was demonstrated that one GSH moiety is sufficient to block the channel activity and S-glutathionylation occurs when the channel is in its open state (55).
S-glutathionylation of ryanodine receptor
The RyR is the first ion channel found to be S-glutathionylated, and S-nitrosylated (4, 118, 119). Expressed on the endoplasmic reticulum (ER), the RyR is responsible for Ca++ release from ER to cytosol. The RyR protein has ∼100 cysteine residues and is highly sensitive to oxidation. Both S-glutathionylation and S-nitrosylation could occur on RyR channels with S-nitrosoglutathione (GSNO) treatment (5). Further study has demonstrated that certain cysteine residues (Cys1040 and Cys1303) are exclusively S-nitrosylated, some (Cys1593 and Cys3193) are selectively S-glutathionylated, and others can undergo both reactions (4).
S-glutathionylation of cystic fibrosis transmembrane conductance regulator
Cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel belonging to the ABC transporter family (43). CFTR regulates salt and water transportation across epithelial membranes in lung and gut. Mutations of the CFTR gene can lead to cystic fibrosis and congenital bilateral absence of vas deferens (108). During inflammation, extensive production of ROS could target the epithelial cells and modulate the plasma membrane-bound CFTR. Indeed, Wang et al., found that several oxidized forms of glutathione including GSSG (10–20 mM), and nitrosylated glutathione/S-nitrosoglutathione (GSNO, 50–200 μM) can inhibit the channel activity markedly in excised patch recordings (129). In addition, the combined application of GSH and diamide (5–100 μM, equimolar) can also cause significant channel inhibition. These reactions could be rescued by the reducing agent DTT (20 mM) and the deglutathionylation enzyme Grx (4 μM). Moreover, biochemical experiments further demonstrate that diamide (100 μM) facilitates the incorporation of biotin-GSH (125 μM) into CFTR protein. Using site-directed mutagenesis, Cys344 located in the NBD2 domain was identified as the primary site for S-glutathionylation. This modulation could potentially affect the nucleotide binding and disrupt the ATP-dependent channel opening. Although a schematic diagram has been provided to explain the structure-function of CFTR S-glutathionylation (129), detailed structural insight has not been reported, presumably due to the lack of an accurate template for modeling.
S-glutathionylation of CRAC channel
Ca++ release-activated Ca++ (CRAC) channels belong to store-operated channels, which involve in a variety of signaling cascades of non-excitable cells, including T lymphocytes and mast cells. Recent studies have identified Orai1 (CRAC modulator) as the pore-forming subunit of the CRAC channel while stromal-interacting molecule 1 (STIM1) serves as the ER Ca++ sensor (15, 91, 95). Hawkins et al. reported that oxidative stress, induced by lipopolysaccharide (LPS) (1.5 μg/ml) and H2O2 (100 μM), alters the Ca++ signaling in B-lymphocytes through the modification of the CRAC channels (47). They further identified by pull-down experiment that the ER-resident STIM1 is S-glutathionylated. A conserved cysteine residue (Cys56) was revealed as the target of S-glutathionylation. The incorporation of GSH to STIM1 was confirmed by MS. The S-glutathionylation of STIM1 results in a constitutively activated CRAC channel.
S-glutathionylation of voltage-gated Ca++ channels and channels for Ca++ homeostasis
Intracellular Ca++ concentration and Ca++ oscillations play important roles in endothelial cells, including the regulation of vascular permeability, inflammation, and so on. The disruption of Ca++ homeostasis has been associated with oxidative stress, whereas the molecules that are directly targeted by ROS are not clear. Lock et al. demonstrated that diamide, a thiol-oxidizing agent, could increase the intracellular Ca++ concentration and oscillations (68). The authors showed that this modulation is related to GSH. It is further identified that IP3 receptors and the plasmalemmal Ca++-ATPase pump are the molecules targeted by ROS via S-glutathionylation using pull-down experiments with BioGEE (69).
Disruption of calcium homeostasis also contributes to cardiac hypertrophy and cardiac failure. Tang et al. found that the L-type Ca++ channel (CaV1.2), a main channel for calcium influx into cardiac myocytes, could be regulated by S-glutathionylation (123). The open probability of CaV1.2 increases with GSSG and biochemical evidence shows that the CaV1.2 channel is glutathionylated both in H2O2 treatment and in ischemic human heart.
S-Glutathionylation of Ion Transporters
Besides ion channels, ion transporters (or ion pumps) are another group of molecules that move ions across cellular membrane, but at the expense of energy consumption (e.g., ATP or the concentration gradient of another ion) (42). Many ion transporters are regulated by S-glutathionylation as well (93).
For example, sarco/endoplasmic reticulum Ca++-ATPase [SERCA], is a calcium ATPase that transfers Ca++ from cytosol to the lumen of the sarcoplasmic reticulum. It has been reported that SERCA can be activated by NO via peroxynitrite-mediated S-glutathionylation at the Cys674 residue (1). Na+/K+-ATPase (sodium-potassium adenosine triphosphatase) is another ion transporter that is found to be modulated by S-glutathionylation (38). Peroxynitrite, among other stimuli, can facilitate S-glutathionylation of β-1 subunit of Na+/K+-ATPase at the Cys46 residue (38). S-glutathionylation also occurs on the catalytic α subunit of Na+/K+-ATPase, affecting the ATP binding (92). The crosstalk between S-nitrosylation and S-glutathionylation has also been observed on Na+/K+ ATPase, indicating that a switch between S-glutathionylation and S-nitrosylation contributes to the oxygen-induced regulation of Na+/K+-ATPase (132).
Ion Channel S-Nitrosylation and Its Relationship with S-Glutathionylation
S-nitrosylation is referred to as the addition of an NO moiety to the thiol group of a cysteine residue (46). Like S-glutathionylation, no enzyme that can facilitate the formation of protein S-nitrosylation has been found yet. However, enzymes like S-nitrosoglutathione reductase (GSNOR) have been shown to mediate the denitrosylation (72). Many ion channels (or their regulators) (46), including the RyR channel (4), voltage-gated sodium channel (97), the voltage-gated potassium channel (6), the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (103, 104), the TRP channel (140), and the KATP channel (21, 58, 64, 67, 120), are found to be regulated by S-nitrosylation or NO.
Recent studies suggest that the NO-mediated pathway and the ROS-mediated pathway may have extensive crosstalk (37, 93, 132); the reaction between NO and superoxide gives rise to peroxynitrite, which can be a potent mediator of S-glutathionylation in the presence of GSH; NO may form protein-SNO, which can serve as a precursor to protein-SSG. And in the case of RyR channels, S-nitrosylation and S-glutathionylation may compete for the same cysteine residues (4).
In addition, S-nitrosylation and S-glutathionylation share lots of common features (72): they are both facilitated by oxidants; they both target the thiol group and attach a moiety to the cysteine residues. Therefore, in experimental design, reactive disulfides can be used to mimic both S-nitrosylation and S-glutathionylation (72, 137, 140). Moreover, studies have shown that for a given protein, both S-nitrosylation and S-glutathionylation could occur depending on the accessibility of the cysteine, specific local environment around the cysteine, the redox state of the proteins, and other unidentified factors. It is still elusive to predict if a given cysteine may be selective for S-glutathionylation of S-nitrosylation. Why S-nitrosylation and S-glutathionylation selectively occur on different cysteine residues is not known. In some cases, S-nitrosylation and S-glutathionylation occur on the same cysteine residues, but how S-nitrosylation competes with S-glutathionylation or serves as a precursor to S-glutathionylation for the same residue is not clear either. Future studies are needed to understand the intertwining relationship between S-glutathionylation and S-nitrosylation.
Ion Channel S-Palmitoylation and S-Sulfhydration
Besides S-glutathionylation and S-nitrosylation, other thiol modulation mechanisms, such as S-palmitoylation and S-sulfhydration, have been reported to modulate ion channels. S-palmitoylation refers to the formation of a reversible thioester linkage of a palmitoyl lipid to cysteine residue. This modification has been shown to control the maturation, trafficking, and regulation of ligand- and voltage-gated ion channels (114).
S-sulfhydration refers to H2S-mediated PTM (81, 89). H2S is a gaseous messenger molecule that is generated in vivo from L-cysteine through the enzymes cystathionine β-synthase and cystathionine γ-lyase (CSE) (127). S-sulfhydration shares many common features with S-nitrosylation, as both are mediated by gasotransmitters (H2S and NO) (82, 106). Endogenous H2S could modulate proteins by converting the −SH group of cysteine to an −SSH group (81, 133). The KATP channel reported elsewhere to be mechanosensitive (23, 27) is regulated by H2S (83, 121, 144). Besides the KATP channel, other channels including L- and T-type Ca++ channel, TRPA/V channels, Cl− channels, and so on, are also subjected to the modulation of H2S (90, 122). Readers who are interested in these modification mechanisms are directed to other excellent reviews covering these topics (89, 90, 122, 128).
Possible Crosstalk Among Different Thiol Modifications
The crosstalk or interplay among various types of cysteine modifications is particularly interesting. The crosstalk could happen in many ways directly or indirectly, including, (i) Different thiol modification mechanisms may compete for the same cysteine residues (Fig. 5A), for example, in the case of RyR channel. (ii) Different modifications may occur sequentially, for example, S-nitrosylation may serve as a precursor to S-glutathionylation in certain conditions (Fig. 5B). (iii) The modification of a cysteine residue by one mechanism may alter the local environment so that the reactivity of a nearby residue (e.g., cysteine, phosphorylation site) may be altered (Fig. 6A). (iv) The modification of a cysteine residue in the protein may have an allosteric effect so that the reactivity of another cysteine residue in a distinct region of that protein or in another protein of a multiple protein complex may be altered (Fig. 6B). (v) Thiol modification of a protein may affect the activity or production of key elements in another thiol modification cascade, for example, S-glutathionylation of certain proteins (CSE, nitric oxide synthase [NOS] or their regulators/transcription factors) may lead to increased activities of CSE or NOS, or lead to the production of more CSE, NOS proteins through gene regulation (Fig. 7).


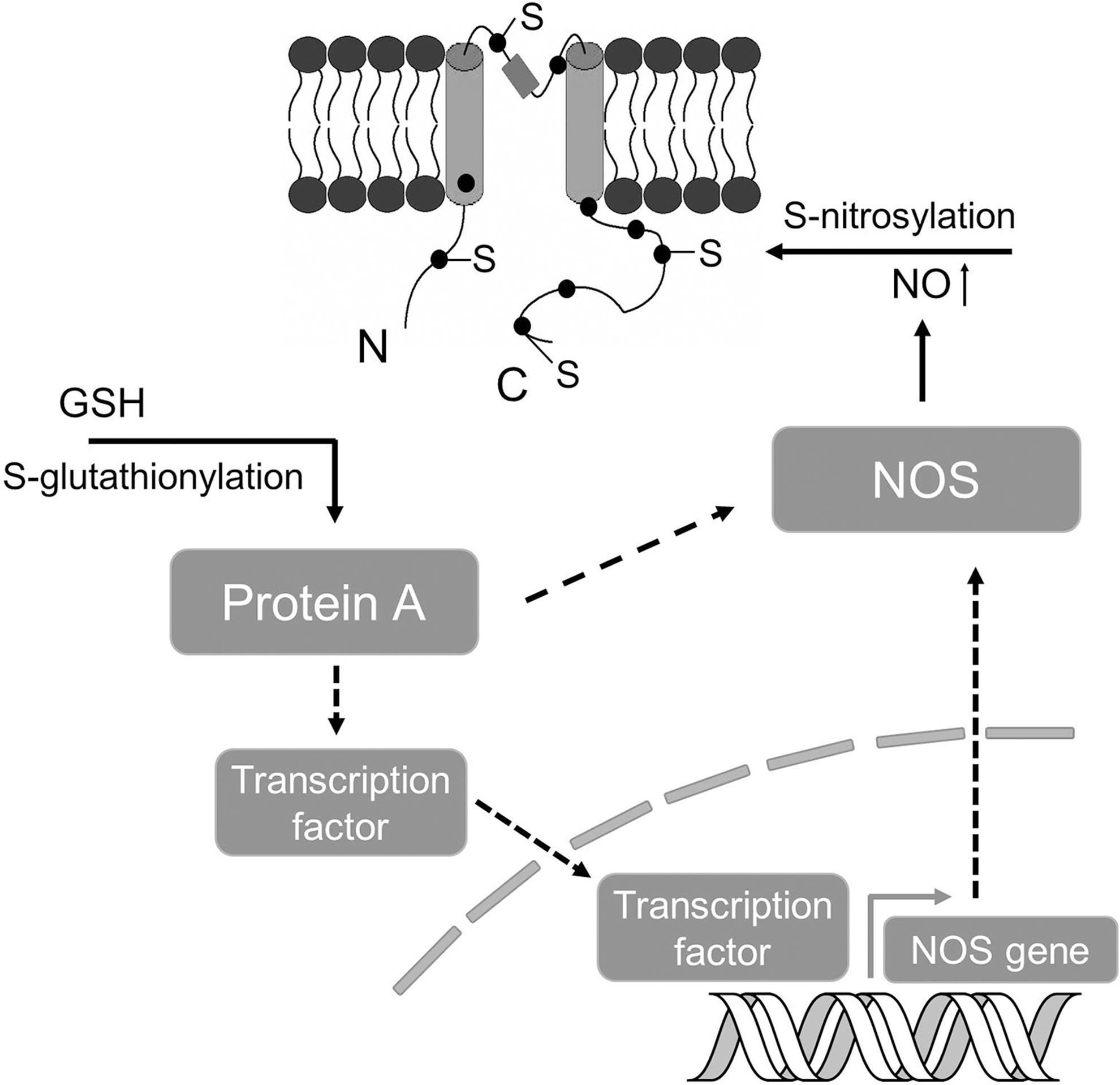
Certain possible crosstalk among different PTMs has been previously discussed in reviews (49, 114). Recent research from many labs, especially Dr. Snyder's group, provided more evidence for some interesting possibilities. Ho et al. reported that S-palmitoylation and S-nitrosylation can regulate the principal protein of postsynaptic densities (PSD-95) via competitive modification of cysteine 3 and 5 residues (52). In another study, Sen et al. found that H2S can lead to S-sulfhydration of the p65 subunit of NF-κB at the Cys38 residue, where S-nitrosylation can also occur in a competitive manner (105). More recently, Vandiver et al. showed that S-nitrosylation and S-sulfhydration (sulphydration) might be reciprocal events for parkin, an E3 ubiquitin ligase involving in Parkinson's disease (126). Sulfhydration occurs physiologically and enhances the catalytic activity of parkin, whereas S-nitrosylation inactivates parkin, suggesting a neuroprotective effect and therapeutic implications of H2S donor (126). AMPA receptors are subjected to many types of PTM (70). S-nitrosylation of GluA1 (at Cys875), a regulatory subunit of AMPA receptors, facilitates phosphorylation (at Ser831) and enhances AMPA receptor channel conductance (104). This type of crosstalk was also seen in endothelial NOS (eNOS) from an earlier study (36). In addition, it is also known that NO may affect H2S level in vascular tissues via increasing CSE activity. As CSE has many cysteine residues, S-nitrosylation of CSE has been the subject of speculation (128). With the exception of the RyR channel mentioned above, whether crosstalk or interplay of these thiol modifications may occur in other ion channel proteins is still unknown and would be an exciting new direction in the field.
Ion Channels in Mechanotransduction and Possible Contribution of S-Glutathionylation
Mechanotransduction is a process through which cells sense altered physical forces and convert them into biological signals (18). The response of endothelium to dynamic fluid shear stress is one example of mechanotransduction (25, 78). Interestingly, the activities of many ion channels could be modulated in response to altered shear stress in this process (18, 27, 131). These channels sense mechanical forces and transduce them into electrical/biochemical signals. In the context of flow and its cessation, K+ channels have been reported to respond to alterations of flow. An inward rectifying K+ channel was initially found to be activated with onset of flow (85) and later studies identified KATP channel as the inward rectifying K+ channel that is closed on cessation of flow (27). The closure of the KATP channel depolarizes the endothelial cell plasma membrane, triggers many downstream signaling cascades (PI3K-Akt), and eventually causes activation of NOX to produce ROS (24, 27). NOX generates superoxide into the extracellular side of the cellular membrane and the extracellular superoxide could flux across the plasma membrane through chloride channel-3, induce intracellular Ca++ release, and activate further mitochondrial superoxide generation (48). The depolarization of endothelial cells could also trigger the activation of eNOS to produce NO in a Ca++-dependent manner (Fig. 8). Collectively, the activation of these signaling cascades thus could lead to vasodilation, cell proliferation, injury, or other consequences (17, 26, 73, 142). As ROS and NO are able to modulate thiol groups of many ion channels (1, 27, 101) including the KATP channel (21, 64, 138), which are potentially involved in membrane potential regulation and downstream signaling, we propose that a feedback loop may exist linking membrane potential, ion channel regulation (by S-glutathionylation or S-nitrosylation), and ROS and/or NO production in mechanotransduction of endothelium (Fig. 8).
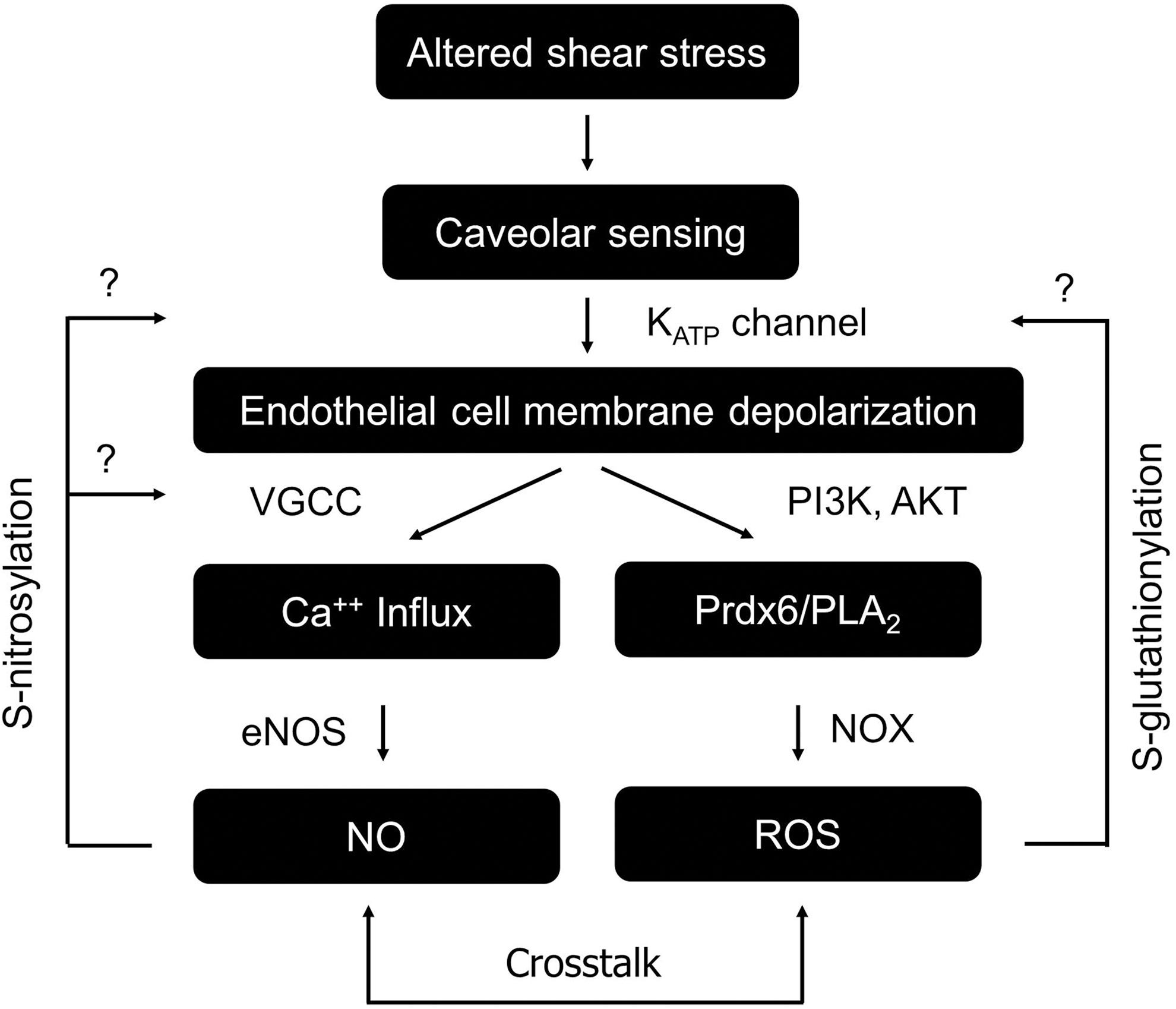
The mechanosensitive cascade initiated by the KATP channel hinges on channel activities and the subsequent S-glutathionylation of KATP channel by ROS would lead to channel inhibition, and presumably affect the ability of KATP channel to sense stopped flow. However, it is not clear if and to what extent deglutathionylation in vivo would restore channel function and mechanosensing. As S-glutathionylation is a relatively new concept in ion channel and mechanotransduction field, the modifications of mechanosensitive channels by S-glutathionylation have not been clearly reported yet. Regardless, based on the strong link between mechanotransduction and oxidative stress, especially in vasculature, we propose that S-glutathionylation could be an important mechanism that regulates mechanosensitive channels during oxidative stress.
Conclusions and Perspectives
Thiol modifications, especially S-glutathionylation, have received increasing recognition as a major mechanism that regulates ion channel structure and function. Especially, in vasculature (93), redox-mediated ion channel modulations seem to be complex phenomena, affecting not merely channel functions but possibly forming feedback loops. On one hand, ROS modify ion channels via S-glutathionylation, S-nitrosylation, and/or other PTMs and affect channel functions and downstream signaling cascades. On the other hand, mechanical and other stimuli may alter ion channel activities, leading to signaling cascades that produce ROS, NO, or other molecules, which in turn, modulate ion channels. A balance among these modulations might be required for vascular homeostasis and other important cellular functions.
Nevertheless, with the advances of research tools and standardized protocols, the list of ion channels that could be regulated by S-glutathionylation would grow rapidly. However, certain outstanding challenges and questions are still awaiting answers from the research community. They are as follows: (i) Although it is pretty clear that oxidative stress could promote S-glutathionylation and S-nitrosylation, the exact physiological or pathological roles of these modulations are not well understood. Especially, the functional consequences of S-glutathionylation in tissue and organ level, as well as in whole animals have not been explored extensively. Would S-glutathionylation of certain ion channels represent a protection mechanism against oxidative stress or simply represent a consequence of oxidative stress? Would S-glutathionylation serve as a critical signaling mechanism similar to protein phosphorylation contributing to signal transduction? (ii) With the growing list of newly crystallized ion channel structures and the improvement of structural modeling tools, more studies may be performed to tackle ion channel thiol modification from a structural perspective, addressing how GSH, NO, H2S, or other molecules may affect the local or global conformation of ion channels to affect channel functions. (iii) As it is known that many PTMs (e.g., S-glutathionylation, S-nitrosylation, S-palmitoylation, and S-sulfhydration) target the thiol group of cysteine residues, investigating the intriguing relationships among these mechanisms would provide critical insights for the understanding of thiol modulation in general. Why are different cysteine residues preferably targeted by specific mechanisms? Could prediction algorithms be developed to provide information regarding which cysteine is sensitive to which modification mechanism under which condition? Indeed, it is very fascinating that cells need such a complicated system to modulate the cysteine residues. Understanding the physiological significance and how the cells fine-tune all these thiol modulations could lead to major breakthroughs.
We anticipate that the understanding of all these basic questions on S-glutathionylation of ion channels may provide a platform to further explore therapeutic approaches to treat many oxidative stress-related devastating diseases.
Footnotes
Acknowledgments
C.J. and X.J. are supported by NIH (1R21HD060959 and 1R01NS073875). Y.Y. was a B&B fellow at GSU and is currently supported by the Connecticut Stem Cell Research Grants Program. We thank Dr. Yun Shi, Dr. Weiwei Shi, Anuhya S. Konduru of Georgia State University, Dr. Mark Estacion, and Dr. Andrew Tan of Yale University for critical reading of the article. We thank Dr. Aron B. Fisher and Dr. Shampa Chatterjee of University of Pennsylvania, Dr. Rui Wang of Lakehead University and Dr. Michael Shipston of University of Edinburgh for comments and discussions. We thank Dr. Solomon H. Snyder and Dr. Bindu D. Paul of Johns Hopkins University for sharing preprint materials and comments on this article.