
Abstract
Introduction
P
Reversible redox regulation of the PTP activity by diamide or H2O2 was first reported by Arnold Stern and his colleagues in the early 1990s (63, 84). In addition to oxidation, reactive cysteines in phosphatases can undergo other important modifications that are mediated by the family of signaling molecules termed gasotransmitters, namely by nitric oxide (NO) and hydrogen sulfide (H2S). Among the first analyses reporting oxidative changes of PTPs in response to NO treatment is the 1994 paper by Giampietro Ramponi and colleagues, describing the formation of a disulfide bridge between Cys12 and Cys17 of the low-molecular-weight PTP (LMW-PTP) active site loop in response to NO treatment (9). In the same report, reversible NO-mediated inhibition of Yersinia enterocolitica PTP activity was reported, which immediately raised the suspicion that reversible NO-mediated inhibition may belong to the general physiological mechanisms of PTP activity regulation. Use of the biotin switch technique and of more recent and more advanced methods broadened the spectrum of PTPs known to be S-nitrosylated, leading to the identification of the first sulfhydrated PTP. Recently, the first proteomic papers on S-nitrosylated PTPs were published (30), but the number of PTPs known to undergo Cys S-nitrosylation or sulfhydration is still very limited. Here, we summarize the state of the current literature on the effects of PTP S-nitrosylation and sulfhydration, and list the major methods utilized to detect the gasotransmitter-induced changes of PTP active site Cys residues.
Cysteine S-nitrosylation
Protein S-nitrosylation is responsible for the vast majority of the physiological signaling by NO and S-nitrosothiols. Both hyper- and hypo-S-nitrosylation are considered to cause human pathologies (31). Here, we will focus on the nitrosylation of the Cys residues, because their modification directly affects the activity of PTPs. Other amino acids may also play an important role; in particular, tyrosine is frequently converted to dityrosine when exposed to reactive nitrogen species (RNS) or reactive oxygen species (ROS). Reversible nitration of tyrosine involves modification of the three-position of its phenolic ring by the addition of a nitro (NO2) group, usually mediated by peroxynitrite (78). On Cys residues, S-nitrosylation results from either the oxidative reaction of a Cys thiol with NO in the presence of an electron acceptor (typically O2 or metal ion) or by transnitrosylation consisting of the NO+ transfer from a donor S-nitrosothiol to an acceptor Cys thiol. The pKa of the target Cys thiol usually determines its susceptibility to the transnitrosylation signal, while the hydrophobicity of the amino acids surrounding the Cys thiol is considered as determining the availability to oxidative S-nitrosylation by NO itself (30).
S-nitrosylation: (1) 2•NO+O2→2•NO2
(2) •NO2+•NO→N2O3
(3) •NO2+PTP-Cys-SH→NO2
−+H++PTP-Cys-S•
(4) RS•+•NO→PTP-Cys-SNO (5) PTP-Cys-SH+N2O3→PTP-Cys-SNO+NO2
−+H+
Transnitrosylation: (6) PTP1-Cys-SNO+PTP2-Cys-SH→PTP1-Cys-SH+PTP2-Cys-SNO (7) PTP1-Cys-SNO+PTP2-Cys-SH→PTP1-Cys-SS-Cys-PTP2+HNO
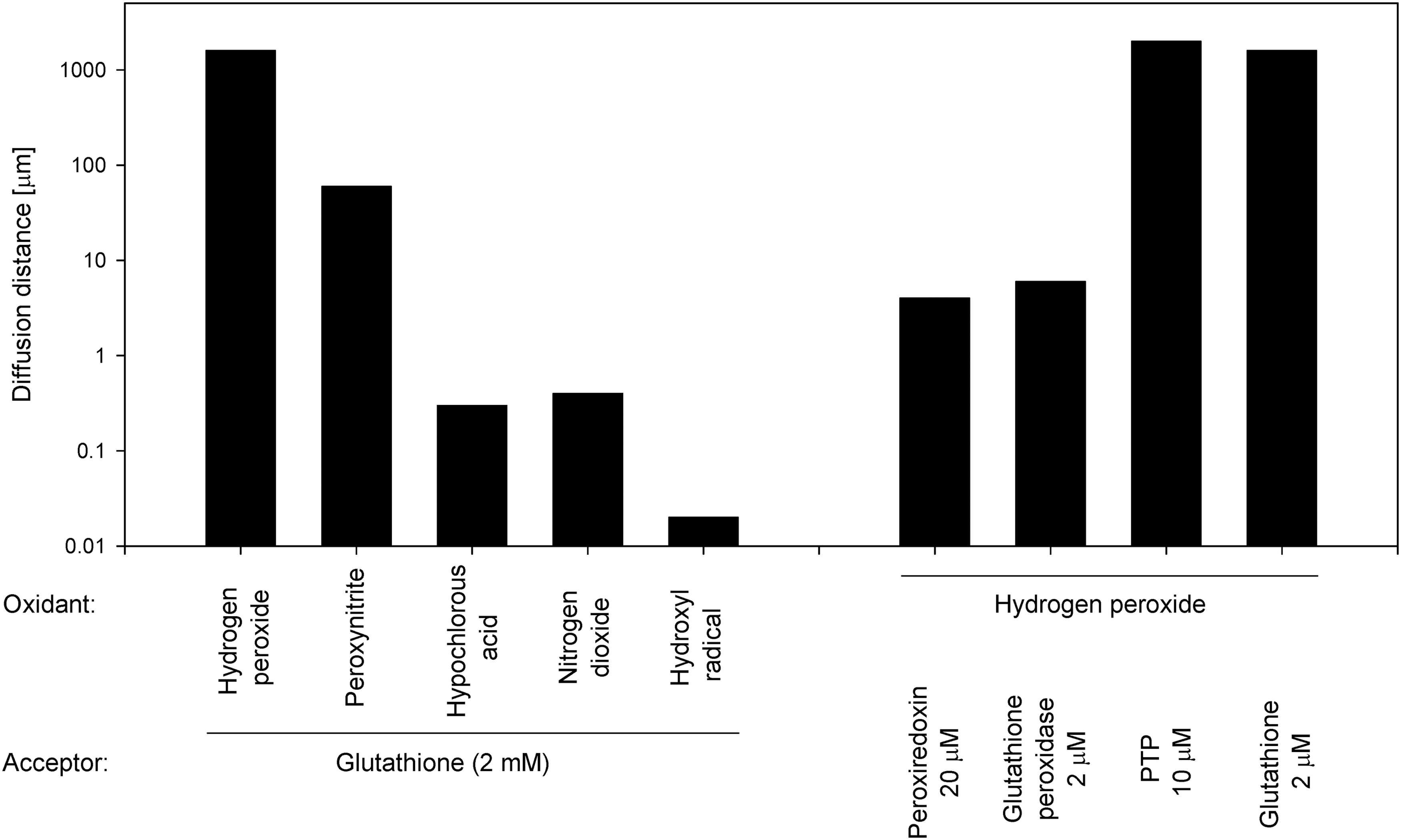
Some authors suggest that a large part of ROS-mediated signaling is, in fact, converted to a nitrosative signal, and, thus, NO is considered an effector molecule (42, 52, 57, 60). NO synthesis is tightly regulated by the arginine- and Ca2+-dependent system, because NO is the product of L-arginine transformation to L-citrulline, and because two of the three isoforms of nitric oxide synthases (nNOS and eNOS) are Ca2+/calmodulin dependent, while only iNOS is insensitive to Ca2+. NO synthesis requires homodimerization and proper phosphorylation status of the NOS enzymes, as well as the presence of three co-substrates (L-arginine, NADPH, and molecular oxygen) and five cofactors (calmodulin, FAD, FMN, heme, and tetrahydrobiopterin) (37). In contrast to ROS, the physiological concentrations of NO generated by Ca2+-activated NOS enzymes are considered nontoxic (2). Importantly, the particular RNS and ROS differ dramatically in their diffusion distances (Fig. 1), further supporting the need for the diversity of RNS and ROS present physiologically in living organisms. In addition to NO, RNS also include nitrogen dioxide (•NO2 −) acting as a free radical, as well as nonradicals, including peroxynitrite (ONOO−), nitrous oxide (N2O), and alkyl peroxynitrites (RONOO−). Details of S-nitrosothiol formation and its role in redox signaling were thoroughly described by Forman et al. (26, 27). Separating the effects of ROS and RNS may be complicated, in part, by chemical reactions commonly underway in living organisms, such as the formation of peroxynitrous acid (from the superoxide radical and NO), the spontaneous decomposition of which may, in turn, lead to the production of hydroxyl radical (33).
Reactions involving peroxynitrite: (8) •NO+O2
•−→ONOO−
(9) ONOO−+H+ONOOH→[NO2 HO•]→NO3
−+H+
(10) ONOO−+CO2→ONOOCO2
−
(11) ONOOCO2
−→[NO2 CO3
i−]→O2NOCO2
−
PTPs are relative newcomers to the field of NO- and RNS-mediated signaling. Many more studies have focused on RNS-mediated modifications of soluble guanylate cyclase and other proteins. One popular hypothesis even claims that S-nitrosylation of most of the protein Cys residues simply serves as an NO reservoir (2, 76, 82), while numerous other studies report important physiological functions of cysteine S-nitrosylation as a critical redox-driven mechanism. Typically, the S-nitrosylated proteins possess a consensus sequence X(K/R/H)C(D/E) (42, 83). Importantly, this conserved motif does not need to be present at the amino-acid chain in a linear form but may result from the protein folding into its tertiary structure (70). The S-nitrosylation occurs preferentially in the hydrophobic regions; the hydrophobicity probably supports the higher concentration of lipophilic molecular oxygen and NO molecules necessary for the generation of the nitrosylating species N2O3 (2).
PTPs are not only the targets of NO signaling. There is also a potential feedback loop consisting of several PTPs that are shown to directly or indirectly regulate the activity of NOSs, regulating either NOS tyrosine phosphorylation (coupled with changes in their activity) or transcriptional factors responsible for the NOS expression (6, 12, 16, 25, 35, 36, 38, 44, 65, 77, 85, 87, 91). The NOSs produce NO by a conversion of one of the terminal nitrogens of the guanidine group of L-arginine to NO, producing L-citrulline (37). NOSs compete for the available pool of cellular L-arginine with arginases. Thus, L-arginine can be metabolized either to NO by NOSs or to urea and L-ornithine by arginases, and proceed further to the polyamine and proline synthesis necessary for tissue repair and wound healing. The balanced ratio of NOSs and arginases is, thus, frequently targeted by pathogens aiming at altering host defense, which frequently use PTPs as their tools to dysregulate the NO production by the host cells. Minimizing the activity of certain PTPs, in particular SHP-1, is thus predicted to contribute to the inhibition of NO synthesis and, thus, to potentiate parasite survival and propagation (80).
Sulfhydration
Although H2S has only recently been considered an important signal transduction molecule, its physiological levels may reach high values, approximately 50–160 μM in the mammalian brain, and 10–100 μM in the peripheral blood (34, 94). At least four independent pathways of H2S synthesis have been identified, acting at different anatomical sites. Cystathionine γ-lyase (CSE) is typically responsible for H2S production in the liver, kidney, enterocytes, and vascular smooth muscle cells; cystathionine β-synthase (CBS) catalyzes H2S synthesis in the brain (CBS can be inhibited by physiological concentrations of CO at Ki 5.6 μM); and 3-mercaptopyruvate sulfurtransferase (MST) is associated with H2S production in the heart (MST activity can be potentiated by Ca2+/calmodulin and S-adenosyl methionine). All the pathways of H2S synthesis cited earlier use cysteine as a precursor. Cysteine can be formed from N-acetyl-cysteine, which is commonly administered as a clinical antioxidant that is considered as serving as a glutathione precursor (34, 99).
The Cys residues subject to sulfhydration usually display a low pK a value (56), and, thus, they can be prevalently found in a form of thiolate anions (S−) at physiological pH. During the sulfhydration, the Cys sulfhydryl group is modified to an–SSH group, which is highly reactive. Alternatively, the sulfhydration may occur at the Cys residue, which had already the thiolate anion oxidized to the sulfenic acid (SOH). A reaction of the sulfenic acid with either HS− or H2S results in the formation of a persulfide bond (23). The third proposed mechanism involves a reaction of the Cys residue-associated sulfenic acid with glutathione, forming a mixed disulfide before reacting with the H2S itself. In PTP1B, the only tyrosine phosphatase known to be sulfhydrated so far, the Cys residue-associated sulfenic acid was proposed to be modified into a sulfonamide intermediate, which may be sulfhydrated when exposed to the H2S (50, 69). Pathologies associated with aberrant H2S levels are probably under-reported. Subjects with trisomy 21 manifesting as Down syndrome produce high H2S levels, because the CBS gene is located at 21q22.3, and the H2S over-production is hypothesized to be responsible for the progressive mental retardation manifesting within the first year of life of Down syndrome subjects (13, 48). Acute H2S intoxication leads to the loss of central respiratory drive, cytochrome c oxidase inhibition, glutathione depletion, reduction of ferric to ferrous ions, and reduction of O2 to ROS (34).
H2S has a pKa of 6.9, suggesting its slightly anionic character at physiological pH. The reduction potential of the HS·/HS− pair is only a moderate 1.08 V (vs. normal hydrogen electrode), while the Cys-S·•/Cys-S− reduction potential is 0.92 V (34). Concentrations of HS− can, thus, reach 38–112 μM in the brain and 7–75 μM in the peripheral blood, which is equivalent to the levels of glutathione considering that the pKa of glutathione is 8.5 and its intracellular concentration is 1–5 mM. H2S is, thus, hypothesized to serve as a major intracellular reductant, with particular importance for cysteines that are inaccessible to larger physiological reductants such as GS−, thioredoxin, or glutaredoxin (34).
Knowledge on direct molecular targets of H2S is far from being complete. In addition to PTPs, strong sulfhydration has been reported for a wide range of proteins, including the highly abundant actin, β-tubulin and albumin, as well as numerous enzymes (64). Sulfhydration stimulates both actin polymerization and glyceraldehyde phosphate dehydrogenase activity.
Methodological Approaches
General approaches utilized to detect protein S-nitrosylation were recently reviewed elsewhere (15, 29). Next, we will summarize the methods utilized for the detection of S-nitrosylated and sulfhydrated PTPs.
Detection of PTP S-nitrosylation using biotin switch technique
NO- or RNS-mediated conversion of protein Cys thiols to S-nitrosothiols plays a role in a vast range of biological processes. In 2001, Solomon H. Snyder's lab developed the biotin switch technique (46), which enables easy quantification of endogenous protein S-nitrosothiols. The method consists of blocking the free Cys thiols by S-methylthiolation mediated by S-methyl methanethiosulfonate, followed by reduction of S-nitrosothiols with ascorbate, and by the in situ S-biotinylation of nascent thiols using biotin-HPDP, where HPDP stands for N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide, a reactive mixed disulfide. The degree of biotinylation obtained equals the degree of S-nitrosylation, and it can easily be detected by streptavidin or by antibody-based approaches, ideally in combination with streptavidin- or antibody-mediated pull-down (28) (Fig. 2). In addition to PTP1B (28), the biotin switch technique was successfully used for the detection of S-nitrosylated SHP-1, SHP-2 (2, 79) and mycobacterial PtpA (21, 59). The biotin switch technique was also utilized by Foster et al. (30) in their protein microarray-based S-nitrosylation analysis of S-nitrosoglutathione (GSNO)-treated cells.

Interpreting results of the biotin switch method is a bit tricky, because obtaining high-quality results requires rigorous blocking of free thiols to minimize the background signal, and as the specificity of the method is dependent on the selectivity of ascorbate to reduce S-nitrosothiols but not the oxidized or glutathionylated Cys residues. False-positive results may be prevented, in part, when the solutions used contain any chelators (typically EDTA, but also DTPA, neocuproine, etc.), because the specificity of ascorbate is limited by the transition-metal-catalyzed reactions. In particular, the outcomes of the biotin switch method are questioned when performed under oxidizing conditions, and when exposed to the sunlight (even indirect), which promotes a strong background, probably via formation of the semidehydroascorbate radical (28). Several studies addressed improvement of the biotin switch method outcomes by changing reaction buffer composition, ascorbate concentration, attempted to modify the content of catalytic metal ions present in the reaction mixture, or attempted to convert S-nitrosothiols to more stable or detectable conjugates by phosphine-mediated reactions (3, 53, 95, 100). Among the particular problems addressed are inefficient S-nitrosothiol reduction (95), sunlight exposure-induced artifacts (28), and potential reduction of disulfides (51).
Detection of PTP S-nitrosylation using the PEO probe
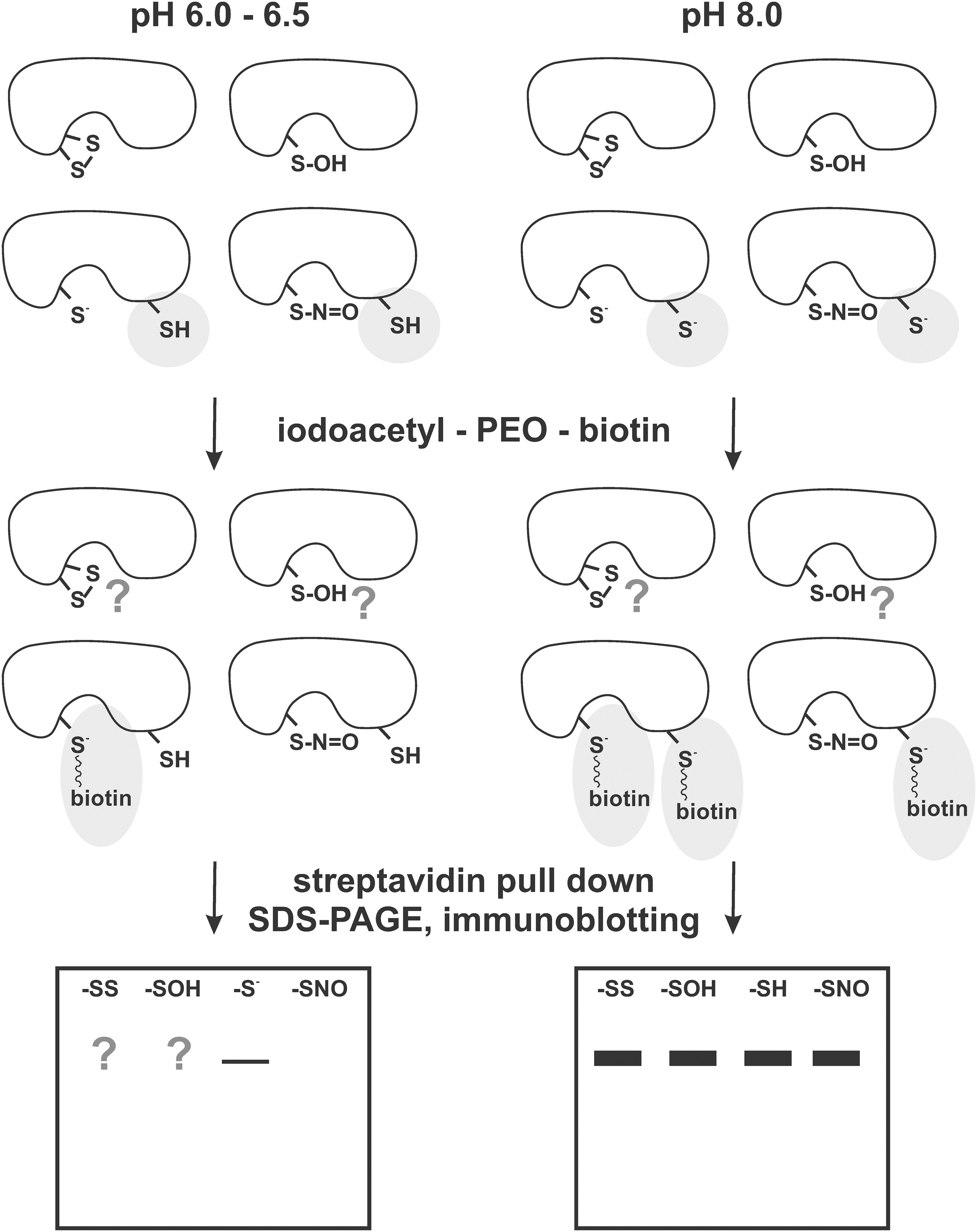
Ming-Fo Hsu and Tzu-Ching Meng criticized the biotin switch method because of its low sensitivity to S-nitrosylated PTPs (44), and they instead proposed an alternative protocol to distinguish between S-nitrosylated and reduced form of PTPs utilizing the PEO probe, which indicates the charge differences between reduced negatively charged active site Cys and the S-nitrosylated active site Cys, which loses its negative charge. At pH 6.0–6.5, the probe binds specifically to the active site Cys-S− and does not bind to the other PTP1B Cys residues present as Cys-SH. At pH 8.0, the method lacks selectivity gained from different pKa of the active site Cys when compared with the other Cys residues, and both the active site Cys and the other Cys residues present in the PTP1B molecule (or at the other proteins present in the reaction mixture) are labeled by the PEO probe, while the S-nitrosylated cysteines and also the active sites of PTP1B C215A mutants remain unlabeled (44) (Fig. 3). Thus, when used at a low pH, this method coupled with streptavidin pull-down followed by immunoblotting for specific PTPs will reveal bands when reduced active sites are present, while no PTPs would be visualized where only S-nitrosylated active sites occur. Since the PEO-probe-based detection of S-nitrosylation is indirect, it remains to be tested how the detection ratios are affected by the presence of oxidized cysteines (sulfenic, sulfinic, and sulfonic acid forms), which was not tested in the earlier pioneering study.
Detection of PTP S-nitrosylation using the DAN assay
Stuart A. Lipton's group recently reported modification of the in vitro chemical assay using 2,3-diaminonaphthalene (DAN) on recombinant S-nitrosylated SHP-2. This fluorimetric method enables measurement of S-nitrosylation on purified PTPs. After their exposure to NO donors, the PTP of interest is applied to a desalting column, and the desalted proteins are incubated with 200 μM HgCl2 and 200 μM DAN for 30 min in the darkness. NO release from S-nitrosylated proteins can be assessed spectrofluorometrically, reflecting the conversion of DAN to fluorescent 2,3-naphthyltriazole product (79).
Detection of PTP sulfhydration
Nick Tonks' lab reported H2S-induced shift in a mass of truncated PTP1B (residues 1–321) to reach 32.62 Da (from 38407.01 to 38374.36 Da) when detected by electrospray ionization mass spectrometry (ESI-MS) using a high-resolution quadrupole time of a flight mass spectrometer. The observed shift was absent in the case of the C215S PTP1B mutant, and it was reversible by treatment with dithiothreitol (50). The difference between the monoisotopic mass of cysteine-sulfinic acid (SO2H, 64.9697 Da) and sulfhydrated cysteine (S-SH, 64.9520 Da) is very minute. To distinguish between them, the use of trypsinized peptides is suggested: The native peptide (725.692) can be clearly distinguished from the sulfhydrated one (736.347, mass error −2.7 ppm) and from the sulfinic acid (736.354, mass error −1.4 ppm) at the MS1 level and by further signature MS2 fragmentation. The earlier technique was successfully used to distinguish changes in PTP1B sulfhydration after the endoplasmic reticulum stress (50).
The measurement of PTP1B sulfhydration by a thiolate anion-directed probe in a cell line has also been published (50). This method involves lysis of H2S-treated cells under anaerobic conditions, followed by a cysteinyl labeling assay utilizing a biotinylated iodoacetic acid probe forming a stable thioether bond due to the nucleophilic substitution of the halide group by the PTP-reactive thiol group (5). Saturation of the response was observed after 30 min of exposure to ∼25 μM H2S and was also detectable only after 2 min of exposure to 100 μM H2S (50).
State-of-the-Art Knowledge on Nitrosylation and Sulfhydration of Particular PTP Molecules
PTP1B nitrosylation
PTP1B probably represents the most intensively examined phosphatase in terms of its reaction kinetics, inhibition, and covalent modifications. It can inhibit insulin signaling, making it a target for type II diabetes and obesity treatment. Later, PTP1B was also reported to act in tumorigenesis, myeloid differentiation, and macrophage activation, in which it can attenuate MAPK and Akt pathways.
Low Mw S-nitrosothiols (S-nitroso-N-acetylpenicillamine (SNAP, Fig. 4,

Takakura et al. reported PTP1B to be inactivated by peroxynitrite in less than 1 s with IC50 ≤900 nM; a similar effect was obtained via cogeneration of superoxide and NO (using SIN-1), leading to peroxynitrite formation in situ. The reaction rate of PTP1B with peroxynitrite was 2.2×107 M −1 s−1, the reaction was irreversible (dithiothreitol treatment restored less than 10% of the original PTP activity), indicating that the effect was mediated by the ROS instead of RNS. The IC50 value for PTP1B inhibition by SIN-1 was 0.64 μM. Contrary to the peroxynitrite, 1 mM GSNO inhibited 50% of the PTP activity after the 30 min treatment, which was completely reversible by the dithiothreitol treatment. In addition, 400 μM DETA NONOate (producing 2 μM NO) was only slightly inhibitory (87).
NO-mediated PTP1B inactivation is reversible by >90%, and the reactivation rate by dithiothreitol or thioredoxin and thioredoxin reductase was shown to be faster by one order of magnitude than those caused by glutathione (Table 1). The reactivation rates are comparable to those achieved when using PTP1B oxidized by H2O2, but they differ from the reactivation rates recorded for sulfhydrated PTP1B (50).
Bolded numbers highlight the differences in response to the compounds used.
Adapted from Krishnan et al. (50).
H2S, hydrogen sulfide; NO, nitric oxide.
NO-induced S-nitrosylation of PTP1B active site cysteine protects PTP1B from irreversible inactivation by ROS (16). This protection can be mediated either by cell treatment with any NO donor (SNAP, GSNO, or DETA-NONOate), or by activating NO synthase.
Physiologically, increased PTP1B nitrosylation is detectable after insulin receptor stimulation (44). The administration of insulin was shown to increase S-nitrosylation of many PTPs (of the tested ones: PTP1B, SHP-1, and SHP-2), but S-nitrosylation of TC-PTP (PTPN2) was reported to be unresponsive to the insulin stimulus. Observed changes in S-nitrosylation were accompanied by changes in the PTP activity; however, the experiments were not treated for changes in the ROS-mediated changes of the same pool of PTPs (44). Importantly, the earlier changes are of pharmacotherapeutic interest, because the inactivation of PTP1B and related PTPs is associated with the enhancement of insulin responsiveness in tissues targeted by insulin. Studies on rodent models showed that eNOS inhibition causes peripheral insulin resistance, hypertension, and hyperlipidemia, and that NO donors are capable of mimicking the activity of insulin by stimulating glucose transport and metabolism in a rodent muscle (44). Thus, utilizing the earlier findings may help overcome the insulin resistance associated with diabetes and prediabetes.
Indirect NO-mediated modulation of PTP1B activity also has physiological importance. For example, caloric restriction stimulates mitochondrial biogenesis by NO-induced up-regulation of sirtuin 1 (SIRT1, NAD-dependent SIR2 deacetylase), which leads to the transcriptional repression of PTP1B in skeletal myotube cells (85) and, thus, to changes in insulin sensitivity in skeletal muscle (45). An indirect link between NO and PTP1B was also indicated by Hassid et al., who observed increased PTP1B activity or expression after SNAP treatment of aortic smooth muscle cells in a cGMP-regulated manner, which led to alterations of cell shape, adhesion, and motility (39).
PTP1B sulfhydration
PTP1B was the first phosphatase shown to be sulfhydrated (50). Sulfhydration affects its active site cysteine (Cys215, but not the other cysteine residues in PTP1B) and inhibits the PTP activity at a second-order rate 22.4±1.8 M −1 s−1. The rate of H2S-mediated PTP1B inactivation is 10±1.4 M −1 s−1, while the rate of NO-mediated PTP1B inactivation under the similar conditions is only 2.1±0.5 M −1 s−1. Further support for the functional relevance of Cys215 sulfhydration comes from the use of substrate-trapping PTP1B C215S and D181A mutants. The Nick Tonks' lab noticed that H2S treatment disrupted the PTP1B complex with its peptide substrate only in the case of the D181A mutant, while the peptide complex with the C215S mutant remained unaffected (50).
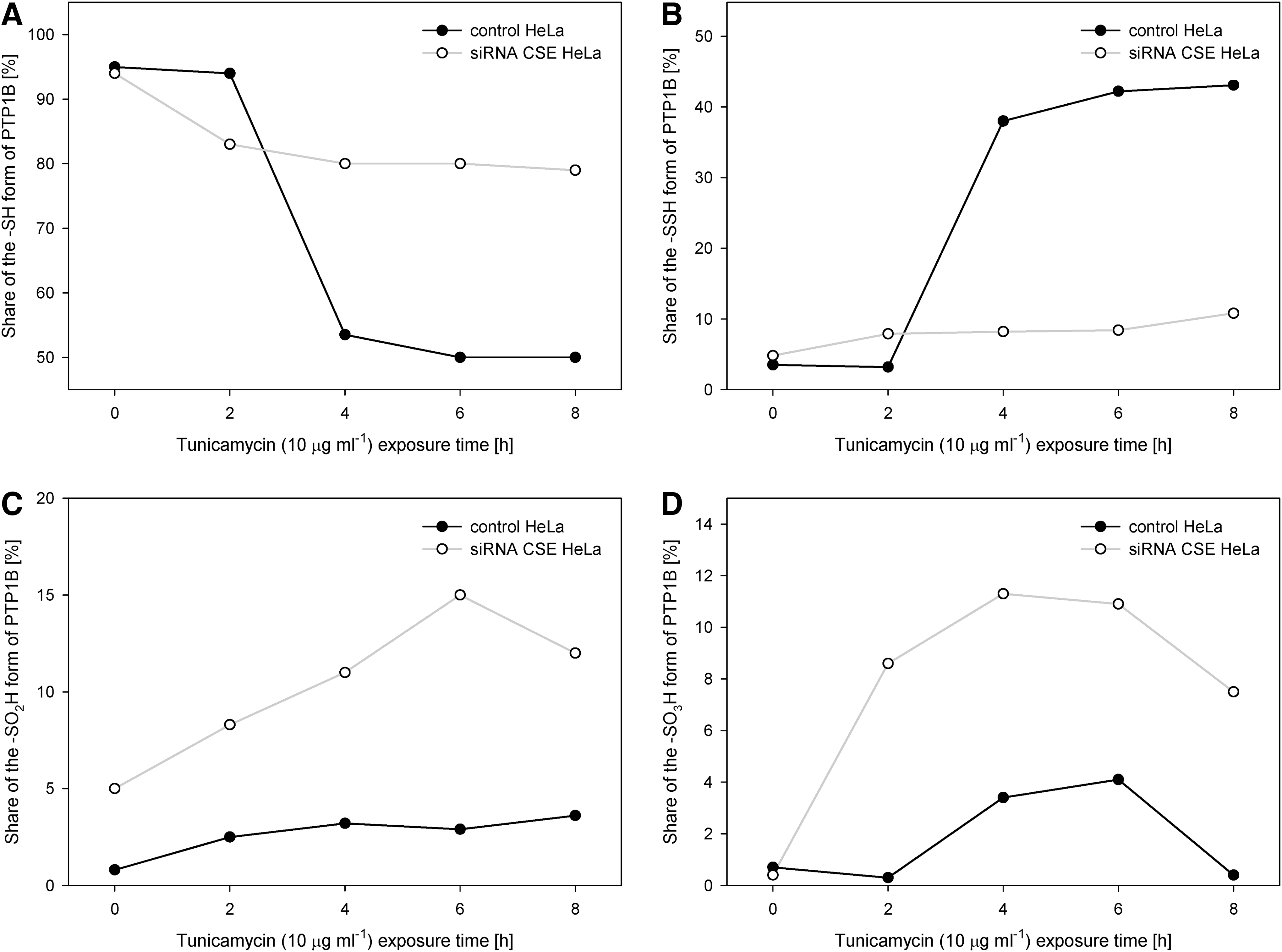
In a model of endoplasmic reticulum stress initiated by tunicamycin, an inhibitor of N-glycosylation and an endoplasmic reticulum stress inducer, a CSE-dependent increase in H2S levels resulted in the sulfhydration of ∼43% of available PTP1B active site cysteines, PTP1B activity decrease, and increased phosphorylation and activation of a direct PTP1B substrate, eIF2α kinase PERK Tyr619 (a major player in the endoplasmic reticulum stress response). The CSE-mediated PTP1B activity decrease was associated with decreased Src kinase activity, as the Src kinase inhibitory C-terminal Tyr527 serves as a direct PTP1B substrate and because the increased phosphorylation of the Src kinase C-terminal Tyr prevents the autophosphorylation of Src Tyr416. Notably, CSE silencing led to a decrease in PTP1B sulfhydration after tunicamycin exposure from 43% to only 8%–11%, while a strong increase in SO2H and SO3H forms of PTP1B was reported (50), and may be related to the competition of H2S, NO, and ROS for the limited number of available reactive cysteines (Fig. 5).
PTP1B inactivation by either H2S or NO is reversible by >90% (Fig. 6). Thioredoxin was identified as a major reducing agent of sulfhydrated PTP1B, instead of glutathione or dithiothreitol, the two widely used reducing agents of oxidized phosphatases. In all three cases, the rate of PTP1B reactivation corresponded to the linear function of the reducing agent concentration, but the thioredoxin and thioredoxin reductase were ∼190×faster than dithiothreitol (Table 1) (50).
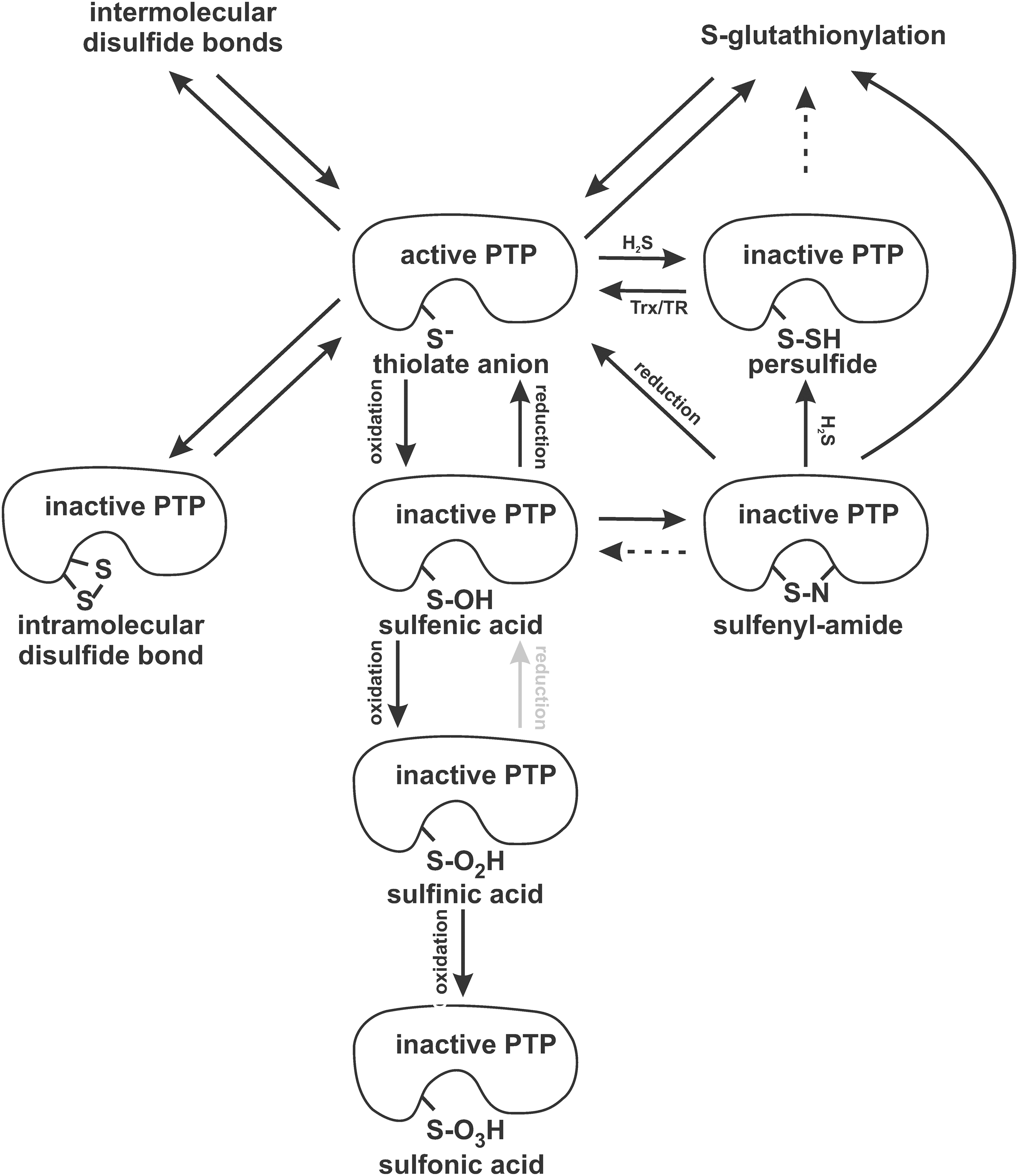
Whether PTP1B sulfhydration involves any intermediates is unclear so far. It is hypothesized that H2S/HS− may interact with H2O2 to generate HSSH, which could only react with the catalytic cysteine (7, 50). In addition, oxidized PTP1B adopting the cyclic sulfenamide conformation is considered a potential H2S target (50, 75).
S-nitrosylation of Y. enterocolitica Yop51
Yersinia Yop51 (YopH) is considered an important effector molecule of these Gram-negative enterobacteria, being injected by the pathogen to the cytosol of the host cells, where it catalyzes a rapid dephosphorylation of host proteins. In fact, the translocated phosphatase Yop51 (YopH) is considered the most active PTP known to date, being highly conserved among Yersinia pestis, Y. enterocolitica, and Yersinia pseudotuberculosis (40). Reversible NO-mediated inhibition of Y. enterocolitica PTP activity was already reported by the pioneering work by Caselli et al. two decades ago (9). Later, Xian et al. addressed the effects of low Mw S-nitrosothiols (SNAP, GSNO, glucose-SNAP-2, and S-nitroso-captopril) and single- and poly-S-nitrosylated human serum albumin on the activity of recombinant Y. enterocolitica Yop51 phosphatase, reporting time- and concentration-dependent inhibition with a second-order rate constant kinact/KI 66–613 M −1 min−1 and 472–1188 M −1 min−1, respectively, as measured in vitro in a solution of 50 mM Tris pH 7.4 and 1 mM EDTA at 25°C. They also noticed the partial prevention of S-nitrosothiol-mediated Yop51 inactivation when an inorganic phosphate was added, obtaining 33% inhibition at a 40 mM concentration of the phosphate (98).
NO-mediated dimerization of LMW-PTP Cys12 and Cys17
LMW-PTPs are 18 kDa enzymes that are identified from a broad range of eukaryotic and even prokaryotic organisms (40). Humans express two active LMW-PTP isoforms (termed IF1 and IF2, or HCPTPA and HCPTPB, respectively) and two catalytically dead variants termed SV3 and SV4 (LMPTP-C). The two catalytically active isoforms differ in the sequence flanking the catalytic site, and, thus, probably possess different substrate specificity and selectivity. Human LMW-PTP is known for its role in cell growth and differentiation, in metabolic responses and tissue sensitivity to insulin (55, 67). The LMW-PTP active site P-loop contains two cysteines (Cys12 and Cys17; the sequence CLGNICRS is conserved among all mammalian LMW-PTPs). These two cysteines can be reversibly oxidized by both NO (in vitro) and ROS (in vitro and in vivo) to form a disulfide bond (9, 17, 55).
Secreted Mycobacterium tuberculosis LMW-PTP, PtpA, is regulated by S-nitrosylation
Secreted mycobacterial LMW-PTPs, PtpA and PtpB, were recently proposed as novel targets of the antitubercular agents. PtpA is pTyr-specific, whereas PtpB acts on pSer/pThr/pTyr residues as well as on phosphoinositide substrates. Both the secreted mycobacterial LMW-PTPs are considered essential virulence factors, being involved in the process of mycobacterial infection and survival, being particularly responsible for interfering with the phagocytic activity of host macrophages by dephosphorylating the vacuolar sorting protein Vps33b (10, 14). Mycobacterial LMW-PTPs are secreted into the cytoplasm of infected macrophages, which supports their pharmacotherapeutic attractiveness, because targeting them requires only entering the macrophage but it does not require the drugs to pass the mycobacterial cell wall and the mycobacterial efflux mechanisms commonly associated with drug resistance (55).
PtpA undergoes unique S-nitrosylation mechanism, which leads to structural instability, but does not affect the catalytic cysteine (59). Although the catalytic site includes the common C(X)5R(S/T) motif, the catalytic cysteine Cys16 does not undergo S-nitrosylation. However, PtpA also possesses two additional noncatalytic cysteine residues, Cys11 and Cys53. Of these two, Cys53 was shown to be S-nitrosylated in response to a wide range of NO donors. GSNO treatment decreases the catalytic activity parameters of PtpA by half, while only Km values remain unaffected. In contrast, the C53A PtpA mutant is insensitive to GSNO treatment (Table 2) (59). The C11A PtpA mutant is inactive (21), while the C16A mutant retains the activity similar to the wild-type enzyme both under reducing conditions and after the 0.1–1.0 mM GSNO treatment. An examination of double- and triple-C/A mutants did not reveal any synergistic effects related to the S-nitrosylation status. The PtpA Cys53 nitrosylation is reversible and decreases the melting temperature by >4°C in course of the thermal denaturation, indicating changes in the stability of the protein after its S-nitrosylation (59). Cys53 is located in the variable loop in the vicinity of residues that are responsible for substrate selectivity and specificity, and, thus, its nitrosylation may affect the spectrum of proteins dephosphorylated by nitrosylated PtpA when compared with its reduced form. So far, hypotheses on the role of Cys53 nitrosylation in substrate specificity or selectivity have not been tested, as the pioneering work reporting Cys53 S-nitrosylation utilized only small-molecule substrates such as pNPP.
Adapted from Matiollo et al. (59).
GSNO, S-nitrosoglutathione.
In contrast to PtpA, PtpB is not modified by NO (21). A similar difference was previously reported in terms of the susceptibility of PtpA but not PtpB to H2O2-mediated oxidation and to oxidation-induced inhibition of their catalytic activity (24).
SHP-1 nitrosylation
SHP-1 is expressed at high levels in hematopoietic cells of all lineages, regulates negatively allergy and asthma development, is necessary for Th2 cell differentiation and Th2 cytokine production in CD4 T lymphocytes, contributes to leukemogenesis, and is suggested to inhibit insulin action and clearance in the liver. Physiologically, increased SHP-1 nitrosylation was detected after insulin receptor stimulation (44). The NO production catalyzed by nNOS was shown to nitrosylate and inhibit SHP-1 in response to low-dose ionizing radiation, H2O2, and the Ca2+ ionophore ionomycin. Mild oxidative stress was, thus, suggested to be converted to the nitrosative signals due to the better signaling properties of NO (2).
SHP-2 nitrosylation
SHP-2 is a cytosolic and nuclear tyrosine phosphatase that is responsible for a signal transduction in response to growth factor signaling, hormones, and cytokines. SHP-2 activation is associated with increased cell survival, predominantly by promoting Erk1/2 signaling.
SHP-2 is S-nitrosylated in response to externally added NO donors, for example, GSNO or S-nitrosocysteine. SHP-2 is also S-nitrosylated in response to NOS stimulation, as shown, for example, for nNOS activated by calcium ionophore A23187, which led to a ∼2.5-fold increase in S-nitrosylation of SHP-2 in HEK293 cells stably expressing nNOS (79).
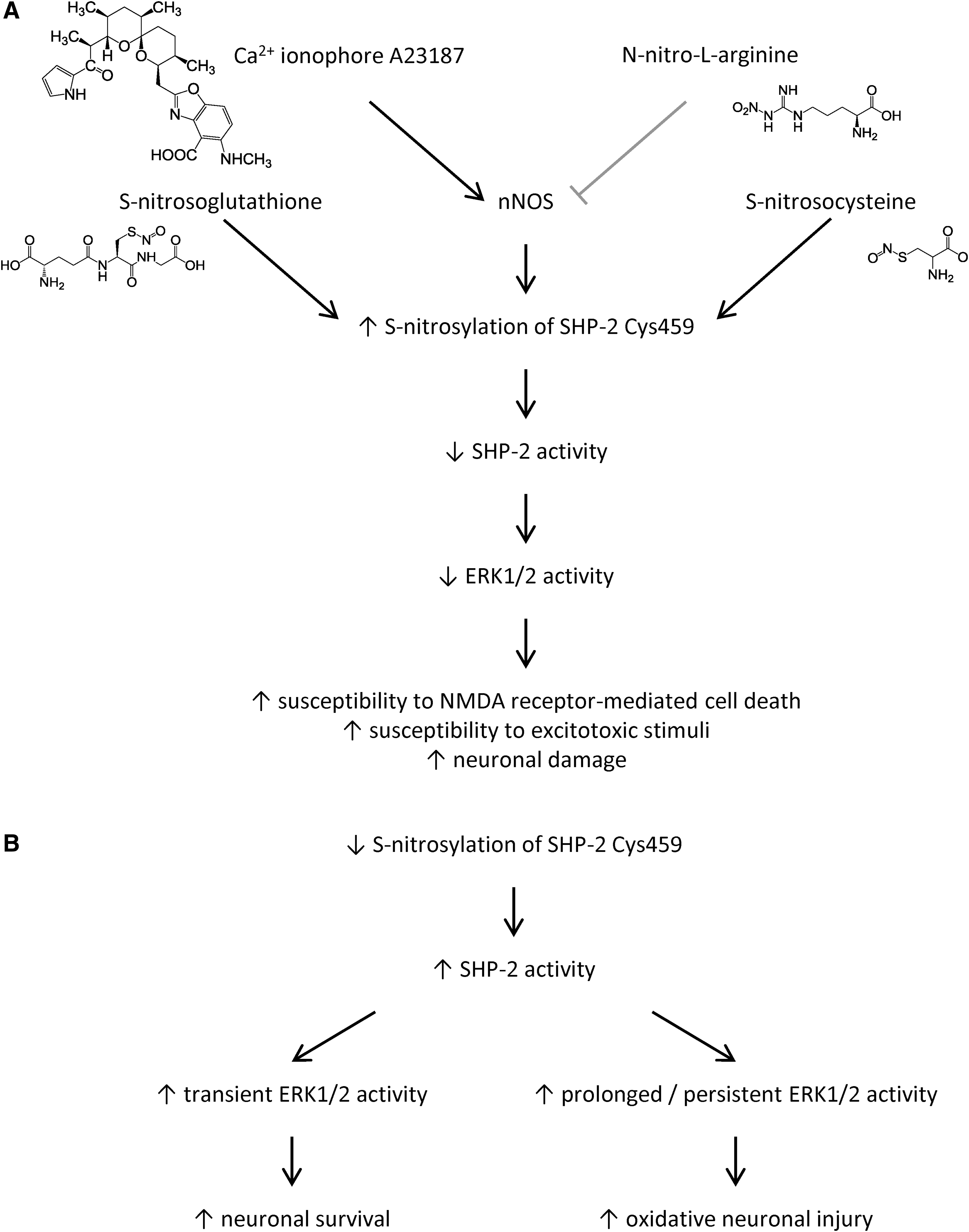
Physiologically, increased SHP-2 nitrosylation at Cys459 was detected after insulin receptor stimulation (44), and in neurons and brain tissue (79). NMDA exposure in vitro and transient focal cerebral ischemia in vivo were shown to increase levels of S-nitrosylated SHP-2, which inhibited its PTP activity, blocked the downstream MEK/Erk pathway, leading to the increased susceptibility to NMDA receptor-mediated excitoxicity (79) (Fig. 7).
LYP (PTPN22) nitrosylation
The LYP phosphatase is an important negative regulator of early-stage TCR signaling and autoimmunity. Among the prominent LYP substrates is Tyr394 of the Src family kinase Lck, which exhibits increased phosphorylation under oxidizing conditions (92). Under the same conditions, the LYP catalytic Cys forms a disulfide bond with another Cys residue that is positioned in close proximity to the core catalytic sequence, reversibly inactivating the enzyme and possibly protecting the catalytic cysteine against irreversible oxidation to sulfinic or sulfonic acid (90). Using sodium nitroprusside (Fig. 4,
The LYP phosphatase possesses nine Cys residues, two to eight of which may undergo simultaneous nitrosylation in response to sodium nitroprusside under the conditions used by Amy M. Barrios and colleagues (49). Predictably, only the catalytic Cys residue was targeted by the peptide, but changes in the ESI-MS peak induced by incubation with the peptide did not correspond with the presence of NO-adduct. Since the mass of the observed peak was 2 Da less than the parent enzyme, it suggests that a disulfide bond was probably formed between the active site and the nearby cysteine residues (49).
CD45 (PTPRC) inactivation by RNS
CD45 is well known as an enzyme that is responsible for Lck pTyr394 dephosphorylation, suppressing CD4 T+ cell lineage commitment and hyperactivity and playing a poorly defined role in myeloma cell proliferation and Alzheimer's disease. CD45 was reported to be inactivated by peroxynitrite in less than 1 s with IC50 ≤900 nM, and a similar effect was obtained via cogeneration of superoxide and NO (using SIN-1), leading to peroxynitrite formation in situ. The reaction rate of CD45 with peroxynitrite was 2.0×108 M −1 s−1, the reaction was irreversible (dithiothreitol treatment restored less than 10% of the original PTP activity), indicating that the effect was mediated by ROS instead of RNS. The IC50 values for CD45 inhibition by SIN-1 was 0.20 μM. Contrary to the peroxynitrite, 1 mM GSNO inhibited 50% of the PTP activity after the 30 min treatment, which was completely reversible by dithiothreitol treatment. In addition, 400 μM DETA NONOate (producing 2 μM NO) was only slightly inhibitory (87).
LAR (PTPRF) inactivation by RNS
LAR phosphatase is associated with insulin resistance, apoptosis, inhibition of nerve regeneration, and breast cancer metastasis. LAR was reported to be inactivated by peroxynitrite in less than 1 s with IC50 ≤900 nM, and a similar effect was obtained via cogeneration of superoxide and NO (using SIN-1), leading to peroxynitrite formation in situ. The reaction rate of LAR with peroxynitrite was 2.3×107 M −1 s−1, and the reaction was irreversible (dithiothreitol treatment restored less than 10% of the original PTP activity), indicating that the effect was mediated by the ROS instead of RNS. The IC50 values for LAR inhibition by SIN-1 was 1.05 μM. In contrast to the peroxynitrite, 1 mM GSNO inhibited 50% of the PTP activity after the 30 min treatment, which was completely reversible by the dithiothreitol treatment. In addition, 400 μM DETA NONOate (producing 2 μM NO) was only slightly inhibitory (87).
PTEN nitrosylation
PTEN is a pTyr and lipid phosphatase, preferentially dephosphorylating phosphoinositide substrates, negatively regulating intracellular phosphatidylinositol-3,4,5-trisphosphate levels, and functioning as a tumor suppressor by negatively regulating the Akt pathway. One of the PTEN isoforms, called PTEN-Long, was recently reported to be secreted as a membrane-permeable phosphatase that is able to enter cells, including tumor cells, and to induce cancer cell death in vitro as well as in vivo (43).
Similar to the LMW-PTP discussed earlier, PTEN may also undergo dimerization of the active-site cysteines after oxidative stress (55). In addition, low NO concentrations were reported to induce S-nitrosylation at Cys83, inhibiting PTEN enzymatic activity and thus stimulating the downstream Akt pathway. Similarly, the administration of GSNO to pulmonary arterial endothelial cells was reported to induce PTEN S-nitrosylation and activity inhibition reversible by dithiothreitol and ultraviolet light (8). Physiologically, the PTEN S-nitrosylation was observed in regions of ischemic mammalian brain with low NO concentrations (core and penumbra regions), while S-nitrosylated Akt was detected only at sites with high NO concentrations (ischemic core). It is hypothesized that PTEN S-nitrosylation (and thus inactivation) prevents Akt S-nitrosylation, improves cell survival by enabling hyperactivity of the Akt pathway, and is associated with neuroprotective effects (58, 66).
PTEN nitrosylation is considered as occurring at a different Cys residue than PTEN oxidation. S-nitrosylation was determined at Cys83, which is allosteric and distinct from the active site Cys124 targeted by ROS (66).
S-nitrosylation of DUSP16 (MKP-7)
DUSP16 is a JNK3 phosphatase known to bind the JNK3 adaptor protein β-arrestin2, which is being considered a prerequisite enabling eNOS-dependent JNK3 activation and subsequent endothelial cell migration. The group of Cam Patterson determined DUSP16 S-nitrosylation and inhibition as a critical step in mediating SDF-1α—induced JNK3 activation (71).
Cdc25 regulation by natural organosulfur compounds from garlic and onion
A large set of organosulfur compounds, produced by the Allium vegetables (Asparagales: Allioideae), was recently suggested to utilize Cdc25 inhibition as one of its major mechanisms of action. These compounds were broadly already utilized in traditional medicine and are known for their antiproliferative effects on various cancer cell types. In garlic and onion, numerous diallyl- and dipropyltetrasulfides are produced, showing inhibitory activity against Cdc25 isoform A and C in vitro and causing G2-M in cellulo. The dual-specificity Cdc25 phosphatases are long-known regulators of the cell cycle. Cdc25A predominantly acts in cell cycle progression into the S phase, while Cdc25B and Cdc25C are required for entry into mitosis and are frequently targeted in cancer onset and progression. The most potent natural organosulfur compounds exhibit activity of approximtaely IC50 1.1 μM (diallyl tetrasulfide from garlic) and 1.4 μM (dipropyl tetrasulfide from onion) against Cdc25C. The IC50 of these compounds against Cdc25A is higher by one order of magnitude, while the IC50 against Cdc25B is higher by more than two orders of magnitude (Fig. 8). The kinetics is relatively slow, with maximal inhibition observed at 90 min after the application of each of the compounds. The reaction can be partially blocked by dithiothreitol or glutathione, but the details of these redox-based effects remain undisclosed (93). Viry et al. claim that the organosulfur-mediated Cdc25A inhibition is irreversible (as tested by dilution of the inhibitory agents from 10 to 0.1 μM), but they did not attempt to add any of the reducing agents to the already inhibited Cdc25A (93). Thus, it is impossible to rule out the possibility that the inhibitory effects of the organosulfur compounds tested may be reverted by altering the redox status, that is, by supplementation of any of the common reducing agents.

Cdc25 serves as an attractive target of redox-oriented pharmacotherapeutic research. Redox regulation of Cdc25 action involves multiple steps, involving more than the direct alterations of the active site cysteine. Let us mention two examples. First, treatment with the S-nitrosothiol S-nitrosocysteine ethyl ester (Fig. 4,
Cdc25 phosphatase belongs to the rhodanese superfamily, which is characterized by the ability to transfer sulfane sulfur from thiosulfate to cyanide to form thiocyanate. Although the primary sequence homology of Cdc25 with the other members of the rhodanese superfamily is very low, Cdc25 possesses a rhodanese-like fork and a conserved active site loop. Similar to the PTPs, the rhodaneses also possess the catalytically essential Cys thiol in the center of their catalytic pocket, and they can be inhibited by S-nitrosothiols (some belong to the most highly S-nitrosothiol-reactive proteins) (30). In contrast to the human Cdc25 phosphatase, its yeast homologue Ygr203wp (Ych1p, yeast Cdc25 homologue 1) was reported to be resistant to S-nitrosylation (while it is sensitive to H2O2), which is considered to be caused by strong alterations in the primary sequence homology, although the catalytic loops of Ych1p and Cdc25 and rhodaneses are superimposable. In contrast to the S-nitrosocysteine, the pTyr mimetic, S-nitroso-4-mercaptophenylacetic acid was capable of inhibiting and S-nitrosylating the yeast Ygr203wp at its catalytic Cys90 (30).
Indirect Effects of NO on PTPs
In vivo experiments focusing on protein redox regulation are always challenging, because the RNS- and ROS-mediated signaling may involve other intermediates. For example, 2–500 μM peroxynitrite is known to deplete cellular glutathione and other reducing agents, and, thus, the peroxynitrite-mediated inhibition observable in vivo was shown to result from the interaction of PTP catalytic Cys residues with H2O2 instead of peroxynitrite itself at concentrations ranging between 2 and 500 μM, while the inhibition was mediated directly by peroxynitrite at the higher concentrations used (62). This is just one of the examples, and since many of the older studies addressed changes of the total redox status of PTP Cys residues rather than the presence of specific redox modifications, some of the previously reported effects claimed to be induced directly by any of the RNS or ROS applies may actually be attributed to some of their reaction intermediates. In addition, the RNS or ROS may act indirectly, affecting the activity, selectivity, or specificity of proteins upstream of the PTPs. Several examples follow.
NO inhibits enterocyte migration in an SHP-2-dependent manner
Sustained release of intestinal NO is typical for diseases of intestinal inflammation such as necrotizing enterocolitis. Inhibition of SHP-2 in the rat small intestine epithelial cell line IEC-6 was found to prevent RhoA activation, to restore focal adhesions, and to restore enterocyte migration, that is, inducing the processes otherwise commonly observed in response to NO. The observed effects occur through SHP-2-mediated activation of the Rho-GTPase-FAK signaling pathway. The exact mechanism of NO involvement in this process remains enigmatic (12).
NO affecting the PTP-PEST activity
The administration of NO increases the activity of PTP-PEST, subsequently decreases tyrosine phosphorylation of its substrate p130cas, and plays a protective role in the ROS-induced motility in cultured rat aortic smooth muscle cells. The observed effects probably do not involve PTP-PEST nitrosylation, but rather indirect NO-mediated modulation of PTP-PEST activity by the pathway involving cGMP, PKG, and Ca2+ (11). Cyclic GMP is required to mediate the effect mentioned earlier (54).
NO-induced and RPTPα-mediated cell death
High concentrations of NO donors, such as sodium nitroprusside or DETA-NONOate, promote cell detachment and cell death of the A431 human carcinoma cell line. Ectopic expression of RPTPα in these cells was shown to switch the mechanism of cell death from apoptosis to necrosis (19). The same group reported that another NO donor, SNAP, stimulated cell proliferation in fibroblasts expressing RPTPα while promoting growth arrest in cells with deleted RPTPα gene (18).
NO-mediated down-regulation of DUSP6 (MKP-3) mRNA levels
DUSP6 is a cytosolic dual-specificity phosphatase that is responsible for apoptosis induction of endothelial cells in response to TNFα, signaling through Erk1/2 dephosphorylation, and Bcl-2 proteolysis. NO was reported to destabilize DUSP6 mRNA and, subsequently, the DUSP6 protein levels, while the activity itself was reported to remain unchanged in response to NO administration (74).
Selected Examples of PTP-Mediated Changes of NO Signaling
PTP-mediated inhibition of NO production in Chagas disease
The etiological agent of Chagas disease (Trypanosoma cruzi) and another trypanosomatid harmless to humans (Trypanosoma rangeli) possess a complicated NO-based system of transfer from the intermediate to their definitive host. While T. cruzi is transmitted by vector feces deposited close to the wound caused to the definitive host by the intermediate host, T. rangeli invades salivary glands of the intermediate host and is inoculated directly into the definitive host's skin. Saliva of the intermediate hosts, such as the blood-sucking triatominae bug Rhodnius prolixus (Hemiptera: Reduviidae), contain a set of NO-binding proteins (nitrophorins) that deliver NO to host vessels and promote vasodilation, enabling efficient blood feeding of the bug. In the epithelium of bug salivary glands, the NOSs are expressed and generate NO that are aimed at entering the mammalian host's blood flow. The NO production in R. prolixus salivary glands is tyrosine phosphorylation dependent, and inhibition of the PTP activity by sodium orthovanadate was shown to suppress NO production, thus being directly linked to changes in survival and transmission rates (35).
PTP1B inhibition stimulates eNOS phosphorylation in chronic heart failure
Indirect evidence supports the inhibitory role of PTP1B in chronic heart failure-induced endothelial dysfunction consisting of decreased NO production in response to the flow-mediated dilatation. In animal models of chronic heart failure, the PTP1B inhibitors AS279, AS098, and AS713 restored flow-mediated dilatation to normal levels. The restoration was inhibited by suppressing eNOS activity. Flow induced a transient eNOS phosphorylation that was absent in chronic heart failure and is, thus, suggested to be caused by changes in PTP1B activity (but not expression) (91). Evidence for the direct involvement of PTP1B in the pathophysiology mentioned earlier is unavailable, and controls for the involvement of other Cys-based PTPs were also undisclosed.
TC-PTP (PTPN2) knock-out leads to an increase in NO production
TC-PTP is recognized as an important negative regulator of cytokine signaling. Deletion of the PTPN2 gene leads to elevated serum levels of IFN-γ and TNF-α, as well as to higher NO production. These changes are associated with higher sensitivity to the lipopolysaccharide stimulation, that is, to bacterial infection (77).
Multiple roles of SHP-1 in NO production
Host SHP-1 mediates inhibition of NO production in a model of Leishmania spp. infection. The causative agents of leishmaniasis, Leishmania spp. (Kinetoplastida: Trypanosomatida), inhibit a number of host signaling pathways in order to establish and survive in their mammalian host. For their survival, it is critical to inhibit IFN-γ—inducible activation of host macrophages, which includes inhibition of the NO production. SHP-1 is activated in course of the Leishmania spp. infection, and its activity is necessary to inhibit NO production in the host macrophages, being associated with the parasite survival and disease progression. The SHP-1–mediated inhibition of NO production occurs through the inactivation of JAK2, Erk1/2, NF-κB, and AP-1, while the STAT1 transcription factor does not contribute to the observed inhibitory effects (25). Conversely, SHP-1 overexpression was shown to inhibit iNOS expression in lipopolysaccharide-activated macrophages (38). The potential feedback loop consisting of low S-nitrosylation of SHP-1 leading to increased activity in course of Leishmania spp. infection was never examined.
The involvement of SHP-1 in iNOS regulation in the course of Leishmania spp. infection was questioned by Gerald F. Späth and his colleagues, who suggested that the footpad infection model generates such a high inflammatory background response that it completely confounded any possible outcomes related to changes in iNOS expression and activity, which then translates into the lack of any effects on intracellular Leishmania spp. survival in SHP-1 deficient mice when using an alternative model of Leishmania spp. inoculation into the rump (81).
In addition to the indirect effects of SHP-1 on NO production documented repeatedly in the course of leishmaniasis, SHP-1 may act directly on the nitric oxide synthases. The platelet eNOS was shown to be regulated by SHP-1–mediated dephosphorylation on its tyrosines, while the same enzyme can be regulated in a phosphorylation-independent way by changing thrombin concentrations. Platelet stimulation by thrombin further increases SHP-1 association with eNOS. Association of SHP-1 with eNOS was confirmed in both resting and thrombin-activated platelets, but it was not detected in the endothelial cells (68). Platelet treatment with 0.1–1.0 mM sodium orthovanadate was shown to increase tyrosine phosphorylation of eNOS, leading to the decrease of basal eNOS activity and the blunting of thrombin-induced guanosine 3′,5′-monophosphate (cGMP) formation; the vanadate-induced changes were abolished when the platelets were preincubated with the Src kinase inhibitor PP2, but not by the PKC inhibitor Ro-31-8220 or PI3K inhibitor wortmannin (61).
In central nervous system glia, SHP-1 was shown to inversely regulate iNOS and arginase I and to control virus infection. In SHP-1–deficient glia, viral mimic dsRNA was reported to suppress iNOS expression and NO production despite the levels of mRNA for iNOS remaining high, while it induced high expression of arginase I. Similarly, IL-4/IL-10-induced expression of arginase I correlated with decreased NO production and increased virus replication in SHP-1–deficient astrocytes (6). Although the mechanisms remain enigmatic, SHP-1 arises as an important regulator of NO production and, thus, of innate immunity in multiple cell types.
SHP-2 decreases NO production
In endothelial cells, SHP-2 affects eNOS expression in response to blood flow and VEGF. The mechanism involves protein kinase A and Akt (20, 47). In response to insulin, endothelial SHP-2 is phosphorylated at one of its C-terminal tyrosines (Tyr542), up-regulates its own expression in a p38 MAPK-dependent manner, and then up-regulates the expression and activity of arginase II, which leads to a decrease in cellular NO levels (36). Although the mechanisms are unclear, several indices suggest that at least some of the observed effects are mediated by the ability of SHP-2 to serve as a scaffold adaptor molecule and are unrelated to the SHP-2 phosphatase activity (20).
DUSP1 (MKP-1) knock-out leads to an increase in NO production
Similar to what was reported for TC-PTP, knock-out of the dual specificity phosphatase DUSP1 stimulates the production of various cytokines and chemokines, including TNF-α, IL-6, IL-10, and MIP-1α and potentiates NO production in response to living or heat-killed Gram-positive bacteria, to the peptidoglycans isolated from them, or to lipoteichoic acid. Bacterial load or survival rates (when infected by Staphylococcus aureus) were reported to remain unaffected by the DUSP1 deletion, but the tissue damage and immune cell activation and recruitment were substantially potentiated (96). Similar to SHP-1, the DUSP1-mediated iNOS inhibition is also tightly synchronized with the up-regulation of competing arginase pathway. In response to endotoxin (lipopolysaccharide) challenge, the DUSP1 was shown to switch the arginine metabolism from NO production to urea production through the regulation of iNOS expression (whereas the arginases were expressed at constant levels independently of the DUSP1 expressions status), but the phosphorylation status of iNOS was not tested (65).
Conclusions and Further Directions
The modulation of PTP signaling by RNS and H2S serves as a basis for the unique and yet underexplored method of tight and flexible regulation of these fundamental enzymes. The specificity and selectivity of the methods used to detect nitrosylation and sulfhydration remains to be corroborated, while further development of robust and straightforward proteomic methods is needed to lift the fog on the full spectrum of proteins that are responsive to Cys S-nitrosylation, sulfhydration, and other RNS- or H2S-induced modifications. Results of the hitherto performed studies on RNS- or H2S-mediated PTP signaling await translation into the clinical medicine and pharmacotherapeutics. In addition to directly affecting the activity of particular PTPs, the use of reversible S-nitrosylation as a protective mechanism against oxidative stress should be of high interest.
Remaining nearly completely unexplored is the possibility that Cys nitrosylation or sulfhydration may not necessarily be associated only with inactivation of the respective PTP, but that it may instead change its catalytic properties or substrate recognition. The first indices of the possible existence of such mechanisms stem from the oxidation of yeast PTP Sdp1, which increases its catalytic activity by enabling substrate recognition (32), and the detection of activity changes in response to oxidation or nitrosylation of other than catalytic Cys residues in M. tuberculosis PtpA (59).
The diversity of covalent cysteine modifications clearly joins those involving other amino acids serving as signal transduction targets, particularly lysine, which undergoes competing reactions involving acetylation, sumoylation, or ubiquitinylation (23). The complexity and clinical relevance of NO-mediated and especially of H2S-mediated signaling is far from being satisfactorily understood, and we may soon face extensive progress similar to that experienced in ROS-mediated alterations of tyrosine (de)phosphorylation during the recent two decades because of the discovery of the essential role of ROS in the growth factor signaling (1, 86).
Innovation
This is the first review addressing the emerging topic of PTP activity modulation by RNS and H2S. Redox modifications of thiols serve as a molecular code enabling precise and complex regulation of PTPs and other proteins. Particular gasotransmitters and even the redox modifications themselves affect each other, of which a typical example is S-nitrosylation-mediated protection against the further oxidation of protein thiols. In addition to directly affecting the activity of particular PTPs, the use of reversible S-nitrosylation as a protective mechanism against oxidative stress should be of high interest.
Footnotes
Acknowledgments
The study was supported by the UNCE Project 204015 and by the PRVOUK Project P31/2012 from the Charles University in Prague, by the Czech Science Foundation Project P301/12/1686, and by the Internal Grant Agency of the Ministry of Health of the Czech Republic Project NT13663-3/2012.