
Abstract
Introduction
T
The ectodomain contains several conserved residues accounting for the formation of three ATP binding sites at the subunit interface and nonconserved residues accounting for the formation of numerous allosteric binding sites. The term “orthosteric sites” is used to describe ATP binding sites on P2XRs, because they are the primary binding sites for the native ligand needed for the conformational changes that allow for the opening of the channels. Consistently, orthosteric agonists other than ATP substitute for ATP in binding and gating and are called full or partial agonists, depending on the efficacy of coupling. Orthosteric antagonists bind the same sites and do not trigger activation of receptors but compete with agonists for binding to these sites; these are also called competitive inhibitors. For their detailed description, see Coddou et al. (35). The “allosteric sites” are topographically distinct from the orthosteric sites that are recognized by ATP. Occupancy of these binding sites alone has little or no ability to trigger P2XR pore opening, but could modulate the channel activity in the presence of ATP. Allosteric perturbation arises not only from the binding of small or large molecules extracellularly, but also from changes in temperature, ionic strength or concentration, and from covalent modification tethering, glycosylation, phosphorylation, and ubiquination, which could take place extracellularly, in the TM region, or intracellularly. Allosteric regulators of P2XRs and the corresponding literature are listed in Table 1.
Biphasic effects.
Additional positive effect in recovery from desensitization.
+, potentiation; −, inhibition; NE, no effect; P2XRs, purinergic P2X receptors; CO, carbon monoxide; ALP, allopregnanolone (3α,5α)-3-hydroxy-pregnan-20-one; THDOC, tetrahydrodeoxycorticosterone (3α,21-dihydroxy-5α-pregnan-20-one).
The short TM domains of P2XRs provide a pathway for cation influx. P2XRs are nonselective cation channels with high calcium permeability and the depolarizing nature of current, leading to additional influx to calcium in cells expressing voltage-gated calcium channels (141). The TM2 plays a dominant role in receptor activity, such as channel assembly, gating, ion selectivity, and the permeability of monovalent and divalent ions. Residues contributing to the formation of the P2XR pore gate are not conserved, further indicating that the same function could be achieved by different structures (52, 93, 109, 110, 155). Both TM domains contain residues that are important for ivermectin action in P2X4R and effects of alcohols on channel gating, which were recently reviewed (35, 94).
The N-terminus of all P2XRs is relatively short (20–30 amino acids), whereas the length of the C-terminus ranges from 26 amino acids (P2X6R) to about 239 residues (P2X7R). Both the N- and C-termini serve as molecular targets for a series of post-transcriptional and post-translational modifications, including RNA splicing and protein–protein interactions (141). The C-termini exhibit only sequence relatedness for the first 25 amino-acid residues, and the C-terminal of P2X7R contains a unique Y358-E375 sequence that contributes to the transition from the open to dilated state (81, 194). The N- and C-termini also contain conserved phosphorylation sites for cAMP-dependent protein kinase A (PKA) and protein kinase C (PKC) and other important regions that are responsible for the regulation of P2XRs. Finally, the C-termini contain several motifs that are involved in trafficking and stabilization of the receptors in the plasma membrane and specific protein interactions (98, 151, 180), as well as motifs which are responsible for the regulation of P2XR activities by lipids, a topic recently reviewed (35).
ATP is synthetized in mitochondria as a key product of the oxidative phosphorylation, which is energetically driven by the electron transport, and is released extracellularly through still not well-characterized mechanism. The byproducts of the electron transport are the reactive oxygen species (ROS). Both ATP and ROS are generated in mitochondria in a simultaneous and concerted manner, and ROS is also generated by other mechanisms (19). ATP and its metabolites and ROS play roles in both intracellular and extracellular signaling; our article summarizes the current knowledge of signal crosstalk between P2XRs and ROS. We also review binding sites in P2XRs for essential trace metals, divalent cations and protons, as well as the available data on intracellular regulation of these receptors by calcium and protein kinases. Moreover, protein–protein interactions with either P2XRs or other ligand-gated ionic channels are briefly covered. The reviewed data indicate that allosteric modulation mechanisms together represent a significant diversification of channel structure and activity depending on time and place of receptor activation and patho/physiological environment.
Tridimensional Organization and Gating Properties of P2XRs
Early biophysical experiments suggested that three molecules of ATP were required for channels gating (10). Biochemical (137), pharmacological (165), atomic force microscopy (8), and electron microscopy (197) have corroborated the trimeric nature of P2XRs. A recent solution of the crystal structure confirmed trimeric organization of zebrafish P2X4R (zP2X4R). To describe the structure of P2XR, the authors compared it with the shape of a dolphin with head domain, body segment, left and right flippers, and dorsal fin, with its tail submerged within the lipid bilayer. The ectodomain projects 70 Å above the plasma membrane, where body segments of three subunits mutually intertwine, forming three vestibular cavities along the central axis of the channel (91). A comparison of the zP2X4R with acid-sensing ion channel structure revealed similarity in pore architecture and vestibule polarity (67). The crystal structure confirmed the existence of all five disulfide (SS) bonds (29), with the first three bonds located in the head domain, the SS4 is located within the dorsal fin, and the SS5 at the ends of two β-sheets in the low body region. Crystallization of zP2X4.1R also confirmed the hypothesis that the TMs adopt α-helical structures forming an hourglass shape (91). The homology model of rat (rP2X4R) shows that the TM2 domains form a triangular iris, which represent the pore structure of the closed channel; whereas the three TM1 domains are located parallel to the central axis of the channel on the periphery of TM2 (163).
Numerous scanning mutagenesis studies resulted in a similar conclusion, identifying seven residues to have the potential to contribute to the formation of the ATP binding site (P2X4R numbering): K67, K69, F185, N293, R295, and K313 (53, 82, 149, 195, 199). All of these residues are also present in distantly related P2XRs (algae, slime mold amoebae, and choanoflagellates) (167). The zP2X4.1R crystal structure in open state confirmed that the key residues accounting for ATP binding are located about 45 Å from the plasma membrane. The K67, K69, N293, R295, and K313 residues are critical for recognition of α, β, and γ phosphates, and the adenine moiety lies deeper in the binding cavity. These interactions provided a rationale as to why P2XRs are specific for ATP rather than cytidine 5′-triphosphate (CTP), uridine 5′-triphosphate (UTP), uridine 5′-diphosphate (UDP), or guanosine 5′-triphosphate (GTP) (70). Recent electrophysiological and fluorimetric studies have addressed in detail the ATP binding site and movements of ectodomains regions on receptor activation using an ATP-derived thiol-reactive agonist, NCS-ATP; a compound that covalently binds to the receptor orthosteric site. It has been shown that ATP may have distinct binding orientations within the binding site (83, 85, 116).
ATP gates rP2XRs in a receptor-specific manner, with the EC50 values ranging from nanomolar to submillimolar concentrations (Fig. 1A). Among receptors, P2X1R and P2X3R have the highest affinity for ATP with an EC50 value in a submicromolar concentration range. ATP is also a full agonist for P2X5R with estimated EC50 values ranging from submicromolar to low micromolar concentration range. P2X2R and P2X4R are also fully activated but less sensitive to ATP, with EC50 values in a low micromolar concentration range. The P2X7Rs is the least sensitive member of the P2XR family to activation by nucleotides, with the EC50 value for ATP in a high micromolar to low millimolar concentration range (35, 94).

The gating of P2XRs usually consists of three phases: a rapid rising phase of inward current induced by the application of agonist (activation phase), a slowly developing decay phase in the presence of an agonist (desensitization phase), and a relatively rapid decay of current after ATP is removed (deactivation phase). The gating kinetics is also receptor specific. Figure 1B illustrates the profile of rP2XR currents in response to supramaximal concentrations of ATP. P2X1R and P2X3R rapidly activate and desensitize, P2X2R and P2X4R slowly desensitize, whereas P2X7R does not show an obvious desensitization but exhibits the secondary current growth (35). Rat P2X5Rs generate low-amplitude nondesensitizing currents, and P2X6R does not express well at the plasma membrane (37). Figure 1 clearly indicates the inverse relationship between the EC50 and desensitization constant values, suggesting that the strength of agonist binding influences receptor desensitization.
Redox Signaling and P2XRs
The redox signaling molecules include both oxygen and nitrogen reactive species, termed ROS and RNS. The oxygen derivate molecules include superoxide anion radical (O2 −), hydrogen peroxide (H2O2), and the highly reactive hydroxyl radical (−OH). The nitrogen derivate molecules include nitric oxide (NO), peroxynitrite, nitrogen dioxide, and dinitrogen trioxide (65). In mammalian cells, the physiological generation of ROS occurs in different subcellular compartments: (i) mitochondria (mainly complex I and III); (ii) endoplasmic reticulum (mainly cytochrome P-450 and b5 enzymes); (iii) peroxisomes (mainly fatty acid breakdown); (iv) cytosol (NO synthases, lipoxygenases, prostaglandin endoperoxide (PGH) synthase, and phospholipase A2); and (v) plasma membrane (NAPDH oxidases, lipoxygenase) (19).
During normal metabolism, mitochondria generates ROS as a byproduct of ATP production, but the amount of ROS produced by this mechanism is controversial (19). Phospholipase A2 and lipoxygenase generate H2O2 from the breakdown of fatty acid (105). Xanthine oxidases catabolize the oxidation of xanthine in uric acid, generating two molecules of H2O2 as a byproduct (121). NO synthase catalyzes the conversion of
ROS at high concentrations react with proteins, lipids, carbohydrates, and nucleic acids, causing irreversible functional changes. All of these molecules can induce transient or permanent changes at target proteins through oxidation or reduction of cysteines' sulfhydryls or disulfides. Thiols can be irreversibly oxidized to sulfinic and cysteic acids (65). These irreversible forms of sulfhydryl oxidations are associated with oxidative stress and cell damage. However, during evolution, organisms develop mechanisms to use ROS in physiological processes. At the cellular levels, ROS regulate growth, apoptosis, and intracellular signaling. Several proteins are known to function as effectors for ROS, which are sensitively and reversibly oxidized by these compounds. This includes phosphatases, transcriptional factors, and ion channels. Such ROS-effector proteins commonly have a highly reactive cysteine residue, which reversible oxidation provides transmission of signaling to downstream targets (124).
The relationship between redox signaling and P2XRs has not been well characterized. Early studies documented that the activation of P2X7R increased ROS production in diverse cell types, such as human neutrophils (166), rat microglia (122, 142), rat submandibular glands (57), mouse erythroid cells (178), primary gingival epithelial cells (23), and cultured cortical neurons (138). In concentrations too low to activate P2X7R, ATP also promotes ROS production in mouse myoblasts (157). Extracellular ATP also evokes NO production in type I spiral ganglion neurons through activation of P2XRs (198). A recent report suggests that the P2X4R can indirectly influence ROS production by regulating the P2X7R functions in mouse macrophages (90).
The NADPH oxidase plays a dominant role in P2X7R-induced ROS formation in macrophages and microglia, as estimated in experiments based on the use of pharmacological inhibitors of the NADPH oxidase (72, 122). Experiments with NOX2 knockout mice also support the role of the NADPH oxidase in P2X7R-induced ROS formation in macrophages (127, 139). Moreover, activation of P2X7R in microglia triggers the translocation of NOX2 to the plasma membrane, which is a critical step in the activation of the NADPH oxidase (142). Other studies have proposed that P2X7R-mediated increase in ROS production in human monocytes depends on the MEK/ERK cascade and likely involves the phosphorylation of NOX2 (104). Role of PKC and MAP kinase was also implicated in stimulation of NADPH oxidase and the subsequent generation of ROS in mouse submandibular cells (158). Although the exact mechanism of P2X7R increase in NADPH oxidase activity and ROS production is not known, it seems that the C-terminal domain of the receptor plays a key role because mutant receptors that do not contain specific sequences in this domain are unable to increase ROS production (139). It also appears that P2X7R can trigger mitochondrial ROS production by overloading cells with calcium (178). On the other hand, the action of P2X7R on NO production in THP-1 cells is indirect, by facilitating NO synthase expression (103).
Redox signaling could play a key role in rapid P2X7R induction of interleukin-1β (73), which appears to be coupled to PGE2 release (7). Moreover, it was shown that P2X7R-mediated apoptosis was mediated by ROS production and subsequent activation of the ASK1-p38 MAPK pathway in mice macrophages (139). The P2X7R activation-associated ROS formation is also suggested to account for cell death in murine EOC13 microglia (9). However, in erythroid cells, the P2X7R-induced ROS production is not coupled to apoptosis (178). In cortical neurons, P2X7R-triggered Ca2+ influx and the coupled mitochondrial dysfunction accounted for cell death independently of ROS signaling (138). It has also been suggested that activated P2X7R eliminate Toxoplasma gondii tachyzoites from infected macrophages by ROS (41) and contributes to tissue factor-dependent thrombosis in mice (61). A general scheme of P2X7R-mediated increase and in ROS and ROS-mediated function is shown in Figure 2.
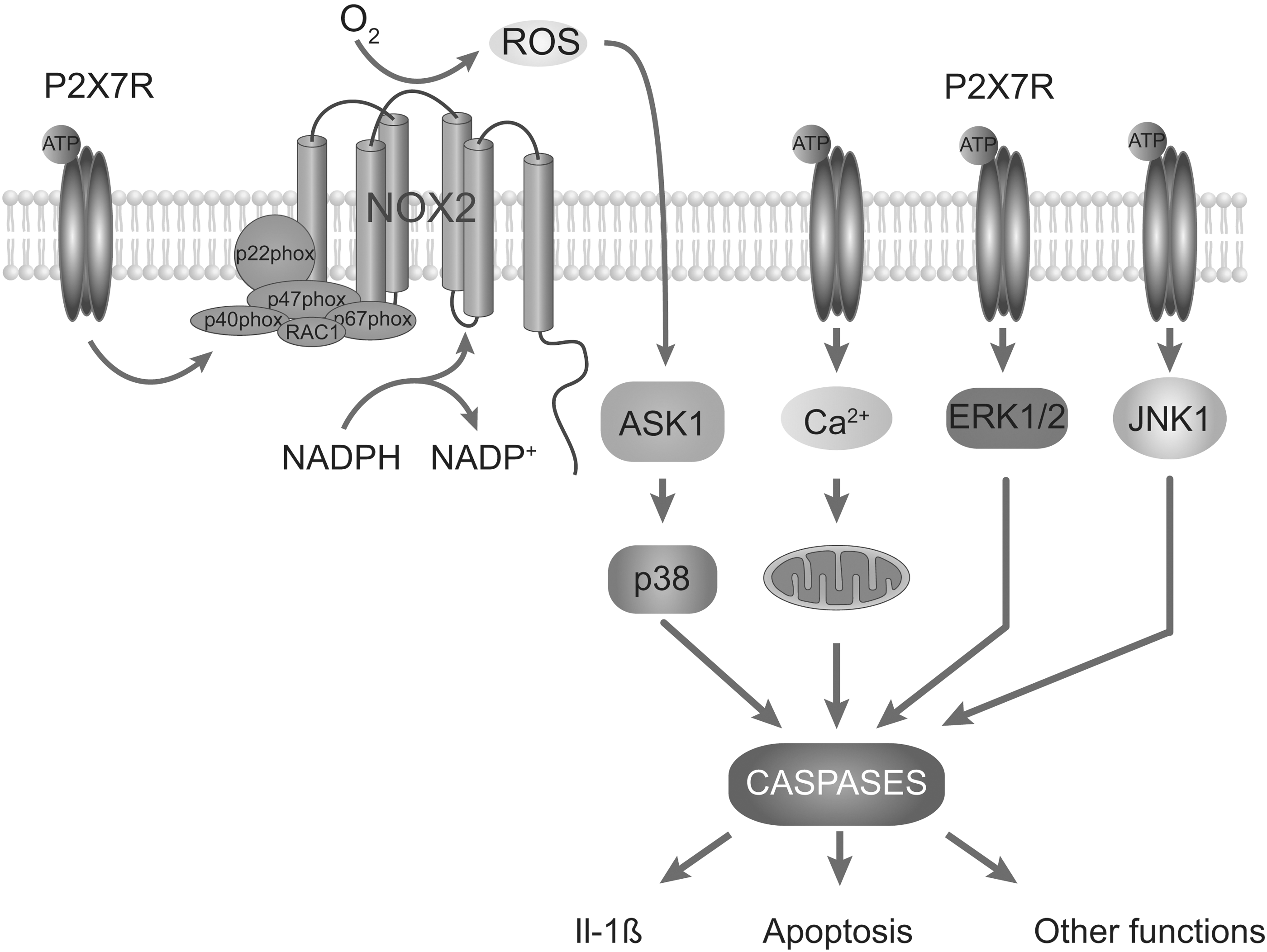
As many other ionic channels (see other articles in this Forum), the activity of P2XRs is affected by changes in the redox balance of cells. Our group reported that H2O2, mercury, and the mitochondrial stress inducers potentiate the activity of rP2X2R. In HEK293 cells, the effect of H2O2 is rapid, reversible, and was prevented with the antioxidant agent N-acetyl-cysteine. Moreover, H2O2 potentiation was only observed in the full-length P2X2aR but not in the shorter P2X2bR. Using site-directed mutagenesis, we further identified a putative intracellular redox sensor, C430. In contrast, the currents mediated by the P2X4R were inhibited by H2O2, suggesting that redox modulation is allosteric and receptor specific (32). These findings indicate that P2XRs can modify their activity depending on the redox state of the cell, as illustrated in Figure 3.
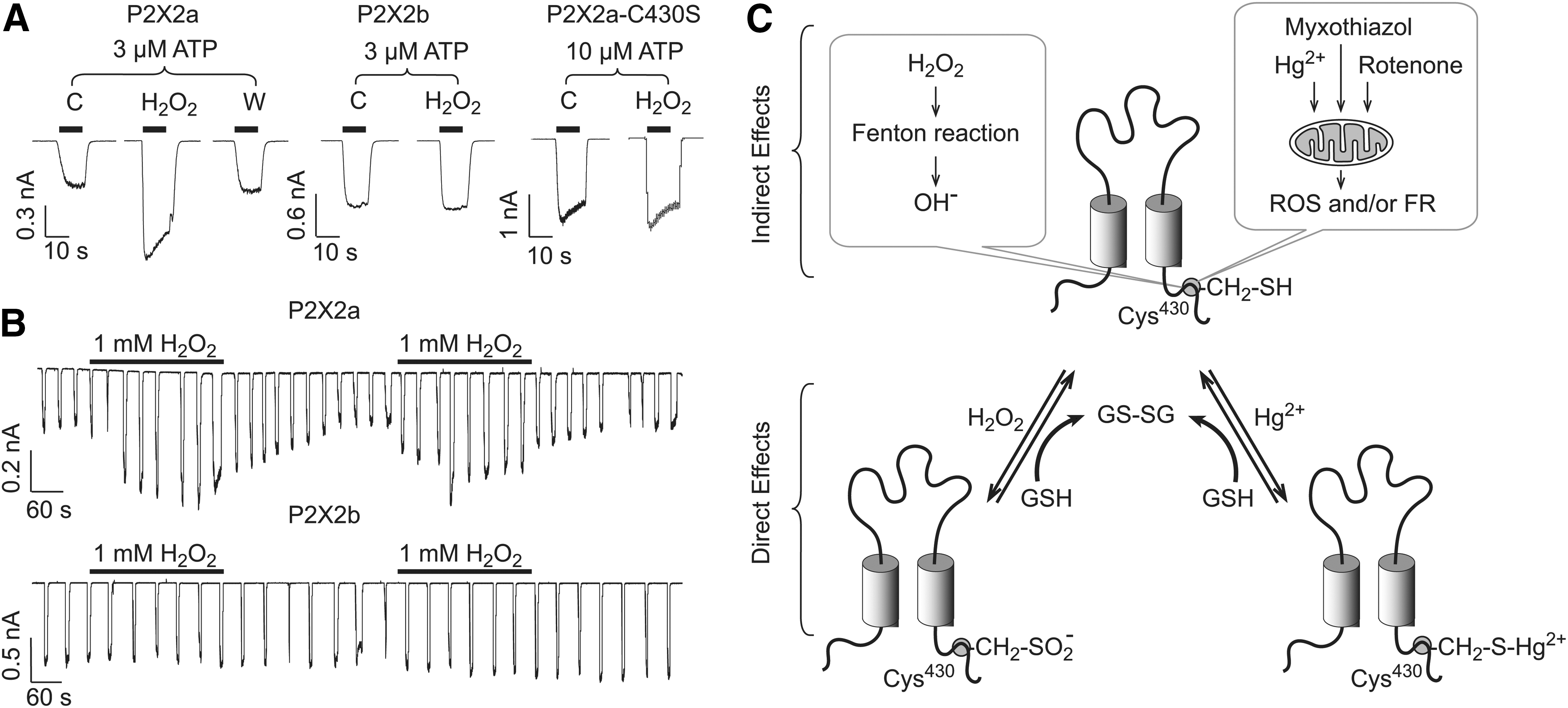
Our results appear to be in conflict with Mason et al. (120), which could reflect experimental differences: (i) we used transient transfected cells, in contrast to stable transfection; (ii) our experiments were done using isolated cells less than 24 h after dissociation, in contrast to nonisolated cells grown in coverslips for 1–5 days; (iii) in contrast to their study, we did not used any pore-forming antifungal in our cell culture to avoid long-term calcium leak or depolarization; (iv) we used divalent and ATP free intracellular buffer, whereas the other group used a different intracellular buffer, which included calcium, ATP, and magnesium, all of which can induce long-term changes in P2X2R activity; (v) our study showed that H2O2, myxothialzol, and rotenone reversible potentiated P2X2R-mediated currents in both HEK 293 cells and Xenopus laevis oocytes; and the Mason et al. (120) study found an initial increase followed by an irreversible decrease in currents after acute application of H2O2, myxothiazol, and rotenone (mitochondrial agents that increase ROS production).
Some reports indicate that ROS and NO can stimulate purinergic signaling in endogenous tissues. For example, in rat vagal lung afferent fibers, H2O2 and hydroxyl radical increase fiber activity and this effect can be prevented with PPADS (154), and ATP-induced NO plays an important role in purinergic signaling in the cochlea (69). When H2O2 was delivered by inhalation to rats, it induced reflex bradypneic responses that are reduced by 50% by PPADS (153). However, it is also possible that ROS have additional effects, increasing ATP release because the bradypneic responses can also be prevented by ATP scavengers (153). Similarly, it has been reported that H2O2 can alter the binding properties of ATP to purinergic receptors cardiac sarcolemmal membranes (129). Recently, an increase of P2X7R-generated current densities in astrocytes treated with rotenone for 48 h (62) has been reported, an effect that could be mediated by ROS. Interestingly, similar effects to that observed with ROS have been described for the gas carbon monoxide; a potentiation of P2X2R-mediated currents; and a modest inhibition or no effect in other subtypes (184). It is known that carbon monoxide can stimulate mitochondrial production of ROS (144), and, therefore, this could explain the similar results between these two studies.
Copper and Zinc as Allosteric Modulators
A common aspect of P2XRs is their regulation by essential trace metals, including copper and zinc (Table 1). In general, the nature of trace metal actions, positive or negative, depends on the subtype of receptor and species differences (Fig. 4). In contrast to the ATP-binding sites, the amino acids involved in P2XR trace metal coordination are not conserved and do not appear to be aligned among P2X subunits. This may provide additional rationale for the opposing effects of copper and zinc on receptor function. Experimental observations also support a hypothesis that there are at least two distinct and separate binding sites for trace metals, at least in P2X2R and P2X4R, and that both sites can be occupied by different elements with variable affinities (35).
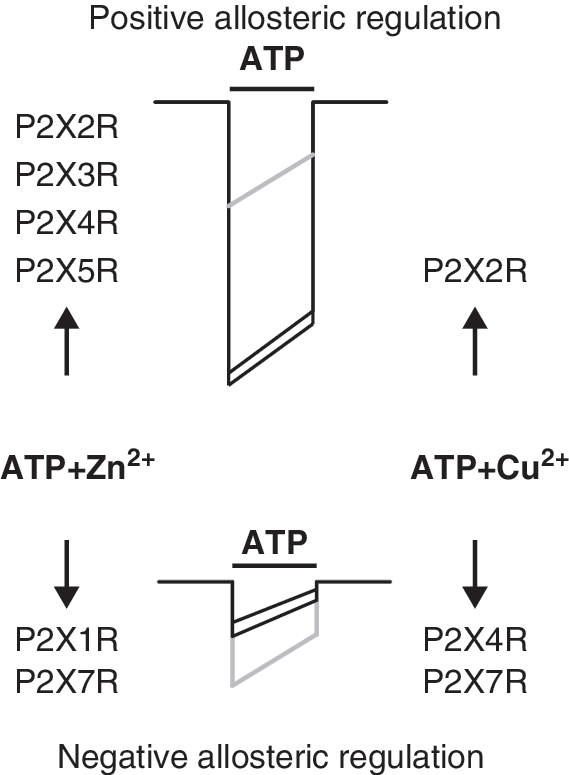
Both zinc and copper exhibit a biphasic effect on rP2X2R function; a potentiating effect at micromolar concentration ranges; and inhibition at millimolar (35). Zinc also potentiates the ATP responses of native rP2X2Rs and rP2X2/3Rs expressed in neurons from parasympathetic ganglia (118), hypothalamus (177), and dorsal motor nucleus (173). Unlike rat and mouse P2X2Rs that are potentiated, human P2X2R (hP2X2R) is inhibited by zinc (169). Site-directed mutagenesis revealed a critical role of extracellular H120 and H213 in the positive modulation by zinc and copper (28, 115). Histidines 192, 245, and 319 are also partially involved, probably contributing to the stabilization of the allosteric site (115). The allosteric sites for zinc and probably copper are formed by H120 and H213 of adjacent subunits (130), a finding confirmed with the crystallization of P2X4.1R (91). The flexibility of the zinc binding sites of rP2X2R was also tested by shifting two essential histidines 13 residues upstream or downstream from their original position. The ability of zinc to potentiate the mutated channels followed the order H120>H121>H119 and H212>H213>H211 (170). In contrast, it has been difficult to identify the residues that compose the zinc/copper inhibitory site at rP2X2R (28, 59). It appears that high-potency zinc modulation of hP2X2R and low-potency zinc modulation of rP2X2R share a common molecular mechanism (148).
The tridimensional structure of the P2X2R homology model indicates that histidine side chains as coordination ligands of trace metals are spatially close to the ATP binding site, allowing us to propose that these metals modify the ligand binding (35). Figure 5 depicts the histidine moiety (H120 and H213) in the proximity of the ATP-binding site, which differs between the open and closed state of the channel. In the closed channel, the distance between the coordination nitrogens of histidine rings is 2.5-fold longer than in the open channel. Moreover, recent studies have shown that in mutant receptors modified with thiol-reactive fluorophores, zinc is able to induce the opening of the channels even in the absence of ATP (48). Similarly, in P2X2Rs containing the T339S mutation that results in spontaneous openings of the channels, zinc is also able to gate currents in the absence of ATP (84) These studies strongly support the idea that the zinc allosteric site at P2X2R is spatially and mechanistically close to the ATP-binding site.

The unique characteristic of P2X4R is its differential modulation by essential trace metals; zinc potentiates receptor activity and copper inhibits it (Fig. 4 and Table 1). Zinc-dependent potentiation is due to an increase in ATP potency, whereas copper decreases the current amplitude without affecting the potency of the agonist. At higher concentrations, zinc inhibits P2X4R function via a reduction in ATP efficacy (2, 183). Among the ectodomain histidines, only the mutation of H140 abolishes the inhibitory action of copper and modifies the pattern of zinc action from a bell-shaped curve, typical of biphasic modulation, to a sigmoid curve, accompanied by an increase in the amplitude of response (34). Subsequent studies revealed that D138 is a second component of the P2X4R inhibitory site, whereas C132 is suggested to account for the positive zinc allosteric effect (31). The homology model of rP2X4R suggested that the H140 residue suggested it is located close to the proposed ATP-binding site which includes N293, R95, and I313 from the same subunit and K67, K69, and F185 from the adjacent subunit (35). Thus, it is reasonable to speculate that D138 and H140 from the same subunit represent the P2X4R-specific region accounting for the inhibitory effects of copper and high concentrations of zinc. The finding that the replacement of C132 abolishes the potentiating effect of zinc also raises the possibility that the SS3 bond is important for zinc potentiation or that this bond is not permanent, but instead breaks and reforms (35). A recent study has indicated additional roles in zinc potentiation for SS1 and SS4; mutations of cysteines that form these bonds resulted in zinc-resistant receptors. Moreover, mutations on cysteines from SS2 resulted in receptors with increased sensitivity to zinc potentiation (108), a similar effect to that observed by our group with the mutation of the inhibitory site (H140, D138). This suggests that the conformational changes induced by mutations in SS1, SS3, or SS4 will disrupt the allosteric site responsible for zinc potentiation, whereas perturbation of SS2 disrupts the inhibitory site for copper and high zinc concentrations. The zinc potentiation is fast and increases with metal preapplication, because it is likely that zinc binds to rP2X4R in the open state in contrast to copper (2), which provides a rationale for why it was easier to predict which residues participate in copper binding based on the crystal structure of the closed receptor (91).
In other P2XRs, there is much less information about trace metal effects and no information about putative allosteric sites. The rP2X7R is inhibited by both copper and zinc, although the latter metal is less potent (1, 114). Copper, but not zinc, also inhibited the mouse P2X7R (128). The mutagenesis studies did not give definitive answers about the binding sites for these metals. One study suggested that the H267A-rP2X7R is a copper-resistant receptor and that H267 and H219 residues account partially for zinc sensitivity (1). The other group has reported a dramatic reduction or almost a complete loss of inhibition by the two metals in cells expressing the H62A and D197A double mutant (114). The function of rP2X1R is inhibited by zinc in a concentration-dependent and competitive manner. In contrast, rP2X3R function is potentiated by zinc, causing a leftward shift in the concentration–response to ATP application, with a maximal effect at 10 μM. However, higher zinc concentrations reduce P2X3R current, suggesting the possible existence of two allosteric sites at this receptor: a high-affinity site with a potentiating effect and a low-affinity site with an inhibitory role (183). The rP2X5R is also modulated in a biphasic fashion by zinc, again suggesting the existence of at least two allosteric sites for this metal (182).
Protons as Allosteric Modulators
Similar to acid-sensing ion channels and the transient receptor potential vanilloid receptor channels, P2XRs can also sense hydrogen ions in the bath medium and respond by modulating the ATP action in a receptor-specific manner (Table 1). In general, P2XRs are inhibited by acidification. The ATP potency for rP2X1R was reduced without altering the maximum current amplitude (164, 183). At rP2X1R, inhibitory effects of extracellular protons and zinc are additive (183). Acidification also reduces the peak amplitude of the rP2X3R current at an EC50 concentration of agonists (164), and it right-shifts the ATP concentration–response curve without altering the peak amplitude of the response (183). Other researchers found an inhibitory effect of acidification that shifts the concentration–response curves to the right and a stimulatory effect which increases the current peak amplitude and activation time constant and accelerates in the recovery from desensitization. The same group also identified the H206 residue as being responsible for these effects (64). It is interesting that fully N-glycosylation of hP2X3Rs is needed to recognize external protons (186). Acidification also decreased the potency of ATP for rP2X4R without a change in the maximum response (71, 164), whereas alkalization enhanced the current amplitude (26) and mutation of H286 removes the extracellular pH sensitivity of this receptor subtype (26, 196). Since this residue is located far from the putative ATP binding site, we may speculate that protons affect gating rather than ATP binding.
Acidification also inhibits the function of P2X5R and P2X7R, but with a pattern of inhibition that differs from other P2XRs; acidification reduced both the potency and efficacy of ATP at rP2X5R (182), whereas it causes a reduction in the peak amplitude of the current without altering the agonist sensitivity at rat and human P2X7R (113). The inhibitory effect of protons on hP2X7R is also visible in single-channel recordings (56). However, changes in the EC50 value of agonists caused by acidification of P2X7R could be masked by the complex biphasic gating properties of these receptors (102, 194) and the sensitization/facilitation of responses during repetitive agonist application (152, 192). It has also been suggested that several residues could contribute to the pH sensitivity of P2X7R, including H130 and D197 (1, 113).
In contrast, for rP2X2R, the potency of all agonists, but not antagonists, is enhanced by acidification 5–10-fold without affecting the maximal agonist response (28, 100, 101, 164). The receptor-specific sensitivity to pH is also obvious in intact cells using single-cell calcium measurements (71). The inhibitory effect is preserved in rP2X2/3R heteromers (164) and is also extremely sensitive to small changes in extracellular pH (about 7.1–7.2) (106, 107); whereas the rP2X1/2R exhibits unique pH sensitivity (20). The pH range is also narrowed for the proton enhancement of the ATP response at the heteromeric rP2X2/6R (99). The basic H319 residue is identified as a putative pH sensor for the rP2X2R (28). This residue is located in a receptor region that has been suggested to operate as a linker region between the ligand-binding domain and the channel pore in other P2XR subunits (150, 195).
The gating of native P2XRs is also altered by changes in pH. Acidic pH potentiates the ATP-gated currents in guinea-pig cochlear outer hair cells (88), mice dorsal root ganglion (DRG) neurons (111), neurons from parasympathetic ganglia (118), and visceral afferent neurons (75). Dictyostelium P2XRs are also modified by acidification, indicating that the pH sensitivity of P2XRs is conserved in evolution (117). These results are consistent with a hypothesis that acidic conditions in the synaptic cleft could modify purinergic transmission, which could be physiologically relevant in certain events, such as pain sensation during inflammation. Finally, ATP-gated receptors can regulate pH-mediated responses, as reflected in the ASIC3 channel sensitization after P2X2R, P2X4R, and P2X5R activation (12).
Extracellular Calcium and Magnesium Affect ATP Gating
There is a large body of information concerning the effects of divalent calcium and magnesium ions on P2XR function. Calcium and magnesium inhibit recombinant channels at millimolar concentrations (141). Native receptors are also sensitive to modulation by calcium (38, 133) and magnesium (126, 171). However, the mechanism of the action of these cations on P2XRs was questioned. The binding of calcium and magnesium to β- and γ-phosphate groups of ATP is a well-established phenomenon (78), but it was unclear as to which form of ATP (ATP4− and/or Mg/Ca-ATP) acts as an orthosteric agonist for this receptor (80). Initially, it was suggested that ATP4− acts as an orthosteric agonist for histamine secretion by mast cells (30). More recently, the effect of ATP on P2X7R currents was studied in the presence and absence of calcium, and the same conclusion was reached (102). In that scenario, effects of divalent cations on P2XR function just reflect change in ratio between ATP4− and Mg/Ca-ATP. Although it has never been supported by direct evidence, this hypothesis has prevailed in the literature during the last two decades.
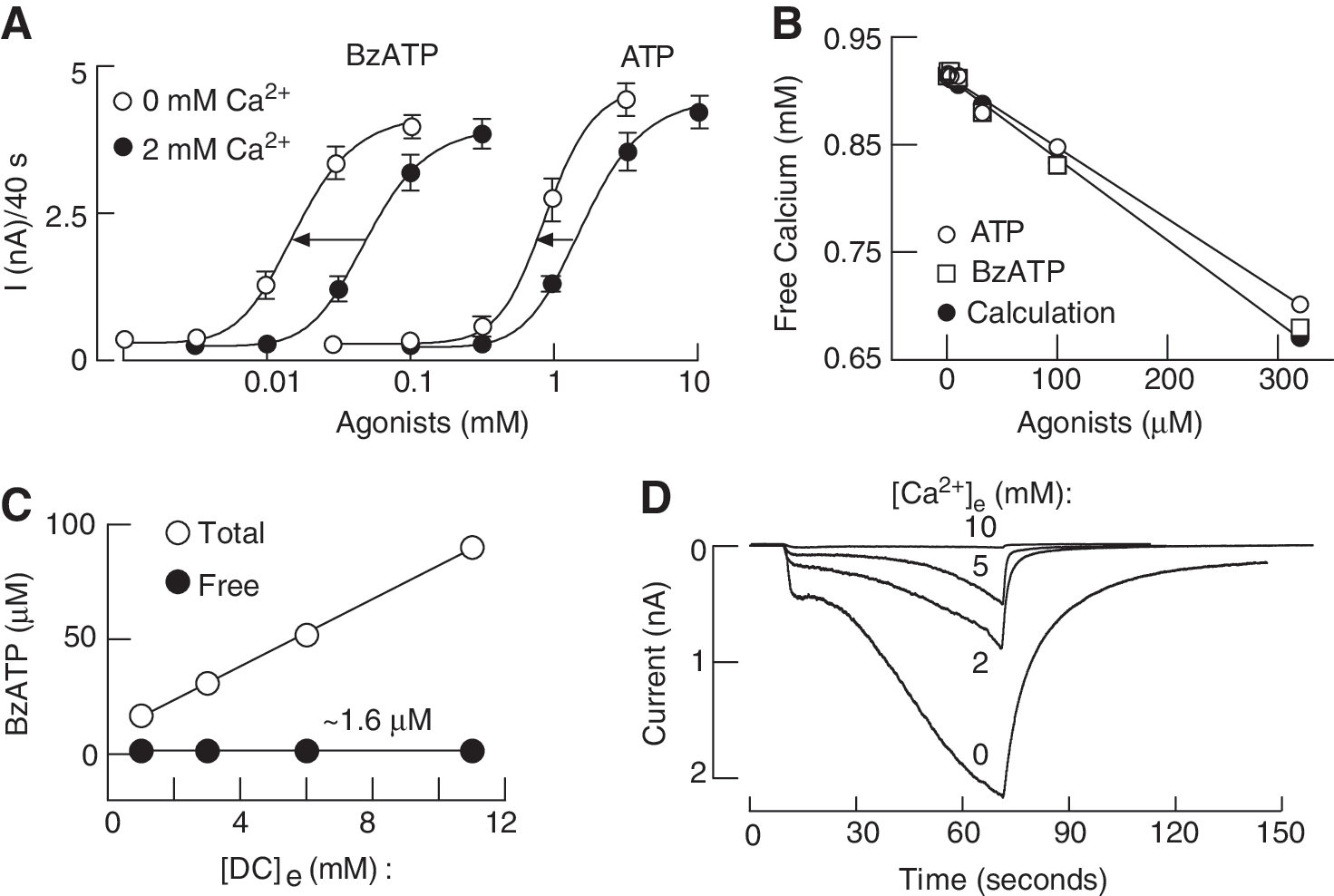
Figure 6A illustrates effects of bath calcium on concentration dependence of BzATP and ATP-induced rP2X7R peak current amplitude reached during 40-s agonist application. For both agonists, there was a leftward shift in agonist concentration of about 0.5 log unit. Figure 6B shows highly comparable concentration-dependent effects of ATP on free calcium concentration obtained by calculations and by measurements for ATP and BzATP, indicating that BzATP also binds calcium. However, these experiments cannot distinguish between the two hypotheses: (i) ATP4− rather than the divalent bound ATP is the native agonist for this receptor subtype or (ii) bath calcium acts as an allosteric inhibitor, and 2 mM calcium causes a rightward shift in the potency of agonists of about 0.5 log unit independent of the concentration of free acid form of agonists. One way to overcome this limitation is to have variable calcium concentrations and identical-free agonist concentrations (Fig. 6C). Under such experimental conditions, the relationship between total agonist concentration and the pattern of current response was lost; practically, the lowest total BzATP concentration was most effective, and the highest concentration was the least effective. Furthermore, although the free acid from of BzATP was identical in all four media, we still observed the full concentration-dependent inhibitory effect of calcium on the peak amplitude of current response (Fig. 6D). This clearly indicates the allosteric nature of action of this cation (193).
Others reported that extracellular calcium inhibits both the rP2X7R cationic current and dye uptake. The inhibitory effect of this cation on receptor function was mimicked by other divalent cations. For each of these metals, the IC50 values for current and dye uptake were similar: Cu2+>Cd2+∼Zn2+>Ni2+>> Mg2+∼Co2+>Mn2+>Ca2+=Ba2+>> Sr2+. Furthermore, this inhibition was voltage independent, a finding inconsistent with the hypothesis that divalent cations cause functional inhibition by blocking the channel pore (168). Furthermore, the major inhibitory effect of divalent cations was observed in relatively low concentrations, a pattern typical for allosteric modulation (175).
The rP2X7R-H130A mutant is resistant to magnesium inhibition, reinforcing the hypothesis that this metal acts through an allosteric mechanism (1), but not to calcium inhibition (193), further suggesting separate binding sites for these metals. A distinct role of calcium and magnesium in the modulation of native P2X3Rs from cultured rat DRG neurons was also reported. Calcium positively modulated these channels, while magnesium had an inhibitory effect (66). Single-channel recordings of P2X4R revealed that magnesium reversibly decreases the amplitude of ATP-evoked single channel currents in a concentration-dependent manner that is independent of the membrane potential. They also found that magnesium shortens the mean open time without affecting the mean closed time, suggesting that magnesium inhibits the function of human P2X4R by means of an open-channel block via a binding site located at the exterior surface of the pore (136).
Calcium also exhibits a strong inhibitory effect on rP2X2Rs, with a half-maximal block of the current at about 5 mM, but not on hP2X1Rs (54). However, rP2X3R was more than 10-fold less sensitive to blockade by bath calcium, whereas heteromeric rP2X2/3Rs were more sensitive. Thus, the rank of order for inhibition by calcium at these receptors is P2X2R>P2X2/3R>P2X3R>P2X1R (176). Single-channel recordings also confirmed the inhibitory effects of calcium and magnesium on recombinant rP2X2R. These effects manifest as a fast block, visible as a reduction in amplitude of the unitary currents (49).
Calcium has additional effects on P2XR gating. It increases the rates of rP2X2R desensitization, an action that is mimicked by magnesium, barium, and manganese, although the calcium-induced desensitization exhibited a higher affinity and co-operativity (50). Our recent experiments revealed very complex effects of bath calcium on P2X2R gating. During sustained agonist application, receptors continued to desensitize in calcium-independent and calcium-dependent modes. Calcium-independent desensitization was more pronounced in P2X2bR and calcium-dependent desensitization in P2X2aR. In whole-cell recordings, we also observed use-dependent facilitation of desensitization of both receptors (92). Figure 7 illustrates development of use-dependent desensitization of P2X2R in cells bathed in Ca2+-containing medium (Fig. 7A) but not in cells bathed in Ca2+-deficient medium (Fig. 7B). A stimulatory role of calcium in P2X3R recovery from desensitization was also reported (39). These effects of calcium are not necessarily a manifestation of the extracellular allosteric action of this cation, but they probably reflect its intracellular actions (for details, see next).

Roles of Intracellular Calcium and Protein Kinases in P2XR Activity
All P2XRs conduct calcium directly through their pores and indirectly by depolarizing cell membrane and activating voltage-gated calcium channels. Intracellularly, calcium binds to various signaling proteins, such as calcineurin or calmodulin (CaM)-kinases, activating its phosphatase or kinase activity. Moreover, CaM alone also modulates channel activity, as has been described for the P2X7R. In cells expressing this receptor, CaM facilitates both current and membrane blebbing. The proposed CaM-binding site is located in the C-terminal sequence I541–S552 (152). Later, it was shown that the hP2X7R is resistant to CaM modulation because it lacks three critical residues of the CaM allosteric site; substitution of these residues with the ones present in the rat receptor resulted in CaM-sensitive receptor (151). In DRG neurons, CaM-dependent protein kinase II potentiates purinergic responses, probably by promoting trafficking of P2XRs (191). In addition, the neuronal calcium sensor VILIP1 was shown to regulate some aspects of P2X2R gating, such as affinity and efficacy of ATP (22).
Calcium, directly or indirectly, can also regulate the activity of PKA, some PKC isoforms, as well as (NO) oxide synthase, leading to generation of NO, stimulation of soluble guanylyl cyclase, and activation of cGMP-dependent protein kinase. PKA has been shown to regulate the rP2X2R; both 8-Br-cAMP and the PKA catalytic subunit inhibit the current amplitudes in cells expressing this receptor and mutation of the C-terminal residue S431 abolishes this effect (25). PKA also potentiates the P2X4R; it has been suggested that this effect is indirect and requires an accessory protein which interacts with the YXXGL endocytosis motif (18). The potentiation of P2X3R currents in DRG neurons mediated by prostaglandin E2 is also PKA dependent (179), as well as the glucocorticoid-induced inhibition of P2XR-mediated currents in HT4 neuroblastoma cells (68). A critical role of the cAMP sensor Epac in the sensitization of native P2X3R has also been reported (179).
Experiments with the PKC activator PMA had shown that this kinase could specifically potentiate the currents mediated by P2X1R and P2X3R currents (17, 143, 174), but not by other subtypes (17). P2X1R-mediated currents are potentiated by serotonin-2A receptor activation, an effect that is probably mediated by PKC (5). Other groups have suggested that PKCγ potentiates P2X7R function in the type-2 astrocyte cell line (76), whereas PMA inhibits P2XR-mediated currents in isolated DRG neurons from adult rats (11).
The N-terminal of all P2X subunits contains a putative binding motif (TXR/K) for PKC phosphorylation. Mutation of the conserved threonine residue results in dramatic changes in receptor activity, altering the current amplitude (P2X1R, P2X2R, and P2X3R) and desensitization profile (P2X2R) (13, 58, 112). However, biochemical experiments failed to show a direct phosphorylation at this conserved threonine residue (17, 58, 174), suggesting that the structural changes induced by mutagenesis and no phosphorylation accounts for the changes observed in receptor function. Similarly, the permeation properties of the P2X7R pore change by replacing T15, and this effect is not related to PKC phosphorylation of this residue (194). An alternative target for ecto-PKC phosphorylation has been proposed for the hP2X3R; point mutations of putative extracellular PKC phosphorylation sites abolished (T134A, S178A) the potentiation of currents observed with UTP (162, 185). It is also possible that the regulation of P2XRs by phosphorylation is indirect. A recent report has shown that P2X3R currents are potentiated by pro-inflammatory agents which activate protease-activated receptor-2, and this effect can be prevented by inhibition of PKA or PKC in DRG neurons (180). Parkin, an ubiquitin E3 ligase, can also regulate the function of P2XRs in PC12 cells by indirectly modulating the activity of PKA and other kinases (156).
Other protein kinases also affect the P2XR function. P2X3R-mediated currents are decreased by both the C-terminal Src kinase and the cyclin-dependent kinase-5 (Cdk5). For the C-terminal Src kinase-dependent regulation, the C-terminal T393 residue has been proposed as a target for phosphorylation, whereas the Cdk5 regulation could be mediated by an accessory protein (42, 131). In mouse trigeminal sensory neurons, phosphorylation of the P2X3R is controlled by nerve growth factors (43).
Regulation of P2XR Activity by Interaction with Other Proteins
There is growing evidence that P2XRs can associate with other proteins and these interactions can modify the gating properties of channels. In addition to the previously mentioned VILIP1 and CaM which regulate the activity of the P2X2R and the P2X7R, it has also been proposed that the beta-amyloid precursor protein-binding proteins Fe65 interacts with the P2X2R, resulting in changes in the permeability properties of this receptor (119). More interestingly, it has been also shown that P2XRs can interact with other ligand-gated ionic channels, as the observed cross-inhibition (the magnitude of the current gated by simultaneous activation is smaller than the sum predicted by activating each one separately) between P2X2Rs and nicotinic AChRs that is observed in both heterologous expressed and endogenous receptors (96). Moreover, cross-inhibition has been observed between RHO1 GABA receptors and both the P2X2R (15) and the P2X4R (188). Similar interactions have been found between these P2XRs and GABAA receptors (16, 86). Another study also points to a mutual cross-inhibition between P2X2 and 5-HT3 receptors in heterologous and native systems (14). Finally, the level of receptor expression also influences the gating properties of the P2X2R, such as ATP affinity (27), ionic permeability, and rectification properties (60), suggesting that receptor-receptor interactions and cooperative mechanisms play a role in P2XR-mediated responses.
Physiological and Pathological Implications of P2XR Allosteric Regulation
After 40 years from the proposal of purinergic signaling by Burnstock (21), there is no doubt that P2XRs are allosterically regulated by a myriad of endogenous and exogenous compounds. The pharmacological relevance of such modulation is obvious. Allosteric modulation of P2XRs may also be of physiological relevance. For example, both zinc and copper are released during synaptic transmission (6, 89) and they can reach micromolar concentrations in the synaptic cleft where they could exert their allosteric actions. In this context, a remarkable example is the zinc potentiation of glycinergic transmission. A knock-in mouse that expresses a zinc-insensitive GlyR instead of the wild type resulted in a hyperekplexia phenotype (74). This mutated receptor had similar expression and agonist affinity compared with the wild type, and the only difference was its insensitivity to zinc, unmasking that endogenous allosteric regulation by this metal is crucial for normal glycinergic transmission. Similar approaches with P2XRs transgenic mice could unmask the role of allosteric modulation in this family of receptors.
On the other hand, some pathological conditions could result in an excessive accumulation of divalent metals, impairing normal synaptic transmission; that is, the case of Wilson's disease in which a nonfunctional P-type copper transporter ATPase ATP7B results in copper accumulation that can lead to neurological disorders, within other symptoms (181) or during the immune response, where the copper levels can drastically change (145). At least a part of these effects may result from constant allosteric modulation of ionic channels, including P2XRs. In this context, the identification of binding sites and mechanisms of action could have relevance in the design of drugs and therapeutic strategies to treat these and other pathologies.
Similarly, allosteric modulation by protons and ROS could be of physiological relevance; as well as in divalent metals, the levels of ROS and pH may not only dramatically change during pathological events, but they can also slightly change during normal physiological processes. In this scenario, P2XRs are polymodal sensors that can change their gating properties not only after binding extracellular ATP, but also after changes in its extracellular and intracellular milieu. Again, knowing and understanding allosteric modulation could provide a valuable tool to treat diverse pathologies, such us pain-related diseases and states. In this regard, the availability of both closed and open crystal of the zP2X4.1R constitutes a significant advantage to achieve this goal.
Conclusions and Future Perspectives
P2XRs have numerous regulatory sites in both the extra- and intracellular domains, which provide the physiological and pharmacological frames for the modulation of the activity of these channels by a complex set of synapse- and/or paracrine-derived native agents, intracellular metabolites, and drugs. In contrast to the ATP-binding sites, the nonconserved residues appear to play a critical role in allosteric modulation of these channels by trace metals, divalent cations, and protons, which explain the receptor specificity of the actions of these molecules. P2XRs are also modulated by second messengers and membrane lipids acting intracellularly. The gating of P2XRs can also be regulated by redox signaling, and the formation and production of ROS can be facilitated by activation of P2XRs, constituting a mutual feedback that is of physiological relevance. Thus, as with other ligand-gated receptor channels, the P2XR affinity for agonists and/or the changes in their maximal responses is not only determined by the orthosteric ligand concentration, but also depends on the availability of allosteric regulators in the receptor milieu. These findings provide a basis for further studies addressing the dependence of the activity of a particular receptor on the metabolic state of cells, tissues, and organs.
In general, allosteric modulators constitute a valuable alternative for future drug design, especially for receptor-specific drugs. The current information regarding the residues involved in orthosteric and allosteric modulation combined with the channel structure provided by crystallization will also lead to a better understanding of the structure–activity relationships that govern the gating of P2XRs. Specifically, the close topological location of the trace metal coordination amino acid residues to the orthosteric ligand sites provides important clues for further structural modeling and investigation into the actions of metals in the receptor ectodomain. Since trace metals can be released to the synaptic cleft and exert effects on P2XR signaling, further studies are needed to clarify the physiological conditions for this action in vivo. Changes in cellular pH caused by ischemia could affect P2X3R activity places allosteric modulation by protons in a more physiological/pathophysiological context. Future experiments should also be directed in order to identify mechanisms and allosteric sites for ROS at P2XRs and to elucidate the signaling pathways that are responsible for P2XR-mediated ROS formation. In this regard, it is also necessary to test whether other P2XRs are regulated by or regulate the redox balance of cells.
Footnotes
Acknowledgments
This research was funded by the Intramural Research Program of the National Institute of Child Health and Human Development, NIH (S.S., E.L.-S., and C.C.) and the FONDECYT Initiation Grant 11121302 (C.C.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.