
Abstract
Introduction
S
B-cell lymphoma-extra large (Bcl-xL) protein is a member of the Bcl2 protein family whose main function is to promote cell survival. Bcl-xL is abundantly expressed during development and in adult neurons and can translocate into mitochondria during death stimuli or neuronal activity (1, 6, 9). Bcl-xL prevents homo-oligomerization of pro-apoptotic family members such as Bax and Bak, directly inhibits activators of Bax and Bak (25, 46, 48), and enhances cell metabolism by interacting with the ATP synthase (1, 6). In addition to its protective roles, Bcl-xL can play a pro-death role during neuronal ischemia by forming a cleaved pro-apoptotic moiety (2, 21, 38), although the anti-apoptotic and neuroprotective functions of Bcl-xL against various insults have been more widely reported (38, 42). In non-cell death-related roles, Bcl-xL enhances synapse formation and plasticity (13, 20, 27, 28) but whether it regulates neurite outgrowth is unknown.
Bcl-xL is a pro-survival protein that is located on the outer mitochondrial membrane and interacts with ATP synthase at the inner membrane. Previously, we reported that Bcl-xL conserves neuronal energy and facilitates synaptic function. We now show that Bcl-xL plays an important role in neurite outgrowth. We identify that DR6 is upregulated by Bcl-xL silencing and by hypoxia using in vivo ischemia and in vitro hypoxia models. This study suggests that Bcl-xL may regulate the formation of neuronal networks and that at least one and maybe more specific molecular pathways involving Bcl-xL and opposed by DR6 produce neuroprotection during hypoxia.
Neurite outgrowth is known to be negatively regulated by death receptor 6 (DR6, TNFRSF21), a member of the tumor necrosis factor (TNF) receptor superfamily that contains an intracellular death domain. DR6 is widely expressed in the brain and is especially abundant in the hippocampus (18). Endogenous DR6 regulates developmental axonal pruning, but not somatic degradation, in a caspase 6 dependent manner, presumably through release of mitochondrial factors (36, 47, 53). In contrast to endogenous DR6, overexpressed DR6 induces cell (somatic) death, and this can be prevented by anti-apoptotic members of the Bcl2 family. Notably, the pro-apoptotic protein Bax is required for DR6-induced apoptosis (53) and for axonal pruning (36). Studies also show increased levels of DR6 in the brain of Down syndrome patients (18) that are predisposed to early Alzheimer's disease through early development of Aβ-containing plaques. In keeping with this observation, it has been recently reported that DR6 interacts with another β-amyloid precursor protein (APP) cleavage fragment (not Aβ) of the APP (36).
Since the full cell signaling pathways both upstream and downstream of Bcl-xL and DR6 are unknown, we studied the role of DR6 in normal neuritic outgrowth in the absence and presence of a death stimulus (hypoxia) in cells depleted of Bcl-xL by siRNA. We found that DR6 levels are increased when endogenous Bcl-xL is silenced, and this is correlated with a decline in neurite outgrowth over many days in culture, which is eventually followed by delayed cell (somatic) death. Depletion of DR6 reverses these changes. Neurite damage in Bcl-xL-depleted cells is accentuated compared with that of controls during hypoxia, dependent on DR6, suggesting that attenuation of neurite outgrowth and subsequent somatic death are under Bcl-xL and DR6 control, but that these changes are rapidly accelerated by a decline in oxygen availability.
Results
Synaptic connectivity and neural network formation are not only important for development and normal synaptic plasticity but may also be neuroprotective (12, 20). We previously reported that Bcl-xL depletion by siRNA or pharmacological inhibition of Bcl-xL caused a decrease in synaptic protein markers (28), and these structural changes were associated with changes in synaptic activity (27) but not with cell death.
Bcl-xL is depleted by siRNA
To test whether Bcl-xL expression can be targeted by RNAi gene silencing, we infected primary hippocampal neurons with recombinant adeno-associated virus (rAAV). Hippocampal cell lysate collected at 1, 2.5, or 4 weeks after rAAV transduction confirmed that Bcl-xL siRNA significantly decreased Bcl-xL expression in primary culture at 2.5 weeks of rAAV post-infection, but not at 1 week (Fig. 1). In analyzing expressing cells, we found that fluorescence appeared strongly by 2.5 weeks and cells remained fluorescent (demonstrating continued successful expression of the rAAV construct) for approximately 5–6 weeks after infection. Since transduction rate is a critical factor in our study, we performed experiments when 70% or more of cells were expressing dsRED usually at 16–19 days after rAAV transduction. We note this time point as 2.5 weeks. Experiments performed at 8 and 29 days after rAAV transduction are noted as 1 and 4 weeks, respectively.

Bcl-xL depletion does not cause immediate cell death
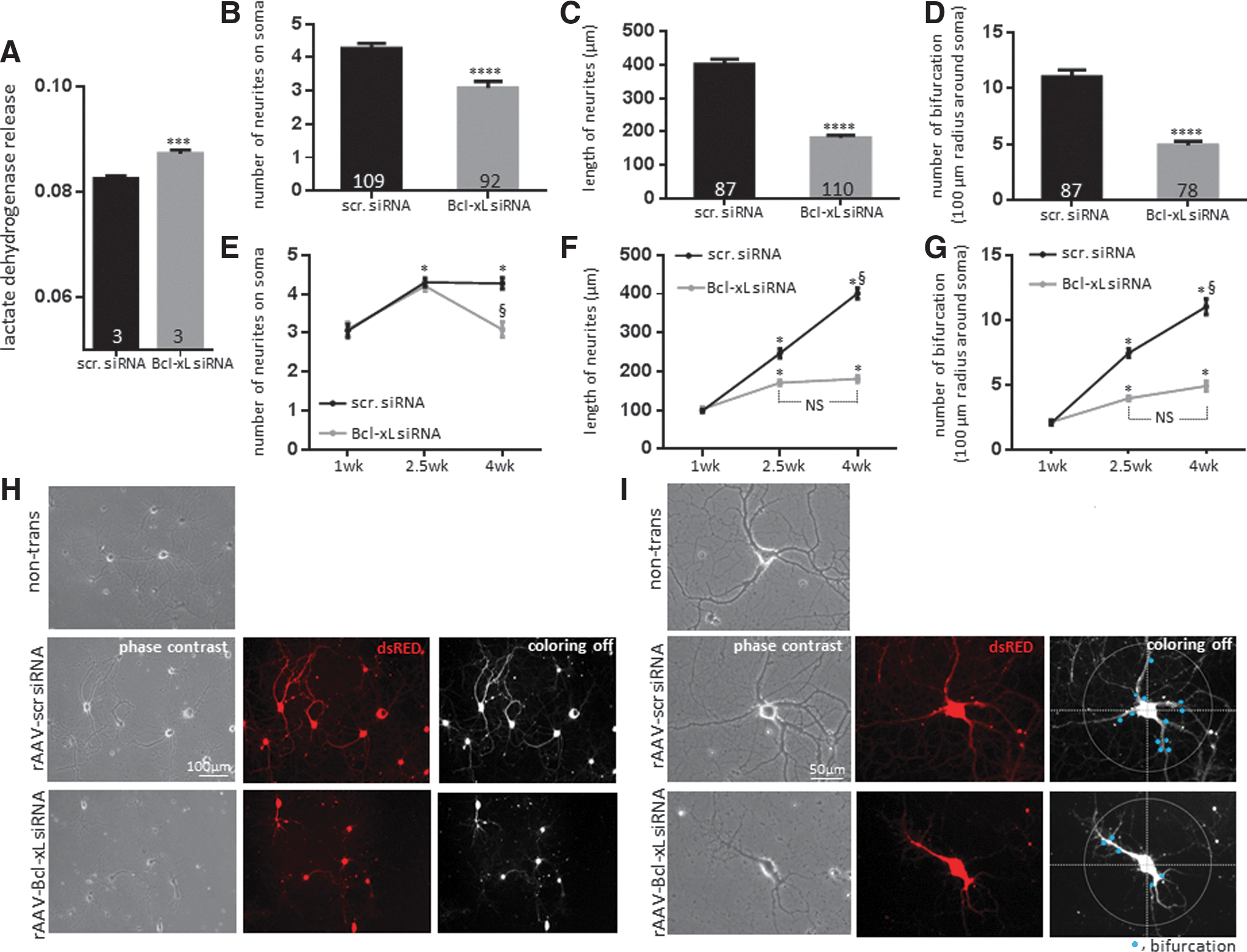
Although Bcl-xL is thought to function as an anti-apoptotic molecule, its depletion did not appear to cause immediate cell death in non-stressed neurons. The activity of lactate dehydrogenase (LDH), a marker for cellular damage, was not significantly different between scrambled siRNA- and Bcl-xL siRNA-expressing neurons at 2.5 weeks after rAAV transduction (Fig. 2A). Although LDH is a widely accepted marker of cytotoxicity, it does not distinguish between necrosis and apoptosis. To test whether Bcl-xL siRNA induced apoptotic death in primary neurons, we stained neurons with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). As a positive control, to ensure the capability of TUNEL to detect fragmented DNA, we treated healthy, non-challenged neurons with nuclease to force DNA breakage before TUNEL staining. We observed that nuclease-exposed neurons express TUNEL-positive signals (Supplementary Fig. S1; Supplementary Data are available online at
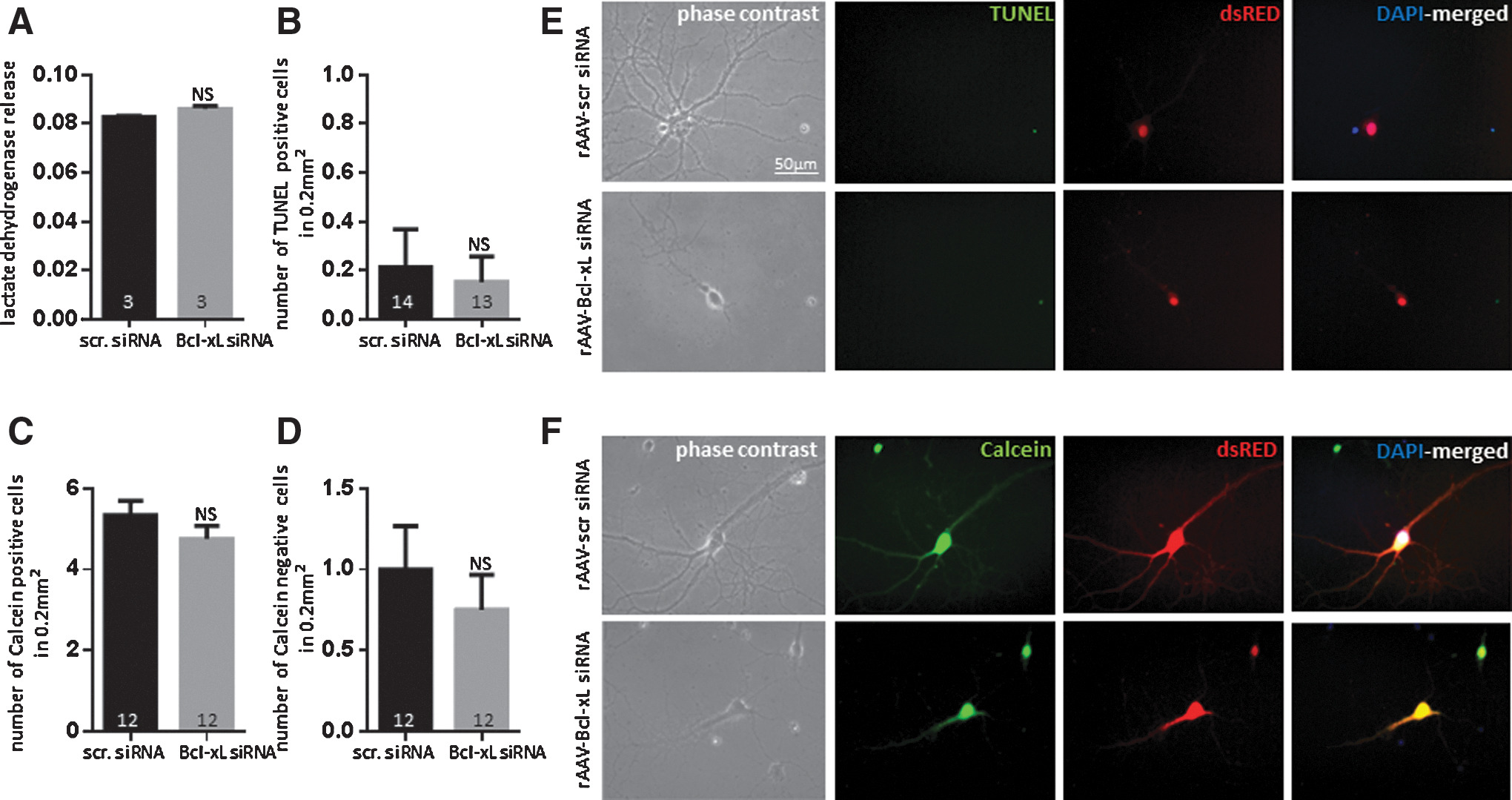
Bcl-xL depletion causes abnormal neurite outgrowth
We obtained images of hippocampal neurons at 1 week after rAAV transduction. Cells did not express rAAV-derived fluorescence at this time point, which indicated that siRNA expression was low (Fig. 3). Western blots also confirmed insufficient depletion of Bcl-xL at 1 week after rAAV transduction (Fig. 1A). Phase-contrast images of a group transduced for 1 week indicated that the number of neurites at the soma, the length of neurites, and the number of bifurcations within 100 μm radius of the soma were not significantly different between the non-transduced, scrambled siRNA-, and Bcl-xL siRNA-expressing neurons (Fig. 3).
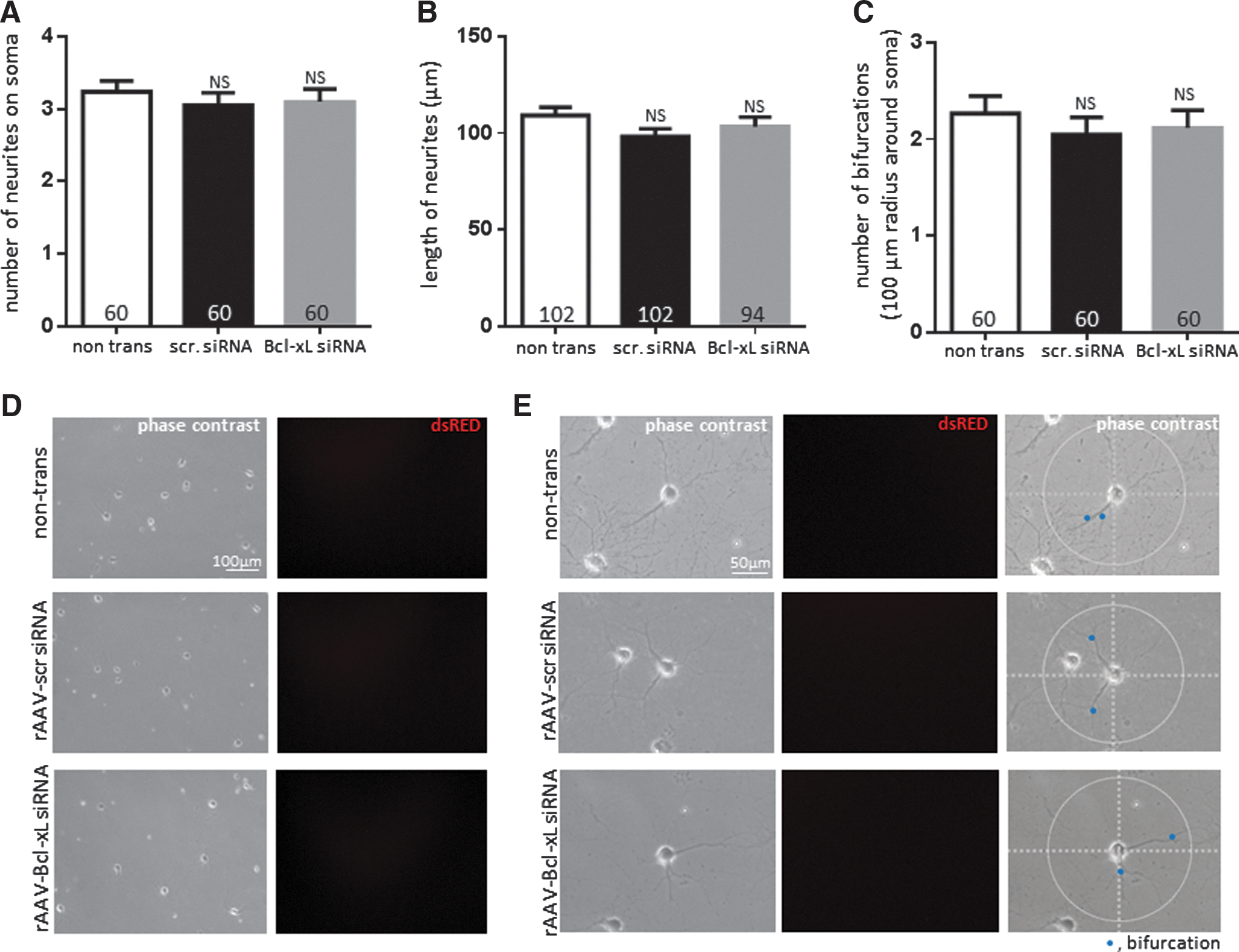
Although depletion of Bcl-xL did not influence neuronal viability at 2.5 weeks, we observed unexpected differences in neuronal morphology of Bcl-xL siRNA-expressing neurons compared with scrambled siRNA-transduced controls. Scrambled siRNA-transduced neurons grew neurites of 246.7±12.12 μm in length on average and some reached approximately 550 μm at 2.5 weeks. However, Bcl-xL-depleted neuronal processes extended only on average 170.8±6.02 μm (Fig. 4B, D). Moreover, scrambled siRNA-transduced neurons formed more complex branching of axons and dendrites. Scrambled siRNA transduced neurons showed 7.45±0.34 bifurcations on average within a 100 μm radius around the neuronal cell body. Bcl-xL siRNA-expressing neurons only showed 3.94±0.18 branches on average within the same area (Fig. 4C, E). There was no significant difference in the degree of proximal neurite outgrowth at the soma (Fig. 4A), which suggests that until the day of introduction of the rAAV, both groups adhered to the subcellular matrix and extended at the same rate. Our results, therefore, show that Bcl-xL-depleted neurons have significantly decreased length and bifurcation of neurites, suggesting failure to form a normal neuronal network.
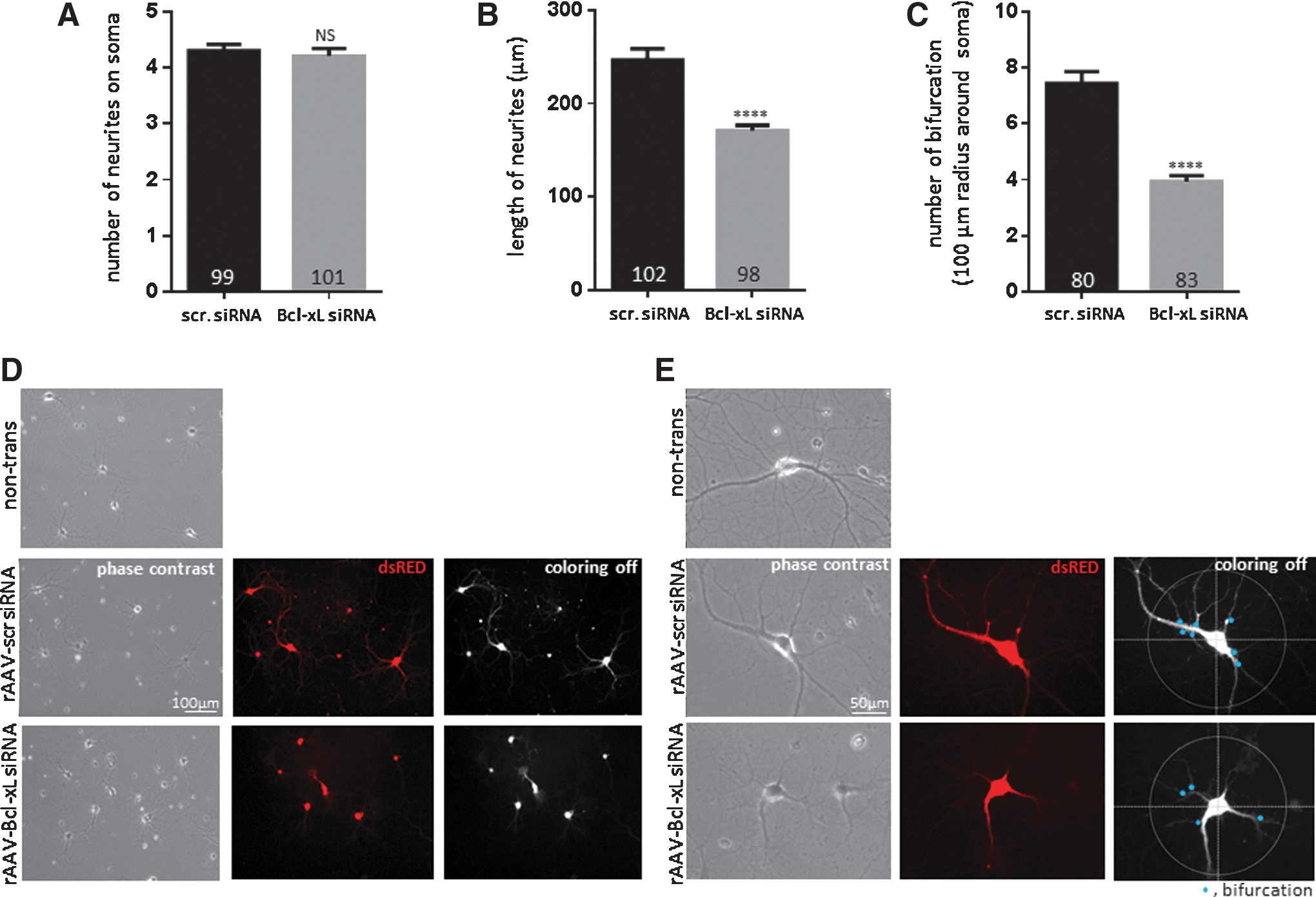
Very delayed cell death of Bcl-xL-silenced neurons follows slowing of neurite growth
We had previously shown and repeated in this study that Bcl-xL functional inhibition or gene depletion was not associated with neuronal death for approximately 2.5 weeks after rAAV introduction (Figs. 1 and 2) (28). To determine whether the changes in neurite morphology observed at 2.5 weeks were associated with delayed subsequent somatic demise after 2.5 weeks, we studied neurons at 4 weeks in culture. We found that Bcl-xL-depleted neurons began releasing significant amounts of LDH at 4 weeks after rAAV transduction (Fig. 5A) and many neuronal somata appeared connected to short, thin, and fragmented neurites at this time; while control neuron neurites stretched a longer distance and had developed increased branching compared with 2.5 weeks (Fig. 5B–I). Therefore, we suggest that, after Bcl-xL depletion, cultured neurons over time fail to form or sustain neuronal networks and this is associated with delayed somatic death.
Depletion of DR6 partially recovers neurite growth arrest of Bcl-xL-depleted neurons
To determine factors that may be associated with neurite outgrowth arrest and/or delayed cell death, we tested candidates known to be implicated in neurite retraction/neurite plasticity or growth arrest (8, 47, 53). We determined that DR6 is a possible downstream target of Bcl-xL regulation. DR6 is reported to be necessary for normal axonal pruning in response to nerve growth factor withdrawal in spinal neurons (36). DR6 is widely expressed in neurons during pro-apoptotic states and is of interest given its role in neuronal growth and repair (35, 36, 44). In our preliminary screening, we measured DR6 expression in cultured hippocampal neurons depleted of Bcl-xL. We found an increased abundance of DR6 protein in Bcl-xL siRNA-expressing hippocampal neurons compared with scrambled siRNA-transduced controls, suggesting that DR6 expression may be suppressed by endogenous Bcl-xL.
To test whether DR6 plays a role in Bcl-xL-mediated neuronal outgrowth, cells were coexpressed with DR6 siRNA along with Bcl-xL siRNA, or equal amounts of corresponding control viral constructs. GFP-tagged lenti-DR6 siRNA-expressing primary hippocampal neurons decreased DR6 protein level with ∼70% knockdown efficiency (Fig. 6A). Depletion of DR6 did not enhance neurite outgrowth in scrambled siRNA-expressing neurons. However, DR6 depletion attenuated loss of neurite outgrowth in Bcl-xL siRNA-expressing neurons measured at 2.5 weeks of transduction. Although depletion of DR6 did not completely recover neuronal outgrowth in cells depleted of Bcl-xL, the length of neurites in the doubly depleted group was ∼35 μm longer, and the number of bifurcations was 1.3 higher than in Bcl-xL-depleted neurons, indicating that depletion of DR6 partially recovers neuronal outgrowth in the condition of Bcl-xL depletion (Fig. 6B–D). These findings support the notion that DR6 is one important downstream effector of Bcl-xL depletion during neurite outgrowth.

Cell death is enhanced by Bcl-xL depletion in in vitro hypoxia
To elucidate the importance of intact neurite morphology in prevention of neurotoxic insult, we exposed primary cultures to hypoxia. To confirm hypoxic conditions, we measured oxygen content in hypoxia-exposed cell culture medium (Supplementary Fig. S2). During preliminary screening, we observed that 1–3 h hypoxic exposure did not induce neurotoxicity, while 6 h hypoxia induced minor LDH release, and 72–96 h hypoxia caused massive irreversible hippocampal injury. Thus, we chose 24 h hypoxia to induce neurotoxicity in our culture system. In addition, we noted that 6 h hypoxia triggered caspase 6, caspase 3, and caspase 7 activation, indicating apoptotic signaling starting at time points earlier than 24 h. Depletion of Bcl-xL enhanced activation of caspases (Fig. 7A, B).

To study the vulnerability of Bcl-xL-depleted cells to hypoxia, hippocampal neurons were placed under hypoxic conditions for 24 h after 2.5 weeks of rAAV transduction. LDH release was significantly higher in Bcl-xL siRNA-expressing cells exposed to hypoxia compared with scrambled siRNA-transduced neurons exposed to hypoxia, indicating increased cytotoxicity after Bcl-xL depletion in the hypoxic cultures (Fig. 7C). We also observed that Bcl-xL siRNA-expressing neurons had significantly increased TUNEL staining after hypoxia (Fig. 7D, G), consistent with cell death. The viable cells were also much reduced. (Fig. 7E, F, H). Our viability and toxicity assays, therefore, showed that although depletion of Bcl-xL by itself did not induce neuronal death (Fig. 2), neurons depleted of Bcl-xL were more vulnerable than controls to hypoxic insult (Fig. 7) and displayed markers of cell death.
Bcl-xL depletion enhances neurite damage in hypoxic neurons
Neurite network damage is known to occur during hypoxic insults (49). To study this in our model, neurites of both scrambled- and Bcl-xL siRNA-expressing neurons were assayed at 24 h after hypoxic exposure. Controls were relatively protected compared with cells depleted of Bcl-xL during hypoxic insult. Scrambled siRNA-transduced neurons only suffered damage to small parts of the neuronal structure such as distal neurite tips. In contrast, the majority of Bcl-xL siRNA-expressing neurons completely lost their axons and neurites (Fig. 8A–E). We found discontinuous tracks of debri around broken somata and countless fluorescent positive fragments around intact somata, indicating severely damaged neurites in these hypoxic Bcl-xL-depleted neurons. Quantitatively, Bcl-xL siRNA-transduced hypoxic neurons displayed fewer somatic branches, greatly shortened neurite length, and many fewer bifurcations than scrambled siRNA-transduced hypoxic cells (Fig. 8A–E).

DR6 depletion protects against neurite damage during hypoxic insult
To understand any role of DR6 in neurite loss during hypoxic insult, hippocampal neurons were challenged with hypoxia for 24 h after 2.5 weeks of cotransduction, and cell morphology was analyzed in DR6 siRNA-expressing neurons (Fig. 9). We found significant protection from hypoxic insult in both scrambled and Bcl-xL-depleted groups. As evident in Figures 7 and 8, depletion of Bcl-xL increased vulnerability of neurons to hypoxic challenge. When Bcl-xL siRNA was coexpressed with DR6 siRNA, however, neurons partially retained neurite morphology, with strikingly improved neurite length and enhanced number of bifurcations compared with Bcl-xL siRNA alone.

Both Bcl-xL depletion and hypoxia increase DR6 levels
Although the earlier experiment supports a role for DR6 in hypoxic injury to neurites, it falls short of determining whether DR6 is a necessary downstream effector of Bcl-xL depletion during hypoxic insult. To further clarify this, we tested whether Bcl-xL depletion or hypoxia affected levels of DR6 expression. We found by immunoblotting that DR6 expression was markedly elevated in Bcl-xL-depleted normoxic neurons (Fig. 10A–C). In fact, depletion of Bcl-xL resulted in an ∼2.9-fold higher DR6 expression compared with scrambled siRNA controls. Hypoxia also produced DR6 protein upregulation to a level not significantly different from that produced by Bcl-xL depletion, suggesting that hypoxia could also induce increased DR6 (compare Fig. 10B, C), perhaps through pro-apoptotic cleaved Bcl-xL formation or Bcl-xL sequestration.
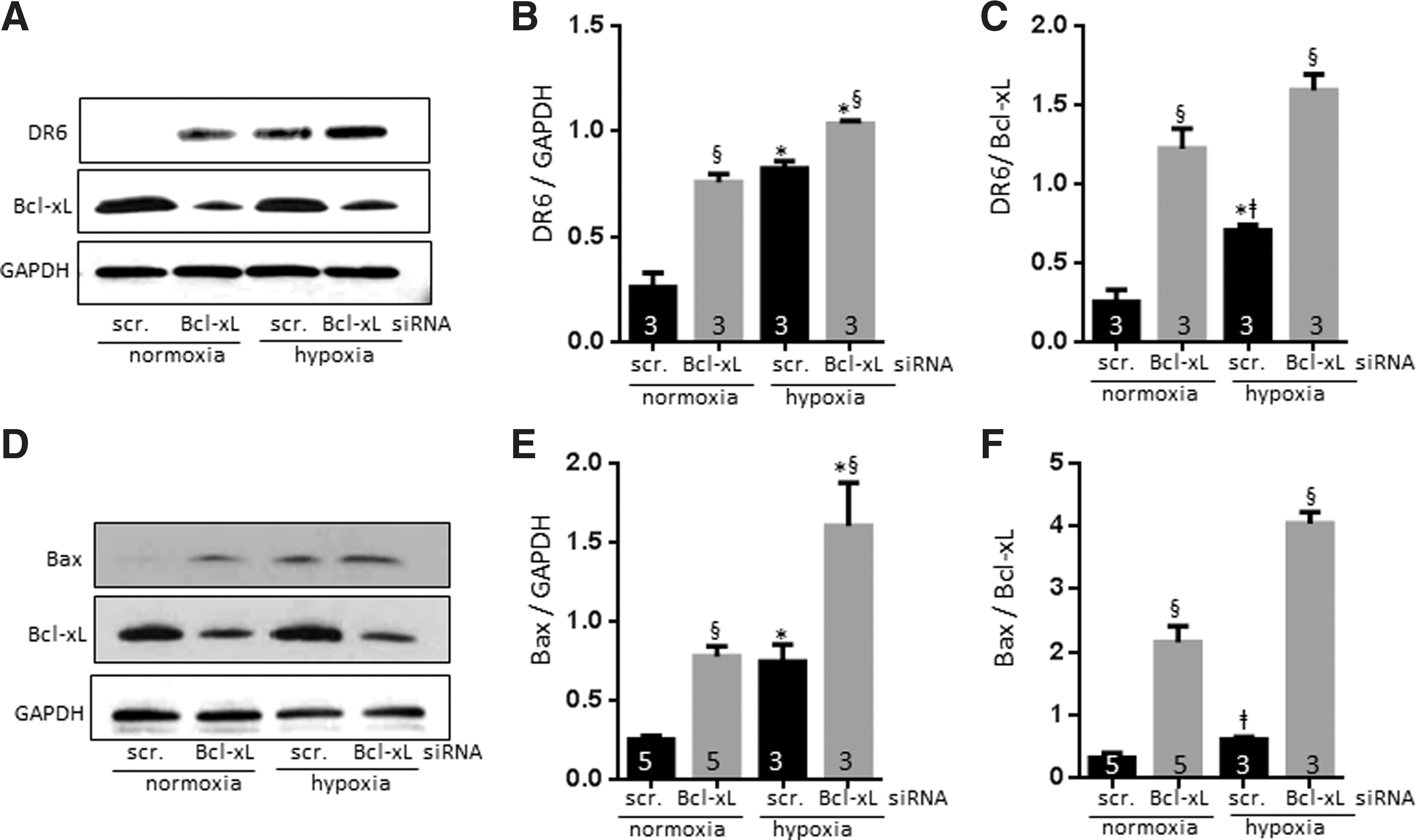
Both Bcl-xL depletion and hypoxia increase Bax activation
One role of Bcl-xL is to sequester BH3-containing molecules to prevent Bax activation (39). Bcl-xL depletion in neurons (Fig. 10D–F) increased formation of active Bax (∼3.1-fold higher than scrambled siRNA-transduced neurons), while other tested pro-apoptotic candidates (PUMA and NOXA) were unchanged (data for PUMA and NOXA not shown). Hypoxia enhanced active Bax to about the same level as depletion of Bcl-xL alone, consistent with the notion that inhibition/sequestration of Bcl-xL during death stimuli activates Bax (14). We observed the highest level of active Bax formation in hypoxic Bcl-xL-depleted neurons, consistent with our findings of increased cell death and neurite loss in this group (Figs. 7 and 8) and consistent with the results for DR6 (Fig. 10A–C). The role of Bcl-xL in Bax activation (39) has been reported previously. We now suggest that there are parallels between Bax activation and DR6 upregulation. Furthermore, we suggest that not only does Bcl-xL depletion/sequestration during normoxia/hypoxia enhance activation of Bax and upregulation of DR6, but there may also be a role for other apoptotic mechanisms such as pro-apoptotic cleaved Bcl-xL formation (Fig. 10E, F).
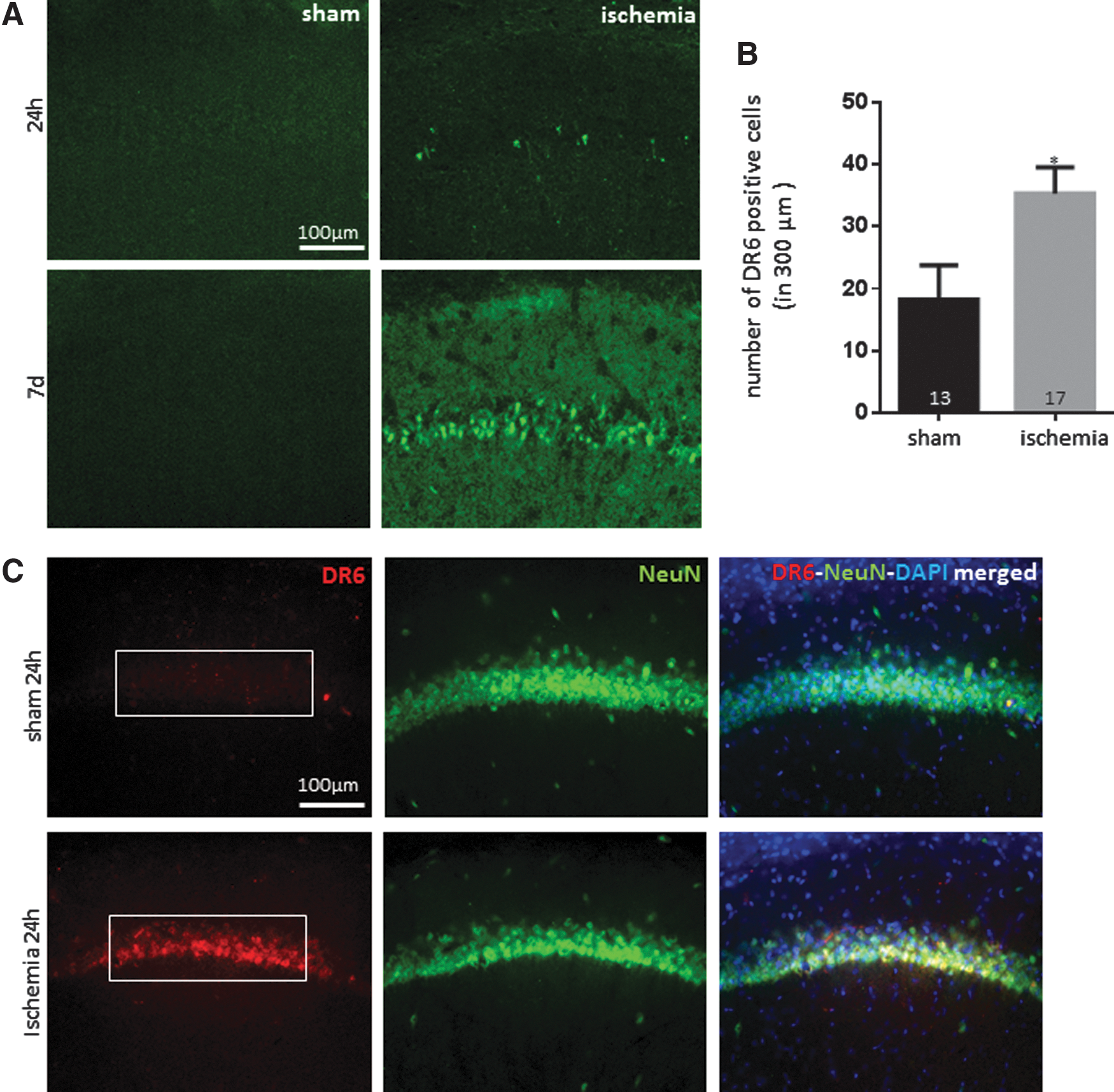
DR6 levels are increased in vivo after transient global ischemia
To determine whether DR6 is important in neurotoxic injury in vivo, we studied transient global ischemia in rodents. We applied global ischemia in rodent brains for 10 min using four-vessel occlusion (4VO). Ten minutes of occlusion does not produce edema or other complications, but results in very delayed apoptotic death in pyramidal neurons in the hippocampal CA1 and only sparsely in other areas (16, 37, 38, 43).
Animals that have undergone 4VO show little Fluoro-Jade positive signal at 24 h, indicating a low level of neurodegeneration at that time point. However, rats express abundant Fluoro-Jade positivity in CA1 hippocampal neurons at 7 days after occlusion (Fig. 11A). We tested DR6 expression in ischemic brain tissue harvested at 24 h after reperfusion when there is little Fluoro-Jade positivity. DR6 was upregulated at 24 h after occlusion compared with sham. The early changes indicate that DR6 could be playing an early role in network damage even before somatic death after ischemia in vivo (Fig. 11B, C).
Discussion
In this study, we show that Bcl-xL depletion by siRNA causes neurite growth arrest followed by delayed cell death. DR6 is necessary for full expression of the process of neurite growth arrest, as depletion of DR6 in Bcl-xL-depleted cells rescues neurite growth arrest. Once neurite arborization or extension is impaired, neurons become more vulnerable to an acute death stimulus. We show this in a model of brief hypoxia in hippocampal culture. Neurons that had been previously depleted of Bcl-xL fared much worse under hypoxic conditions, demonstrating frank neuritic damage and loss as well as enhanced cell death. DR6 depletion protected from neurite loss during hypoxia, suggesting that Bcl-xL processing or sequestration during hypoxia stimulates the activities of DR6. Indeed, we discovered that DR6 was upregulated either after Bcl-xL depletion in normoxia and/or after hypoxia, suggesting its involvement to enhance neurite degeneration under these conditions.
The pro-apoptotic molecule Bax was also activated, suggesting a role for the mitochondrial apoptotic pathway in neurite degeneration. Previously, we have reported that Bcl-xL enhances synaptic activity and induces synapse formation via dynamin-related protein 1 (DRP1) (26), a large GTPase which is involved in dendritic outgrowth (10). Post-translational modification of DRP1 is reported to change mitochondrial function and regulate electron transport activity (32, 34). We also found that Bcl-xL enhances energy metabolism in hippocampal neurons (1, 6). The ability both to meet metabolic demand and to control energy production is tightly regulated in growing cells (51), and previous studies have emphasized the importance of mitochondrial energy metabolism in neurite outgrowth (4, 26).
We suggest based on our data that failure to produce neurite outgrowth eventually leads to neuronal death, perhaps because unconnected synaptic endings signal apoptosis of the cell soma in a delayed fashion. We find that depletion of Bcl-xL produces shortened neurite length and a reduction in bifurcations which impairs normal neuronal network development over time. Neurites may be more susceptible than somata to altered energy metabolism given their role in neurotransmitter release and the sensitivity of this system to metabolic change (45). Impaired neurite growth is a noted pathological feature of many neurodegenerative diseases, including Alzheimer's and Parkinson's disease (7, 52), and an inverse relationship between neurite outgrowth and apoptosis has been previously reported (33, 50). We find here that failure of neurite growth may not only result from pathologic conditions in the nervous system but when stimulated de novo, may also serve as a potential trigger of cell death.
We were particularly interested in the role of DR6 in this phenomenon, because DR6 is involved in axonal degradation and mitochondrially mediated apoptosis (36, 53). We found that neurons depleted of Bcl-xL showed a higher abundance of DR6 protein, which may contribute to the slowing of neurite outgrowth at 2.5 weeks of rAAV incubation. In addition, we noted DR6 upregulation during hypoxia. If, indeed, endogenous Bcl-xL suppresses DR6 expression, and hypoxia upregulates DR6, this predicts that endogenous Bcl-xL may suppress DR6 expression during normoxia or hypoxia. Under resting conditions, Bcl-xL and other anti-cell death molecules bind activating BH3-only molecules, preventing their interaction with, and activation of, Bax (23). After a death stimulus, however, inactivating BH3 molecules sequester Bcl-xL, releasing activating BH3 molecules to activate Bax (24). We have found that Bax is activated either by Bcl-xL depletion or during hypoxia, suggesting that Bcl-xL may become sequestered under hypoxic conditions. Therefore, Bcl-xL sequestration and Bax activation during hypoxia could lead to upregulation of DR6 downstream of the hypoxic death stimulus (Fig. 12).
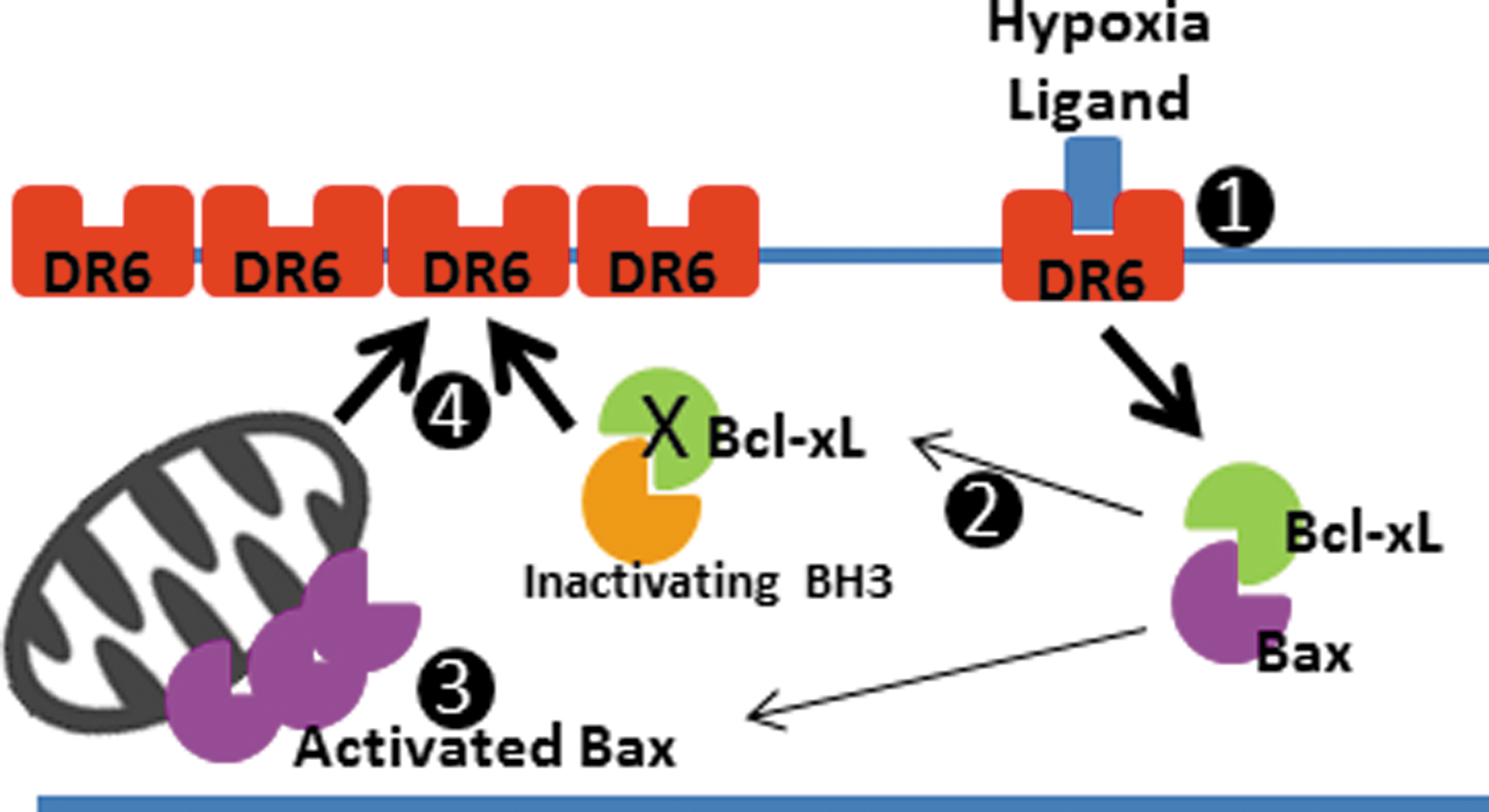
We also find that both hypoxia and Bcl-xL depletion increase activated caspase 6, placing Bcl-xL upstream of procaspase 6 proteolysis (Supplementary Fig. S3). Previously, the role of caspase 6 in axonal degeneration was reported (36, 47). Knocking down caspase 6 prevented axonal damage in these studies. In another work, overexpression of Bcl-xL inhibited cleavage of procaspase 6 under tetracycline-induced apoptotic conditions (53), which is consistent with our findings.
It is likely that Bcl-xL depletion mimics growth factor withdrawal (5, 31), possibly activating binding of endogenous ligands to DR6 (36). The possible scenario is that Bcl-xL depletion produces Bax translocation to mitochondria in neurites, initiating low-level early caspase 3 activation (30) that may be required for DR6 upregulation, enhancing neuritic degeneration via a positive feedback loop. Although our luminescence assay for caspase 3 levels is not positive in Bcl-xL-depleted cells, many reports indicate that caspase 3 activity occurs soon after an acute stimulus for neurite degeneration (30), which is impossible to measure under our conditions of slow decline in Bcl-xL levels. Either DR6 or caspase 3 may be required for caspase 6 activation. Hypoxia also activates Bax and produces caspase activation, feeding into the same pathway. One role of Bcl-xL is to heterodimerize with Bax or BH3-only activator molecules, preventing Bax activation downstream of the DR6-ligand interaction (36). It is now clear from our study that Bcl-xL is also upstream of DR6 upregulation, as Bcl-xL depletion induces an upregulation in DR6 protein levels and depletion of DR6 protects against neuritic degeneration in Bcl-xL-depleted cells. It is, therefore, possible that Bcl-xL is upstream of a transcriptional or translational mechanism that suppresses DR6 protein production.
In addition to loss of full-length Bcl-xL, pro-apoptotic Bcl-xL proteolysis products may coordinate with Bax and release pro-apoptotic factors from mitochondria, further activating downstream caspases (38). In future studies, it will be important to elucidate whether Bcl-xL regulates DR6 via caspase activation, or whether Bcl-xL is involved in DR6 transcriptional/translational regulation in an independent pathway. Taken together, our findings indicate that Bcl-xL mediates neuronal growth by regulating multiple signaling pathways, including one or more involving DR6, and it also specifically protects neuronal processes under hypoxic conditions. Therapeutic endeavors to activate Bcl-xL in the brain may protect neurons against hypoxia and neurodegeneration by enhancing neuritogenesis or neurite protection.
Materials and Methods
Primary cultures of rat hippocampal neurons
Primary rat hippocampal neurons were prepared from rat fetuses (Sprague–Dawley, day 18 of gestation; Harlan, Indianapolis, IN) as previously described (3, 22). After isolation of hippocampi from prenatal brains, neurons were dissociated and seeded (0.07×106 cells/35 mm plate) onto plates containing medium with 5% fetal bovine serum. After 2–4 h of incubation, cells were maintained in neurobasal medium supplemented with B-27, glutamine, and antibiotics (Invitrogen Gibco Life Technologies, Carlsbad, CA). Neurons were grown at 37°C in 5% CO2 and 20% O2 in a humidified incubator. Neurons were transduced with rAAV constructs (25,000 viral particles/ml) at 7 days in vitro (DIV), and experiments were performed at 1, 2.5, or 4 weeks after rAAV introduction.
Construction and preparation of rAAV
The DNA fragments containing U6 promoter followed by shRNA sequence against rat Bcl-xL or scrambled siRNA were cloned into rAAV vector, in front of the ubiquitin promoter-DsRED expression cassette. The control siRNA was against EGFP sequence, thus lacking endogenous mRNA targets (11). rAAV2/1 viral stocks were produced as previously described (15). Briefly, human embryonic kidney (HEK293) cells were transfected with rAAV expression plasmid and pDp1 and pDp2 helper plasmids using Lipofectamine reagent (Invitrogen). Cell pellets were collected at 3 days post-transfection, lysed in three freeze-thaw cycles in a dry ice/ethanol bath. Next, collected lysates were filtered using 0.4 μm gauge filters, and viruses were purified using HiTrap heparin HP affinity columns (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Viral titers were determined in rat hippocampal primary cultures. The scrambled siRNA sequences were as follows:
scrambled up: 5′-tttgcaagctgaccctgaagttcttttt-3′
scrambled down: 5′-cgaaaaagaacttcagggtcagcttg-3′
The Bcl-xL shRNA sequences were:
Bcl-xL up: 5′-TTTggagtcagtttagcgatgtcgGTGAAGCCACAGATGcgacatcgctaaactgactccTTTTT-3′
Bcl-xL down: 5′-CGAAAAAggagtcagtttagcgatgtcgCATCTGTGGCTTCACcgacatcgctaaactgactc-3′
Neuronal growth analysis
Primary hippocampal neurons were transduced with rAAVs or a combination of lenti-DR6 shRNA or lenti-copGFP control (Santa Cruz, Dallas, TX) at DIV 7. Neuronal images were taken with a Zeiss Axiovert 200 microscope (Zeiss, Oberkochen, Germany) and processed using AxioVision 4.8 at the time point described in the relevant figure legends. All fluorescent images were taken consistently (RED, exposure time: 2000 ms; Green, exposure time: 2000 ms). Data were analyzed under the fluorescent channel, using the color-off setting. We found that fluorescent-based images were more sensitive to monitor neuronal growth compared with phase-contrast images (Supplementary Fig. S4). Although we obtained consistent data sets from phase-contrast images, these images failed to detect fine extensions of neurites. Therefore, all data in the main manuscript figures represent fluorescent images except the 1 week group.
Definitions
Number of neurites on soma: The number of neuritic extensions that directly connect to somata of hippocampal neurons.
Length of neurites: Largest distance from one end of the cell to the other end of the cell (neurite tip to neurite tip) measured using AxioVision 4.8.
Number of bifurcations: The number of branches of somatic neurites within 100 μm radius of the soma. The center point of a circle is placed on the center of the soma. Poorly visualized extensions or neurites shorter and 20 μm were excluded.
Induction and confirmation of hypoxia
Primary hippocampal neurons were challenged with hypoxia at 2.5 weeks after transduction. On the day of the experiment, hypoxic gas (5% CO2 and 95% N2) was flushed through a modular incubator chamber (Billups-Rothenberg, Inc., Del Mar, CA) for 5 min; then, the chamber was sealed and placed in an incubator maintained at 37°C. Cells were assayed at 24 h after hypoxic exposure as described in the corresponding figure legends. Composition of cell culture medium containing 25 mM glucose remained unchanged during hypoxic exposure. Ten to 20 ml sterile water was placed inside of the chamber to prevent evaporation of culture media. Oxygen content in the hypoxia-exposed cell culture medium was measured using a Clark-type electrode (Supplementary Fig. S2). Liquid-phase calibration was performed using an Oxygraph respirometer (Hansatech Instrument, King's Lynn, United Kingdom, kindly provided by Prof. Sabrina Diano) according to the manufacturer's protocol. Briefly, oxygen content in temperature-controlled, air-saturated, deionized water was analyzed and set as 100%. Zero-oxygen was obtained using sodium hydrosulfite (Na2S2O4). One milliliter of either hypoxic or normoxic cell culture media was added into the chamber. Oxygen concentration was recorded at ten times per second using Oxygraph software at 37°C from three or four independently prepared plates and normalized to the value for aerated water generated by the oxygen calibration.
Immunoblots
After protein extraction, protein concentration was determined using BCA protein reagents (Thermo Scientific, Rockford, IL). Samples (30–120 μg of protein/lane) were separated on a 4%–12% sodium dodecyl sulfate–polyacrylamide gel (Bio-Rad, Hercules, CA) and probed with anti-Bcl-xL (1:1000 dilution; Cell Signaling Technology, Danvers, MA), anti-caspase 6 (1:500 dilution; Cell Signaling Technology), anti-DR6 (1:500 dilution; NOVUS Biological, Littleton, CO), and anti-active bax (1:300; Enzo Life Science, Farmingdale, NY). To evaluate loading efficiency, membranes were probed with anti-GAPDH antibody (Sigma-Aldrich, St. Louis, MO). We confirmed that secondary antibodies do not produce false positive chemiluminescence signals (not shown). Images were analyzed using ImageJ software.
Immunocytochemistry
Primary hippocampal neurons fixed in 10% buffered formalin were blocked in 10% goat serum for 1 h, then incubated with anti-NeuN (1:100 dilution; Millipore, Billerica, MA), anti-Bcl-xL (1:100), anti-dsRED (1:100; Santa Cruz Biotechnology, Dallas, TX), and anti-RFP (1:100; Abcam, Cambridge, United Kingdom) overnight at 4°C. Cells were washed and incubated with Alexa-fluor 488 antibody or Alexa-568 antibody (1:200 dilution; Invitrogen, Molecular Probes, Carlsbad, CA) for 1 h at room temperature and mounted on glass slides. Images were taken with a Zeiss Axiovert 200 microscope (Zeiss) and processed using AxioVision 4.8.
Viability assay
LDH assay
The viability of primary hippocampal neurons was assayed by measuring leakage of LDH using an in vitro toxicology assay kit (Sigma-Aldrich) as previously described (40, 41). Briefly, the culture media were collected from individual plates after treatment of neurons as described in the relevant figure legends; then, cells were lysed and collected separately. LDH assay mixture was made according to the manufacturer's protocol and added to each sample. After 20 min of incubation, the reaction was terminated by adding 1N HCl. LDH activity was spectrophotometrically measured with a VICTOR3 multilabel reader (PerkinElmer, Waltham, MA) with absorbance set at 490 nm.
TUNEL assay
After treatment of neurons as described in the corresponding figure legends, cells were fixed in 10% buffered formalin. Quantification of DNA fragmentation was measured using an in situ apoptosis detection kit according to the manufacturer's protocol (Trevigen, Gaithersburg, MD). Terminal deoxinucleotidyl transferase incorporated nucleotide was labeled with strep-fluorescein, and 1 μg/ml DAPI (Invitrogen) was used for nuclear counterstaining. All data were analyzed within dsRED-positive neurons, and dsRED-negative cells were neither collected nor analyzed in this study. To obtain data from randomly collected images, we drew a square at the bottom of the glass or plate, and imaged an entire field of magnification just inside each corner of the square, leaving out the lines of the square so that the lines do not appear in the micrograph. Four corners together equal 0.2 mm2 at 32× magnification.
Calcein-AM
Calcein-AM is a neutral, hydrophobic, non-fluorescent, cell permeable compound that is a substrate for intracellular esterases and is hydrolyzed to membrane-impermeable, hydrophilic, and fluorescent Calcein in viable cells. After treatment of neurons as described in the corresponding figure legends, 25 nM Calcein-AM (Invitrogen) and 1 μg/ml DAPI were added to the culture medium for 30 min. Images were taken using a Zeiss Axiovert 200 microscope and analyzed using AxioVision 4.8. All data were randomly collected and analyzed within dsRED-positive neurons. dsRED-negative cells were neither collected nor analyzed in this study.
Caspase assay
After treatment of neurons as described in the corresponding figure legends, caspase activity was measured using Caspase Glo 6 assay kit and Caspase-Glo 3/7 assay kit (Promega, Madison, WI) based on the manufacturer's protocol. Briefly, plates were washed with sterile phosphate-buffered saline (PBS), and incubated with substrates (Ac-VEID-pNA or Ac-DEVD-pNA) for 30 min. Active caspases cleave luminogenic substrate and produce bioluminescence. Caspase-induced luminescence was measured with a VICTOR3 multilabel reader (PerkinElmer).
Four-vessel occlusion
Transient global ischemia was induced in 6–7 week-old male Sprague–Dawley rats as previously described (38, 43). Briefly, animals were anesthetized by isoflurane; then, left and right vertebral arteries were permanently occluded by electocauterization on the day before occlusion. After 24 h, both common carotid arteries were occluded with aneurysm clips and reperfused after 10 min. Animals were placed in their home cages, and sacrificed at 24 h or 7 days after 4VO. This surgical protocol enables oxygen glucose deprivation in the forebrain, but typically results in neurodegeneration in pyramidal neurons in the hippocampal CA1 and only sparsely in other areas of the brain. Animals suffering from complications (e.g., pain, bleeding, or death) during or after 4VO were excluded. Sham animals were subjected to the identical procedure, minus carotid artery occlusion. Rats were maintained in a temperature- and light-controlled environment. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Yale University, New Haven, CT.
Histology
Animals were euthanized and fixed by transcardial perfusion with ice-cold 4% paraformaldehyde (wt/vol) 0.1 M PBS, pH 7.4. Brains were removed and immersed in fixative. Coronal sections (20 μm) were cut and stained with 0.0001% Fluoro-Jade®C, anti-NeuN (1:100; Millipore), and anti-DR6 (1:100; Santa Cruz Biotechnology). Images were taken using a Zeiss Axiovert 200 microscope and analyzed by using AxioVision 4.8.
Statistical analysis
Data are reported as mean±SEM of at least three independently conducted experiments. Difference in means was tested using one-way ANOVA with Tukey's test or two-tailed Student's t-test. p<0.05 was considered statistically significant. p values are provided in figure legends.
Footnotes
Acknowledgments
The authors appreciate very much the help and guidance they received from Dr. Naoki Kaneko and Dr. Suzanne Zukin of Albert Einstein College of Medicine in preparation for performing the in vivo studies.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.