
Abstract
Introduction
T
Diverse and tumor-specific genetic and epigenetic upheavals underlie the occurrence of many yet unknown oncogenic mechanisms. Part of these oncogenic events comprises ectopic activation of normally tissue-specific genes (30). One of these genes is prolactin (PRL) reported here to be illegitimately activated in very aggressive lung tumor and associated with particularly poor prognosis. In breast and prostate cancer, oncogenic PRL acts through its receptor (PRL-R) following an autocrine or paracrine pathway (16). In this study, we show that a combination of genetic alterations and ectopic activation of PRL is at the heart of a completely unexpected receptor-independent oncogenic mechanism that can be counteracted by histone deacetylase (HDAC) inhibitors.
Prolactin (PRL) and its receptor have now been convincingly shown to be involved in breast and prostate tumorigenesis (10), and consequently targeting the corresponding signaling systems could be considered as a reasonable therapeutic strategy in different cancers, especially in prostate cancer (16). To date, the tumor-promoting action of PRL has been thought to be dependent on its receptor (PRL-R), which is also expressed by cancer cells and enables them to respond, through paracrine or autocrine loops, to circulating PRL from the pituitary gland or to local PRL produced by the cancer cells themselves (10).
In this study, we report the investigation of the out-of-context expression of PRL in lung cancer to explore its relevance as a potential biomarker for prognosis and as a guide in orienting a particular therapeutic approach.
Results
Ectopic PRL activation in a subset of very aggressive lung tumors
Recently, we reported transcriptomic data from a cohort of 293 lung tumors (30). These data were used to monitor the expression of PRL, which was found to be expressed in 27 lung tumors out of our series of 293 (9.2%) (Fig. 1A, B). A search of expressed sequence tag (EST) database (Supplementary Fig. S1A; Supplementary Data are available online at

We became particularly interested in the functional consequences of the ectopic activation of PRL in lung tumors following the observation that most of the patients with PRL+ tumors died rapidly (p-value <0.0001; Hazard ratio=3.23) (Fig. 1D). This association between the ectopic PRL expression and adverse clinical outcome was confirmed in a completely independent study (transcriptomic data publically available under the GEO reference GSE19188), where the patients with PRL-expressing tumors (10.9%) also exhibited a very poor prognosis (Fig. 1E). Since neuroendocrine lung tumors are more prone to express neuronal genes (13, 35), we wondered whether the ectopic PRL activation could only occur in tumors of the neuroendocrine subtype. The analysis of the lung tumors of our series demonstrated that the majority of the PRL-expressing tumors indeed belonged to the neuroendocrine types of LCNE or small cell lung cancers (SCLC). However, we also found that not all LCNE and SCLC tumors expressed PRL and that, within each histological subtype, the ectopic activation of PRL was associated with a particularly aggressive behavior of the tumors (Fig. 1F). These observations, together with a multivariate analysis quantifying the implication of various parameters (histological classification, tumor node-metastasis [TNM] parameters, PRL expression) in the survival (Supplementary Fig. S1D), suggested that PRL activation was not simply a phenomenon associated with neuroendocrine lung tumors, but could itself be a strong predictor of poor prognosis.
Receptor-independent action of ectopically activated PRL
Since PRL is believed to act through a specific receptor, we wanted to know if the PRL-expressing lung tumor cells also expressed the corresponding receptor (PRL-R). The analysis of our series of 293 lung tumor transcriptomes showed that 59 tumors ectopically activated PRL-R. However, only 5 of these tumors also expressed PRL, so that 22 out of the 27 PRL+ tumors did not show any expression of PRL-R (Fig. 2A). We were also able to demonstrate that PRL-R expression is not associated with tumor aggressiveness (Fig. 2B). According to the EST databanks (Supplementary Fig. S2A), and consistent with our RT-qPCR data (data not shown), the receptor is normally expressed in the breast, placenta, and uterus, but not in the lung.

These observations suggested a possible and previously undetected PRL-dependent, but receptor-independent oncogenic mechanism and prompted us to investigate the issue. Accordingly, to identify PRL+ cell lines, we screened the available transcriptomic data from lung cancer cell lines (data not shown) and from various cancer cell lines available to us (Fig. 3A). Following these approaches, we chose four cell lines, all from SCLC, three (H146, H524, and H69) expressing PRL at variable, but low amounts (not exceeding the placental expression level), and one (H526) showing no PRL expression (Fig. 3B, C). The analysis of the expression of PRL-R showed that none of our three PRL+ cell lines expressed the receptor (Fig. 3B). In this experiment, to make sure that we confidently defined the state of PRL-R expression, a PCR primer set was chosen in a region present in all PRL-R encoding mRNAs (2, 20) (Supplementary Fig. S2B) and confirmed the absence of PRL-R expression in the nonpathological lung and in our three PRL+ cell lines (Supplementary Fig. S2C). These cell lines appeared therefore as good model systems to investigate the molecular basis of PRL activity in lung cancer.

Ectopically expressed PRL mRNAs in lung cancers bear 5′ truncations and systematically lack the signal peptide-encoding region
To better understand the possible receptor-independent cellular functions of the ectopically activated PRL in lung cancers, we first addressed the question whether an important functional determinant of the PRL protein, its signal peptide, was present in the ectopically expressed PRL mRNA. The absence or the presence of a nonfunctional signal peptide would lead to the production of a nonsecreted PRL and the accumulation of the protein in the cells, which would have the potential to affect various cellular functions as opposed to a secreted protein that, in the absence of PRL-R, would not have any effects.
We designed a series of primers to better characterize PRL transcripts expressed in our PRL+ lung cell lines. We first focused on the two known transcript variants of PRL, differing only in the 5′UTR and encoding the same protein, which are expressed either from a proximal promoter in the pituitary gland (variant 1) or from a distal promoter used in the extrapituitary tissues (variant 2) (15). Although we could detect the expected transcripts in the pituitary gland and placenta, the transcripts detected in our lung cancer cell lines lacked the 5′ regions, which are normally present in transcripts produced from either of the promoters (Fig. 4A).
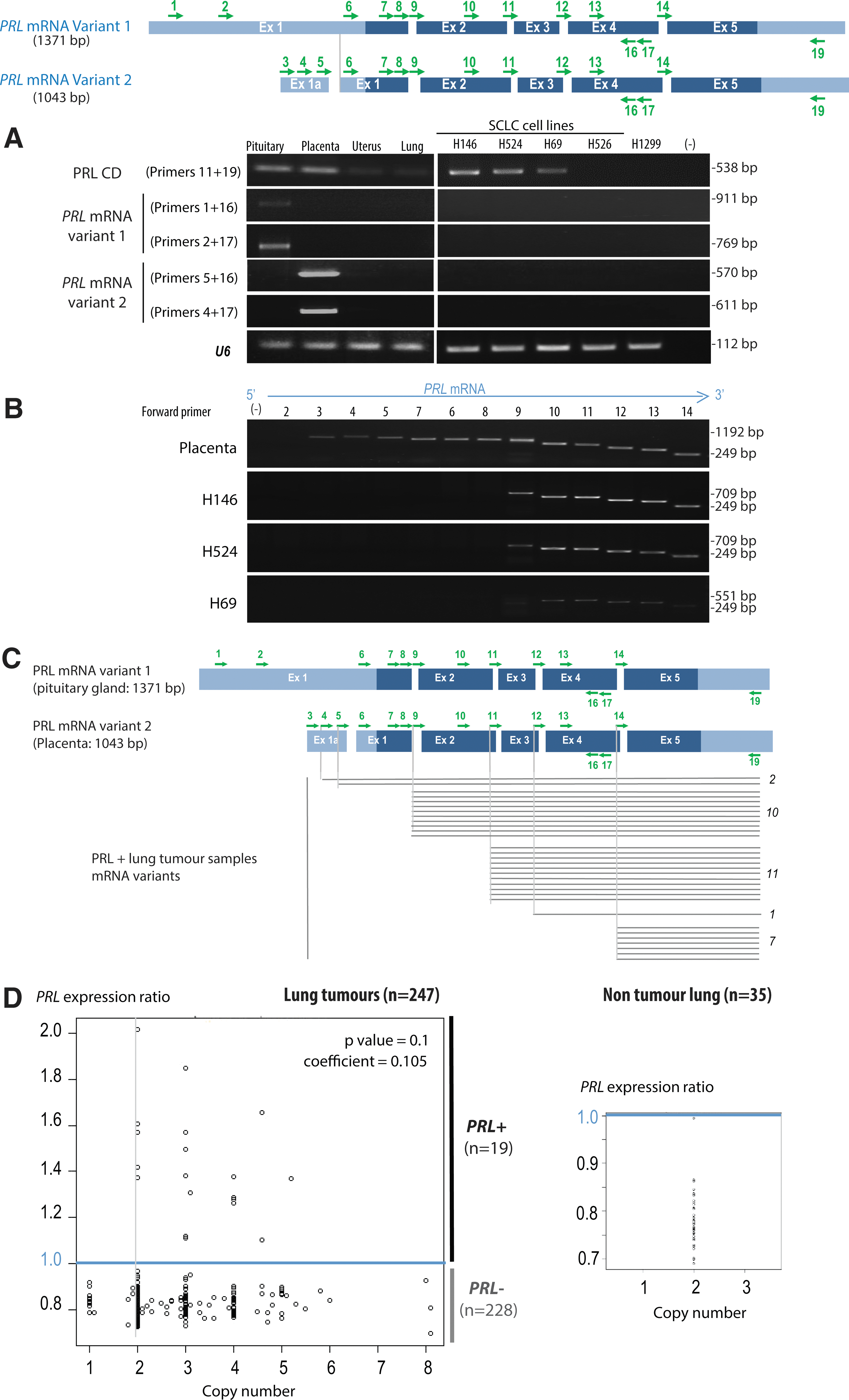
A more detailed analysis of these transcripts with sets of primers spanning the PRL transcript from a fix 3′ position toward the 5′ end showed that in all the three PRL+ cell lines (H146, H524, and H69), PRL transcripts lacked its 5′ exons (exon 1 and even a portion of exon 2) (Fig. 4B). Since the beginning of the signal peptide is encoded by exon 1, all the PRL transcripts produced in the three studied cell lines lack at least a portion of the signal peptide-encoding region.
Following this observation, we also tested 50 lung tumors, including 31 PRL+ and 19 PRL− samples. The 31 PRL+ group included 23 PRL+ tumors detected by the affymetrix transcriptomic analysis as well as 9 additional PRL+ tumors, which were identified by RT-PCR among the tumors originally classified as PRL− following affymetrix analysis, thanks to the sensitivity of PCR approaches.
Scanning PRL transcripts with some of the above-mentioned primers showed that 29 out of the 31 PRL+ tumors were missing regions encoding the signal peptide, with various extents of 5′ deletions (Fig. 4C and Supplementary Fig. S3). These data therefore indicate that in lung tumors and cell lines, not only PRL is ectopically activated but also genetic alterations almost systematically affect the genomic region encoding the 5′ part of the transcript that covers the signal peptide.
We also wondered whether the ectopic expression of PRL could be associated with amplification of the gene or its genomic region. Data from comparative genomic hybridization (CGH) arrays, obtained for 247 of our 293 lung cancer patients, demonstrated that, although the genomic region corresponding to the PRL gene was amplified in a proportion of patients, there was no correlation between amplification and expression (Fig. 4D). Moreover, survival comparisons between groups of patients according to the presence or absence of amplification within this region demonstrated that there was no association between the amplification of this region and prognostic (data not shown).
The ectopically expressed truncated PRL is a potent regulator of gene expression
Our data show that all the PRL+ lung tumor samples and cell lines express a truncated form of a PRL transcript lacking the 5′ part of the mRNA to various extents. Since this almost systematic 5′ deletion includes the translation initiation ATG codon, it is unlikely that PRL is produced. Nevertheless, we decided to investigate the consequences of the ectopic expression of these transcripts. This was achieved through a siRNA knockdown in two PRL+ cell lines (H146 and H524) followed by a whole genome transcriptomic analysis. With a threshold of 1.2 for mRNA variations between control siRNA and the anti-PRL siRNA, the number of genes affected was found to be proportional to the level of PRL expression. In the H146 cell line, expressing the highest level of PRL mRNA (Figs. 3C and 6A), PRL silencing resulted in the upregulation and downregulation of 2559 and 2402 genes, respectively, whereas in the H524 cells, which express about half the level of PRL mRNA (Figs. 3C and 6A), 800 and 763 genes were, respectively, upregulated and downregulated (Fig. 5A). Interestingly, the majority of the genes affected by PRL silencing in the H524 cell line were also PRL-dependent genes in the H146 cells. Indeed, 74% and 68% of genes, respectively, upregulated and downregulated in H524 cells were included in the corresponding gene lists from the H146 cell line after PRL knockdown (Fig. 5A). These data strongly suggested that PRL affects the same regulatory pathways in these two different cell lines and that the extent of these effects depends on the amount of PRL expressed. Within the genes affected in both cell lines, we chose 12 for validation by RT-qPCR using H146 RNA from cells treated with control or anti-PRL siRNAs, and the data confirmed the robustness of the analysis (Fig. 5B). Hence, these results reveal an important gene regulatory function for these truncated PRL RNAs in both cell lines.
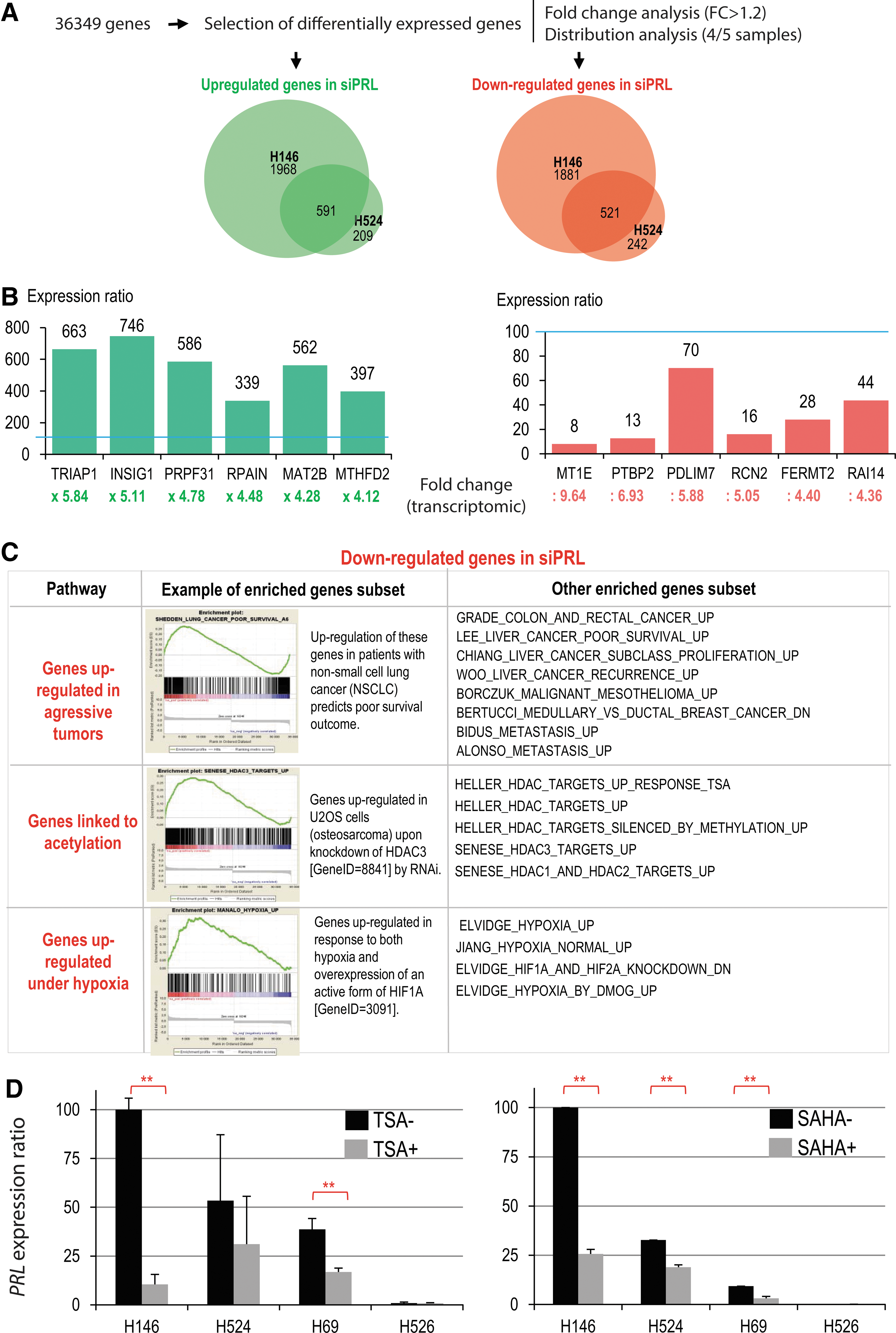

In an attempt to identify the cellular regulatory circuits controlled by PRL, a Gene Set Enrichment Analysis (GSEA) was performed on the list of genes differentially regulated after PRL knockdown in the H146 cell line. This analysis showed that the group of genes requiring PRL to be expressed (downregulated after PRL knockdown) significantly overlaps with previously established lists of genes upregulated in non-small cell lung tumors as well as in a variety of unrelated cancers (Fig. 5C).
Interestingly, the GSEA also revealed that in these cells, which are here maintained under normoxic culture conditions, the ectopic PRL expression activates a group of genes that were found to be also expressed in response to both hypoxia and HIF1α (7) (Fig. 5C).
Remarkably, this analysis also revealed a highly significant overlap with genes that are upregulated either after treatment with the HDACi trichostatin A (TSA) or after the knockdown of HDAC1, HDAC2, or HDAC3 (18, 33) in independent studies and cell systems (Fig. 5C). These observations suggested that in PRL+ lung cancers, PRL somehow controls cellular regulatory circuits involving histone acetylation-dependent transcriptional activities, since both PRL knockdown and HDAC1–3 inhibition or knockdown affect a significant number of common genes. To better understand the relationship between PRL transcripts/peptides and HDACs, we treated our three PRL+ and one PRL− cell lines with TSA, as well as with the FDA approved HDAC inhibitor, SAHA, and measured the level of PRL mRNA by RT-qPCR. Interestingly, and consistent with the GSEA results, we observed that treatment with both TSA and SAHA leads to a remarkable decrease in the steady-state amounts of PRL mRNA (Fig. 5D), indicating that by downregulating PRL, a treatment by HDAC inhibitors could also affect a significant number of common genes.
PRL downregulation sensitizes lung cancer cells to genotoxic treatments
Since in many cases, cancer cell treatment with HDAC inhibitors restores cell cycle check points and apoptotic cell responses (22, 26), we wondered if PRL downregulation could also render cells more sensitive to a cytotoxic treatment. Our three PRL+ and one PRL− lung cancer cell lines were treated with either a control or the two anti-PRL siRNAs (Supplementary Table S1A), leading to a significant knockdown of PRL expression in all PRL+ cell lines (Fig. 6A).
We then assessed the effect of PRL silencing on cell apoptosis. We first observed that the loss of PRL expression does not affect the basal mortality. We then subjected SCLC cells to genotoxic stress, through a treatment with actinomycin D (Acti D). In the three PRL+ cell lines (H146, H524, and H69), PRL knockdown increased the proportion of apoptotic cells following the Acti D treatment, while the PRL siRNA transfection in the PRL− cell line did not significantly modify the proportion of apoptotic cells. The latter cells appeared to be more responsive to a genotoxic treatment, which is consistent with the absence of PRL expression (Fig. 6B). This figure also shows the original FACS data for one experiment with the H524 cell line treated with control or anti-PRL siRNA (siRNA PRL #1) (Fig. 6C). These data show that PRL downregulation in this cell line under genotoxic treatment almost doubled the number of dying cells, which approached the totality of the population. It is of note that the dosage of Acti D used to induce genotoxic effects is below the amount required to block the PRL gene activity (Supplementary Fig. S4).
We concluded that the PRL ectopic expression in SCLC cells is involved in their resistance to cell death following a genotoxic treatment.
Discussion
Genome instability and subsequent gross and subtle genome alterations associated with a profoundly modified epigenetic signaling, systematically accompany malignant cell transformation (34). Growing and invading cancer cells harbor new states of gene expression involving not only activation and repression of normal genes but also de novo creation of genes that never existed in normal cells. Such genes result from chromosomal translocations, mutations, loss and gain of specific genomic regions. The combined action of these created new functional factors, with gene silencing and ectopic activations of hundreds of tissue-restricted genes, gives birth to cells that acquire new properties (9, 24, 31) allowing them to sustain growth and dissemination, while avoiding destruction (17). In this study, we show that PRL, a gene never expressed in normal lung cells, becomes ectopically activated in lung cancer tumors, essentially in a fraction of the neuroendocrine subtype. This activation of PRL is strongly associated with adverse clinical outcomes, not only in our population of lung cancer patients from the Grenoble university hospital but also in a completely unrelated group of patients. Unexpectedly, we also found that, in both groups of patients, the majority of PRL+ lung tumors did not express detectable amounts of PRL-R transcripts. However, our present knowledge of PRL biology suggested that, in the absence of a receptor, the produced PRL should have no effects on the PRL-expressing tumor cells, since most of the synthesized proteins would be secreted out of the cells and no signaling could be initiated in the absence of the receptor. This was inconsistent with our clinical data, showing a systematic association between PRL expression and rapid fatal outcome, which suggested that the produced PRL should have an effect on the cells and somehow promote their aggressive behavior. Therefore, we made the hypothesis that, in the absence of its receptor, the only way PRL expression could have an impact on cells would be that the protein somehow would lose its signal peptide, and hence becomes trapped inside the cells. Our investigations in the selected PRL+ cell line models and tumors revealed genome-altering events, which almost systematically affected the first exon of PRL. In fact, in most of the tested cases, the transcript did not contain the translation initiation codon ATG. Even in the five cases of tumors expressing PRL-R and PRL mRNA, the PRL transcripts were truncated.
The important question following this observation is whether these mRNAs could give rise to a polypeptide. The use of an antibody able to detect large amounts of PRL in the pituitary gland, did not allow us to visualize the protein neither in lung tumors nor in our lung cancer cell lines (data not shown). Therefore, at this point, the absence of the translation initiation codon ATG in the transcripts, suggests that actually no easily detectable peptides are expressed. Nevertheless, the detailed investigation of our PRL+ cells revealed clear and characteristic molecular responses to the knockdown of PRL mRNA. Most remarkably, PRL mRNA silencing was associated with very consistent alterations in gene expression in two different cell lines. This gene expression response was proportional to the level of PRL expression: a higher number of genes were affected in the cell line expressing the highest amount of PRL mRNA. Additionally, the majority of the genes affected in the cell line expressing a lower amount of PRL mRNA were included in the list of differentially expressed genes in the first cell line.
This observation suggests that the truncated PRL mRNAs could titer out other regulatory molecules, and hence, the higher the concentration of PRL mRNAs, the stronger the effect. One very attractive hypothesis could be that PRL mRNAs act as endogenous competing RNAs (ceRNA) and titer out specific microRNAs [for review see de Giorgio et al. (5)]. However, important investigations are required to unravel the exact molecular mechanism underlying the oncogenic activity of truncated PRL mRNAs.
GSEA also indicated a tight link between the genes repressed following PRL knockdown in lung cancer cell lines cultured under normoxic conditions and genes that become expressed following hypoxia or a pharmacological stabilization of HIF1α by a 2-oxoglutarate-dependent dioxygenase inhibitor (7). It has also been shown that HIF1α contributes to important gene expression programs affecting processes, such as DNA repair, and that a hypoxic microenvironment could enhance genetic alterations in apoptosis-deficient cells (39). By regulating such a specific hypoxia-dependent gene expression program, PRL ectopic activation, could confer to cells under normoxic conditions, specific oncogenic properties that normally appear only under hypoxia.
GSEA also revealed a clear and significant overlap between genes upregulated after the knockdown of PRL and genes upregulated after a HDAC inhibitor (TSA) treatment or after a targeted knockdown of HDAC3, HDAC2, and HDAC1 (18, 33). This observation is reminiscent of the results reported by Schwartz et al., where the knockdown of BRD4-NUT in NMC led to a gene activation that was comparable to the effect observed after treatment of cells with HDAC inhibitors (32). In the case of NMC, the molecular dissection of the underlying mechanism showed that the fusion protein BRD4-NUT traps CBP/p300 in nuclear foci that can be released after BRD4-NUT knockdown (29). Although we have not here been able to identify the molecular basis of the control of gene expression by PRL, it is possible to speculate on a similar direct or indirect effect of PRL on the activity of major cellular HATs, so that PRL knockdown could restore normal acetylation and the underlying gene expression. Additionally, we showed that a TSA treatment can directly downregulate PRL. This could also account for the overlap between HDACi gene responses and our cell line response after PRL knockdown. The fact that PRL is a strong and reliable predictor of poor clinical outcome in the case of lung cancer and that HDAC inhibitors could downregulate PRL or activate a shift of the PRL-associated aggressive gene signature toward that observed in tumors with better prognosis, suggests that the treatment of PRL+ lung tumors with HDACi alone or in combination with a more conventional chemotherapy, should be a reasonable therapeutic strategy for this category of PRL+ lung tumor patients.
Finally, this work highlights once more that cancer cells use many yet unknown and unexpected molecular mechanisms to ensure their malignant states and that a comprehensive understanding of cancer cannot be approached without a global consideration of all the oncogenic events.
Materials and Methods
Material
H146, H524, H69, H526, and H1299 cell lines were obtained from the American Type Culture Collection (ATCC). H146, H524, H69, and H526 are derived from small cell lung carcinomas from Caucasian males, respectively, 59, 63, 55, and 55 years old. H1299 is a non-SCLC from a Caucasian 43-year-old male.
Human tissue RNAs were purchased from the Clontech society (Human Total RNA, Master Panel II), except for placenta RNAs, which were a kind gift from Dr Nadia Alfaidy-Benharouga (CEA).
The Grenoble lung cancer cohort, established by Pr. Elisabeth Brambilla and Christian Brambilla (CHU), includes 293 patients who underwent surgery at the University Hospital of Grenoble. For over 10 years, clinical data (patient age and gender, smoking status, date of diagnosis, histological and TNM classifications of the tumor, treatment received, and date of death) and biological samples (biopsies, tumor sections, and RNAs) were collected. For each patient, a transcriptomic study was performed on a tumor sample collected before implementation of treatment, and in some cases, on a sample of adjacent noncancerous lung tissue. Detailed clinical and biological information relative to these patients is available in Rousseaux et al. (30).
PRL+ versus PRL− cell line transcriptomes
All transcriptomic data in lung cancer cell lines were obtained using the Illumina HumanHT-12V4.0 expression beadchip and were processed exactly as described in Refs. (14, 28). The raw and processed data from the array are deposited in the GEO databank under the accession GSE49544.
Transcriptomic and gene copy number variants from Grenoble lung cancer tumor cohort
Transcriptomic data from 293 pulmonary tumors of the lung cancer cohort were obtained on the Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2) as described earlier (30). These data are available on the GEO website (reference number GSE30219).
For 247 of the same tumor samples, gene copy numbers were also calculated thanks to parallel hybridization of genomic DNAs on Illumina SNP HumanCNV370 chips (Illumina). The GISTIC version 2.0 algorithm (
Analysis of PRL and PRL-R expression in tumors
The expression data of PRL and PRL-R genes in lung tumors were extracted from the Affymetrix RMA normalized signal values corresponding to probe sets 205455_at (PRL) and 205455_at (PRL-R). For each gene, a threshold value was defined corresponding to a positive expression, by using data from a series of normal human somatic tissue samples, as described previously (30). For the PRL gene, the threshold above which it was considered expressed was the mean value of the 205455_at probe set signal in 109 samples of normal human somatic tissues (excluding the pituitary samples). For the PRL-R gene, the threshold above which it was considered expressed was the mean+2 standard deviations of the 205455_at of the signal values in 112 normal human somatic tissue samples.
RNA extraction and reverse transcription
SCLC cells growing in suspension were harvested by pipetting 2 days after seeding (and eventual transfection of siRNA or drug treatment). The extraction of total RNA was performed with the NucleoSpin RNA II (Macherey-Nagel) kit according to the supplier's recommendations. The RNA content and purity were assessed using NanoDrop 1000 (Thermo Scientific). The reverse transcription reaction was performed on 1 μg of total RNA using the kit SuperScript III RT First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen) according to the supplier's recommendations.
PCR reactions
Primers were designed with the help of the three following online software: Universal Probe Library Assay Design Center (Roche Applied Science), Primer 3 (
The PCR reactions were carried out in a final volume of 25 μl containing 0.5 μl dNTPs (1 mM, 5′), 0.5 μl of each primer, forward and reverse (10 μM), 2.5 μl of 10× buffer, 0.5 μl of TaqPol enzyme (5 U/μl, 5′), and cDNAs (RT product diluted 1/10). The PCR program was as follows: denaturation at 95°C for 1 min, then 35 cycles (denaturation 95°C 30 s, annealing 30 s, elongation 68°C), and finally 5 min of terminal extension at 68°C. The elongation time varied depending on the length of the PCR product expected (1 min/kb). The annealing temperature was chosen according to the Tm of primers (often 65°C, sometimes down to 55°C). A negative control was achieved by replacing the cDNA by DEPC H2O. The PCR products were analyzed by electrophoresis in a gel containing 1% agarose (Invitrogen) and 5 μl of BET (10 mg/ml; Sigma-Aldrich) in 100 ml of 1× TAE buffer. DNA (25 μl of PCR product) was mixed with 3 μl of loading buffer (bromophenol blue, 40% sucrose) and loaded onto the gel, together with a size marker (DNA Ladder, Fermentas or 1 kb-Plus; Invitrogen). Migration was performed in 1× TAE at 100 V for about 30 min. The reading was done under a UV lamp.
qPCR reactions
qPCR analysis was performed using Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MiQE) guidelines (3). Primers were designed so that the length of the amplicon is between 70 and 200 bp. qPCR was performed in 96-well plates in a final volume of 20 μl. Each well contained 0.03 μl of each primer, forward and reverse (100 μM), 10 μl of Brilliant SYBR Green 2× III (Agilent Technology), and 4 μl of cDNA (RT product diluted 1/10). For each condition, a technical duplicate and a control point where the cDNA is replaced by the extracted RNA (no RT) were added. The reaction was carried out in the Mx3005P qPCR cycler (Agilent Technology) with the following program: 3 min of initial denaturation at 95°C, then 40 cycles with 20 s of denaturation at 95°C, and 20 s of hybridization and elongation at 60°C.
The qPCR analysis was performed using the MxPro software (Agilent Technology). After verifying that a given pair of primers gives a single peak of dissociation, the value of the threshold cycle (Ct) was given by the number of cycles necessary for the measured fluorescence value to exceed the threshold value (threshold=235). When the Ct exceeded 35, we considered that there was no expression. When the Ct value obtained for the no RT control point was too close (NoRT-RT <2), we did not take the value into account. The analysis was performed on the average of the two Ct values obtained for the two technical replicates (difference <1). Expression values were normalized with the selected control genes (Actin, U6, RELA, and/or AUP1). The expression values were given as a percentage of the value corresponding to a reference condition (reference tissue such as pituitary or placenta or breast, or nontransfected control condition for cell lines) by the method of Delta Ct (18): 2ΔCt gene of interest (reference condition−condition of interest)/2ΔCt control gene (reference condition−condition of interest).
Cell culture and transfection
H524 and H526 cells were cultured in the Roswell Park Memorial Institute medium (RPMI 1640; Gibco) supplemented with 10% fetal bovine serum (PAA), 2 mM
siRNAs targeting human PRL and nonspecific control siRNAs were designed with the DSIR software (
Two days after transfection, the culture medium was complemented with 250 nM Acti D (Sigma-Aldrich) for 12 h or 100 ng/ml TSA (Sigma-Aldrich) for 6 h.
Quantification of apoptotic cells
Cells were harvested and washed in PBS. Active caspase-3 was detected using the Active caspase-3 antibody kit (BD Pharmingen) following the manufacturer's instructions. Briefly, cells were fixed and permeabilized for 30 min, and then incubated with the phycoerythrin-conjugated antiactive caspase-3 antibody. Analysis of active caspase-3-positive cells was performed by counting 10,000 events on a Becton Dickinson FACScan flow cytometer and data were fit using DIVA6 software (Becton Dickinson).
Footnotes
Acknowledgments
The authors wish to thank Dr Nadia Alfaidy-Benharouga (CEA, Grenoble) for her kind gift of human placenta RNA samples. The work in S.K. and E.B. laboratories is supported by INCa, ANR EpiSperm, and ARC Subvention libre programs. A.D. has been fully supported by INCa, AGIRDOM, and ANR grants. The clinical research on lung cancer was funded by PNES POUMON INCA 2005 and BIOMARKSCAN PHRC 2003. The transcriptomic analyses were performed thanks to the program Carte d'Identité des Tumeurs supported by the Ligue NATIONALE CONTRE LE CANCER.
Author Disclosure Statement
The authors declare that there is no competing financial interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.