
Abstract
Cellular glutathione peroxidase 1 (GPx1) is an important antioxidant selenoenzyme. Due to the presence of selenocysteine (Sec) encoded by UGA, normally recognized as a stop codon, the expression of mammalian GPx with traditional recombinant DNA technology is extremely difficult. In this study, a series of human GPx1 (hGPx1) mutants with significantly high catalytic activities were produced for the first time in an Escherichia coli BL21(DE3)cys auxotrophic strain using the single-protein production system. Cys residues in hGPx1 were mutated to Ser in turn because untargeted substitution of Sec in place of Cys resulted in the decline of recombinant selenoenzyme activity. The results of this work showed that the catalytic activities of the mutants increased progressively with decreasing number of noncatalytic Sec residues. Seleno-hGPx1-C2/78/115/156/202S with all Cys residues changed to Ser showed the highest activity (21,268 U/μmol), which was more than 10-fold higher than bovine liver GPx. This increase could be explained by structural analysis of hGPx1 mutants based on homology modeling and binding site analysis. These results lead to the hypothesis that the conversion of noncatalytic Sec residues to Ser may optimize the structure of seleno-GPx in this expression system and consequently increase the catalytic efficiency. Antioxid. Redox Signal. 20, 1524–1530.
Introduction
G
Glutathione peroxidase 1 (GPx1) is an important antioxidant selenoenzyme. This work was the first time that several seleno-human GPx1 (hGPx1) mutants with significantly high catalytic activities were produced in Escherichia coli. Substitution of Ser for noncatalytic selenocysteine optimized the structure of the selenoenzyme and resulted in a marked increase in the catalytic activity. This research lays the foundation for expressing mammalian selenoprotein in E. coli, overcomes the problem of limited sources of natural GPx, and contributes to study of the structure and properties of GPx. More importantly, recombinant seleno-hGPx1 has potential to be an excellent antioxidant agent.
Recently, a novel expression system called the single-protein production (SPP) system for the production of a single protein in E. coli has been developed. This system can produce a protein of interest in the absence of other cellular proteins through the introduction of MazF to host cells. MazF is an E. coli toxin possessing single-stranded RNA- and ACA-specific endoribonuclease activity (2). At the same time, SPP dramatically reduces the cost of protein production without sacrificing protein yield by condensing the culture (3). Using this system, it is feasible to obtain high yield of recombinant selenoenzyme from E. coli BL21(DE3)cys auxotrophs.
Here, we prepared a series of seleno-hGPx1 mutants with high catalytic activities using the efficient expression system. The Cys residues in hGPx1 were mutated to Ser (individually or totally) to improve their activities, and the impact of structure on the catalytic activity was studied.
Results and Discussion
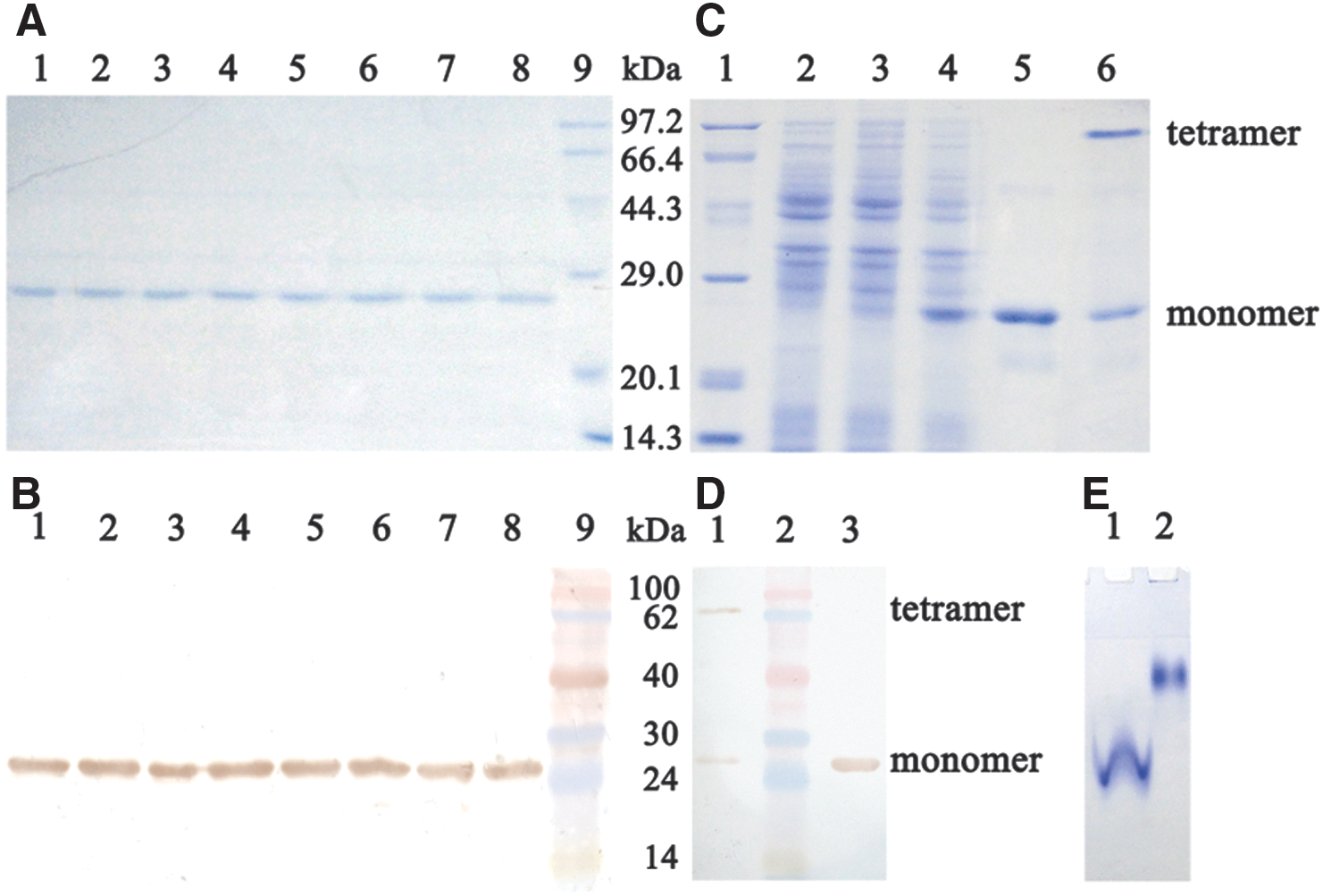
To express hGPx1 proteins, full-length hGPx1 cDNA was generated by polymerase chain reaction and cloned into the pColdI expression vector. The unusual Sec codon was mutated into a codon for Cys using site-directed mutagenesis. Sec residues could efficiently substitute for Cys in the Cys auxotrophic strain when Cys was omitted from the medium. Considering that all five Cys residues in hGPx1 would be changed to Sec in this expression system and such substitution could affect the catalytic activity of hGPx1, we prepared a series of mutants by changing different Cys residue(s) to Ser. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis of hGPx1 mutants showed a single band with a molecular mass of ∼24.2 kDa in each lane (Fig. 1A), and the same band was detected by western blot analysis (Fig. 1B). Together, these results suggested that the purified hGPx1 mutants were successfully obtained. The yield of soluble recombinant selenoprotein was ∼17 mg/L of the culture. Denatured seleno-hGPx1-C2/78/115/156/202S migrated as a single band of ∼24.2 kDa (Fig. 1C, Lane 5), whereas a sample composed of nondenatured and denatured seleno-hGPx1-C2/78/115/156/202S migrated as two bands of ∼97 kDa (at the top of Lane 6) and 24.2 kDa (at the bottom of Lane 6), and the same bands were also observed by western blot analysis (Fig. 1D). Native-PAGE (Fig. 1E) analysis suggested that seleno-hGPx1-C2/78/115/156/202S migrated as a single band under both denaturing and nondenaturing conditions but showed different migration rates. These results indicated that seleno-hGPx1-C2/78/115/156/202S exists in a tetramer similarly to native hGPx1.
We then determined the activities of the hGPx1 mutants, and the results are listed in Table 1. From this analysis, it could be concluded that the activities of hGPx1 mutants vary with the number of noncatalytic Sec residues. Seleno-hGPx1-U49C displayed a low GPx activity of 2448 U/μmol following the conversion of all native hGPx1 Cys to Sec. Mutant seleno-hGPx1-C2/78/115/156/202S with noncatalytic Sec residues converted to Ser residues displayed the highest activity (21,268 U/μmol) of all investigated mutants; this level of activity was not only prominent among the mimics (7, 8) but also ∼10-fold higher than that of bovine liver GPx (5).
GPx, glutathione peroxidase; hGPx1, human GPx1.
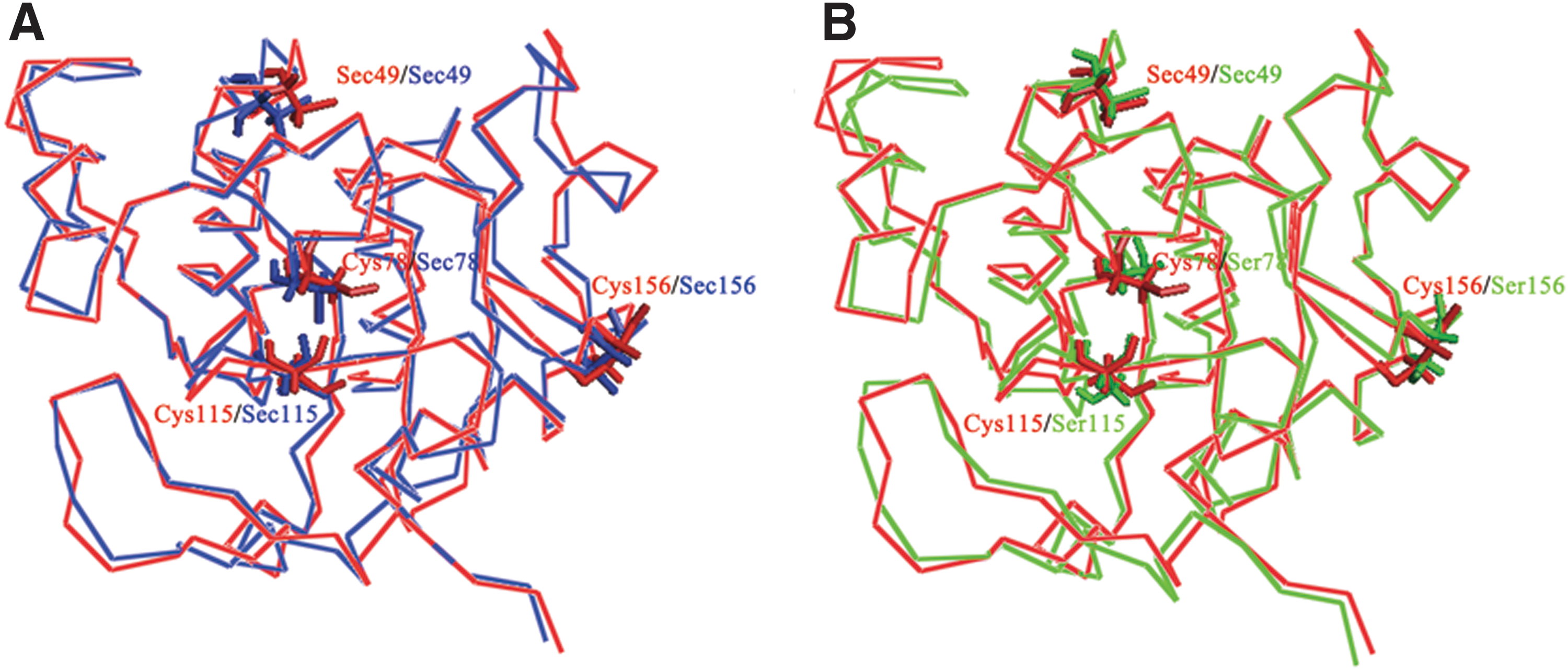
To analyze the effect of mutation on catalytic efficiency, molecular modeling was used to compare the structures of seleno-hGPx1 mutants. Putative structures of natural hGPx1, with Gly49 converted to Sec49, and the mutants were built using the X-ray crystallographic structure coordinates of hGPx1 (PDB ID code 2F8A) as a template. Molecular structures of putative natural hGPx1 and its mutants in complex with glutathione (GSH) are shown in Figure 2. There was little change in the distances between Sec49, Gln84, and Trp162; however, the distance between the Se atom of Sec49 and the S atom of GSH varied with the number of noncatalytic Sec residues. From exploration of numerous GPx mimics, the GSH binding site is considered to be pivotal in eliminating peroxides, in addition to the Sec within the catalytic center. In the GPx catalytic cycle, GSH is involved in the regeneration of the catalytically active selenol of Sec by forming a glutathionylated intermediate of GPx. Thus, the rapid reaction of GPx with GSH is very important for maintaining the catalytic efficiency of the enzyme. Although the structure of this intermediate has not been solved to date, the distance between the Se atom of Sec in the catalytic center and the S atom of GSH is believed to be a key factor for its formation. In seleno-hGPx1-U49C, the distance between Se and S atoms is 9.72 Å, and it is the longest distance among the mutants. The long distance between the two atoms may hinder the redox reaction, which may be a reason why this mutant showed the lowest activity (Table 1 and Fig. 2). It is worthwhile to note that the replacement of noncatalytic Sec with Ser could shorten the distance between the Se and S atoms, and mutants having a shorter distance displayed the higher activity. Although models with Cys2 and Cys202 could not be built because they are not contained in the crystal structure model, it is inferred that the substitution of Ser for these two Cys residues is probably associated with the optimization of the structure of catalytic center due to the high activities of seleno-hGPx1-C2/78/115/202S and seleno-hGPx1-C2/78/115/156/202S. Thus, it is reasonable to postulate that a relatively short distance between the Se atom in the catalytic center and the S atom of GSH may facilitate the catalytic reaction, thereby increasing the activity.

Structural comparison between putative natural hGPx1, seleno-hGPx1-U49C, and seleno-hGPx1-C78/115/156S is shown in Figure 3. These comparisons clearly suggested that the structure of the catalytic center of seleno-hGPx1-C78/115/156S is closer to that of the putative natural hGPx1 compared with seleno-hGPx1-U49C. These results indicated that replacement of Cys residues caused a conformational change in the active center and had an effect on enzyme binding to GSH.
To further analyze the enzymatic properties of seleno-hGPx1-C2/78/115/156/202S, the GPx activity of seleno-hGPx1-C2/78/115/156/202S was investigated across a temperature range of 20°C–60°C and across a pH range of 5.8–10.2. As shown in Figure 4A, the selenoenzyme exhibited high activity between 30°C and 50°C, with the maximum activity at 30°C. The optimum pH for the mutant was ∼9.3 (Fig. 4B), and the activity at pH 7.4 was ∼33% of its maximum activity at pH 9.3. Sufficient stability of enzymes is usually a major factor limiting usefulness in clinical application, which is mainly associated with the structure. Rational protein engineering is regarded as an efficient way to fulfill the stability requirement. To compare the stability of seleno-hGPx1-U49C and seleno-hGPx1-C2/78/115/156/202S, they were stored at 4°C, and their activities were measured at a series of time points. Some clear trends were noted (Fig. 4C): storage of seleno-hGPx1-U49C for 14 days resulted in almost complete loss of activity, whereas seleno-hGPx1-C2/78/115/156/202S still retained half of its activity after storage for 30 days. This finding is important for the development and application of selenoenzyme.

To determine whether the catalytic mechanism of seleno-hGPx1-C2/78/115/156/202S was similar to that of native GPx, the initial rate of catalysis was determined at several concentrations of the two substrates. Double-reciprocal plots of initial rate versus substrate concentration led to a family of parallel lines for both substrates (Fig. 5). These results indicated that seleno-hGPx1-C2/78/115/156/202S followed a ping-pong mechanism, in analogy with those of natural GPx. That is to say, the catalytic reaction comprises two independent events: oxidation of the reduced enzyme by hydroperoxide and reduction of the oxidized enzyme by GSH. Apparent kinetic parameters were deduced from a fit of the experimental data to Equation (1) and are listed in Table 2. The apparent second-order rate constants k
cat/K
H2
O2
m and k
cat/K
GSH
m were found to be 5.4×107 and 1.0×107 M
−1·min−1, respectively. The value of k
cat/K
H2
O2
m was lower than that of natural hGPx1 (4), but much higher than those of most GPx mimics (8). And k
cat/K
GSH
m was in the same order of magnitude as that of natural hGPx1 (4), which means that seleno-hGPx1-C2/78/115/156/202S and natural GPx have similar affinities for GSH. This may be mainly attributed to the structural similarity of the active site and GSH binding site in the mutant compared with that of natural GPx, which is not observed in most GPx mimics.
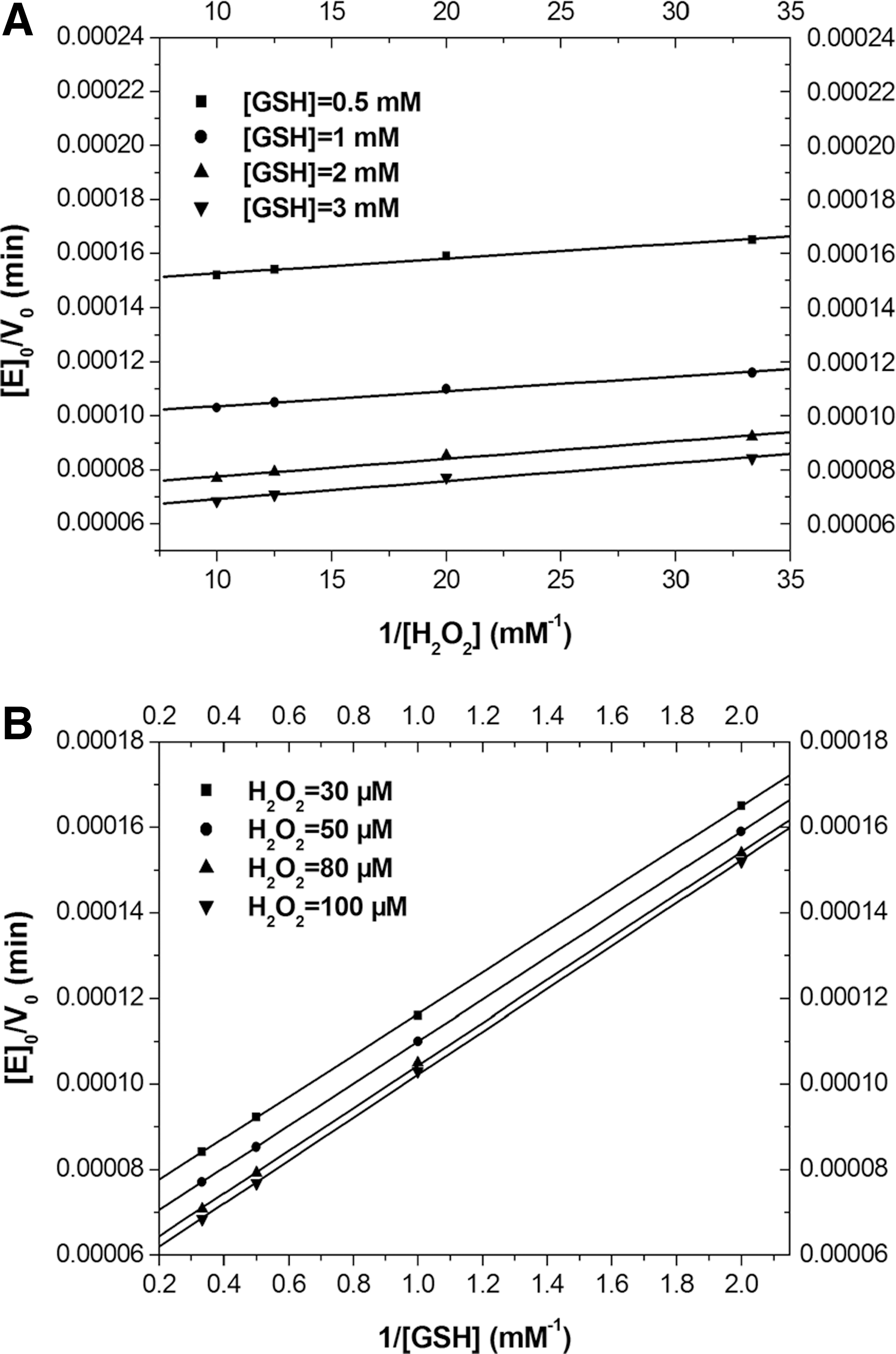
GSH, glutathione.
In conclusion, we optimized a system for heterologous expression of human selenium-dependent GPx1 and produced an hGPx1 mutant with significantly high catalytic activity. Compared with the mutant having all Cys replaced by Sec, the substitution of Ser for noncatalytic Sec could improve the catalytic activity of the selenoenzyme. The distance between the Se atom of Sec in the catalytic center and the S atom of GSH was regarded to be associated with the catalytic efficiency by computer simulation analysis. In addition, the mutant with all Cys converted to Ser exhibited a ping-pong mechanism similar to natural GPx. The creation of the new hGPx1 mutant lays the foundation for developing antioxidant agent, and it is of great importance to deepen the study of structure and catalytic mechanism of GPx.
Notes
Strains, plasmids, and growth conditions
E. coli DH5α, used for plasmid propagation, was grown at 37°C in the LB medium. The medium for expression of selenoenzyme was a modification of M9 minimal medium (MM medium) as described previously (9), supplemented with 250 mg/L of
Construction of plasmids
The human GPx1 gene was amplified from the cDNA library of the human liver hepatocellular carcinoma cell line Hep G2. Primers were designed to amplify a 612 base pair product. Primer sequences were hGPx1-F 5′-GGAATTCCAT ATGTGTGCTGCTCGGC-3′ and hGPx1-R 5′-CCCAAGCTT CTAGGCACAGCTGGGC-3′. Amplified fragments were cloned into the NdeI and HindIII sites of the pColdI expression vector, yielding pCGPx1. Positive clones were verified by enzyme digestion.
ACA sequences in the hGPx1 gene were replaced by site-directed mutagenesis, retaining the amino acid sequence. Point mutations were generated using a QuikChange® Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's recommendations. Plasmid pCGPx1 was used as the template for the site-directed mutagenesis reactions. Following digestion with DpnI restriction enzyme, mutant products were transformed into E. coli DH5α and plated on the LB medium containing 100 μg/ml ampicillin. Constructs were sequenced to confirm that no other mutations had been introduced.
Overexpression and purification of proteins
Production of selenoprotein was performed according to the previously described protocol with slight modification (1). E. coli BL21(DE3)cys cells were transformed with mutant plasmids together with pMazF and grown in a 200-ml culture of the MM medium at 37°C in the presence of Cys (50 μg/ml), ampicillin (100 μg/ml), kanamycin (25 μg/ml), and chloramphenicol (25 μg/ml). When the A600 value reached 0.5, the culture was chilled in an ice-water bath for 5 min and incubated at 15°C for 45 min to acclimate the cells to cold shock conditions. Cells were harvested by centrifugation (5000 g for 5 min at 15°C) and washed twice with 0.9% NaCl to remove Cys added to the MM medium. Cells were resuspended in 5 ml of the MM medium supplemented with ampicillin (100 μg/ml), kanamycin (25 μg/ml), chloramphenicol (25 μg/ml), and Sec (600 μM) (Sigma). Isopropyl β-
PAGE and western blot analysis
Cells containing recombinant proteins were collected by centrifugation and subjected to SDS-PAGE followed by Coomassie Blue staining. Nondenatured protein samples diluted 1:1 with nondenaturing loading buffer [50 mM glycine (pH 8.0), 20% glycerol (v/v)], and the mixture was electrophoresed using a 10% native PAGE gel (pH 8.0) with Tris/glycine running buffer. Protein samples were also separated on a 12% (w/v) SDS-PAGE gel and electrophoretically transferred to nitrocellulose membrane. The membrane was blocked with TBS (Tris-borate-NaCl) containing 5% nonfat dry milk and incubated with mouse anti-His monoclonal antibodies (Pharmacia) (1:1000). Horseradish peroxidase conjugated secondary antibody (Pharmacia) was used at a dilution of 1:5000. Reacting proteins were visualized by DAB-H2O2 substrate that gave brown color.
Assay of enzyme activities
GPx activities of the hGPx1 mutants were assayed according to the method described previously (7). The reaction was carried out at 37°C in 500 μl of solution containing 50 mM sodium phosphate buffer (pH 7.4), 1 mM EDTA, 1 mM GSH (Sigma), 0.25 mM NADPH (Sigma), 1 U of GSH reductase, and 20–50 nM of sample protein. The sample was pre-incubated with GSH, NADPH, and GSH reductase for 5 min at 37°C. The reaction was initiated by the addition of 0.5 mM H2O2. The enzyme activity was determined by measuring the decrease of NADPH absorption at 340 nm. Activity units are defined as the amount of the protein utilizing 1 μmol of NADPH per min, and the specific activity is expressed in U/μmol.
Characterization of seleno-hGPx1-C2/78/115/156/202S
The activity of seleno-hGPx1-C2/78/115/156/202S was measured as above. The pH of the buffer ranged from 5.8 to 10.2 to measure the initial rates of reaction to determine the optimal pH condition for seleno-hGPx1 catalyzed reactions. Similarly, catalytic reactions were carried out at different temperatures ranging from 20°C to 60°C to determine the optimal temperature for the GPx activity of seleno-hGPx1-C2/78/115/156/202S.
The effect of storage time on stability of seleno-hGPx1-U49C and seleno-hGPx1-C2/78/115/156/202S was investigated. Purified seleno-hGPx1-U49C and seleno-hGPx1-C2/78/115/156/202S were incubated in phosphate-buffered saline (50 mM, pH 7.4) at 4°C for durations ranging from 1 to 30 days, and activities were measured using the method described above.
Steady-state kinetics of seleno-hGPx1-C2/78/115/156/202S
The assay of kinetics of seleno-hGPx1-C2/78/115/156/202S for reduction of H2O2 by GSH was similar to that of seleno-hGSTZ1-1 (7). Initial rates were measured by observing the decrease of NADPH absorption at 340 nm at several concentrations of one substrate, whereas the concentration of the second substrate was held constant. GPx activities were measured using the as described above at 37°C and pH 7.4.
Molecular modeling
All computations were performed with Insight II package, version 2000 (Accelrys) using the method described previously (9). The X-ray crystallographic structure of hGPx1 from Protein Data Bank (PDB ID code 2F8A) was used for the starting coordinates for calculations. The 3D structures of hGPx1 mutants were constructed using the Homology program. The obtained structures were refined by molecular dynamics simulations using DISCOVER module. After the optimization procedure, the structures were checked with Profile-3D and Procheck. GSH was docked within each predicted mutant models by using Affinity program. The complexes were used as the original conformation for further energy minimization and geometrical optimization before the final models were achieved. The 3D-predicted structures were carried out using molecular visualization system PyMOL (open-source software published by DeLano Scientific LLC).
Footnotes
Acknowledgments
The first two authors contributed equally to this study. The authors thank Prof Marie-Paule Strub and August Böck for providing the E. coli cysteine ausotrophic strain, BL21(DE3)cys. This work is supported by the National Natural Science Funds, China (nos. 30970633 and 31270851).