
Abstract
Introduction
P
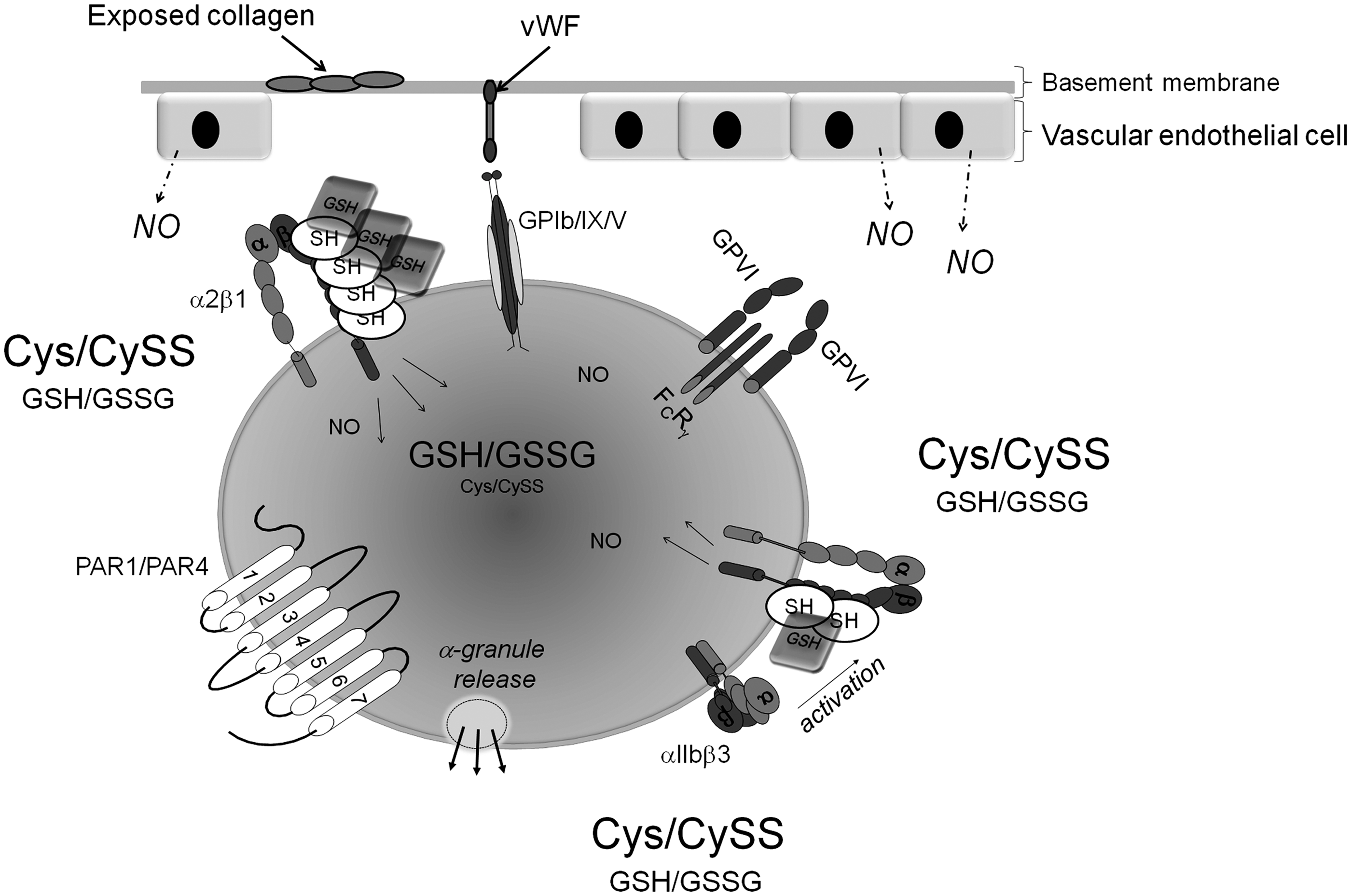
The first stage involves the capturing and activation of platelets by von Willebrand factor (vWF) and exposed collagen at the site of trauma in the vessel wall (62, 128). When an injury to a blood vessel occurs, endothelial cells lining the vessel are damaged, leading to the exposure of the platelet activating agent, collagen, in the subendothelium. vWF is a large glycoprotein that is found circulating in the plasma. It is secreted from the α-granules of activated platelets and released from damaged endothelial cells (4). Immobilized vWF assists in capturing and tethering platelets to collagen at the site of vascular injury under high shear rates (>600 s−1). The glycoprotein Ib-IX-V complex (GPIb-IX-V), in addition to integrin αIIbβ3, plays a key role in the interaction between platelets and vWF. GPIb-IX-V, the second most abundant receptor on platelets, is a receptor complex that is composed of four membrane-spanning polypeptides: GPIbα, GPIbβ, GPIX, and GPV (88, 100). vWF binds to GPIbα through its A1-domain and to collagen through its A3-domain, thereby indirectly implicating GPIb-IX-V in collagen adhesion (109). Several collagen receptors have been identified on the platelet surface, most notably glycoprotein VI (GPVI), a member of the immunoglobulin superfamily, and the integrin α2β1 (69). As the platelets become tethered at the site of injury, they form a monolayer with subsequent generation of thrombin and further activation of the platelets (101). It has been long assumed that the main function of the interaction between vWF and GPIbα is to slow down platelets and enable the interaction of collagen with its platelet receptors. The engagement of these receptors by their ligands initiates a series of signaling cascades within the platelet (28).
In the second stage of activation, additional platelets are recruited to the site of the platelet monolayer. At this point, the platelet secretes additional activators that include adenosine diphosphate (ADP) and thromboxane A2 (106). Each of these activators, in turn, interacts with specific receptors on the platelet surface, resulting in an increase in intracellular calcium (119). The signals resulting from these interactions lead to the activation of the platelet integrin αIIbβ3, the binding site for fibrinogen on the platelet surface (Fig. 1) (89). vWF, fibrinogen, and fibronectin bind to activated αIIbβ3, leading to firm platelet adhesion, the recruitment of additional platelets, thereby resulting in platelet aggregation. In the third stage of platelet activation, the platelet plug is stabilized. This occurs as a result of the close proximity of the platelets to each other. It involves outside-in signaling through the surface integrins and receptor tyrosine kinases. These three stages, ultimately, lead to the consolidation of the platelet plug (49).
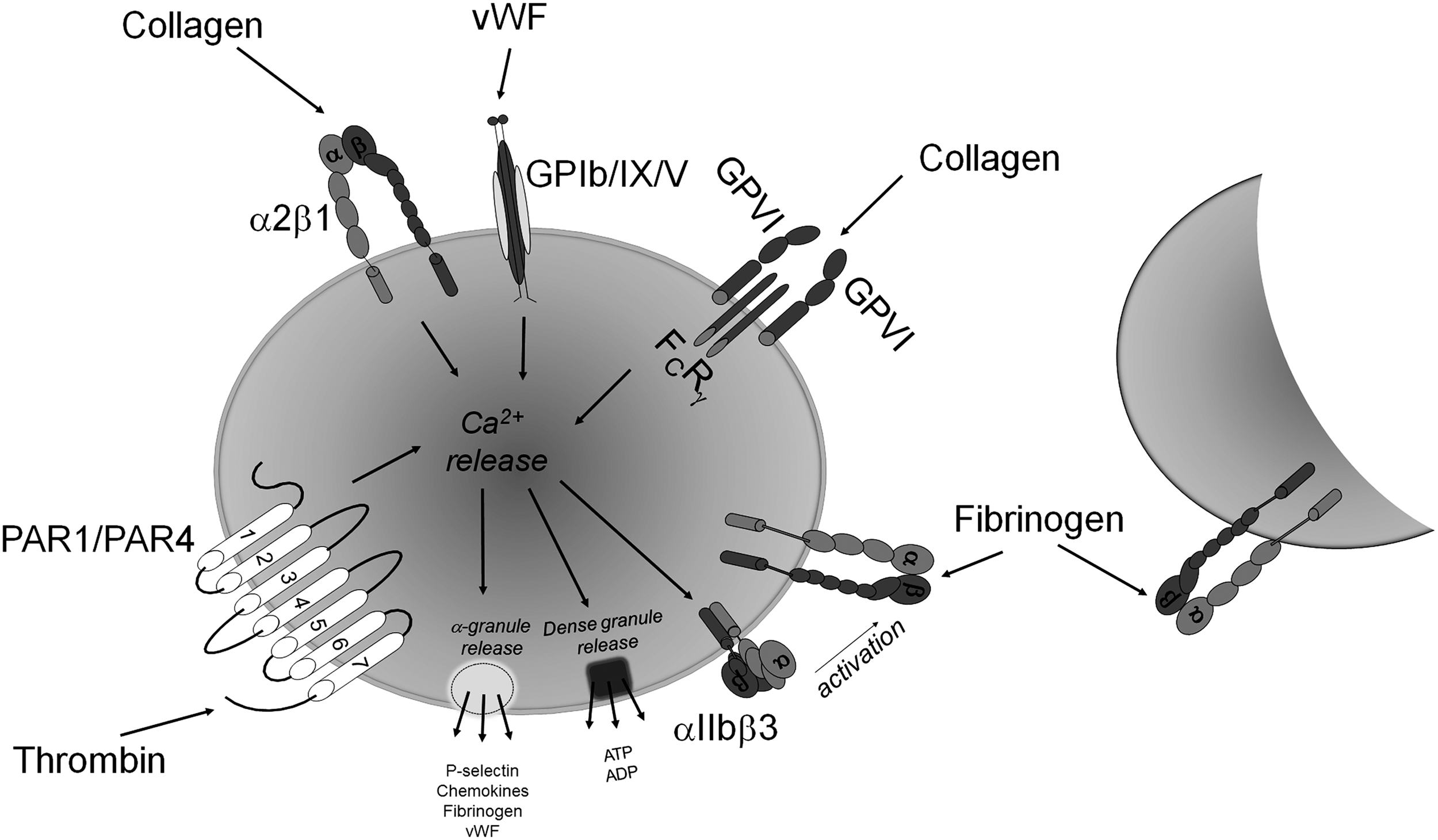
A number of free thiol-containing proteins have been identified to be associated with platelets (42). Some known thiol-containing platelet proteins include protein disulfide isomerase (PDI), GPIbα (15), and P2Y12, the ADP receptor (35). Thiol/disulfide groups have also been found to be a crucial component of the platelet integrins αIIbβ3 (113, 155) and α2β1 (56). There is overwhelming evidence which suggests that cysteine residues and thiol/disulfide bonds are critical players in the complex conformational changes of a myriad of proteins that are associated with platelet activation. In this review, therefore, we aim at exploring how modifications of platelet protein thiol/disulfides regulate platelet activity.
Thiols and Disulfides
Cysteine residues are the most conserved residues in functionally important sites within proteins. These residues have been assigned the following functions (44): 1. Catalytic redox-active cysteine residues are directly involved in catalytic reactions such as oxidation, reduction, and isomerization of disulfide bonds. They are found in thiol-oxidoreductase enzymes, including PDI (110) and thioredoxin (Trx) (135). The highly conserved CXXC motif is commonly present (23). 2. Regulatory cysteine residues regulate or modulate protein activity by changing their redox state, but are not catalytic themselves. Disulfide bond formation, S-glutathionylation, and S-nitrosylation are the signaling mechanisms that are utilized by regulatory Cys residues (1, 13). They are found in many proteins, including kinases (125), phosphatases (130), and transcription factors (10). 3. Structural cysteine residues participate in protein structure through the formation of stable intramolecular and intermolecular disulfide bonds (141). 4. Metal-coordinating cysteine residues coordinate metal ions such as Zn2+ and are frequently in the form of the highly conserved CXXC motif, making a distinction between catalytic redox cysteines and metal-binding cysteines difficult (54, 58). 5. Catalytic non-redox cysteine residues participate in catalysis but do not change their redox state, are highly conserved, and act as nucleophiles. Such residues are found in glyceralderhyde-3-phosphate dehydrogenase (158).
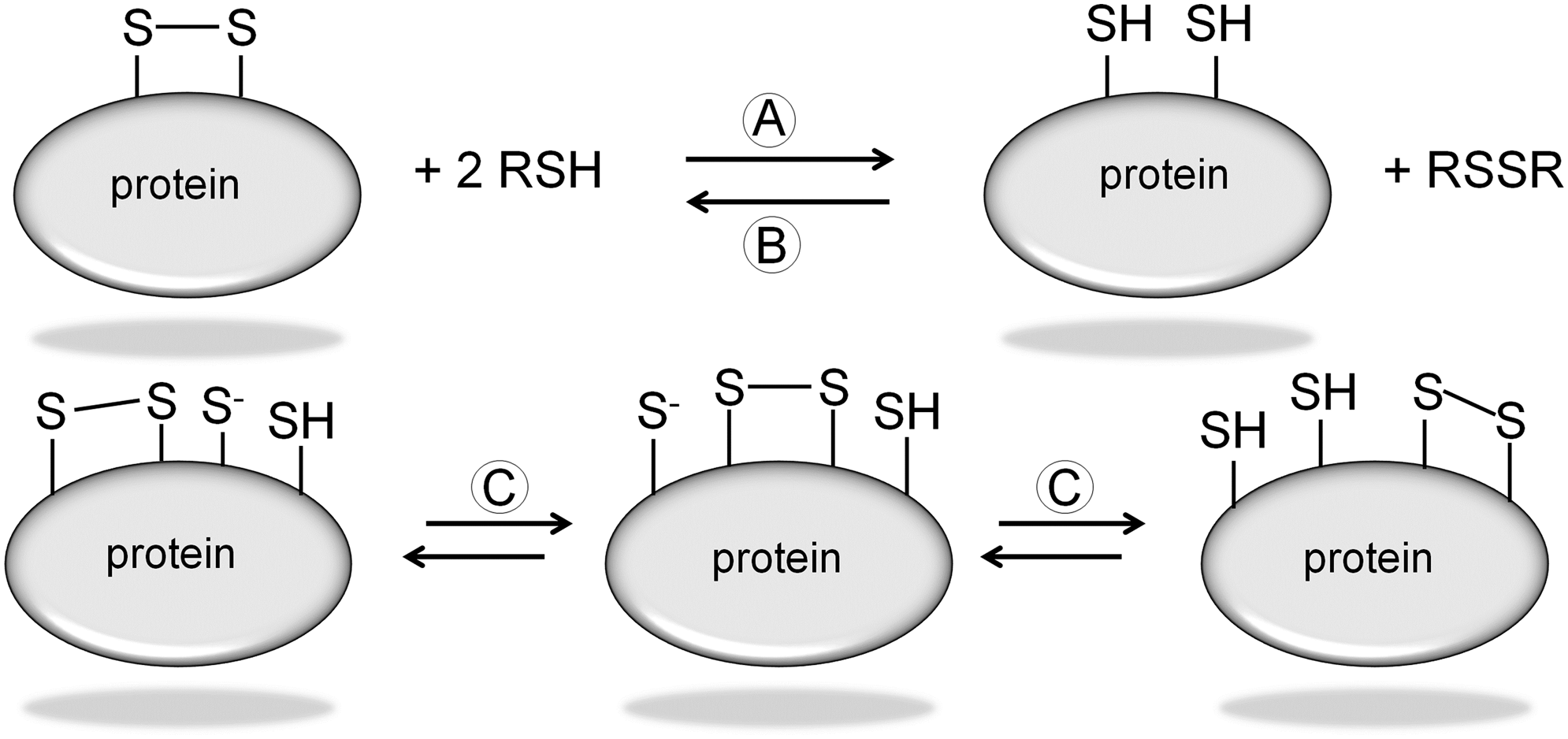
A disulfide bond is a covalent bond that is formed between two cysteine amino-acid residues and is referred to as cystine. It is composed of two α-carbons, two β-carbons, and the two sulfur atoms of both cysteine amino-acid residues. Rotation about the bonds between these six atoms forms the five χ angles. Each χ angle can be either positive or negative, which equates to 20 possible disulfide bond configurations. The bond is right handed if the χ3 angle is positive and left handed if the angle is negative (Fig. 2A) (134). An allosteric disulfide bond is defined as a disulfide bond that controls protein function by mediating conformational changes on reduction. There are three disulfide bond configurations: hooks, spirals, and staples, each of which may be left handed (LH) or right handed (RH) (Fig. 2B) (21). Disulfide bonds are classified as follows: 1. Structural disulfide bonds having a −LH spiral configuration. 2. Catalytic disulfides are +/− RH hook configuration. 3. All known allosteric disulfide bonds are −RH staple configuration.

Thiol Isomerases and Platelet Function
PDI catalyzes several reactions, which involve disulfide bonds and free thiol groups. These include thiol/disulfide exchange, isomerization (intra-molecular switching of two disulfide bonds), and disulfide bond reduction or disulfide bond formation (oxidation) (Fig. 3). It has a primary role in the correct formation of disulfide bonds in nascent proteins in the endoplasmic reticulum. PDI is one of the most abundant residents of this organelle and is so named, because it catalyzes thiol/disulfide interchange reactions that can lead to the “shuffling” or isomerization of intramolecular disulfide bonds. It is believed to accelerate the folding of proteins by catalyzing this interchange reaction, a rate-limiting step during protein folding (46). It exhibits a broad specificity and has been used to catalyze the in vitro folding of a variety of proteins (64). Model-building studies show that the Trx-like domains of PDI, the canonical CXXC motifs -Cys-Gly-His-Cys-, can fold in a similar manner to those of Trx. These domains can function as an independent oxidoreductase with an interconvertible dithiol/disulfide group. Since PDI contains two such domains and is a homodimer in solution, this implies there are four active groups per PDI molecule (47). Both catalytic sites have been shown to act independently of one another. Site-directed mutagenesis was used to determine whether one or both of the -Cys-Gly-His-Cys- sites acted as catalytic sites. Three expression plasmids of human PDI varying in their signal sequences were constructed and expressed in Escherichia coli. In plasmid number one, the first Cys of the N-terminal site was mutated to a serine; in plasmid number two, the first Cys of the C-terminal was mutated to a serine; and in plasmid number three, the first Cys of both sites was mutated to a serine. The PDI activity of plasmids one and two was found to be 50% of wild-type PDI, while that of plasmid three had no activity. This implies that the two -Cys-Gly-His-Cys- sites of PDI represent the catalytic sites for isomerase activity and that each acts independently of one another (145).
PDI has been shown to be present on the cell surface of various tissues and involved in various aspects of cell surface biology (37). There are approximately 21 members in the broader PDI family (12). Recently, a family of thiol isomerase enzymes called endoplasmic reticulum proteins 5, 29, 44, 57, and 72 (ERP5, ERp29, ERp44, ERp57, and Erp72) as well as transmembrane thiol isomerase 3 (TMX3) were identified in platelets and megakaryocytes (66, 75). PDI activity in the form of ERP5 is known to be secreted by activated platelets and has been localized to the platelet surface, playing a role in αIIbβ3 activation. The other members of the ERp family are also released by platelets and relocated to the cell surface on activated platelets. TMX3 was also detected on the platelet surface but did not increase with activation. Platelet PDI mediates platelet aggregation, secretion, and fibrinogen binding to integrin αIIbβ3 as well as plays a role in platelet adhesion to fibrinogen and collagen by αIIbβ3 and α2β1, respectively (39, 80, 126). Thiol labeling studies with 3-(N-maleimidylpropionyl) biocytin (MPB) in agonist-stimulated aggregating platelets exhibited increases in sulfhydryl labeling in 11 platelet proteins, with further evidence of thiol/disulfide rearrangement playing an important role in platelet activation (15).
In addition to the role that PDI plays in modulating the response of platelets, it is surmised that it also plays a role in thrombus formation and may indirectly de-encrypt tissue factor (TF) (127). The binding of cell surface TF to the serine protease factor VIIa is one of the first steps in the activation of coagulation. This binding activates factor VIIa and the TF/VIIa complex, subsequently recruiting factor X to form the TF/VIIa/X coagulation-initiation complex. This complex generates factor Xa and, eventually, thrombin, which is required for normal hemostasis (91, 111). It is considered that TF exists in at least two conformations on the cell surface. The first is the coagulation inactive or encrypted form and the second is the coagulation active or decrypted form (83, 148). Most TF on the cell surface appears to be encrypted, but the de-encryption mechanism remains controversial. A number of studies show that encrypted TF is due to the reduction of the membrane proximal disulfide bond (2, 20, 123, 142). This disulfide bond is Cys186-Cys209 and has an–RH staple configuration that is characteristic of allosteric bonds. Such bonds break and form in a precise manner that ultimately changes the function of the protein (21, 59, 134). Furthermore, the redox potential of this particular disulfide bond in TF is −278 mV, falling into the range of other known allosteric disulfides (87). Indeed, thiol labeling experiments reveal the presence of free thiols in a sub-population of cell surface TF, and the oxidation of these thiolates is associated with increased TF coagulation activity (2, 20). Mutation of either Cys186 or Cys209 to either serine or alanine resulted in reduced TF coagulation activity. Taken together, these studies suggest a thiol/disulfide exchange mechanism which is involved in the activation of TF and that disulfide-bonded TF is the active conformation of TF. However, there is controversy over the activation of TF. A number of studies show that TF activity in a number of cells requires the presence of certain receptors and/or phospholipids (17, 116). Furthermore, cellular knockdown of PDI resulted in an increase in TF coagulation activity (72). While there is evidence suggesting a role for PDI in thrombus formation, a direct link to TF has not been proved in vivo. In mouse models of thrombosis, inhibition of PDI activity reduced fibrin formation and thrombus accumulation (24, 71). When exogenous encrypted TF was introduced into the mouse circulation, active PDI was required for full fibrin formation. Moreover, blocking the active site of PDI, by either mutation or alkylation of the two catalytic cysteines, resulted in the loss of PDI-mediated increase in TF coagulation activity (124). PDI increases the release of TF-positive pro-coagulant microparticles from monocytes through ATP stimulation of the surface P2X7 receptor (78). This effect is lost when the free thiols in PDI are blocked. In a separate study, PDI is shown to modulate the lipid environment of the cell surface. Inhibition of PDI increased phosphatidylserine exposure on the surface of an endothelial cell line. This resulted in an increase in prothrombinase activity (118). Overall, these studies indicate that PDI has multiple roles to play in thrombus formation and that targeting of PDI is a viable anti-thrombotic strategy.
Platelet Integrin Thiols
Platelets possess a large number of integrins. The principle integrins are αIIbβ3 and α2β1. αIIbβ3 receptors are expressed solely on platelets, numbering about 50,000 to 80,000 copies per platelet. The main ligand is fibrinogen (68). α2β1 is the second most abundant platelet integrin, with 2000 to 4000 copies per platelet (30). Exposed collagen is bound by both the α2β1 integrin and the GPVI receptor on the platelet surface (70). However, α2β1 has an increased affinity for soluble collagen compared with fibrillar collagen (132). All integrins contain an unusually high number of cysteine residues. An analysis of integrin cysteine patterns reveals that there are four highly conserved cysteine-rich repeats in each beta integrin subunit. The patterns of the cysteine residues are not only similar in both α2β1 and αIIbβ3 but also similar in all integrin subunits. The alpha integrin subunit of αIIbβ3 contains 18 cysteines and the beta integrin subunit has 56 cysteines, 31 of which are located in an extracellular cysteine-rich domain in the four epidermal growth factor-like domains and eight are located in the carboxyl-terminal β-tail domain, with a similar pattern in α2β1 (113). These highly conserved cysteine residues are believed to form intra-molecular disulfide bonds. However, unpaired free thiols are known to be present in the β3 subunit of αIIbβ3, which exhibit the properties of a redox site that is directly involved in the activation of this integrin (155). Moreover, the acquisition of a ligand binding conformation is dependent on extracellular free thiols and enzymatically catalyzed disulfide bond exchanges (80). Certain cysteine residues within αIIbβ3 form disulfide bonds with neighboring cysteine residues that are considered critical in maintaining the structure of the inactive or “bent” conformation which is seen in the non-ligand bound integrin, while disulfide bond reduction and a reshuffling of disulfide bond pattern with an increase in the number of free thiol groups (-SH) have been reported in the activated ligand-binding conformation of this integrin (102, 156). Indeed, the central role for thiol/disulfide exchange within αIIbβ3 during platelet activation was further confirmed by the discovery of an endogenous thiol isomerase activity that was predicted from the presence of nine -CXXC- sequences in the β3 integrin subunit (113). This is the same motif of the active sites of several thiol oxidoreductase enzymes, including PDI, Trx, and glutaredoxin (8).
Reduction of integrin disulfide bonds and subsequent thiol/disulfide exchange is known to play a large part in the switching from an inactive conformation to the activated ligand-binding conformation (92, 93, 102). It is not surprising, therefore, that several sulfhydryl reagents are known to affect platelet aggregation and adhesion. Membrane impermeant reagents such as p-chloromercuribenzenesulfonate (pCMBS) and 5, 5′-dithiobis (2-nitrobenzoic acid) (DTNB) inhibit collagen-induced platelet aggregation (43). Platelet adhesion to collagen, fibrinogen, and fibronectin is inhibited by the thiol-blocking agents N-ethylmaleimide (NEM), pCMBS, and thiolyte monobromotrimethyl-ammoniobimane (qBBr). In addition, pCMBS completely abolishes the adhesion of platelets that are treated with the universal integrin activator manganese (Mn2+) (43, 79). The role of vicinal thiols in αIIbβ3 activation was demonstrated using the vicinal thiol-blocking reagent phenylarsine oxide (PAO) that inhibited platelet aggregation and free thiol labeling in the integrin (93). Reduced glutathione (GSH) alone and in combination with nitric oxide (NO) reversed PAC-1 binding to Mn2+ and thrombin-activated platelets, further indicating redox regulation of integrin activation (147). Blocking of free thiols with DTNB or pCMBS inhibited platelet adhesion to both Gly-Phe-Hyp-Gly-Glu-Arg (GFOGER) and monomeric collagen via α2β1 (80). In the same study, specific anti-PDI antibody mouse monoclonal anti-PDI (clone RL-90), Fab fragments of rabbit anti-PDI, and the PDI inhibitor bacitracin inhibited platelet adhesion to these substrates, but not to the GPVI-specific peptide collagen-related peptide (CRP).
In αIIbβ3, the Cys 663–687 disulfide bond in the beta integrin subunit has been identified as −RH staple. A feature of allosteric disulfide bonds is the short Cα-Cα′ distance between adjacent cysteine residues, 4.3 Å compared with 5.6 Å for all other disulfide bonds. Such allosteric bonds link adjacent strands in the same anti-parallel β-sheet. This causes puckering of the strands, deforming the sheet, thereby imparting high torsional energy on the bond (21). Mutations disrupting the Cys 663–687 disulfide bond resulted in a constitutively active integrin (18). Thiol/disulfide exchange that is mediated by PDI likely occurs during integrin activation. Essex has proposed a model for PDI-mediated αIIbβ3 activation by which agonist stimulation of platelets causes inside-out signaling, resulting in low-affinity binding of fibrinogen to αIIbβ3. A PDI-catalyzed event, possibly a thiol-disulfide exchange or disulfide bond reduction, occurs in the integrin. This leads to a high-affinity, high-avidity state. External glutathione, cysteine, or other low-molecular-weight thiols may also generate thiols in both PDI and αIIbβ3. Cytoplasmic-reducing equivalents are supplied by a platelet membrane NAD(P)H system (38). The role of free thiols and disulfide bond reduction in integrin activation is well established. Disruption of the Cys5-Cys435 disulfide bond within the beta integrin subunit of αIIbβ3 by introduction of the mutations C5A and C435A in co-transfected CHO cells resulted in an integrin conformation that was capable of binding soluble fibrinogen and the fibrinogen mimetic antibodies PI-55 and PAC-1 (138). Blocking of free thiols on the platelet surface with the membrane-permeable NEM resulted in inhibition of adhesion to collagen, fibrinogen, and fibronectin, showing a role for free thiol groups in integrin-mediated platelet adhesion (79). The affinity of integrin α4β1 for the specific ligand LDV-FITC-containing small molecule increases dose dependently when human monocyte (U937) cells are treated with increasing concentrations of the membrane-impermeable reducing agents dithiothreitol (DTT) and 2, 3-dimercapto-1-propanesulfonic acid (DMPS). This suggests a sequential reduction of disulfide bonds generating distinctive conformations of the integrin (22). Integrin activation by ultra-violet C irradiation was demonstrated to be a direct result of disulfide bond reduction and increased in free thiol groups in β1, β2, and β3 integrins on washed human platelets and HL-60 cells (143). Thus, the conformational changes that are required to change from an inactive to a fully activated integrin are known to be redox sensitive and are strongly dependent on thiol/disulfide interactions.
In a recent publication, Kim et al. demonstrate the role that platelet PDI plays in regulating αIIbβ3 activation without affecting P-selectin exposure, Ca2+ mobilization, protein phosphorylation, beta 3 integrin subunit/talin-1 interaction, or platelet spreading on immobilized fibrinogen (77). Moreover, the isomerase activity of platelet PDI has been shown to be important in the regulation of integrin αIIbβ3 activation. In addition, other isomerases such as ERp57 may regulate the distinct phases of αIIbβ3 activation. It is speculated that platelet PDI may isomerize sulfhydryl groups on the integrin while ERp57 may oxidize or reduce the same or different sulfhydryl groups. This is supported by the finding that the isomerase activity of PDI is greater than that of ERp57 (48) but is in contrast to extracellular PDI, by which the inhibition of extracellular PDI results in a strong inhibition of platelet thrombus formation, fibrin generation, and hemostasis in mice models (24). The authors conclude that platelet PDI is essential for thrombus formation but not for hemostasis in mice. More recently, Wang et al. describe considerable expression of ERp57 in activated platelets. Their data suggest a major role for platelet-derived ERp57 in the activation of αIIbβ3 and platelet incorporation into a growing thrombus (149). In a separate study, Swiatkowska et al. describe how endoplasmic reticulum oxidase 1α (Ero1α) co-associates with both platelet PDI and αIIbβ3 in activated platelets. Ero1α provides oxidative equivalents to platelet PDI and in doing so, assists in changing αIIbβ3 from an inactive to an active conformation (139).
Thiol Exchange in vWF
vWF is a complex multimeric glycoprotein that is composed of 2813 amino acids, which encompasses a signal peptide of 22 residues, a large pro-peptide of 741 residues, and a mature subunit of 2050 residues (129). vWF consists of a number of repeated conserved structural domains or motifs that are arranged in a specific sequence: D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK. It is a cysteine-rich protein containing 234 cysteine residues, composing approximately 8% of the entire protein. These Cys residues play an important role in the assembly of vWF, a complex process requiring a number of separate steps. In the endoplasmic reticulum, “tail-to-tail” dimerization of vWF pro-peptide (pro-vWF) subunits occurs though disulfide bonds involving one or three of the Cys residues: Cys2008, Cys2010, or Cys2048 near their carboxyl-termini in the CK (cystine knot) domain (76). After pro-vWF dimer formation, these dimers are transported to the Golgi, where additional “head-to-head” disulfide bonds are formed near the amino-termini of the subunits, involving Cys379 and one or more of the Cys residues: Cys459, Cys462, or Cys464 (36). Formation of these interdimeric disulfide bonds results in large vWF multimers, a process that is known as multimerization. Multimers can vary in size, up to a maximum of approximately 20,000 kD, termed ultra-large vWF (ULvWF). ULvWF multimers are those that are stored in the α-granules of platelets or Weibel–Palade bodies in endothelial cells. When released from these storage granules, some ULvWF multimers are rapidly cleaved by the metalloprotease, ADAMTS-13. Large multimeric forms of vWF have a greater affinity for its ligands, such as collagen and activated platelets, than monomeric vWF fragments, and, consequently, ULvWF multimers that are not cleaved by ADAMTS-13 are highly adhesive in binding and agglutinating platelets (6, 50, 99).
vWF multimer size is regulated by the formation and breakage of disulfide bonds. Reduction of multimer size is associated with the generation of new thiol groups, indicating the reduction of disulfide bonds. A protein disulfide bond reductase, thrombospondin-1 (TSP-1), has been found to be responsible for reducing the average size of vWF multimers that are secreted from endothelial cells (154). Similarly, ADAMST-13 has also been shown to exhibit disulfide bond-reducing activity that regulates thiol-disulfide exchange in vWF (157).
Thiol groups have also been demonstrated to be essential for the interaction between vWF and platelets. Some plasma vWF multimers contain surface-exposed free thiols which are involved in a shear-induced thiol-disulfide exchange mechanism that enhances vWF binding to platelets (25). This study also demonstrated that the increased levels of vWF binding to platelets under shear stress are inhibited by the thiol-blocking agent, MPB, again emphasizing the importance of thiols in the structure and function of vWF. The free thiols were found to be localized to the N-terminal D3 and C-terminal C domains (25, 85, 157).
The structure of vWF is strongly influenced by shear forces. Shear stress extends vWF from a globular to an elongated conformation to facilitate inter-chain disulfide bonds among vWF multimers. This leads to lateral, rather than linear, self-association among multimers, resulting in a meshwork of vWF fibers. Free thiol groups within the C2 domain are critical mediators of lateral self-association, with interactions at the disulfide bonds Cys2431-Cys2453 and possibly, Cys2451-Cys2468 proving to be particularly important (51).
vWF meshwork is considered superior at capturing platelets from flowing blood at a site of vascular injury compared with individual molecules. Due to the importance of thiol/disulfide exchange events in the self-association and subsequent formation of vWF fibers and mesh, the external redox environment may be considered critical in the regulation of these processes. Therefore, imbalances within the redox environment, that is, oxidative or reductive stress, may have a significant impact, potentially leading to impaired lateralization of vWF. Similarly, an altered redox environment may also have an impact on the thiol-/disulfide-mediated interaction between vWF and platelets. Ultimately, disruption to vWF structure and function linked to redox environment alterations may result in defects in patients.
Cellular and Plasma Thiols
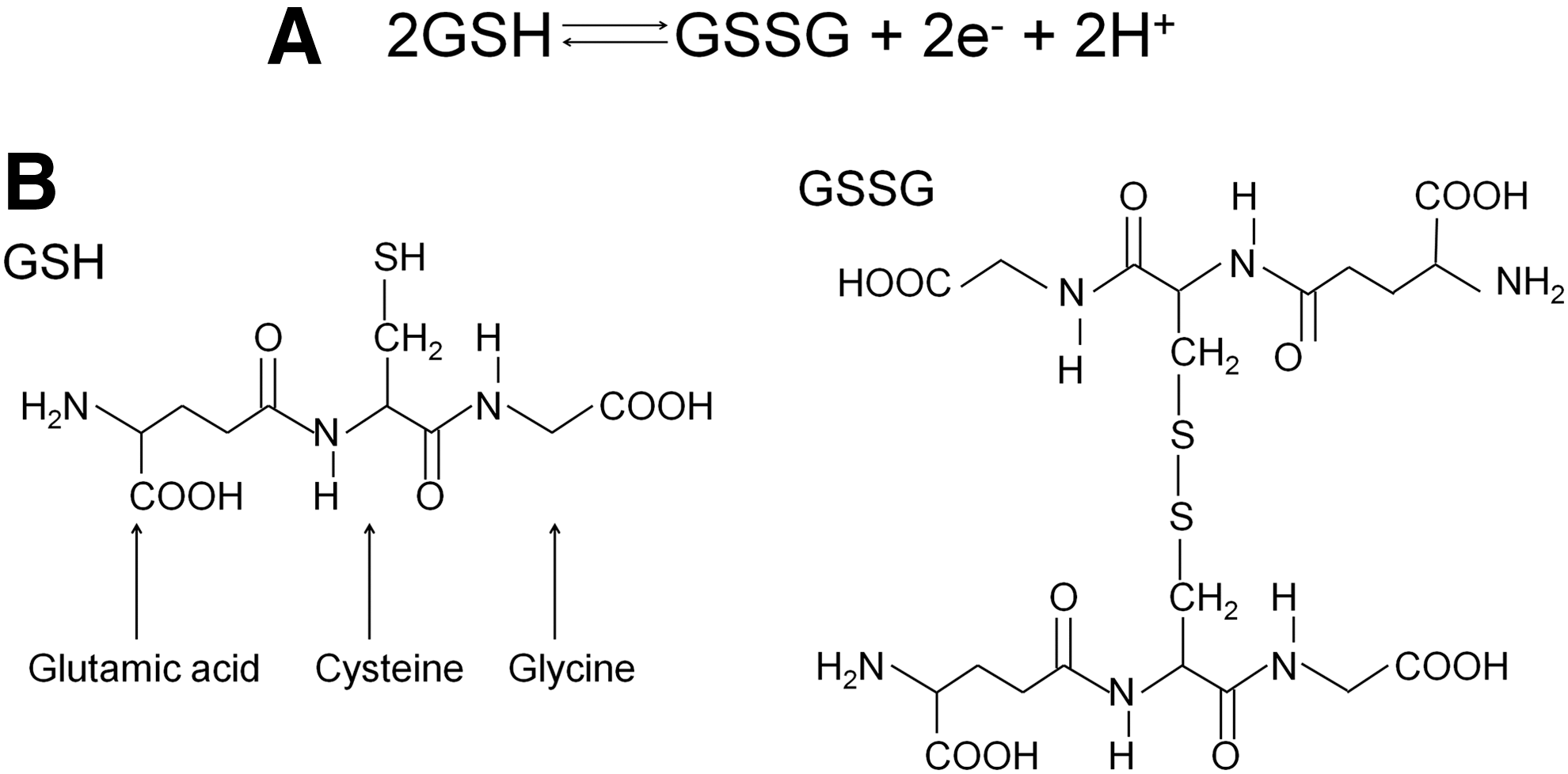
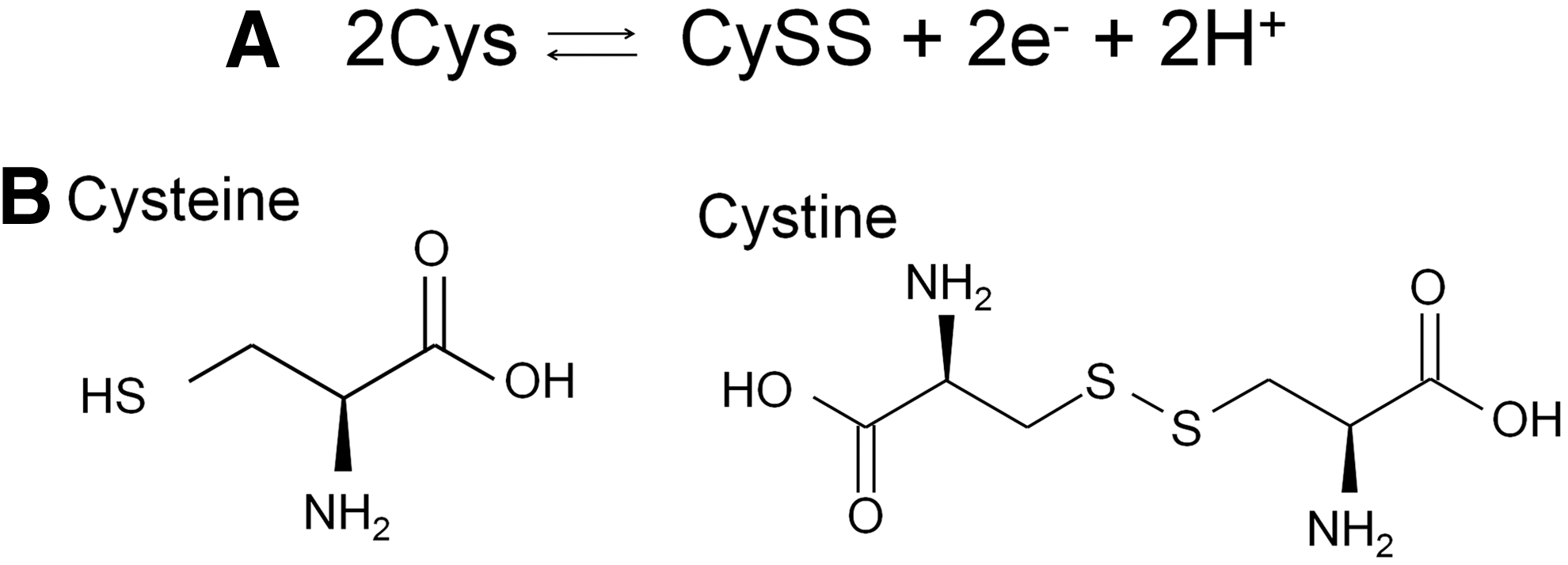
All cells produce a variety of antioxidant molecules, chief among which are the low-molecular-weight thiols GSH and Cys. Both of these thiols exist in reduced (GSH, Cys) and oxidized forms (GSSG, CySS). Each reduced/oxidized (redox) pair forms a discreet redox couple (GSH/GSSG and Cys/CySS), with the GSH/GSSG redox couple predominating within the cell and Cys/CySS predominating in the plasma (55). GSH has a concentration range of 0.5–10 mM with the majority found in the cytosol (90%) and the remaining distributed between the various organelles, including the nucleus and mitochondria. GSH is the reduced form of glutathione, and its cysteine thiol may become oxidized, losing an electron and a hydrogen ion in the process. Two of these oxidized GSH molecules combine to form the disulfide-linked GSSG, which is often referred to as oxidized glutathione (Fig. 4).
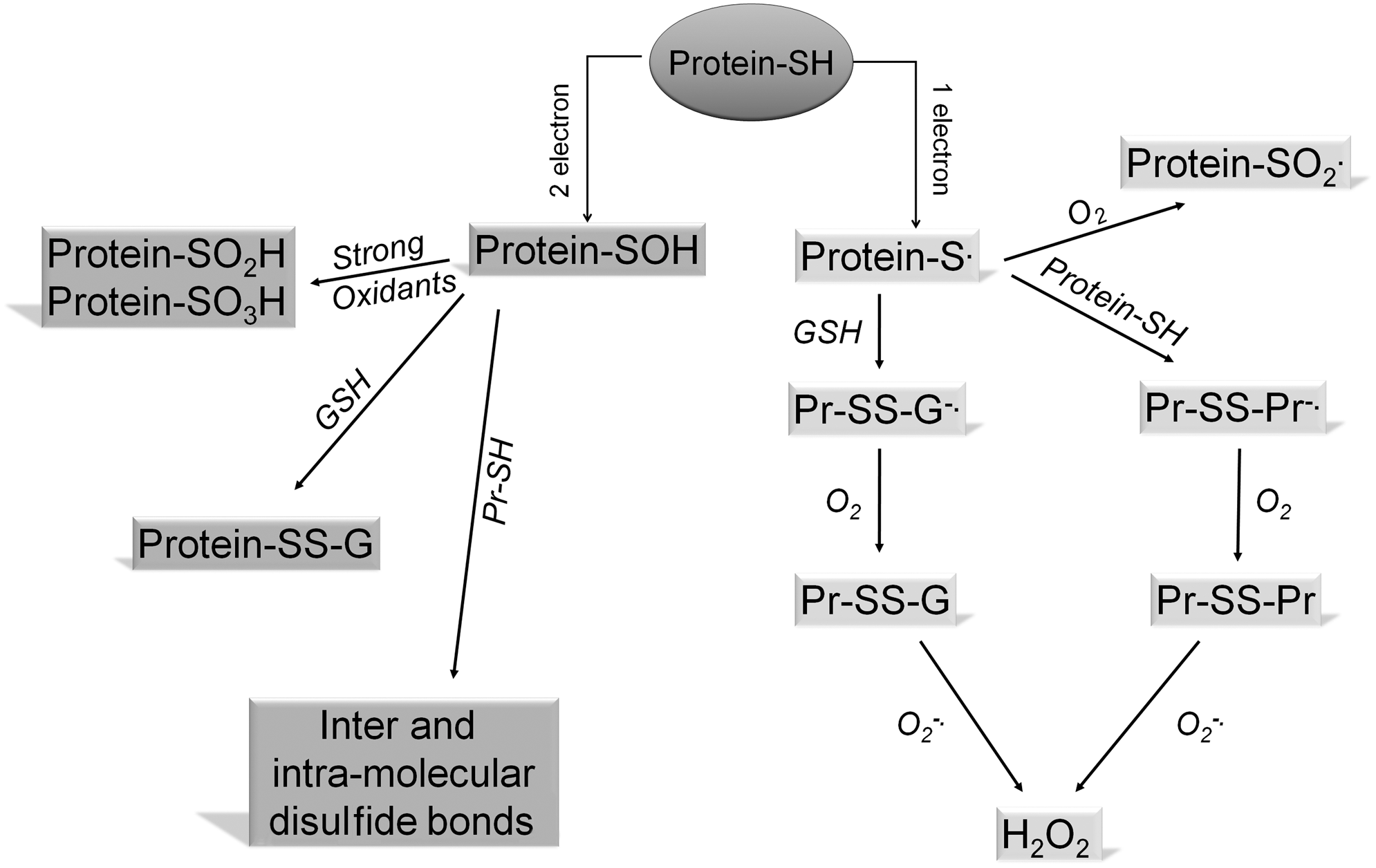
Such oxidation of GSH may occur enzymatically, as in the redox reaction with hydroperoxides that are catalyzed by glutathione peroxidase (GPx), and GSSG may be converted back to GSH by glutathione reductase. GSH may also react in a non-enzymatic manner with a wide range of substances to yield GSSG. GSH is the major source of reducing capacity within the cell. It contributes to anti-oxidant metabolic pathways by acting as a strong reducing agent (electron donor) or as a cofactor of electrophilic conjugates. Almost all physiological oxidants react with thiols (151). Several examples of redox reactions involving GSH are illustrated in Figure 5. Thiols may participate in either one- or two-electron transfer reactions depending on the oxidant species involved. One-electron reactive species include superoxide (O2 −·) and hydroxyl (OH·) radical, which, when reacting with thiols, yield the thiyl (S·) radical. Under aerobic conditions, the most favored subsequent reaction is with GSH or a protein thiol group to form a mixed disulfide (45). Two-electron oxidants such as hydrogen peroxide (H2O2), hypochlorous acid (HOCl), or peroxynitrite (ONOO−) react with thiols to generate sulfenic acids (R-SOH) (107). The products of one- and two-electron oxidation of protein cysteine residues are shown in Figure 6. Sulfenic acids are unstable and highly reactive intermediates that may be further oxidized to higher oxidation states such as sulfinic and sulfonic acids (RSO2H and RSO3H). Sulfenic acids may be reduced by cellular reductants such as GSH or the enzyme Trx (122). Sulfinylation of protein cysteines may only be reversed by the action of the recently discovered ATP-dependent enzymatic sulfinic acid reductase known as sulfiredoxin (14).

Plasma glutathione levels vary from 2–25 μM or higher, mostly occurring in the reduced form (GSH) (3). Glutathione is present in all animal cells, including platelets, with a concentration range of 0.5–10 mM (153). There is, thus, a very large GSH concentration gradient across the cell membrane. GSH is negatively charged, ionized to thiolate (GS−), at physiological pH and there is a large negative potential (−35 to −66 mV more negative relative to the extracellular redox potential) within the cell (57). These large electro-chemical gradients have the potential to drive GSH transport out of the cell. However, there is ambiguity as to whether or not specific GSH membrane transporters exist. The study by Gukyasyan proposes that an Na+/GSH symport may exist in certain pathology scenarios, such as cystic fibrosis, by which the movement of GSH with Na+ out of the cell is thermodynamically favored. In a separate study, members of the multidrug resistance-associated protein/cystic fibrosis transmembrane conductance regulator family of proteins (MRP/CFTR) and members of the organic anion transporting polypeptide (OATP) family of proteins have been identified as putative GSH transporters (11).
The most abundant thiol compound is cysteine, existing in both the reduced form cysteine (Cys) and the oxidized disulfide cysteine (CySS) (Fig 7). Total plasma cysteine is approximately 200 μM with the predominant form being CySS. Within plasma, low-molecular-weight thiols have been reported to be disulfide linked to plasma proteins. The concentrations of the protein bound forms are higher than the free thiols. Cys is more rapidly incorporated into plasma proteins than GSH, possibly due its lower pKa value (8.3 vs. 8.8), and more readily oxidized at physiological pH (82). This process has been suggested to occur by non-enzymatic protein thiol/disulfide exchanges (94). Plasma protein-thiol binding is considered a thiol-buffering system that protects against transient thiol-disulfide changes within the plasma.
The Extracellular Redox Environment
In living cells, energy is captured during the oxidation of glucose and other organic molecules, resulting in an overall reducing environment within cells and tissues. This environment is maintained by several redox couples that are responsive to electron flow and, hence, to changes in the reducing/oxidizing (redox) environment. The redox environment is considered a combination of the tendency of such a redox couple to reduce another species (reduction potential), and the number of electrons that are available for reduction (reducing capacity). Reducing capacity is determined from the concentration of the reduced species in a redox couple. The redox state of a couple is quantified by its redox potential that measures both the reduction potential and the reducing capacity. The redox potential can be calculated from the Nernst equation and is expressed in millivolts (133). The major physiological redox couples include the glutathione system (GSH/GSSG), the cysteine system (Cys/CySS), the nicotinamide adenine dinucleotide phosphate system (NADPH/NADP+), and the thioredoxin system [Trx (SH)2/TrxSS], where the first species in each couple is the reduced species and the second is the oxidized form. Both the GSH/GSSG and Cys/CySS redox couples are present in blood plasma and may undergo reversible oxidation/reduction as shown in Figures 4 and 7. Each of these reactions forms an electrochemical cell comprising two half-cell reactions that are in equilibrium with each other. The electromotive force from each redox pair is ΔE=E2 − E1, where E2 is the half-cell reduction potential for the reduced species, and E1 is the half-cell reduction potential for the oxidized species. This electromotive force is a measure of the electrical potential energy driving the flow of electrons in a particular direction. If ΔE=0, there is no net electron flow but this is never the case in living organisms. In biological systems, there is, therefore, always a net half-cell reducing potential (Eh) that is equal to ΔE for a given redox couple. The Nernst equation is used to calculate the reducing (redox) potential of any redox couple once the absolute concentrations of both reduced and oxidized species are known (133). The Nernst equation is written as follows:
where R is the gas constant, 8.314 JK−1 mol−1, T is temperature in Kelvin, F is the Faraday constant, 9.6485×104 Cmol−1, n=number of electrons, and Q=[Reduced species]/[Oxidized species]. The value of the calculated redox potential is expressed in millivolts. The natural log (ln) term may be replaced by log10 using 2.303 as a conversion factor. Since redox potential is pH dependent, it is necessary to modify the equation to allow for biological pH ranges. The term E° designates non-standard conditions, such as pH=7.4, for plasma at 37°C. Following these modifications, the Nernst equation becomes
for the GSH/GSSG and Cys/CySS redox couples, respectively. It should be noted from the equations in Figures 4 and 7 that n=2 for each of these equations, representing two electrons available for reduction, and E0=−264 mV for GSH and −250 mV for Cys at pH=7.4 and T=37°C.
Oxidative stress is associated with a more oxidizing redox potential and reductive stress with a more reducing redox potential (121). A recent work by Go and Jones determined the redox potentials of both the GSH/GSSG and Cys/CySS redox couples in plasma samples from more than 700 randomly selected subjects. The mean±SD plasma redox potentials (Eh) are −130.9±22.9 mV and −72.4±12.8 mV for the GSH/GSSG and Cys/CySS redox couples, respectively (55). The redox state of each of these redox couples has been shown to regulate a range of biological processes such as enzyme catalysis (27), cell proliferation (9), apoptosis (26), gene expression (5), and atherosclerosis (67). The redox state or redox potential (Eh) of each redox couple varies among individuals, ranging from Eh=−200 to −50 mV for GSH/GSSG and from Eh=−130 to −20 mV for Cys/CySS (55). Much evidence shows that the redox status of the plasma becomes more oxidized in smokers, type 2 diabetics, and patients with cardiovascular disease (60, 73, 114). It is established that the redox potentials of both the GSH/GSSG and Cys/CySS redox couples also become measurably more oxidizing with an increase in age, with GSH/GSSG Eh changing by +0.7 mV/year and Cys/CySS Eh by +0.16 mV/year (74). Among smokers, the mean plasma GSH/GSSG and Cys/CySS redox potentials were more than 6% and 15% more oxidizing than those of non-smokers, respectively (103).
An imbalance between pro-oxidant species and physiological redox buffers by which pro-oxidant species levels are diminished while reducing redox buffering levels are increased is defined as reductive stress. This is the opposite scenario to oxidative stress. This physiological phenomenon of reductive stress was first reported by Rajasekaran et al. (121). In this study, mice expressing the human mutant αB-crystallin protein developed higher levels of GSH, increased expression of heat shock proteins (Hsp) and GPx, and decreased myocardial levels of reactive oxygen species (ROS). Clinical symptoms were reported as increased protein aggregation and cardiomyopathy. More recently, the specific role of Hsp27 in this reductive stress-induced cardiomyopathy was demonstrated. Over-expression of the antioxidant Hsp27 in transgenic mice resulted in elevated GSH/GSSG ratio and decreased levels of ROS, with cardiac hypertrophy, dysfunction, and reduced lifespan (159).
Changes in the cellular redox status due to variations in GSH/GSSG or Cys/CySS redox potentials induce reversible formation of mixed disulfides between protein thiol groups and glutathione/cysteine (S-glutathionylation/S-cysteinylation) in a range of cellular proteins (32). S-glutathionylation and S-cysteinylation may be viewed as modes of redox regulation of protein function, serving both to maintain normal protein activity and to protect critical protein cysteine thiols from oxidative damage. Although relatively little is known about the regulation of platelet function in a varying external redox environment, it is reported that some concentrations of GSH and ratios of GSH/GSSG potentiate platelet aggregation (41). In addition to exposure of sub-endothelial matrix proteins, variations in the redox status of the blood plasma are also believed to be involved in platelet activation and thrombotic events. An altered plasma redox state may be brought about through variations in the levels of physiological redox buffers such as GSH/GSSG and Cys/CySS (7) or by the addition of exogenous antioxidants such as α-tocopherol. α-tocopherol dose dependently inhibits the platelet aggregation response to ADP, collagen, and epinephrine (137). In addition, in this laboratory, it has been shown that integrin αIIbβ3 exists in an activated state in subjects with elevated plasma homocysteine levels (98). The generation of highly oxidizing free radicals, including superoxide (O2 −·), the hydroxyl radical (OH·), and the non-radical species H2O2, readily targets protein thiols. Such oxidative damage may be reversible as in the formation of cysteine sulfenic or sulfinic acids, or irreversible as with the formation of cysteine sulfonic acid (52). Cysteine sulfenic acid (Cys-OH) may be recycled back to the original thiol (Cys-SH) via disulfide bonded intermediates by cellular reductants such as Trx, glutaredoxin, cysteine, or glutathione themselves (117). Resultant S-glutathionylation or S-cysteinylation of cysteine thiols can modulate protein function by inhibiting or enhancing protein function such as the reversible inactivation of the enzyme α-ketoglutarate dehydrogenase by H2O2-induced S-glutathionylation in response to mitochondrial GSH redox status (112). A large number of cellular proteins have been identified as targets for S-glutathionylation, including endothelial nitric oxide synthase (19), the platelet and fibroblast cytoskeletal protein actin (33, 115), hemoglobin (16), and the mitochondrial enzyme NADH oxidoreductase (140). S-cysteinylated proteins include the bacterial proteins argininosuccinate synthase and cobalamin-independent methionine synthase (65), human signal transducer gp130 (105), and human L-xylulose reductase, where S-cysteinylation of Cys138 resulted in a 10-fold decrease in catalytic efficiency (160).
Platelet Targets for S-glutathionylation
Thiol groups differ in their reactivity due to the variability of the pKa of the cysteine thiol, depending on the redox environment of the thiols or on the electrostatic interactions of the surrounding amino-acid residues. The pKa values of cysteine thiols may be decreased sufficiently by charge interactions with positively charged, basic amino acids that are adjacent to the cysteine thiol (144, 146). Such thiols can exist as thiolate (S−) groups that are highly reactive and are considerably better nucleophiles than their protonated forms, giving rise to the term “reactive cysteine” (97). Proteins with low pKa cysteine residues include the protein tyrosine phosphatases (PTPs). All PTPs contain a reactive cysteine that is essential to their catalytic function, having a pKa value of 4.7 to 5.4, compared with the typical thiol pKa value of 8.5 (125). Cysteine pKa values for various proteins have been determined by a variety of methods, including Raman spectroscopy (84), the measurement of pH dependence of 240 nm absorbance, the reactivity with fluorescein iodoacetamide across a range of pH values, and the determination of functional pKa by pH dependence of competition with HRP for H2O2 (108). The reactive cysteine of PTP is contained within a signature motif, His-Cys-X-X-Gly-X-X-Arg-Ser/Thr, where X denotes any amino acid and the thiol group exists as a thiolate anion at neutral pH (34). The basic residue histidine (His) is believed to stabilize the thiolate anion of the cysteine through electrostatic interactions.
An analysis of potential reactive cysteine residues within a broad spectrum of proteins has indicated that in addition to the linear amino-acid sequence and the proximity of basic amino acids to a particular cysteine residue, other factors also contribute to the reactivity of cysteine thiols (96). The three-dimensional tertiary structure of the protein may bring basic amino acids into close contact with otherwise distant cysteine residues, bringing electrostatic stabilizing influences to bear on the cysteine thiolate. In addition, the solvent accessibility of the cysteine residues is important, as deeply buried cysteine residues are inaccessible to solvents and, thus, unlikely to play a role in redox-regulated thiol modifications. Isolated, surface-exposed cysteines have been calculated to be more reactive with oxidants and thiol-containing reagents due to their lower pKa values: approximately 7.5 for isolated exposed cysteines compared with approximately 9.5 for isolated, buried cysteines. Surface-exposed cysteine thiols may have lowered pKa values due to interactions with H-bond partners such as water molecules and other titratable groups of polar residues, which are abundant on the protein surface (95). Another determinant of cysteine reactivity is the proximity to another cysteine residue. A strong predictor of cysteine susceptibility to oxidation occurs when the cysteine–cysteine distance is ≤6.2 Å. The oxidation product most likely here would be cysteine disulfide (131). The β1 integrin subunit contains several such closely spaced (vicinal) cysteines that are also adjacent to basic amino acids. Reactive cysteine thiols may well be exposed on the surface of α2β1 in the unactivated platelet. They may also become exposed during the large conformational changes occurring during integrin activation. A comparison of the cysteine-rich beta integrin subunits found on human platelets showed almost twice as many potential reactive cysteine residues on the β1 compared with the β3 integrin subunit (Fig. 8). There were three cysteine residues in the β1 integrin subunit having a basic amino-acid residue on either side: Cys477 (K-C-H) and Cys576 (K-C-R) in the first and third cysteine-rich repeat regions and Cys691 (H-C-K) while this is not seen at all in the β3 integrin subunit. The β1 integrin subunit also has four cysteines with two basic amino acids immediately preceding or following the cysteine: Cys415 (R-K-C), Cys500 (R-H-C), Cys516 (C-R-K), and Cys538 (C-R-K). Only two of these motifs, Cys474 (H-R-C) and Cys643 (C-K-K), are seen in the β3 integrin subunit. Eighteen β1 cysteine residues are adjacent to at least a single basic amino acid compared with only 10 in β3, showing a much greater number of potentially redox reactive cysteines in this integrin.

. There are three -XCX- motifs in the β1 integrin subunit only. They are KCH, KCR, and HCK and are highlighted in the oval shapes  .
.
In the presence of the thiol-specific oxidant diamide, a concentration-dependent increase in the S-glutathionylation of platelet cytoskeletal proteins was observed (31). Actin proved to be the most readily S-glutathionylated protein. It is the most abundant cytoskeletal protein that is found in close association with the platelet membrane, and may be disulfide linked to other membrane proteins (120). More recently, the N-acetyl cysteine-mediated reduction of surface-exposed β-actin in human peripheral blood mononuclear cells detected by the cell-impermeable thiol detection reagent N-(biotinoyl)-N-(iodoacetyl) ethylendiamine (BIAM) has been reported (81). Human β-actin possesses a single reactive cysteine (Cys374) that is immediately preceded by three basic amino-acid residues (H-R-K-C-F) (Uniprot Protein Data Bank, Accession number P60709). Therefore, exofacial platelet membrane protein thiols respond to local conditions of reductive stress or oxidative stress created by the presence of either the reducing or oxidizing redox potentials.
Conclusion and Perspectives
Platelets and all of the other blood components are continuously exposed to the redox-buffering systems of the plasma. These redox buffers comprise chiefly the glutathione and cysteine redox couples and are mainly considered the first-line defense against excess reactive oxygen or reactive nitrogen-free radical production during times of oxidative stress (61). In this role, both glutathione and cysteine are considered as acting as either potent antioxidants or simply ROS scavengers neutralizing the pro-oxidant effects of reactive oxygen and nitrogen species directly. Such a role could influence platelet activation, which is known to be more pro-aggregatory under oxidizing conditions. Platelet thiol/disulfide interactions play a crucial role in the activation of the integrin αIIbβ3. The studies that demonstrated this used non-physiological disulfide bond reducing agents such as DTT (156), the free-thiol blocking agents DTNB and NEM (43) or employed the generation of specific mutations of cysteine residues within the integrin β integrin subunit itself (102). The requirement for free thiols and disulfide bond formation/disruption within αIIbβ3 during its transitions from the inactive to the active ligand-binding receptor conformation is clear. The underlying mechanism behind this thiol/disulfide exchange process remains unexplained, but there is much evidence supporting a role for the intrinsic thiol isomerase activity of the integrin itself or for platelet PDI in these conformational changes (39, 40, 113, 147).
There is growing evidence which suggests that the redox state of thiol/disulfide redox couples influences the outcomes of redox-based cell signaling events (104). Protein thiols offer a great deal of flexibility with regard to the types of modification they can undergo and the range of chemical signals they can perceive. Different modifications of the same protein can create multiple activation states and, thus, deliver discrete regulatory outcomes. Cysteines act as “redox switches,” sensing and reacting to changes in the surrounding environment, and, therefore, represent a key target for signaling cascades (53). Modification of cysteine residues by GSH and NO is well known to alter protein function. The concept of redox regulation as a dynamic signaling system in both mammalian and bacterial cells has been emerging over the past number of years. In a previous work, we established that integrins exhibited an endogenous thiol isomerase activity. Furthermore, we demonstrated that redox modulation of the integrin αIIbβ3 involved an allosteric regulation of this enzymatic activity. Our confirmation that S-nitrosylation of αIIbβ3 can also modulate the activation state of the integrin in the intact platelet revealed one aspect of the interplay between redox and NO in regulating platelet function. However, the precise mechanisms involved in redox modification of proteins are not clear. It is noted that the clinical role of oxidative stress in platelet function and thrombosis is unclear and complex. Nonetheless, redox regulation of the platelet is crucial to the modulation of its function. Redox sites are present on the surface of the platelet in the form of thiols and disulfides. The redox state of such sites most likely impacts platelet function, as it does in other biological processes. These include integrin-mediated adhesion, human immunodeficiency virus (HIV) entry into the cell, and receptor shedding. The implications, therefore, are that the regulation of platelets by redox and NO is an important biomedical issue (Fig. 9). A thorough understanding of these mechanisms and how they interact with other platelet signaling events is of the utmost importance for the development of novel therapeutic targets so that we can protect against inappropriate thrombus formation.
Footnotes
Acknowledgments
S.O'N. was funded by the RCSI Research Committee Fund, Health Research Board Ireland (HRB), Science Foundation Ireland (SFI), and the National Biophotonics Imaging Platform Ireland (NBIPI). N.M. was funded by the HRB, SFI, and NBIPI.