
Abstract
Introduction
E
The human genome encodes for 109 PTPs (3, 4, 80, 85, 142), which have been organized into several classes (Fig. 1). PTPs are characterized by a conserved amino acid sequence

As for other classes of enzymes, universal cellular, molecular, and biochemical approaches have been applied to the study of PTPs and have provided tremendous progress in understanding their regulation and function. However, the advent of PTP-tailored technologies, including (i) substrate trapping and profiling, (ii) activity-based probes and fluorogenic substrates, and (iii) redox-sensing probes, has been invaluable in the investigation of this intriguing family of enzymes. Approaches to study in vitro PTP enzymology have already been discussed in several excellent reviews (9, 39, 43, 64, 72, 107, 128). In this study, we will focus on approaches to study intracellular PTP biochemistry and physiology.
Identifying Intracellular PTP Substrates
In 2007, Tiganis and Bennett proposed three criteria for the classification of a tyrosine-phosphorylated protein as a PTP substrate (118), which were adapted from the 1979 Krebs and Beavo guidelines for assignment of phosphorylation and dephosphorylation events as physiologically significant (63). These PTP substrate criteria consist of (i) demonstration of interaction of the substrate with a PTP substrate-trapping mutant, (ii) modulation of substrate tyrosine phosphorylation levels in a cellular context, and (iii) dephosphorylation of the substrate by the enzyme in vitro. Identification of physiologically relevant PTP substrates has proven to be a challenging, yet possible task. In this section, we will describe two methodologies that are currently available to identify proteins to be proposed as candidate intracellular PTP substrates, substrate trapping and substrate profiling.
Substrate-trapping methods
Coprecipitation of PTPs with their substrates can be difficult due to the transient nature of the PTP enzyme–substrate complex (145). The concept of PTP substrate trapping involves mutation of one or more residues in the catalytic domain of the phosphatase so that substrates can bind the catalytic cleft, but catalysis cannot be completed. This leads to formation of a stable enzyme–substrate in which the substrate is trapped by the enzyme. A sufficiently stable PTP-substrate complex will allow for biochemical isolation and detection of the substrate. An ideal substrate-trapping mutant will have a low specific activity (low k cat), high affinity of binding to the substrate (low K M), and low rate of dissociation from the substrate (low K d) (31, 133).
PTP catalytic mechanism
To aid in understanding of the available PTP-trapping approaches, we will first provide a brief overview of the PTP catalytic mechanism. PTPs are Cys-dependent hydrolytic enzymes and their mechanism of action has been thoroughly reviewed (17, 143, 144). The general catalytic mechanism involves nucleophilic attack at the phosphate of pTyr by the catalytic Cys residue followed by protonation and loss of the Tyr substrate assisted by the general acid catalysis of a nearby Asp residue. The phosphoenzyme intermediate is then hydrolyzed by a water molecule, with the neighboring Asp residue serving as a general base.
There are two motifs in the PTP catalytic domain critical for catalysis. Amino acids in the
Substrate-trapping mutants
The first attempts at substrate trapping involved mutation of the catalytic Cys to Ser or Ala. Replacement of this residue allows binding of the phosphotyrosyl substrate to the PTP catalytic pocket, however, nucleophilic attack on the phosphate cannot occur. This should lead to a stable enzyme–substrate complex. This approach was used for, among others, YopH (10), PTP1B (52, 86), SHP-1 (120), SHP-2 (136), MKP-1 (60, 110), VHR (122), PTEN (90), and CD45 (34).
Use of an alternate mutation was pioneered by the Tonks group (31, 35). Substitution of the invariant Asp in the WPD loop (Asp-181 of PTP1B or Asp-199 of PTP-PEST) to Ala created mutants that retained substrate binding, but were dramatically reduced in the catalytic activity (both mutants exhibited ∼105 less activity compared with the wild-type proteins). These mutants were used to precipitate the epidermal growth factor receptor (EGFR) as a substrate of PTP1B, and p130cas as a substrate of PTP-PEST. In both reports, the PTP1B-D181A and PTP-PEST-D199A mutants were much more effective at substrate trapping than the corresponding PTP1B-C215S and PTP-PEDT-C231S mutants. It is likely that for some PTPs, removal of the Cys impairs the affinity for the target substrate such that the stoichiometry of the complex falls below the sensitivity threshold of detection methods. Among some of the PTPs whose substrates have been identified via the D/A approach are mouse Ptpn22 (Pep) (24), TC-PTP (119), SHP-1 (120), RPTPɛ (121), and DEP-1 (46, 92).
Since these early reports, modifications to the original single point mutations have been reported. The D425A/C459S double mutant was introduced to identify the EGFR and Gab1 as SHP-2 substrates. The SHP-2-D425A mutant retained some catalytic activity and displayed no trapping capabilities (1). Although the SHP-2-C459S mutant did trap the EGFR, the double mutant precipitated notably more protein. It is likely that the double mutant abolishes residual catalytic activity retained by SHP-2-D425A. However, another group was able to use SHP-2-D425A to isolate major vault protein as a substrate (61). The double D195A/C227S mutant was also used to identify substrates of human PTPN22 (LYP) (132).
The comparable D/A-C/S double mutation cannot be utilized for all PTPs. The Zhang group characterized a series of mutants of PTP1B and found that the D181A/C215S double mutant was not more effective than the single D181A mutant; interestingly PTP1B-D181A/Q262A was the most effective PTP1B substrate trap (133). This highly conserved catalytic site Gln aids in stabilizing the water molecule that attacks the phosphocysteinyl intermediate during catalysis. The D425A/Q506A approach was also effectively used to identify p190-B RhoGAP as a substrate for SHP-2 (62).
Other unique mutations have enhanced the trapping abilities of particular PTPs. Most classical PTPs possess a Thr or Ser residue immediately after the invariant Arg residue. Both the single SHP-2-T466A and double C459S/T466A mutations effectively trapped the EGFR, and it was proposed that the catalytic Thr of SHP-2 functions to maintain the sulfhydryl group of the catalytic Cys in a reduced state (84). Another example is the KIM-PTP T106D/C270S substrate-trapping mutant of HePTP (25). HePTP is one of three members of the kinase interaction motif (KIM) subfamily of PTPs. These PTPs contain a 15 aa sequence, called the KIM, which confers high-affinity binding to their mitogen-activated protein kinase substrates. It was found that mutation of a Thr residue to Asp in the KIM domain of HePTP in combination with the prototypical C270S mutation was essential for generating a substrate-trapping mutant sufficiently stable to support cocrystallization of HePTP with an Erk2 substrate peptide. In certain cases, post-translational modifications, such as phosphorylation, need to be taken into account for the development of an effective trapping agent. For example, due to phosphorylation on a Tyr residue (Y676) in the catalytic domain that prevented substrate access, the PTPH1-D811A mutant was reported to be incapable of trapping substrates when expressed in cells. Mutation of this residue to Phe restored substrate access to the mutant, and the PTPH1-D811A/Y676F mutant was used to isolate VCP as a substrate in NIH3T3 cells (140). Another report showed that incorporation of a Y46F mutation of a catalytic Tyr on the PTP1B-D181A mutant strongly enhanced trapping of the insulin receptor (IR) precursor compared with the single D181A mutation (13).
Taken together, these examples demonstrate the significance of specific structural elements in each PTP catalytic site that participate in substrate recognition. Employment of the crystal structure data that have become increasingly available for PTPs (2, 113) can aid in identification of residues to be modified for generation of additional trapping mutants. These examples also highlight the importance of empirical testing when deciding which mutations to use for a PTP whose substrate-trapping mutant is not yet available.
Substrate-trapping experimental design
The classical means by which substrate trapping is used to identify PTP substrates (Fig. 2A) has been to treat cells with pervanadate, an irreversible inhibitor of PTPs that oxidizes the catalytic Cys (49), to increase global tyrosine phosphorylation levels (31, 35). Cells are then lysed in a buffer containing low millimolar concentrations of ethylenediaminetetraacetic acid (EDTA) and iodoacetic acid (IAA). IAA inhibits any nonoxidized PTPs that would retain the catalytic activity in the lysate by alkylation of the active-site thiol. Dithiothreitol (DTT) is then added to the lysate to inactivate any residual IAA and to convert pervanadate to vanadate. EDTA present in the lysis buffer subsequently chelates the vanadate. After clarification to remove the insoluble fraction, lysates are then incubated with affinity-tagged (often glutathione-S-transferase [GST]-tagged) recombinant PTP substrate-trapping mutants, typically isolated from bacteria. The enzyme–substrate complex is subsequently precipitated with resin to bind to the affinity tag (glutathione-Sepharose beads in the case of GST-tagged proteins). The precipitated substrates can be identified by western blotting if an antibody for a candidate substrate under consideration is available, or by mass spectrometry to uncover the identity of an unknown precipitating protein.

The advantages of using this approach are that (i) pervanadate treatment induces abundant tyrosine phosphorylation of intracellular proteins, enhancing the possibility that the substrate can be trapped and subsequently detected, and (ii) it is relatively easy to generate large amounts of recombinant protein and cell lysates; thus, it is feasible to precipitate large amounts of enzyme–substrate complex to facilitate detection. Disadvantages are that (i) the pervanadate-induced phosphorylation may encourage the enzyme to bind to phosphorylated proteins that it would not recognize in a physiological context, and (ii) incubation of recombinant protein in the cell lysate abolishes subcellular protein compartmentalization, and thus may expose the enzyme to proteins it would not normally have access to. Several alterations are available that can be used in varying combinations with the above-mentioned experimental design. One is to overexpress an affinity-tagged fusion of the PTP-trapping mutant in the cells of interest and coprecipitate the interacting substrate either by immunoprecipitation (IP) of the PTP or substrate (if a candidate is known) or by precipitation of the affinity tag. In the case of IP, it is recommended to avoid the use of excessive concentrations of DTT, which can destabilize the disulfide bonds of the antibody and lead to reduced IP yields. To increase trapping yields, cells subjected to knockout or knockdown of the phosphatase of interest can be used, which will reduce competition between the trapping mutant and endogenous PTPs for substrate binding. Additionally, cells can be treated with pathway-specific stimuli to induce phosphorylation, such as signaling through antigen or growth factor receptors. Immunofluorescence microscopy can be performed to determine if the PTP mutant colocalizes with the candidate substrate.
Further improvements to the classical method have been proposed. One report used fluorescence resonance energy transfer (FRET) to demonstrate that PTP1B-D181A interacts with both the EGFR and the platelet-derived growth factor receptor on the surface of the endoplasmic reticulum (ER) (40). Another group coupled substrate trapping with bioluminescence resonance energy transfer (BRET) to establish an interaction between PTP1B and the IR in HEK293 cells (14). Cells were transfected with plasmids encoding Renilla luciferase (Rluc)-fused IR and yellow fluorescent protein (YFP)-fused PTP1B-D181A. The Rluc was excited by a substrate, coelenterazine. When the Rluc and YFP were less than 100 Å apart, luminescence of the Rluc excited the YFP, and fluorescent emission from the YFP could be detected in live cells by fluorescence microscopy. Whereas the interaction between wild-type PTP1B and the IR was too transient to detect BRET, the interaction was stabilized by use of the substrate-trapping mutant, and the authors were able to follow real-time dynamics of interaction of PTP1B and the IR following insulin treatment of the cells. A cell-permeable trapping version of STEP was developed, which contains the C300S mutation and is fused to a cell-penetrating TAT peptide (94, 141). Cell-permeable STEP-C300S could be incorporated directly into primary neuronal cells and hippocampal slices and was used to identify Erk1/2 (94) and glutamate receptor 2 (141) as substrates.
PTP substrate trapping has also been combined with the yeast two-hybrid system. In this adaptation, cDNA library products were phosphorylated by conditional expression of the tyrosine kinase v-Src, and the PTP substrate-trapping mutant of interest was used as bait. This approach was used to identify as putative PTP substrates, among others, the G-protein-coupled receptor kinase-interactor 1 for PTPζ (59), p250GAP for RPTPσ (20), and the p85 regulatory subunit of phosphoinositide 3-kinase for CD148 (126).
A question to be resolved whenever a substrate-trapping experiment is performed is whether the coprecipitating proteins represent substrates or mere interactors (35). A well-validated approach is the vanadate competition experiment. Low millimolar concentrations of vanadate can be added to the recombinant trapping protein before exposure to the substrates. Vanadate acts as a catalytic site inhibitor, mimicking pTyr, and thus will compete for binding with molecules that bind to the PTP catalytic site, but not with other interactors that bind to different areas of the protein. It should be noted that vanadate was found to be a better competitive inhibitor for binding to PTP-PEST-D199A (35) and SHP-1-D419S (120), rather than the respective C231S and C453S mutants, presumably because vanadate interacts with the thiolate anion of the Cys residue. Additonally, when performing substrate-trapping experiments, EDTA should be omitted from the lysis buffer to avoid chelation and inaccessibility of the vanadate. Another means to validate a putative substrate is to determine if the tyrosine phosphorylation status of the candidate is lowered by overexpression of the PTP and/or increased by knockdown or knockout of the phosphatase in the cells of interest.
Substrate profiling methods
Another means to identify candidate PTP substrates is substrate profiling, in which a substrate consensus sequence is applied in a bioinformatics search for putative proteins containing the sequence (Fig. 2B). Candidates are then prioritized based upon known expression patterns or functions. Two different methods will be discussed here, based upon the generation of the consensus sequence. In the first, the sequence is built around known substrates for the particular enzyme in question. In the second, an ideal peptide substrate sequence is constructed from enzymatic screening of combinatorial peptide libraries.
Consensus sequence of known substrates
The Tonks group identified TYK2 and JAK2 as PTP1B substrates based upon the consensus shared with the IR (89). It had previously been shown that the motif surrounding the IR phosphorylation sites, Gln/Asp-pTyr-pTyr-Arg-Lys, served as an excellent substrate for PTP1B in vitro (101). Through a bioinformatics search, they identified this motif in the activation loop of the tyrosine kinases TYK2 and JAK2. Substrate-trapping experiments with the PTP1B D181A mutant revealed a stable interaction of both of these kinases with PTP1B. Interestingly, another kinase in the same family, JAK1, which does not contain this consensus motif, was not precipitated with the mutant. Further functional assessment validated TYK2 and JAK2 as PTP1B substrates. It should be noted that the double pTyr motif is not limiting in identifying PTP1B substrates, as it is absent in several other reported substrates (74).
The Noda group constructed a consensus sequence from known substrates to identify a new candidate PTPζ substrate (33). They had previously identified three substrates of PTPζ using the yeast two-hybrid substrate-trapping system (59). Sequence examination surrounding the pTyr within these proteins revealed the consensus motif, Glu/Asp-Glu/Asp-Glu-Asp-Xaa-Ile-Val-pTyr-Xaa (where Xaa is not an acidic amino acid). A bioinformatics search revealed that the protein paxillin shared this consensus motif. They then demonstrated that Tyr phosphorylation of paxillin within this motif was decreased upon coexpression of wild-type PTPζ, but not PTPζ-C1934S, suggesting that paxillin is a putative PTPζ substrate.
Inverse alanine scanning
The Zhang group identified SKAP-HOM as a LYP substrate based upon a consensus sequence identified through an inverse alanine scanning peptide library approach (137). A substrate consensus sequence was first constructed in vitro with enzymatic assays. Each Ala residue in the parent peptide Ala-Ala-Ala-Ala-pTyr-Ala-Ala-Ala-Ala was separately and sequentially replaced by the 19 other amino acids. The LYP-mediated dephosphorylation of each of these peptides by LYP was assessed and used to identify preferred amino acids at each position of the peptide. Two consensus sequences were constructed: Tyr-Gly-Glu-Glu-pTyr-Asp-Asp-Leu-Tyr and Tyr-Gly-Tyr-Glu-pTyr-Asp-Asp-Glu-Tyr. Bioinformatics search of the first sequence yielded SKAP-HOM, which was validated by substrate trapping with both the LYP-C227S and LYP-D195A. A homologous protein, SKAP-55, which does not contain this consensus sequence, was not precipitated.
Assessing Intracellular PTP Activity
Chemical and biological methods to examine the intracellular enzymatic activity of individual PTPs hold promise to offer a wealth of insight into the physiological functions of these enzymes. PTP functions are in large part subjected to PTP catalytic activity, which is finely tuned by post-translational modifications, including oxidation, phosphorylation, nitrosylation, and sumoylation. Approaches to directly monitor PTP activity can provide broader insight into the function and regulation of PTPs than assays relying solely on protein expression levels. This section will describe recent developments in the methods to investigate intracellular PTP activity. Specific detection of PTP redox status will be discussed in the Monitoring Intracellular PTP Redox Status section.
In-gel methods
Burridge and Nelson proposed the first in-gel PTP activity assay, in which dephosphorylation of 32P radiolabeled glutamic acid/tyrosine copolymer (4:1 ratio of Glu:Tyr) by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE)-resolved PTPs was detected by autoradiography (18). In this assay, the probe is Glu4Tyr phosphorylated with 32P-labeled phosphate by a kinase and incorporated into a polyacrylamide gel before polymerization. PTPs or cell lysates were resolved by SDS-PAGE, further denatured by soaking gels in 6 M guanidine hydrochloride, and then renatured in buffers containing nonanionic detergents or reducing agents. PTP-mediated hydrolysis of the substrate was detected by loss of 32P from the region in the gel where a PTP was located by autoradiography. This form of assay can be used to assess the PTP activity from complex proteomes when used alongside Coomassie blue staining or western blotting to identify the PTPs of interest. Since then, several modifications have been proposed, including coupling with two-dimensional (2D) electrophoresis (78) and use of radiolabeled peptide substrates to increase enzyme specificity (57). Further developments shifted toward the use of nonradioactive substrates and methods that do not require denaturation/renaturation steps, which are not suitable for all PTPs (e.g., transmembrane PTPs are not renatured efficiently). The small molecules 4-methylumbelliferyl phosphate (MUP) and 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) can be used as fluorogenic phosphatase substrates for in-gel assays (55, 56). These substrates can stain the gel after electrophoresis, without the need of earlier incorporation in the gel. MUP has also been used as a substrate in nondenaturing 2D electrophoresis (5). Although this method does not provide selectivity for PTPs (these substrates are readily dephosphorylated by Ser/Thr phosphatases and alkaline phosphatases), it permits the avoidance of radioactivity. A further improvement has been the use of phosphorylated coumaryl amino propionic acid (pCAP)-containing peptides as fluorogenic substrates, which are only dephosphorylated by Tyr phosphatases and can be used to increase selectivity for individual PTPs by modification of the peptide sequence (107).
In-lysate methods
Several series of PTP activity-based probes have been developed for detecting active PTPs in cell lysates/mixtures of complex proteomes. Such probes bind to the catalytic site of PTPs, forming a covalent adduct and irreversibly inactivating the enzyme in a time-dependent fashion. Although different routes have been taken to develop the chemistry of the probes, several common features are shared in the tools developed thus far, which include (i) a reactive group that binds covalently to the PTP-active site, (ii) a linker region, and (iii) an affinity tag or reporter used for purification and/or visualization. The labeled PTPs can subsequently be detected and identified by a variety of methods, most commonly by western blotting, in-gel fluorescence imaging, or mass spectrometry. The different probes utilized to study PTP activity in cell lysates are described below.
Electrophilic pTyr-mimic probes
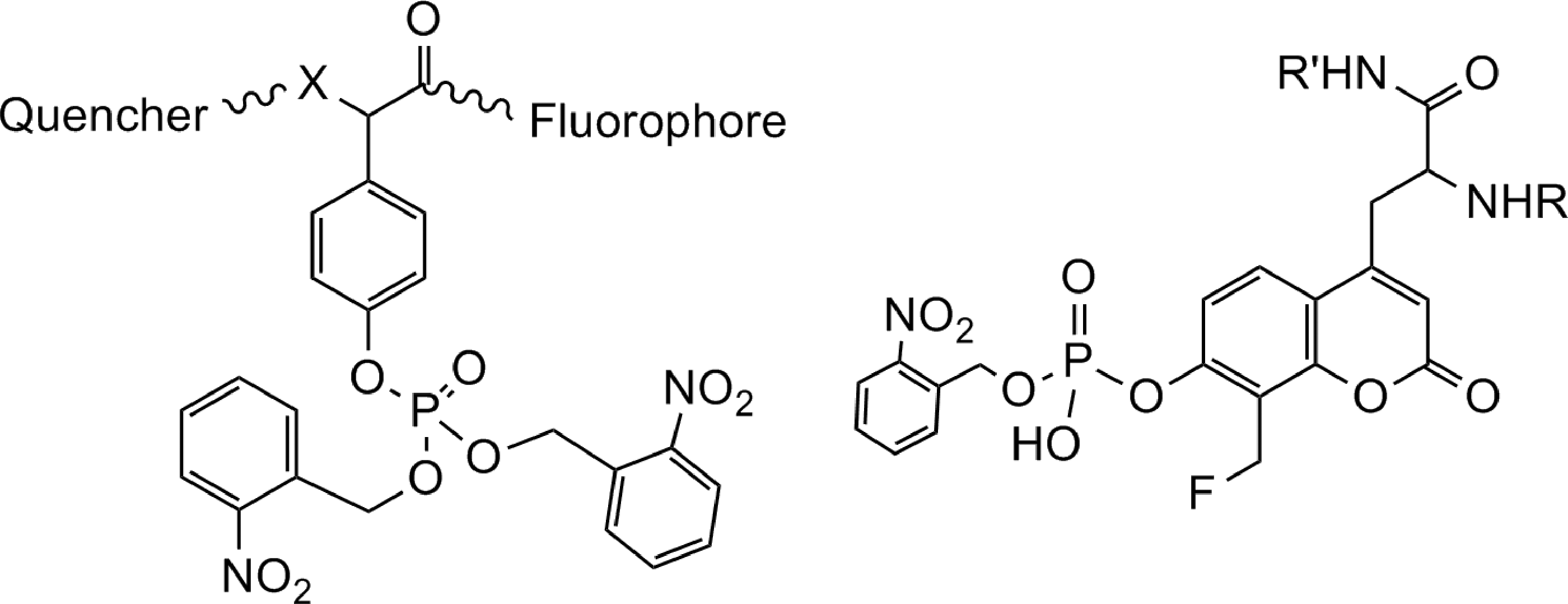
The Zhang group introduced several series of activity-based probes for PTPs for use in cell lysates/complex proteomes. The α-bromobenzylphosphonate (BBP) series contain a phenylphosphonate group, which binds to the PTP catalytic site as a pTyr mimic, with an alpha-bromide that serves as a latent leaving group that reacts only upon binding to the PTP-active site (Fig. 3A). These probes have been linked to either a biotin tag (66) that allows for purification and/or visualization, or to fluorescent lissamine rhodamine B (67). BBP probes irreversibly inactivate a variety of PTPs, including cytosolic PTPs YopH, PTP1B, HePTP, PTPH1, and SHP-2, transmembrane PTPs LAR, PTPα, and DEP-1, dual-specificity PTPs (DUSPs) VHR, PRL-3, and CDC14, and the low-molecular-weight PTPs (LMPTP or LMW-PTP), but do not bind to alkaline phosphatases, prostatic acid phosphatase, Ser/Thr phosphatases, SH2 or PTB domains, SRC kinase, serine proteases, metalloprotease thermolysin, the cysteine proteases calpain and papain, glyceradehyde-3-phosphate dehydrogenase, GST, lysozyme, or the entire E. coli proteome (66, 67). Covalent binding of the probes to PTPs was confirmed by western blotting for biotinylated PTPs or by in-gel imaging of PTP fluorescence, and by mass spectrometry. Site-directed mutagenesis has confirmed that these probes bind to the catalytic Cys residue of the PTP-active site. The biotinylated derivative offers the advantage of purification/enrichment of labeled PTPs, while the fluorescent counterpart allows greater sensitivity of detection.

The primary utility of these probes thus far has been the assessment of global changes in PTP activity in varying biological states, such as in cancer or exposure to oxidants. For example, BBP probes were used to demonstrate that four PTPs showed anomalous activity/expression in lysates of the MCF-7 breast cancer cell line compared with lysates of the human telomerase reverse transcriptase immortalized mammary epithelial cell line (67). Interestingly, when a side-by-side comparison of a fluorescently imaged versus a Coomassie blue-stained gel of the same lysates treated with the fluorescent BBP probe was performed to assess PTP activities in a panel of cancer cell line lysates, differences in PTP activity were revealed that were not evident in the Coomassie blue staining of total protein in the crude lysates. These probes are also capable of monitoring oxidation-induced decreases in PTP activity in lysates of hydrogen peroxide (H2O2)-treated cells. In an extension of the BBP probes, another group synthesized the amino acid derivative,
Another activity-based series of PTP probes are aryl vinyl sulfonates and sulfones (73). These probes also act as pTyr mimetics and bind to PTP-active sites through the Cys residue. Both phenyl vinylsulfonate (PVSN) and phenyl vinyl sulfone (PVS) (Fig. 3A) irreversibly inhibited all PTPs tested, including cytosolic PTPs YopH, PTP1B, and HePTP, the transmembrane DEP-1, the DUSPs PRL-1 and VHR, and LMPTP, but were highly inefficient at inhibiting alkaline phosphatases or the cysteine proteases papain and cathepsin B. Cocrystallization of YopH with PVS revealed a covalent thioether linkage between the Cys403 thiol and the terminal carbon of the vinyl group, and indicated additional amino acids involved in hydrogen bonding with the probe. In the reported assay, PVSN and PVS were synthesized with an azide tag, added to cell lysates, and subjected to in-lysate click chemistry to biotinylate the azide group. The biotin tag thus allowed for isolation/visualization of the PTP-bound probe. Although the potency of these probes is similar to the BBP probes, they offer two advantages for cellular assays—resistance to solvolysis and cell permeability, which allows for labeling of the azide-tagged PTPs directly in cells, and then conjugation to the biotin tag upon cell lysis. Direct treatment of COS-7 cells with low millimolar concentrations of PVS or PVSN increased western blotting-detected Tyr phosphorylation of intracellular proteins to levels comparable to Tyr phosphorylation after cells had been treated for the same length of time with 1 mM sodium orthovanadate.
Quinone methide-generating probes
Several groups have developed activity-based PTP probes involving generation of a highly reactive quinone methide intermediate that alkylates nucleophiles present in the PTP active site (54, 75, 104). These probes contain a pTyr mimetic moiety, which serves as a PTP recognition site, with a fluoride leaving group. When the probe is dephosphorylated by a PTP, it undergoes elimination of the fluorine atom, forming an electrophilic quinone methide. A subsequent addition reaction between a nucleophilic residue on the PTP and the quinone methide forges a covalent bond between the probe and the PTP (Fig. 3B).
The Lin group generated a biotinylated probe containing the 4-fluoromethylphenylphosphate (FMPP) moiety (Fig. 3B) (68, 75). When applied to a cell-based assay, lysates are incubated with a biotinylated probe, followed by IP of the PTP of interest. Detection of biotin by western blotting with an anti-biotin antibody or labeling with a reporter-conjugated avidin-based molecule reveals the amount of labeled, active PTP. This approach was used to show that treatment of the human lung carcinoma cell line, H1299, with the antitumor agent cisplatin increased the pool of active SHP-2, but did not affect the activity of TC-PTP or PTP1B. Treatment of cells with other antitumor agents 5-fluorouracil, etoposide, or 5-azacytidine had no effect on the SHP-2 activity. The probe did not label beta-galactosidase or trypsin. Similar to the above-mentioned in-lysate probes, this probe was also used in the low millimolar concentration range.
An activity-based probe for PTP1B was developed based upon the peptide incorporation of an unnatural amino acid, a 3-difluoromethyl analog of pTyr, which also acts as a quinone methide-generating activity-based probe (FMPPaa, Fig. 3B) (104). This amino acid was incorporated into a miniature peptide substrate presumably selective for PTP1B as it contains a secondary motif to enhance binding. A lack of labeling of recombinant PTP1B-C215S confirmed the activity-based nature of the probe. The authors showed selectivity for PTP1B versus PTP4A3, but selectivity was not shown for any other PTPs. However, incubation of Jurkat cell lysate with a 0.8 mM probe for 5 h caused substantial labeling of only PTP1B and one other protein. Interestingly, coincubation with 6 mM BBP eliminated labeling of the additional protein, indicating that nonspecific reduction of PTP activity can decrease the background binding of the probe. This assay highlights a unique option that can be employed if the probe exhibits higher affinity for a particular PTP versus others; by lowering both the signal and the background of the assay, an increased signal:background ratio can be achieved. The Yao group reported the design and synthesis of 2-FMPT, an unnatural amino acid that can be incorporated into peptides that is very similar to FMPPaa (Fig. 3B) (54). This moiety is identical to pTyr with the exception of the inclusion of 2-fluoromethyl on the aromatic ring, which allows for generation of the reactive quinone methide intermediate to covalently label the PTP as mentioned above. The incorporation into peptides improved the affinity and selectivity for particular PTPs and overcomes the diffusive nature of other quinone methide probes, which could potentially cross-react with other nearby proteins rather than the targeted PTP-active site. These 2-FMPT-based probes were conjugated to rhodamine or biotin tags. A peptidic substrate probe for PTP1B was designed based on enzymatic screening of a peptide library. This probe labeled recombinant PTP1B in vitro, but not after H2O2 treatment. Incubation of HEK293T cell lysates with a low micromolar concentration of biotinylated probe showed labeling of many endogenous proteins, including a specific band for PTP1B that was not evident in lysates of 3T3 cells (which do not express detectable levels of PTP1B).
F2PMP-based photoaffinity probes
Skorey et al. reported the use of a cell-permeable, irreversible photoaffinity tag to label PTP1B in HepG2 cell lysates (105). This probe was developed by attaching a trifluoromethylphenyl diazarine photophore to the previously reported [difluoro(phosphono)methyl]phenylalanine (F2PMP)-containing peptide inhibitors of PTP1B and TC-PTP (Fig. 3C) (29, 117). This photophore can be photolyzed with UV light to generate a carbene intermediate that is highly reactive toward hydrocarbons, including amino acids. The nonphotolyzed probes behaved as competitive reversible inhibitors of PTP1B with low nM IC50 and high selectivity compared with CD45. By LC-MS, the authors demonstrated that the photolabeled probe formed a 1:1 covalent adduct with 8% of PTP1B. Successive rounds of incubation with fresh probes followed by photolysis increased the stoichiometry of labeling to 50% of PTP1B. The probe likely labeled the PTP1B-active site, as it was competed by the active site inhibitor BzN-EJJ-amide; however, the probe also labeled the PTP1B-C215S mutant. Neither the H2O2- nor the heat-inactivated wild-type enzyme was labeled. Although the efficiency of labeling of PTP1B in cell lysates or intact cells was low (maximal efficiency of labeling of PTP1B in HepG2 cell lysate occurred with a 4 nM 125I-radiolabeled probe, however, with an efficiency of only 1%–3%), the photolysis of the probe was quite effective with nearly 80% complete photolysis after 5 min of irradiation; this study suggests that coupling the photoaffinity approach with other chemical PTP inhibitors will likely provide a useful method for probing PTP activity.
In-cell methods
Although information about the activity of a given PTP in a cell lysate can provide insight into the biological roles and regulation of that PTP, methods that can be used to monitor and, ultimately, quantify PTP activity directly in living cells are critical. This section will describe the progress that has been made on this front by applying the principles of both biology and chemistry.
Indirect methods with small-molecule substrates
The first intact cell-based assay for PTP activity was proposed by the Kennedy group in 1999 (26). The fundamental design of this assay was to incubate Spodoptera frugiperda insect cells overexpressing the PTP of interest (in this case, PTP1B or CD45) with the diffusible cell-permeable PTP substrate p-nitrophenyl phosphate (pNPP). The hydrolysis of pNPP was measured spectrophotometrically by OD405 of the treated cell supernatants. The authors demonstrated increased pNPP hydrolysis in cells overexpressing either CD45 or PTP1B compared with mock-transfected cells, and generated IC50 curves of the general PTP inhibitors H2O2, PAO, vanadate, and pervanadate. Although all of these inhibitors showed IC50 values in the low or submicromolar concentration range in cells overexpressing PTP1B or CD45, only vanadate was compared in the mock-infected cells (IC50 0.29 μM for PTP1B overexpressing cells, 0.06 μM for CD45 overexpressing cells, and 1.6 μM for mock-infected cells). They also showed that the Na-K-ATPase inhibitor ouabain and the Ser/Thr phosphatase inhibitor okadaic acid did not inhibit pNPP hydrolysis (up to 1 mM and 6.7 μM, respectively). Whereas the assay suffered from the limitations of the need for an overexpression system, in which the PTP was not evaluated in its physiological context, and the reliance upon the diffusion rates of the substrate into and out of the cells, this innovative report provided the first conceptual evidence of the feasibility of detection of intracellular PTP activity in live cells, and proposed the basis for an assay that could, at the very least, be used to screen for cell-permeable PTP inhibitors.
Fluorogenic pTyr mimic probes
Two reports proposed the use of fluorescein diphosphate (FDP)-based substrates for direct detection of PTP activity in live cells. FDP emits very low fluorescence, but is converted to the highly fluorescent fluorescein (maxex=490 nm; maxem=515 nm) upon dephosphorylation. In 2002, 1-(2-nitrophenyl)ethyl protected FDP (NPE-FDP) (Fig. 4A) was used as a cell-permeable substrate for PTPs in live cells (131). The PTP substrate FDP was derivatized to a photolabile protected form, NPE-FDP, which increases the cell-permeability of the probe and cages the phosphate groups to allow activation of the substrate in a temporal manner. Upon exposure to UV light, the probe becomes uncaged and available for dephosphorylation by the PTP. The authors were able to detect fluorescence by microscopy of Sf9 cells incubated with 20 μM substrates only after uncaging with UV light. However, when using the probe to detect the CD45 activity in Jurkat cells (Jurkat cells express abundant levels of CD45), there was no difference in the rate of hydrolysis detected beyond that of the CD45-null Jurkat derivative, J45.01 cells.
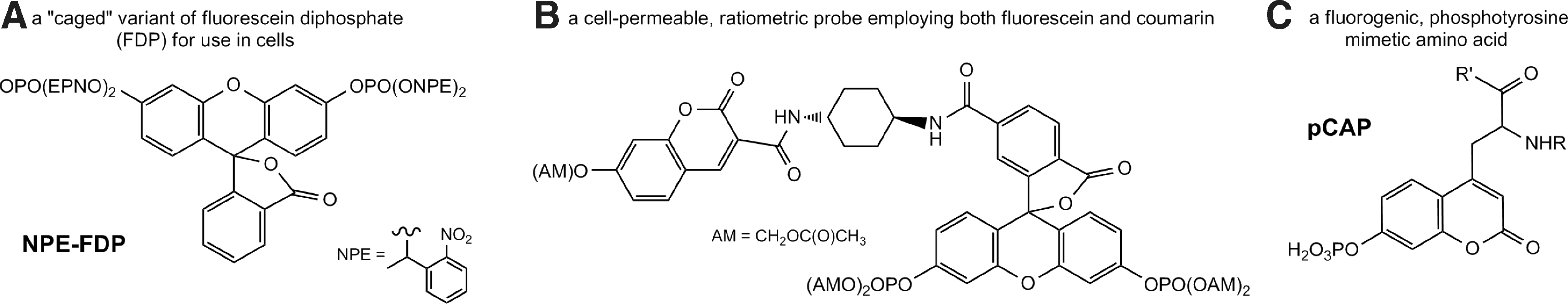
In 2003, a modified FDP-based substrate for ratiometric detection of PTP activity in live cells was reported (114). This FRET-based substrate consisted of a coumarin moiety donor (maxex=402 nm; maxem=445 nm) linked through a cyclohexane group to an FDP latent FRET acceptor (Fig. 4B). This substrate was dephosphorylated in vitro by both PTP1B and CD45, as demonstrated by a time-dependent increase in fluorescent emission at 515 nm and decrease at 450 nm. Cell-permeability of this probe was achieved by incorporation of acetoxymethyl residues on the phosphate and hydroxyl groups, which would theoretically be cleaved by esterases in the cell cytosol. The effectiveness of this approach was demonstrated by a time-dependent increase in FRET fluorescence detected by fluorescence microscopy for 20 min following incubation of human umbilical vein endothelial cells with 5 μM substrate, which was abolished by coincubation with 1 mM sodium orthovanadate. Although the FDP-based assays offer the advantages of cell-permeable, caged, and potent substrates, the lack of selectivity for PTPs (FDP is also dephosphorylated by other classes of phosphatases) limits their utility for assessment of individual intracellular PTPs in the absence of further modification.
Whereas phosphocoumarin derivatives have been well utilized as fluorogenic phosphatase substrates in vitro (88), the introduction of coumarin-based substrates as phospho-Tyr mimetics into peptides has recently afforded the development of cell-based assays to directly monitor intracellular PTP activity at the single-cell level. Mitra and Barrios reported the incorporation of pCAP (Fig. 4C) into peptides by standard Fmoc-based solid-phase peptide synthesis, and showed that when incorporated into peptides, pCAP behaves as a close mimic of phospho-Tyr (87). The nonphosphorylated (CAP) form of these peptides exhibited greater than 104-fold fluorescence intensity compared with an equimolar solution of the corresponding pCAP peptide (maxex=334 nm; maxem=460 nm).
We recently reported the development of a single-cell pCAP peptide-based assay for PTP activity, which is amenable to either high-content microscopy or flow cytometry (108) (Fig. 5). We established that pCAP peptides could be exploited to selectively assess intracellular PTP activity over other classes of enzymes, as evidenced by complete lack of dephosphorylation by Ser/Thr phosphatases, and as intracellular dephosphorylation of these peptides was entirely eliminated by the PTP inhibitor sodium orthovanadate. This assay was validated in RAW 264.7 macrophages and Jurkat T cells. To achieve selectivity for an individual PTP, we (i) took advantage of the variability of PTP-active site surface charge and hydrophobicity and employed highly positively charged lipid polyarginine cell-penetrating peptide tags for intracellular peptide delivery and (ii) used available PTP substrate profiling information and developed peptidic substrate sequences for CD45 based on the activation loop of SRC family kinase LCK (EDNE-pCAP-TARE) and for TC-PTP based on an optimized sequence reported by the Yao group (REGLN-pCAP-MVLAT) (111).
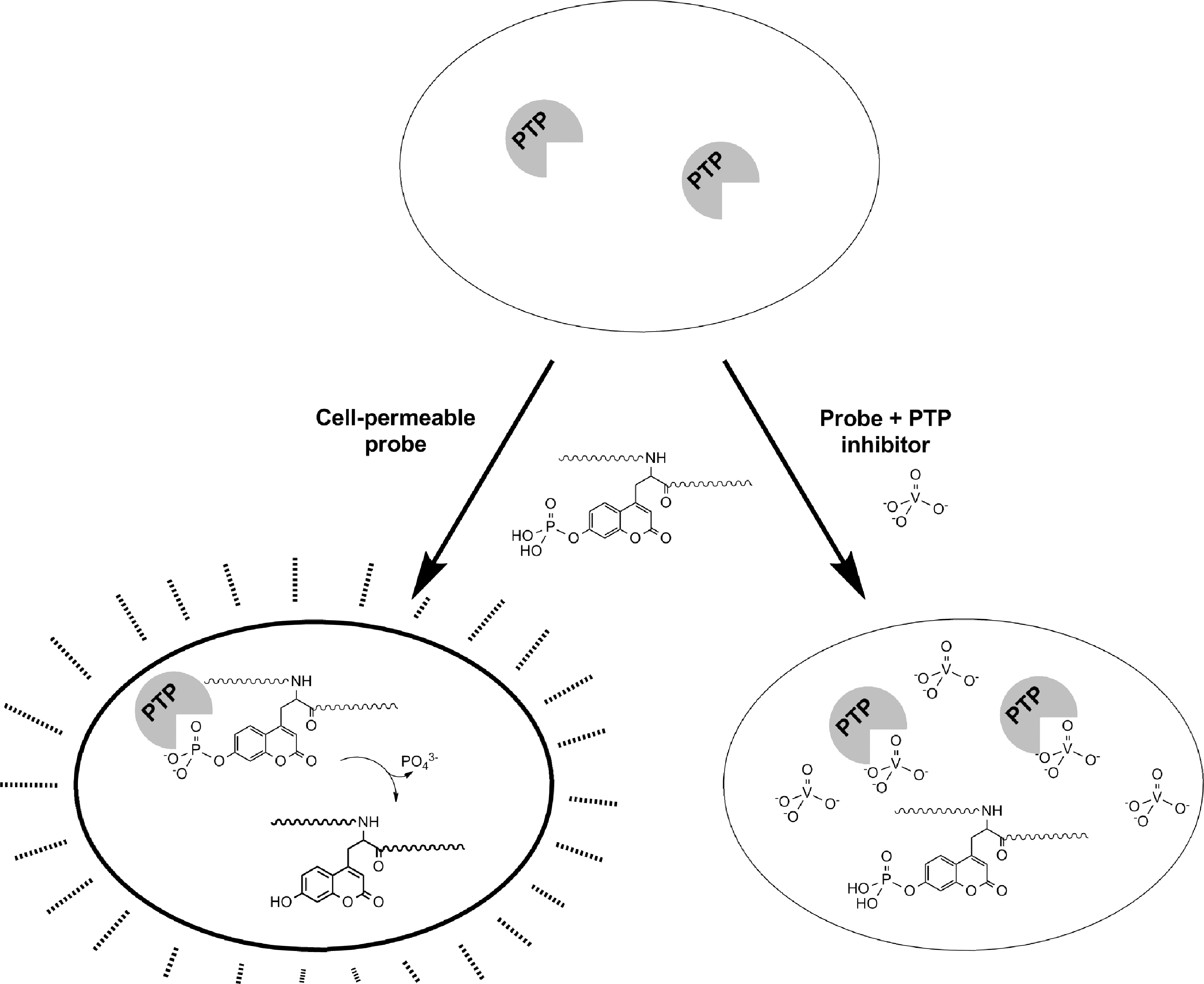
Selectivity for an individual endogenous PTP was achieved by optimization of pCAP peptide sequence, concentration, time of incubation, and choice of cell type. The CD45 assay was validated in CD45-null J45.01 cells, in Jurkat cells subjected to CD45 knockdown with cell-permeable antisense oligonucleotide, and in Jurkat cells treated with a previously reported cell-permeable inhibitor of CD45 (93). The TC-PTP assay was also validated with a previously described chemical inhibitor (139).
The intracellular CD45 pCAP peptide-based assay was challenged to the identification of inhibitors of CD45 in Jurkat cells. Screening of two small chemical libraries, one based on analogs of the CD45 inhibitor NSC 95397 (93), and the other composed of FDA-approved compounds, yielded four efficacious compounds. The effectiveness of these inhibitors was confirmed in follow-up cell phenotypic assays for CD45. This assay thus provides evidence that fluorogenic peptides can be used to assess the intracellular enzymatic activity of a single PTP, given that the conditions of substrate sequence, intracellular delivery, and kinetics of dephosphorylation are appropriately tuned.
Caged quinone methide-generating probes
The Yao group developed modular quinone methide-generating probes (Quinone Methide-Generating Probes section) for utility in direct detection of PTP activity in live cells (48). These probes incorporate the quinone methide system with a quenched one- or two-photon dye system. They contain an enzyme substrate warhead [in the case of PTPs, a photolabile 2-nitrobenzyloxy-caged phosphate (99)], a fluorescence reporter (one- or two-photon dye), and a quencher (dabcyl). Upon uncaging with UV light, the warhead can be activated by PTP-mediated dephosphorylation. This initiates a 1,6-elimination reaction releasing the quencher. The highly reactive quinone methide intermediate is then generated, fixing the probe near the site of the reaction and allowing fluorescent emission of the unquenched dye (Fig. 6). In vitro, such probes were dephosphorylated after UV-mediated photolysis by PTP1B, TC-PTP, LMPTP, and PTPβ. PTP covalent labeling was demonstrated by fluorescence of SDS-PAGE-resolved proteins. To assess intracellular PTP activity, live HeLa cells were viably permeabilized by short incubation with digitonin, incubated with 20 μM probe, and UV irradiated for 10 min. The cells were then fixed and cytosolic fluorescence was evident by microscopy. Interestingly, cells transfected with ER-localizing PTP1B and subjected to the same assay showed intense fluorescence in the ER, as confirmed with an ER-tracking dye and immunofluorescent staining for PTP1B. A cell-permeable derivative of the one-photon probe was synthesized by conjugation of nine Arg residues to the probe peptide backbone. Live imaging of HeLa cells subjected to incubation with the probe and subsequent UV irradiation showed punctate fluorescence throughout the cytosol, likely due to the endocytic uptake of the poly-Arg-tagged peptide.
An alternative approach was the coupling of quinone methide-generating chemistry with a caged, fluorogenic, unnatural pTyr mimetic to generate a UV-photoactivatable peptidic probe for PTP activity (Fig. 6) (36). This probe series comprised a 2-nitrobenzyl-caged, fluorinated derivative of coumaryl amino propionic acid (Fluorogenic pTyr Mimic Probes section), linked to a cell-penetrating peptide to target the probe to either the intracellular membrane (K[palmitoyl]-NH2), ER (KDEL-NH2), or mitochondria (
FRET-based method to image enzyme–substrate interaction
An elegant approach to monitor the PTP1B-substrate interaction in live cells was developed by Bastiaens and colleagues (138). They developed a FRET-based assay to detect the spatial regulation of PTP1B activity in live cells. A phosphorylated peptide substrate (Asp-Ala-Asp-Glu-pTyr-Leu) based on the EGFR-Y992 phosphorylation site was prepared with a caging photocleavable protective group and an N-terminal lissamine rhodamine B. PTP1B protein was transiently expressed in COS-7 cells as a green fluorescent protein (GFP) fusion. The caging moiety on the peptide prevented its binding to PTP1B until removal of the moiety by exposure of the peptide to UV light. Upon interaction of the enzyme–substrate complex, GFP fused to PTP1B served as a FRET donor to excite lissamine rhodamine B on the peptide. FRET was abolished once the substrate was dephosphorylated and released by PTP1B. The authors microinjected COS-7 cells with the peptide substrate and measured interactions of the complex by fluorescence microscopy. Intriguingly, they demonstrated that the dephosphorylated version of the peptide was rephosphorylated by intracellular kinases, and thus, a steady state enzyme–substrate complex was maintained that could be continuously monitored by FRET, and showed that a higher concentration of enzyme–substrate complex was observed at the cell periphery than in the perinuclear region. This suggests that spatial regulation may be a cellular mechanism to control the strength of signaling in discrete subcellular compartments, and indicates that studying PTP activity in a spatiotemporal context will be essential for developing a complete view of the function of these enzymes.
Yeast growth method
A report to measure PTP activity in living cells demonstrated a yeast growth-based assay as a biological readout for PTP activity (42). Saccharomyces cerevisiae was subjected to toxic levels of overexpression of v-Src, which inhibited cell growth. Overexpression of a counteracting PTP, namely PTP1B, TC-PTP, PTP-PEST, SHP-2, or PTPα, rescued the yeast growth, as detected spectrophotometrically at OD600. Overexpression of the catalytically inactive mutants of PTP1B, TC-PTP, or PTP-PEST did not rescue the cells, although they did induce a slight increase in growth, presumably due to substrate-trapping effects.
Single-cell capillary electrophoresis method
A single-cell capillary electrophoresis method to detect intracellular PTP activity was recently proposed by Phillips et al. (96). A Tyr-phosphorylated peptide, shown to be dephosphorylated in vitro by PTP1B and TC-PTP, was microinjected into single A431 epidermoid carcinoma cells, and the percentage of peptide retaining phosphorylation was measured by capillary electrophoresis. This assay was used to demonstrate that incubation of A431 cells with 1,2-napthoquinone, zinc pyrithione, and pervanadate inhibited intracellular PTP activity.
Monitoring Intracellular PTP Redox Status
Redox modulation of PTP activity is emerging as a key aspect of the biological regulation of this family of signaling enzymes (58, 124). The activated catalytic Cys residue is highly susceptible to oxidation in vitro and many PTPs are sensitive to oxidative inactivation in cells (6, 22, 38, 82). PTPs can be oxidized by several biological oxidants, including nitric oxide, H2O2, and the hydroxyl radical (27, 47, 81, 124). Initially, an S-nitrosocysteine or sulfenic acid is formed (32). As shown in Figure 7, these adducts are reversible and the S-nitrosocysteine can be converted into a sulfenic acid by hydrolysis (32, 115). Some PTPs form a relatively stable, observable sulfenic acid adduct. Further oxidation can result in the formation of a sulfinic or sulfonic acid species, resulting in irreversible inactivation of the phosphatase (97, 115). Several PTPs have evolved mechanisms to evade irreversible oxidative inactivation. For example, upon oxidation, the catalytic Cys residue of PTP1B forms a cyclic sulfenyl amide with the backbone nitrogen atom of a neighboring serine residue (100, 129, 135). The presence of a neighboring Cys residue facilitates the formation of a disulfide bond upon oxidation of the catalytic Cys in several PTPs, including SHP-1, SHP-2, LMPTP, Cdc25, PTEN, PRL-1, PRL-3, and MKP3 (15, 19, 21, 23, 69, 91, 103, 106, 112). In addition to oxidative regulation in the presence of reactive oxygen or nitrogen species, the PTP activity is modulated in the presence of hydrogen sulfide (65).

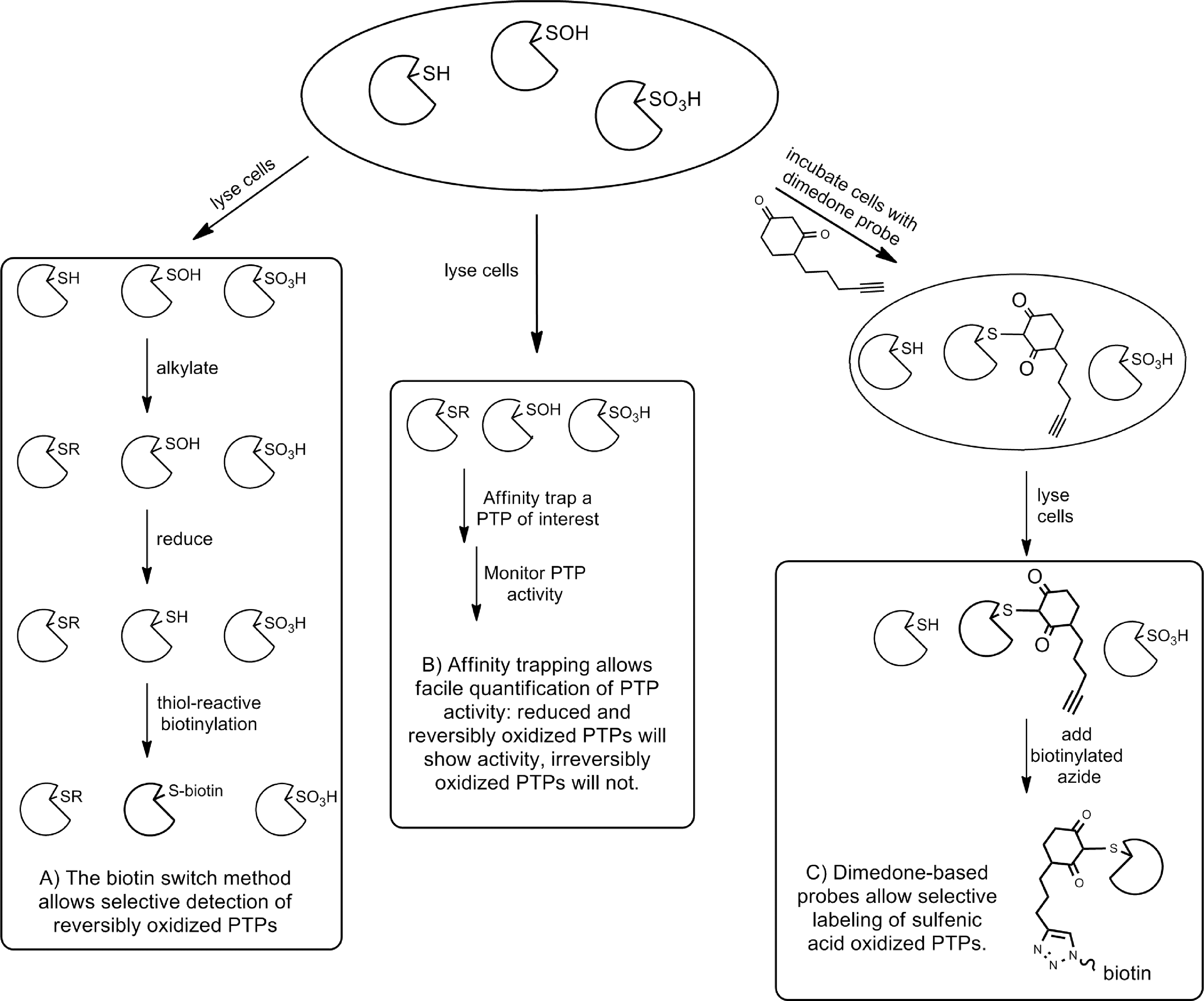
Biotin switch method
The most widely used and adapted approach is the biotin switch method (Fig. 8A) (50). Initially developed for use in identifying S-nitrosylated proteins (50), the approach was adapted also for use in identifying sulfenic acid-modified proteins (102). The technique is based on the concept that an oxidized thiol group in a protein will be protected against alkylation by a thiol-reactive alkylating agent such as IAA (16). Cells can be incubated with an oxidizing agent or stimulus (or left untreated, as desired). Cells are then lysed under anaerobic conditions and exposed to a thiol-reactive alkylating agent. Any PTP that was oxidized in the cell before lysis will maintain an oxidized catalytic Cys residue, and any free Cys residues should be alkylated. After removal of excess alkylating agent, the oxidized Cys residues in the lysate can be selectively reduced to the free thiol. Addition of a thiol-reactive biotinylation reagent results in biotinylation of any PTP that was reversibly oxidized and subsequent affinity-based purification and identification of the modified proteins can be carried out. This approach has been widely utilized and extensively modified, utilizing alternative alkylating agents (16), reducing agents (16, 71), and biomolecular tags (16). A variation of this approach helped to identify the hydrogen sulfide adduct of PTP1B (65). Although this approach has proven quite powerful in identifying oxidized PTPs, limitations in sensitivity and selectivity necessitate further validation of the results obtained.
Affinity trapping method
To monitor the activity of an individual PTP in the presence or absence of oxidizing agents, a method for affinity isolating a PTP from a cell lysate and directly quantifying enzymatic activity was developed (Fig. 8B). An individual PTP in a cell lysate was adhered to the wells of a 96-well plate via a specific antibody coated in the plate. The activity of the immobilized PTP was then measured by monitoring hydrolysis of a substrate, such as the fluorogenic small-molecule DiFMUP. Finally, the amount of PTP in each well was quantified using an ELISA assay. To determine the susceptibility of the intracellular PTP to irreversible oxidative inactivation, cells were incubated with varying amounts of H2O2 before lysis and the differences in enzyme activity between treated and untreated cells were quantified. This approach was initially applied to CD45, which was found to be quite susceptible to irreversible H2O2-mediated oxidative inactivation (98). Later, the approach was optimized to achieve the higher sensitivity necessary for monitoring PTPN22 activity in lysates (7). PTPN22 was susceptible to oxidation, but less sensitive to irreversible oxidation than CD45 (7). It should be noted, however, that it is difficult to separate direct oxidation of the PTP from upstream interference with a pathway that affects PTP activity. For example, if the activity of a PTP is modulated by phosphorylation, oxidative inhibition of the kinase responsible for this phosphorylation would also modulate PTP activity.
In-gel method
As mentioned in the In-Gel Methods section above, in-gel assays to monitor PTP activity and regulation on a global level have been employed (123). An important extension of this approach was developed to characterize PTPs that were reversibly oxidized intracellularly (83). Cells were pretreated with the desired stimulus (H2O2, cytokines, growth factors, etc.), lysed under anaerobic conditions in the presence of a Cys-reactive alkylating agent (such as IAA). Any PTPs whose catalytic Cys residues had been reversibly oxidized would be protected from alkylation, while the PTPs in the sample that were active would be irreversibly alkylated. The lysate was subjected to SDS-PAGE, where the reversibly oxidized PTPs were reduced and activated in the presence of DTT, while the irreversibly alkylated PTPs did not regain activity. A release of radioactive phosphate from the gel indicated the presence of an active PTP, hence, a PTP that had been reversibly oxidatively inactivated, intracellularly. This approach has been used successfully to monitor global PTP oxidation (82, 83). As described in the Affinity Trapping Method section, the results obtained from these experiments can be confounded by possible oxidative interference upstream of the PTP.
Antibody methods
The use of antibodies targeting oxidized PTPs has been employed to quantify the proportion of oxidized and reduced PTPs in a given cellular sample. First, an antibody selective for the hyperoxidized sulfonic acid state of the PTPs was created. Although the antibody was raised against hyperoxidized PTP1B, it is cross-reactive with most of the classical PTPs in a sulfonic acid state. To quantify the oxidized PTPs in a cell type, cells were subjected to a stimulus, resulting in reversible oxidation of some PTPs, irreversible oxidation of others, and no oxidation of the remaining PTPs. The cells were then lysed under anaerobic conditions and exposed to a thiol-reactive alkylating agent, resulting in irreversible alkylation of any active, nonoxidized PTP. Cell lysates were then treated with DTT, followed by pervanadate, to reduce and then oxidize to the sulfonic acid form, the reversibly oxidized PTPs. SDS-PAGE of the treated lysate followed by immunoblotting with the anti-sulfonic acid antibody provided a profile of the oxidized PTPs in the sample (58). By omitting the step in which active PTPs are alkylated, this approach also provides a profile of the entire PTPome in a given cell type (58). Coupling of the antibody enrichment and quantitative mass spectrometry techniques facilitates direct quantification of the PTPome and the PTPredoxome (58). The approach works well for almost all of the classical PTPs, in several different cell types.
The Tonks group pioneered a breakthrough oxidized intrabody approach, in which an antibody selective for a rationally designed PTP1B mutant that mimics the oxidized form of the enzyme was created (41). The resulting PTP1B-OX selective antibody was used to identify oxidized PTP1B as well as to stabilize the oxidized form of PTP1B, providing an innovative approach to inhibit the enzyme activity. As the described antibody is exquisitely selective for PTP1B, this method offers the currently rare circumstance of targeting/probing a specific PTP (41).
Dimedone probe method
Finally, chemical probes that react selectively with the oxidized forms of the PTPs have been designed (Fig. 8C). Such probes allow the direct detection of sulfenic acid species rather than the indirect detection of the biotin switch and sulfonic acid antibody methods. The basis for this approach is the selective alkylation of a sulfenic acid by dimedone (8). Dimedone-based probes can be modified to target PTPs by employing a module that directs binding to the active site (70), or can be quite generic and react with almost any sulfenic acid moiety (95). In any case, the addition of a biomolecular handle, such as an azide, greatly facilitates the identification of the labeled proteins. These probes have been used to identify PTEN, PTP1B, and SHP-2 as phosphatases that are oxidized upon stimulation of the EGFR (95). Interestingly, the best characterized oxidized form of each of these PTPs is not a sulfenic acid; PTP1B forms a sulfenylamide and PTEN and SHP-2 form disulfides, as discussed previously (95). It is not clear whether the sulfenic acid form or the sulfenylamide or disulfide form of these enzymes is the predominant form in vivo.
Innovation
Innovative approaches for studying the protein tyrosine phosphatase (PTP) family of enzymes in living cells have shaped the field. The tools of both chemistry and biology have been combined to provide insight into the biological roles and regulation of these critical signaling enzymes. Significant innovations in this area include approaches for profiling the substrate selectivity of the PTPs and trapping their biological substrates, synthetic substrates, and inhibitors that can be used in cells, and probes that can report on the oxidation state of the enzymes.
Conclusions and Perspectives
Recent innovative approaches to study PTPs have provided much progress in understanding their intracellular functions. Substrate trapping and substrate profiling have shed much light on the biological substrates of PTPs. Activity-based chemical probes have allowed identification of active PTPs in the cellular milieu, and a variety of chemical biology techniques have been devised to monitor intracellular oxidation of the PTPs. Whereas the field has advanced considerably far in the past three decades, much remains to be discovered. There are several critical questions that need to be addressed, including (i) how can we identify all of the biological substrates of each PTP, (ii) what is the chemical and biological basis for the spatial and temporal regulation of the PTPs, and (iii) how do the complex regulatory mechanisms of the PTPs and PTKs combine to form requisite cellular signaling networks? The further development of novel approaches to address these questions will greatly expand our understanding of this important family of enzymes. These approaches will need to be coupled with other cellular and in vivo approaches to fully appreciate the biological significance of each PTP-substrate interaction and to pave the way for the development of clinically successful PTP-targeted therapeutics.
Footnotes
Acknowledgments
This work was partially supported by NIH grants R21 GM079386 to A.M.B. and R01 DK080165 to N.B. and A.M.B. S.M.S. is supported by a postdoctoral fellowship from the Juvenile Diabetes Research Foundation. This is manuscript #1627 from the La Jolla Institute for Allergy and Immunology.