
Abstract
Introduction
C
Intracellular accumulation of ROS and RNS can be triggered by both exogenous and endogenous factors (e.g., irradiation, inflammation, or environmental toxins) (162) (Fig. 1). ROS are generated in the mitochondria by both complexes I and III within the electron transport chains as well as by NADPH oxidase (Nox) and additional enzymes, which are tightly associated with the mitochondria, including the monoamine oxidases (157). In the presence of mitochondrial superoxide dismutase (SOD), O2 − can be converted to H2O2, which can then diffuse out of the mitochondria into the cytoplasm. In the presence of high iron concentrations, H2O2 can form the highly reactive HO− through the Fenton reaction. Superoxide can also react with NO and form the highly reactive ONOO−, which is a potent trigger of oxidative protein and DNA damage, including DNA strand breakage and base modification (Fig. 2). Other sources of ROS and RNS include the endoplasmic reticulum and peroxisomes (61, 155).

As a general rule, the main function of redox molecules is intracellular signaling (32). In fact, they modulate a multitude of redox-sensitive signaling pathways, such as those downstream of growth-factor receptors, cytokine or interleukin (IL) receptors. Hydrogen peroxide is the predominant intracellular redox-signaling molecule, and this is due to the fact that it is small, highly ubiquitous, and can diffuse easily (142). The O-O linkage of H2O2 renders the molecule relatively stable compared with radical species, enabling H2O2 time to encounter and react with specific targets that it oxidizes at discrete sites. Well-characterized targets of ROS are the redox-sensitive catalytic cysteine residues of protein tyrosine phosphatases (PTPs), the oxidation of which reversibly abolishes enzymatic activity (24). Other oxidant-regulated signal pathways include tyrosine (e.g., Src kinase) and serine/threonine (e.g., extracellular signal-regulated kinase [ERK], c-jun-NH2-terminal kinase [JNK], p38) kinases and nuclear transcription factors (e.g., activating protein-1 [AP-1], nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB], p53, the nuclear factor of activated T cells, and hypoxia-inducible factor-1 [HIF-1]) (32, 172, 178).
A great number of physiological functions are controlled by redox-responsive signaling pathways such as oxidative burst, monitoring of oxygen tension, regulation of vascular tone and cell adhesion, and immune responses (38). All beneficial effects of ROS/RNS occur at low/moderate concentrations. Over-production of these molecules results in oxidative stress, a deleterious process that can damage cellular constituents, such as lipids, proteins, and DNA (172), and also interferes with cell function, contributing to the development and progression of a wide range of diseases, including autoimmune disorders (129, 133). Therefore, all cell types have a complex machinery of antioxidant compounds (vitamins C and E, glutathione [GSH], β-carotene and uric acid) and enzymes (SOD, catalase [CAT], glutathione peroxidase [GSHPx], glutathione transferase, glutaredoxin, peroxiredoxin and thioredoxin) that combat the over-production of ROS/RNS (20).
Notably, men and women have a different ability to maintain their redox status during aging (105). In addition, different susceptibility in terms of redox molecules formation and different propensity to undergo apoptosis were reported, with vascular cells from men being more susceptible to the effects of redox alteration than those from women (108).
Here, we briefly review the role of redox molecules in innate and adaptive immunity. Next, we focus on the relationship among ROS/RNS, cell fate (i.e., apoptosis and autophagy), and autoimmunity. In particular, representative autoimmune diseases characterized by inflammatory oxidative conditions are considered. We conclude by summarizing the current knowledge with regard to the role of antioxidant supplements in therapeutic clinical applications.
Redox Molecules and Innate Immunity
ROS have been shown to be produced by immune system-associated enzymes, the Nox family of enzymes, designed to combine NADPH and oxygen to actively generate O2 − (98). Phagocytes migrate to sites of tissue damage in response to chemotactic factors such as IL-8, complement component 5a, and produce ROS within the phagosomal membrane for efficient killing of invading pathogens (88). On the one hand, chemotactic factors can trigger Nox-dependent ROS production; on the other hand, bacteria can trigger ROS production directly through distinct types of receptors on leukocytes. For example, lipopolysaccharide (LPS) interaction with toll-like receptor 4 (TLR4) has been shown to facilitate the binding of TLR4 to Nox4 and, subsequently, the release of ROS (124).
The protection afforded by the phagocytes is crucial, but not without side effects. The production of highly permeable ROS (i.e., H2O2) causes leakage of these molecules from phagocytes and, therefore, unwanted effects on bystander cells. In an environment that is high in oxidative stress, these bystander reactions drive increased activation of the immune system, cell damage, and progression to disease. As stated earlier, ROS can act as signal transduction molecules. Hydrogen peroxide has been suggested to inactivate PTP (24), as well as to activate protein tyrosine kinases (28) and metalloproteases (121, 185) through the oxidation of critical cysteine residues. Phosphatases, such as Src homology region 2 domain-containing phosphatase-1 (SHP-1) (198), serve to decrease inflammation by inhibiting tyrosine kinase activity, although this type of regulation is lost on cysteine oxidation. Similarly, latent metalloproteases require oxidation for activation and, in the presence of HOCl and H2O2, the secretion of chemotactic mediators (L-selectin and proinflammatory tumor necrosis factor-α [TNF-α]) is highly increased (121, 185), thus enhancing inflammation. In addition, H2O2 has been demonstrated to activate NF-κB (116), which plays a major role in promoting inflammation by the induction of proinflammatory cytokines (e.g., IL-1β and TNF-α) (179). It should be noted that the physiologic role for oxidants in the activation of NF-κB has been questioned by studies demonstrating that redox activation is cell-type specific (17), and that Nox-induced ROS do not mediate NF-κB signaling, but lower the magnitude of its activation (60). These observations suggest that the redox regulation of NF-κB and perhaps of other transcription factors and proteins involved in inflammatory and immune responses is extremely complex, and there is strong evidence that this process is cell-type dependent and it is also influenced by ROS/RNS concentration and localization.
The effects of redox molecules on inflammation induction are schematized in Figure 3.

Redox Molecules Regulating Adaptive Immune Response
Phagocytic cells are also necessary for activation of the adaptive immune response. After antigen recognition by antigen-presenting cells (APC), an adaptive immune response is acquired in secondary lymphoid organs and innate-derived ROS play a role as enhancers of this response (88). T cells also produce ROS when activated through the T-cell receptor (TCR) (74). In particular, during early T-cell activation, the Nox dual oxidase 1 (DUOX1) generates H2O2 that transiently inactivates TCR-associated phosphatases and acts in a positive feedback loop to enhance and sustain further TCR signaling (96). Similarly, B-cell receptor (BCR) engagement triggers ROS production that potentiates signal transduction (182). Thus, ROS may have a profound regulatory effect on lymphocytes through the reversible oxidation and consequent inhibition of PTP which are known to modulate TCR and BCR signaling (141). Some of these PTP can contribute to autoimmunity either by enhancing lymphocyte activation or by disabling tolerance induction. In addition, T-cell-derived H2O2 activates NF-κB, which leads to the production of IL-2 and T-cell proliferation (104). On the other hand, TCR-stimulated production of H2O2 negatively feeds back and dampens antigen-induced ERK activation (95). There are numerous other targets of oxidation in T cells, such as the linker for activation of T cells (LAT), calcium channels, potassium channels, integrins, CD2, zeta-associated protein 70 (ZAP70), or lipids in the membranes that could be regulated and contribute to the downstream effects of ROS secreted by APC and those produced by T lymphocytes (66). From these findings, it can be summarized that ROS act as second messengers modulating lymphocyte effector function. However, ROS-mediated effects on lymphocytes are likely to be complex and highly context dependent. In vitro studies with selected cells and a high oxygen pressure could be different from in vivo results in which the tissue context and timing of operations also play an important role.
Role of Redox Regulation in Autoimmunity
Throughout the years, a remarkable number of theories have been proposed to explain how the immune system recognizes itself. On the basis of a large variety of epidemiological, clinical, and experimental evidence, it has been suggested that an unfortunate interplay of genetic susceptibility and environmental factors may play an important role in generating an abnormal immune response. Excessive oxidative stress is considered as playing an important role in the pathogenesis of autoimmune diseases by enhancing inflammation, modulating cell death, and breaking down the immunological tolerance (129). Several genetic/epigenetic studies, such as human disease susceptibility studies and redox gene knockout mouse studies, support the crucial role of redox imbalance in the pathogenesis of autoimmune disorders. In particular, single nucleotide polymorphisms and mutations in antioxidant genes may lead to the modulation of the cellular redox state, affecting lymphocyte activation and/or proliferation (85). Oxidative stress is one of the major events causing protein structural modifications, thus inducing the appearance of neo/cryptic epitopes that could trigger an autoimmune response against the original antigen through molecular mimicry (82, 93). In addition, lipid peroxidation, a process of oxidative degeneration of polyunsaturated fatty acids caused by ROS, leads to the formation of highly reactive aldehydes, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which can bind covalently to proteins and, thus, cause structural protein modifications, changing antigenic profile and enhancing the antigenicity of proteins (5, 56, 75). Next, we focus on the role of redox status in modulating cell fate in autoimmunity, taking into consideration two fundamental cellular pathways: apoptosis and autophagy. It is important to underline that redox molecules have also been reported to contribute to necrotic cell death (200). Morphologically, necrosis is quite different from apoptosis. During necrosis, cells first swell, then the plasma membrane collapses, and cells are rapidly lysed. Cytosolic constituents that pour into the intercellular space through the damaged plasma membrane provoke a strong inflammatory response which may play a crucial role in autoimmunity. However, the discussion of this topic is beyond the scope of our work and readers interested in this item could refer to recent reviews (85, 136, 180).
Oxidative stress and apoptosis in autoimmunity
Apoptosis is a form of cell death in which a programmed sequence of events leads to the elimination of cells without releasing harmful substances into the surrounding area. It plays a crucial role in developing and maintaining the health of the body by eliminating old, unnecessary, and unhealthy cells. Apoptosis has long been known to orchestrate the maintenance of peripheral lymphocyte homeostasis, avoiding unregulated clonal expansion of autoreactive immune cells (50, 90). Increasing evidence provide support that oxidative stress and apoptosis are closely linked physiological phenomena (81) and are implicated in the pathogenesis of some chronic diseases, including autoimmune disorders (129). In general, low levels of ROS/RNS may function as signals to promote cell proliferation and differentiation, whereas a severe increase of ROS/RNS can induce cell death (172). The role of redox molecules in apoptosis is summarized in Figure 4. Increased cellular levels of ROS promote mitochondrial dysfunction, triggering the intrinsic apoptotic pathway (27). During mitochondrial dysfunction, several essential players of apoptosis, including procaspases, cytochrome c, and apoptotic protease-activating factor-1 (APAF-1), are released into the cytosol (147). The multimeric complex formation of cytochrome c, APAF-1, and caspase 9 activates downstream caspases, leading to apoptotic cell death (81). In the extrinsic apoptotic pathway, ligand-death receptor engagement induces lipid raft formation, Nox recruitment/activation, and ROS generation that signal acid sphingomyelinase activation, ceramide production, and receptor clustering (39). These combined processes constitute lipid raft-derived signaling platforms that mediate death receptor activation and induction of apoptosis (196). T lymphocytes display a difference in their susceptibility to ROS-induced apoptosis depending on their differentiation stage, although contrasting results have been reported on this issue. Takahashi et al. (169) found that exposure to H2O2 induced apoptosis preferentially in memory T cells, in particular the CD8+ effector memory T-cell subset, as compared with naive T cells. The mitochondrial pathway appeared to be the primary cell death pathway for memory T cells exposed to H2O2. GSH is one of the antioxidants that protect cells from oxidative stress-induced apoptosis (194). According to the results by Takahashi et al. (169), a higher expression of the GSH molecule has been found in the naive T-cell subset as compared with the memory T-cell subset (46). Different results were reported by Gupta et al. (57), who observed that H2O2 induced apoptosis preferentially in naive T cells and central memory T cells as compared with effector memory T cells via a reduction in GSH levels. Overall, these data, although contrasting, show that distinct T-cell subsets have differential susceptibility to oxidative stress-induced apoptosis, and this susceptibility is associated with intracellular GSH. Further investigations considering the vast heterogeneity (e.g., naive, central memory, effector memory) that exists in T-cell lineage are needed to better understand the differential ROS susceptibility of T-cell subsets. Information generated by these studies could be important from a clinical point of view, as manipulation of the cellular redox state could be a strategy that is used to modulate T-cell subset distribution in a broad spectrum of disorders characterized by a skewed T-cell phenotype, including autoimmune diseases.
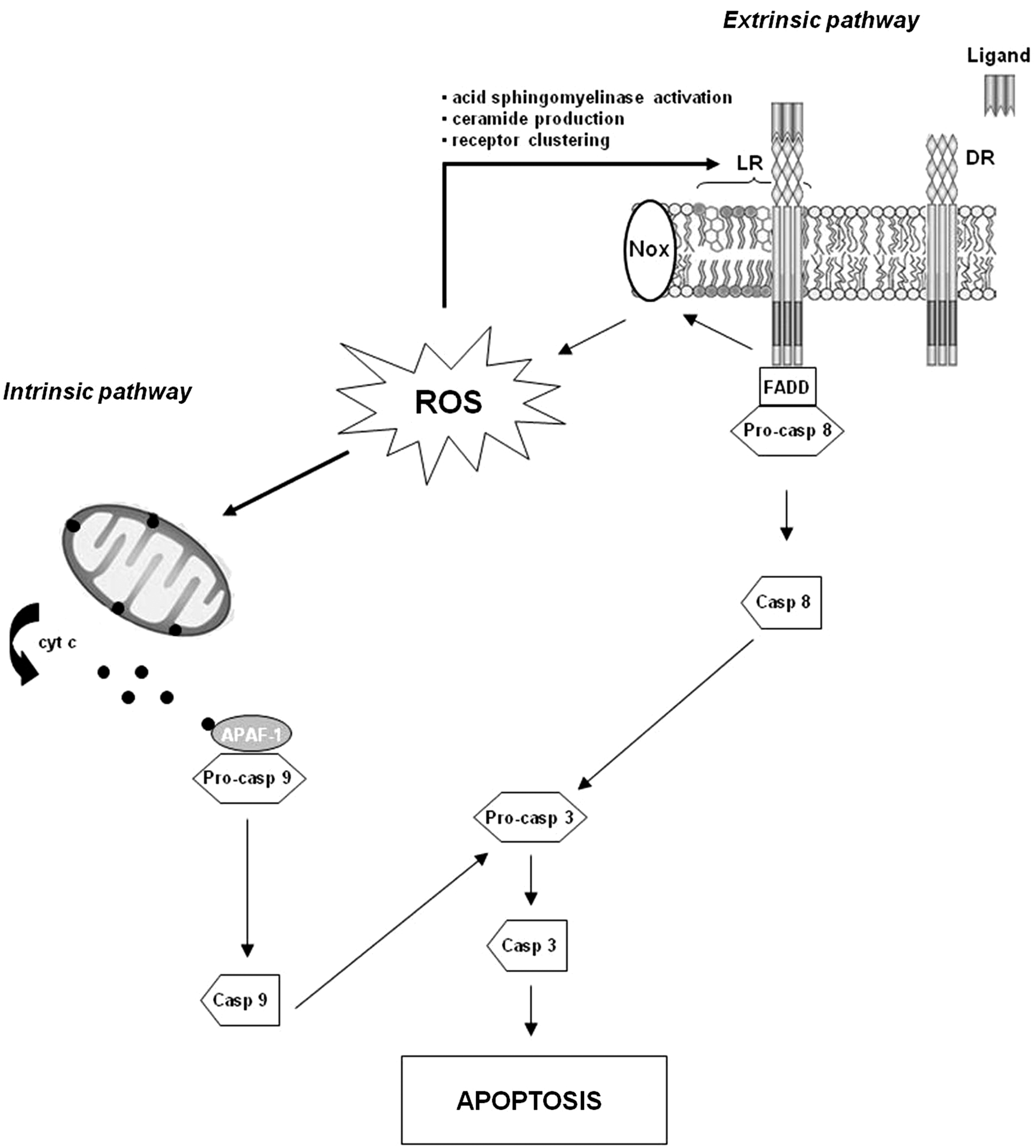
In addition to the stimulation of the apoptotic apparatus, ROS can directly affect the function of caspase proteins. The reduced state of cysteine at the active site is required for the catalytic activity of caspases, and, thus, its function is redox sensitive. Caspases can be activated or inhibited by the redox system, depending on the degree of the ROS stress (172). Thus, apoptotic and antiapoptotic mechanisms may be induced by redox molecules, and the biological consequences depend on the specific cell subtypes and organs involved in the process.
During apoptosis, modification of cellular antigens through proteolysis, changes in the phosphorylation state, and citrullination may give rise to potentially immunostimulatory forms of intracellular or membrane-associated autoantigens. Generally, the efficient clearance of apoptotic cells results in the exposure of intracellular self-antigens to the immune system under noninflammatory conditions, leading to tolerizing of these antigens. However, under a proinflammatory environment, these modified autoantigens, which may also expose cryptic epitopes, may be processed by APC and presented to either naive T cells, which have not been tolerized against the cryptic epitopes, or autoreactive CD4+ and CD8+ that escaped deletion due to defects in T-cell apoptosis (54, 139).
Oxidative stress and autophagy in autoimmunity
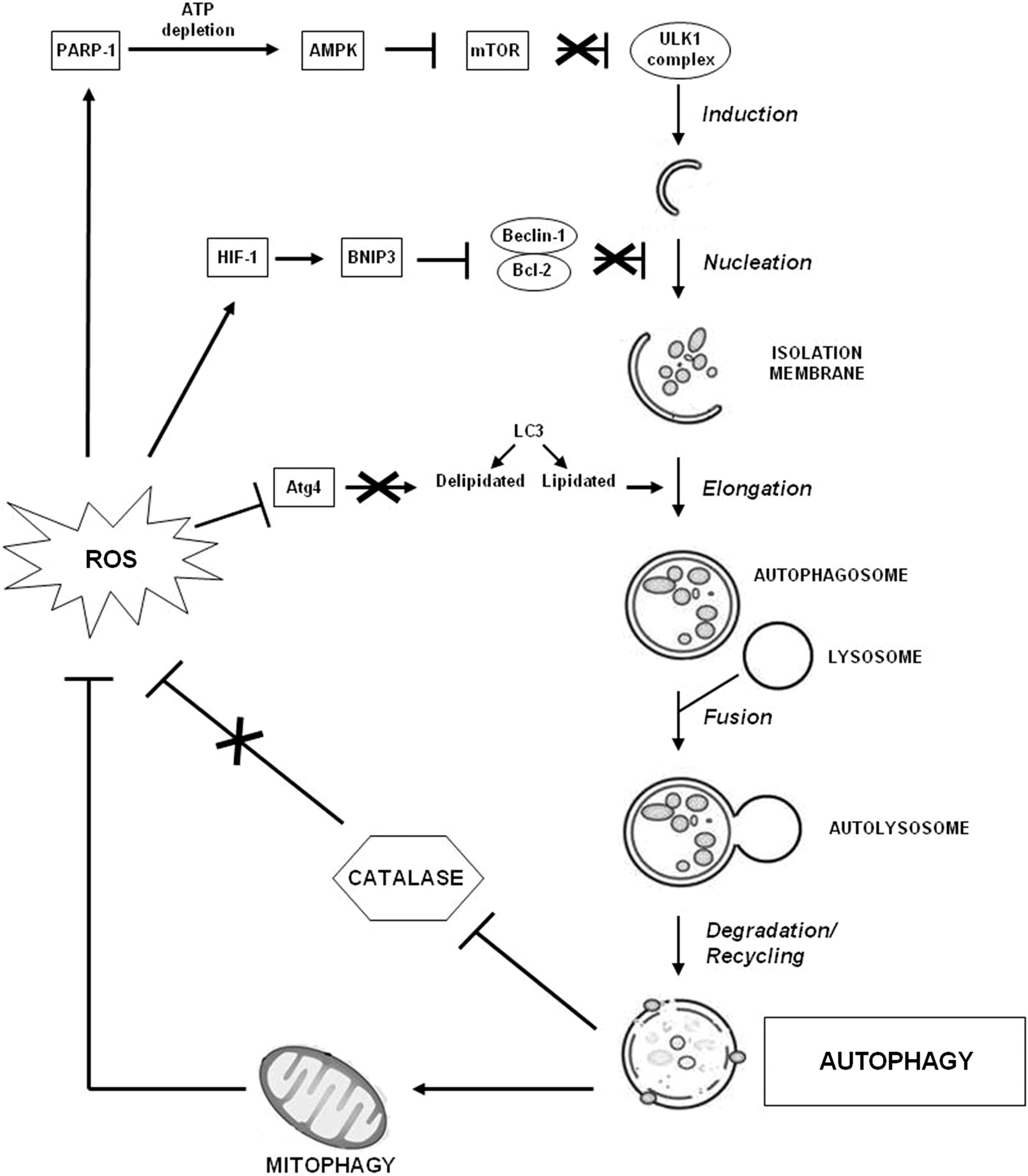
Macroautophagy (hereafter referred to as autophagy) is a highly conserved lysosome-mediated catabolic process that enables cells to degrade, in a regulated manner, unwanted cytoplasmic constituents, and to recycle nutrients (25, 191). It is morphologically characterized by the formation of double-membrane vesicles (autophagosomes) that sequester cytoplasmic components and deliver them to lysosomes for degradation and recycling. Autophagy is controlled by the autophagy-related genes (Atg) extensively reviewed elsewhere (13, 114). Briefly, the induction of autophagy is initiated by the activation of the UNC-51-like kinase 1 (ULK1, Atg1 in yeast) complex. Autophagosome formation requires phosphatidylinositol 3-phosphate (PI3P). The class III phosphatidylinositol 3-kinase (PI3K) hVps34 generates PI3P in a complex with multiple proteins, including Beclin-1/Atg6. Several regulating factors control the production of PI3P by the class III PI3K complex. Particularly, B cell lymphoma-2 (Bcl-2) protein family negatively controls autophagy and, in baseline conditions, Beclin-1 is inhibited by these molecules (62). Vesicle elongation and completion are mediated by two-ubiquitin-like systems: the microtubule-associated protein 1 light chain 3 (LC3, Atg8 in yeast) and Atg12-Atg5. LC3 precursor is proteolytically processed to form LC3-I, which is diffusely distributed in the cytosol. As the isolation membrane matures, the protein LC3-I becomes covalently lipidated into LC3-II and incorporated into the membrane as a crucial scaffolding protein (80). Constitutive, or basal, autophagy contributes to the physiological turnover of proteins and to the removal of old and/or damaged organelles, maintaining cellular homeostasis. As the principle site of ROS generation, mitochondria are particularly susceptible to oxidative damage. Damaged mitochondria generate even more ROS than fully functional organelles (175); thus, autophagy-dependent degradation of mitochondria (mitophagy) plays an important role in reducing oxidative stress. Mitophagy is controlled by a complex array of proteins that are constantly being revised and enhanced. Recent research points to two proteins as essential mediators of selective autophagic mitochondrial degradation: phosphatase and tensin homolog-induced putative kinase 1 (PINK1), a mitochondrially targeted serine/threonine kinase, and Parkin, a cytosolic E3 ubiquitin ligase. Recent studies demonstrated that PINK1 is stabilized in the mitochondria in response to lowered membrane potential, recruits Parkin, and induces mitophagy (78).
The basal level of autophagy increases in response to different extracellular and intracellular stresses, such as nutrient and growth factor deprivation, energy depletion, hypoxia, and bacterial and viral infections. The serine/threonine kinase mammalian target of rapamycin (mTOR) plays a major role in the regulation of autophagy (192). mTOR is the catalytic subunit of two macromolecular complexes, mTOR complex 1 (mTORC1) and mTORC2. In response to growth factors that stimulate the class I PI3K-Akt pathway, or other nutrient-related signals, mTORC1 negatively regulates ULK1 complex. Conversely, nutrient depletion or energy exhaustion inhibits mTORC1-inducing autophagy. In particular, a decrease in the normally high intracellular ATP:AMP ratio leads to the activation of monophosphate-activated protein kinase (AMPK), which facilitates the inhibition of mTOR by activating the mTOR repressor tuberous sclerosis complex (TSC)1/2 as well as by directly phosphorylating the regulatory-associated protein of mTOR (Raptor). Growing evidence supports the importance of autophagy in maintaining T-lymphocyte homeostasis (101, 183). Autophagy may be critically important in T cells for several reasons associated with their activation (183). For example, TCR stimulation leads to an oxidative burst that may produce massive levels of toxic ROS (64). In this context, autophagy may be used to limit the production of ROS, whose overload triggers apoptotic cell death (64).
Although autophagy is induced in the setting of oxidative stress and favors the removal of ROS, in some conditions, this process can contribute to abnormal ROS accumulation by selectively promoting the degradation of the major enzymatic ROS scavenger, CAT. The resulting accumulation of ROS in the cell leads to membrane peroxidation with loss of membrane integrity and cell death (195). Notably, many studies have identified ROS as an autophagy trigger (153). Treating cells with H2O2 or inhibiting SOD-mediated ROS detoxification induces autophagy favoring cell death (23). In addition, H2O2-dependent activation of poly(ADP-ribose) polymerase-1 (PARP-1) triggers autophagy by depleting the ATP level, which, in turn, leads, via AMPK, to mTOR inhibition (67, 117). ROS induce HIF-1-dependent expression of Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), which promotes the dissociation of Beclin-1 from its Bcl-2 inhibitors, thus promoting autophagy (197). Moreover, ROS can inhibit phosphatase and tensin homolog (PTEN), which activates autophagy via down-regulation of PI3K/Akt/mTOR signaling (100). ROS can also directly regulate autophagy proteins, such as Atg4 (152). When LC3 becomes lipidated, a deconjugation event occurs in order for the molecule to be properly recycled. This deconjugation is mediated by Atg4. Thus, ROS-mediated inhibition of Atg4 stimulates autophagy favoring LC3 lipidation.
In sum, there is a complex reciprocal relationship between autophagy and oxidative stress, which is essential to determine cell fate (Fig. 5). This is especially relevant in the context of autoimmune disorders in which autophagy has been suggested to play a crucial pathogenic role (15, 138, 199).
In conclusion, redox balance, apoptosis, and autophagy are three highly integrated networks that work together to maintain cellular homeostasis under basal conditions. Functional defects within any one of these networks results in compensatory responses from the other networks. These responses may contribute to the onset and/or progression of several pathologies such as autoimmune diseases. Next, we highlight key discoveries and controversies on how an imbalance within the redox status, apoptosis, and autophagy networks may underlay the pathogenesis of four paradigmatic autoimmune diseases: systemic lupus erythematosus (SLE), systemic sclerosis (SSc), rheumatoid arthritis (RA), and antiphospholipid (aPL) syndrome (APS).
Systemic lupus erythematosus
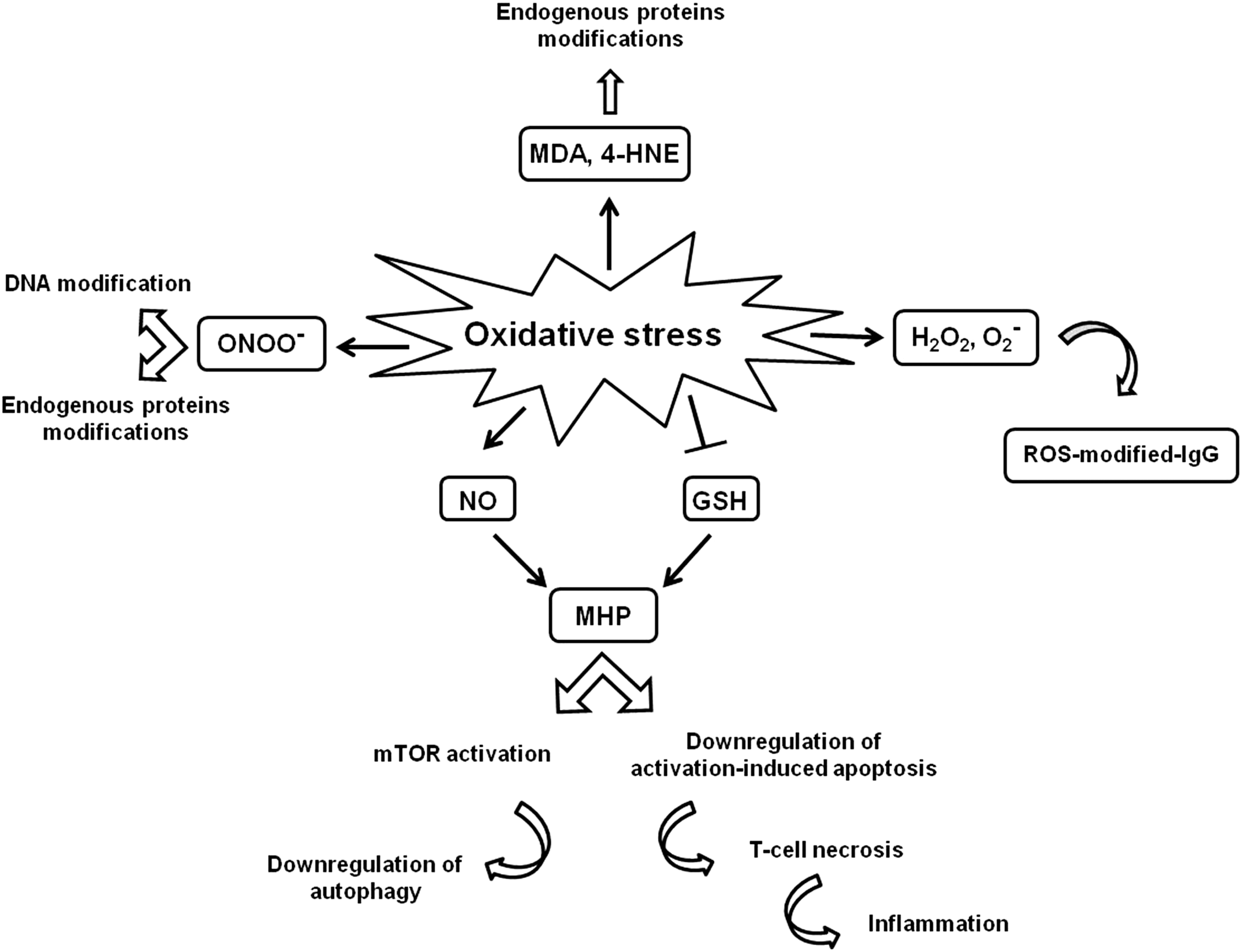
SLE is an autoimmune disease that is characterized by acute and chronic inflammation of various tissues, prominently the kidneys, skin, joints, or the central nervous system. SLE is characterized by autoantibody production by dysregulated B cells, target organ infiltration by inflammatory T cells, and aberrant immune cell activation due to abnormal APC function. The autoantigen-autoantibody interaction triggers the formation of immune complexes that, once deposited, cause tissue injury (140). An altered redox status has also been related to the pathogenesis of SLE (161). Polymorphism of nuclear factor-erythroid 2-related factor-2 (Nrf2), a key transcription factor that plays a central role in cellular defense against oxidative stress by basal and inducible expression of detoxifying and antioxidant genes, could confer a risk for developing kidney malfunction in SLE-affected individuals (29). Nrf2-deficient female mice develop lupus-like autoimmune nephritis and suggest that Nrf2 is one of the genes determining susceptibility to this autoimmune disease (193).
A significantly decreased activity of SOD, CAT, and GSHPx has been reported in SLE patients (161). Interestingly, ROS-modified IgG have been associated with the induction of circulating SLE autoantibodies where neoepitopes elicit an immune response in SLE patients (3). Moreover, the formation of MDA- and 4-HNE-modified proteins are associated with SLE (33, 184). Several studies, both in animal models and in humans, suggest that the overexpression of inducible NO synthase (iNOS) may contribute to pathologic processes of SLE by altering multiple signaling pathways in T cells and leading to glomerular and vascular manifestations (120, 125, 186). The potential role of NO in disease pathogenesis is due to the formation of the potent nitrating and oxidizing agent ONOO− (2). ONOO−-mediated modifications of endogenous proteins and DNA may enhance their immunogenicity, leading to the generation of neoepitopes and to the breakage of immune tolerance (58, 128). Moreover, the persistence of modified autoantigens could be favored by the defective clearance of apoptotic cells observed in SLE where abnormal death of T lymphocytes has been extensively documented (49). In particular, the enhanced spontaneous apoptosis of circulating T cells has been linked to chronic lymphopenia (41) and compartmentalized release of autoantigens (49), whereas the decreased T-cell death induced by activation has been suggested to contribute to the persistence of autoreactive cells (89). Abnormal T-cell activation and death are crucially dependent on the controlled production of ROS and ATP in mitochondria. Oxidative stress, in particular depletion of GSH and increased NO, has been suggested to induce persistent mitochondrial hyperpolarization (MHP) (135). SLE T cells exhibit persistent MHP as well as depletion of ATP and GSH, which decrease activation-induced apoptosis and instead predispose T cells for necrosis, thus stimulating inflammation (119, 136). This process is associated with increased mitochondrial mass, which may account for altered calcium handling, which is a central defect in the abnormal activation of SLE T cells (119). The activation of mTOR has recently emerged as a key sensor of MHP and a crucial factor in the abnormal activation of SLE T lymphocytes (45). Accordingly, a deregulation of autophagy has been described in T cells from lupus-prone mice and from patients with SLE (4, 55). In particular, T lymphocytes from SLE patients resulted in being resistant to autophagic induction (4). Interestingly, the blockade of mTOR with rapamycin improved disease activity in patients with SLE (44). The effects of redox molecules on SLE pathogenesis are summarized in Figure 6.
Systemic sclerosis
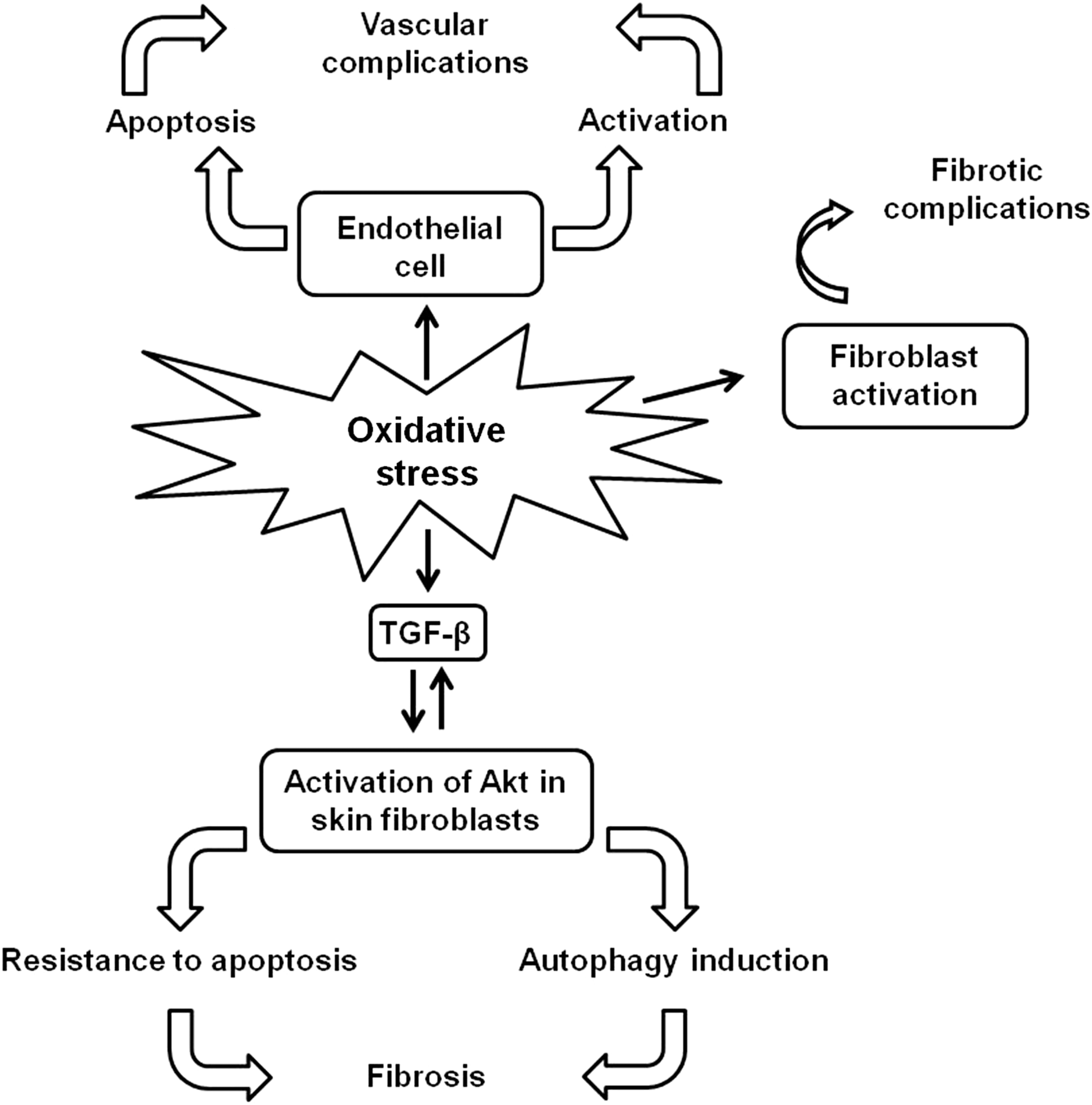
SSc is a rare disorder of the connective tissue that is characterized by fibrosis of the skin, blood vessels, skeletal muscles, and visceral organs. Additional manifestations include activation of the immune system with autoantibody production and vascular injury (47). The disease consists of multiple overlapping and poorly defined clinical subsets. It is typically divided into two major subsets based on the extent of affected skin: limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc). At present, the pathogenesis of SSc remains to be fully elucidated, but it is commonly believed that it is triggered in genetically susceptible individuals by exposure to yet undetermined environmental agents. Several studies, both in humans and in animal models of SSc, support the relationship between increased oxidative stress and the pathogenesis of this disease (48). In an epigenetic study, Selmi et al. (156) reported that the gene specific for ecto-NOX disulfide-thiol exchanger 2 (ENOX2), a membrane copper-containing enzyme that catalyzes electron transfer from hydroquinone or NAD to molecular oxygen, playing a role in the induction of ROS, is a hypermethylated gene in SSc, representing a potential prognostic marker of this disorder. An excessive oxidative stress in SSc was reported by Ogawa et al. (127), who observed increased levels of total antioxidant power in SSc patients, and this finding was interpreted as a global response to enhanced oxidative stress. Tikly et al. (170) showed increased levels of MDA in SSc patients, inversely related with disease duration. An in vitro study by Servettaz et al. (158) revealed that SSc sera induced the production of H2O2 and NO that, respectively, activated fibroblasts, and endothelial cells, leading to fibrotic or vascular complications. In another study, Servettaz et al. (159) showed that ONOO− induced skin fibrosis and serum anti-centromere protein autoantibodies, which characterize lcSSc; whereas hypochlorite or HO− radicals induced cutaneous and lung fibrosis and anti-DNA topoisomerase 1 autoantibodies, which characterize dcSSc. Furthermore, oxidative stress contributes to the release of mediators implicated in fibrosis such as transforming growth factor-β (TGF-β) (11, 14). On the other hand, TGF-β can (i) increase ROS production, (ii) suppress the expression of antioxidant enzymes (e.g., CAT, SOD and GSHPx), and (iii) decrease the intracellular concentration of GSH (48). On the whole, there is evidence of a positive interaction between TGF-β and ROS. The source of ROS in SSc is the membrane Nox system, which is stimulated in all cell types within or surrounding the vessel wall in response to injury (48). Monocytes and skin fibroblasts from SSc patients spontaneously produce large amounts of ROS (148, 149). Differently, SSc neutrophils, without stimulation, generate lower levels of ROS than controls; whereas in response to chemotactic stimuli, ROS generation is increased (12). Dysregulation of cell fate has been reported in SSc, contributing to disease pathogenesis, and oxidative stress could underlie this defect, although no direct evidence has been described yet. SSc fibroblasts are resistant to apoptosis, and TGF-β has been suggested to be responsible for this resistance (77). In fact, TGF-β can activate Akt, a kinase with potent antiapoptotic effects, and skin fibroblasts from SSc patients showed increased amounts of activated Akt as compared with control fibroblasts (79). Interestingly, activated Akt could also lead to a resistance of fibroblasts to autophagy, contributing to excess in the production of extracellular matrix by altering the turnover of proteins such as collagen (35). Increased apoptosis of endothelial cells has been detected in early inflammatory disease stages of SSc, acting as a primary pathogenic event underlying skin lesions (160), and ROS-generating triggering agents could be involved in this process (47). Up to date, contrasting data regarding a possible dysregulation of SSc lymphocyte apoptosis have been reported. Stummvoll et al. (167) showed normal levels of lymphocyte apoptosis whereas decreased apoptosis was observed by Cipriani et al. (26). In this latter study, lymphocyte resistance to apoptosis was supposed to be due to an increased NO generation, which was described in early-stage dcSSc patients (37, 168). In fact, NO is known to prevent apoptosis inhibiting caspase 3 activation by S-nitrosylation of the catalytic cysteine residue (26, 176). Differently, increased levels of CD8+ T lymphocyte apoptosis associated with low levels of NF-κB were found by Kessel et al. (86). To note, NF-κB is able to activate iNOS that generates NO, which, in turn, inhibits caspase activity (176). More recently, Giovannetti et al. (51) detected increased levels of both CD4+ and CD8+ T-lymphocyte apoptosis in SSc patients as compared with controls. In particular, patients with diffuse and active disease displayed higher values of apoptosis than those with limited and inactive disease. GSH content in lymphocytes was also examined, but no significant differences could be demonstrated between patients and controls. The effects of redox molecules on SSc pathogenesis are schematized in Figure 7.
Rheumatoid arthritis
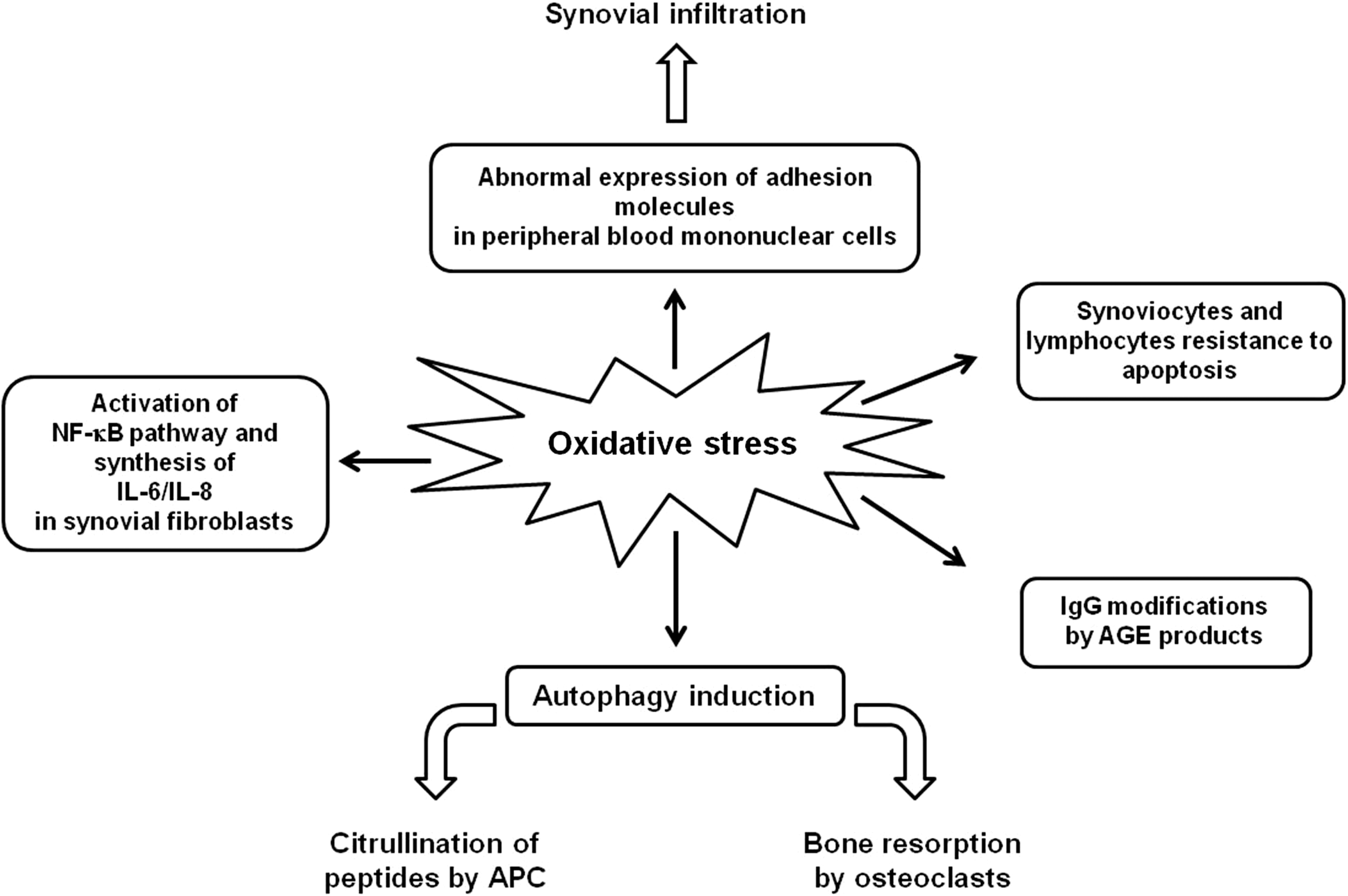
RA is an autoimmune disorder with both articular and systemic manifestations. This disease leads to synovial inflammation and destruction of the joint architecture. The synovial lining of the inflamed joints is invaded by T and B lymphocytes, neutrophils, monocytes/macrophages, and dendritic cells. Citrullinated peptides represent major targets of autoantibodies and autoreactive T lymphocytes. The exact pathogenesis is still unknown, and treatment is noncurative (52). Redox gene knockout mouse studies support a role for redox imbalance in the pathogenesis of RA. For instance, mice knocked out for NAD(P)H:quinone oxidoreductase 1 (NQO1) and dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2), cytosolic enzymes that catalyze metabolic reduction of quinones and derivatives, showed increased susceptibility to collagen-induced arthritis (72). In addition, several studies suggest that oxidative stress contributes to the joint tissue damage observed in patients with RA (6, 130, 137). Although several antioxidative functions are present in the synovial tissue of RA patients (i.e., metallothioneines, thioredoxin reductase, and GSH reductase), they do not fully counterbalance local oxidative stress (154). NO is increased in the synovial fluid of RA patients, and it mediates many different cell functions at the site of synovial inflammation, including cytokine production, signal transduction, mitochondrial functions, and apoptosis (102). In addition, thioredoxin, which is known to be induced in response to oxidative stress, is over-expressed in synovial cells and tissues of RA patients (109), co-stimulates the TNF-α-induced synthesis of IL-6 and IL-8 by synovial fibroblast-like cells, and activates the NF-κB pathway (154). A shift in the prooxidant/antioxidant balance in favor of lipid peroxidation has also been described in RA. In fact, elevated blood MDA levels and significantly lower levels of blood concentrations of total thiols, GSH, and vitamin C were found in patients with RA compared with controls (76). The basal levels of total ROS, O2 −, and HO− have been shown to be significantly raised in neutrophils sourced from both peripheral blood and synovial infiltrate (91). Since ROS generated in neutrophils correlated positively with the markers of disease activity, their evaluation has been suggested to be useful as an indirect measure of the degree of inflammation in RA patients (91). The infiltration and persistence of hematopoietic immune cells within the RA joint result in increased ROS/RNS species generation. On the other hand, the induction of redox-sensitive signaling pathways mediates abnormal expression of several adhesion molecules on lymphocytes and monocytes favoring their migration into RA synovium (181). This loop feeds a continuous self-perpetuating cycle of inflammation and destruction.
As in SLE, oxidative stress has been suggested to be the cause of IgG modification in RA in which advanced glycation end (AGE) IgG has been described (123). AGE results from nonenzymatic glycation of proteins and AGE-modified IgG have been shown to correlate with RA disease activity (94). Other potential contributing factors of RA are represented by circulating IgM anti-AGE-modified IgG (123).
Interestingly, in the context of RA, it has been suggested that cells of the immune system, exposed to increased levels of oxidative stress, become refractory to growth and death stimuli, thus contributing to the perpetuation of the immune response (53). In accordance with this hypothesis, there is accumulating evidence for impaired T- and B-lymphocyte apoptosis in the rheumatoid synovium contributing to joint inflammation (9, 171). In addition, apoptosis is decreased in RA synoviocytes and it is responsible for the massive synovial hyperplasia (131). Multiple mechanisms may contribute to this reduced apoptosis. In particular, Migita et al. (112) observed that NO inhibited caspase 3 activation in rheumatoid synovial cells. Apoptotic resistance in synovial tissues was confirmed by Xu et al. (189), who also demonstrated an inverse correlation between apoptotic and autophagic level in RA synovium. Interestingly, Lin et al. (103) provided evidence for a central role of autophagy in joint destruction. Autophagy has been demonstrated to be activated in RA in a TNF-α-dependent manner and to regulate osteoclast differentiation and bone resorption. Moreover, the role of autophagy in citrullination of self-antigen by APC has been reported to contribute to RA pathogenesis (71). The role of oxidative stress in autophagy induction has not yet been investigated, but it is conceivable. The effects of redox molecules on RA pathogenesis are schematized in Figure 8.
aPL syndrome
APS is a clinical disorder that is characterized by thrombosis and pregnancy morbidity associated with the persistent presence of aPL antibodies, including anti-β2 glycoprotein I (β2GPI), lupus anticoagulant, or both, and complement factors. In addition, aPL antibodies could enhance the risk of atherosclerosis. Procoagulant cell activation, accompanied with tissue factor (TF) expression and TF pathway up-regulation, are key events to be considered while explaining the pathophysiology of thrombosis in patients with APS (31, 69). The relocation of cardiolipin (CL) and its metabolites to the cell surface during apoptosis may represent an in vivo trigger for the generation of aPL antibodies (164). Various studies have evidenced that oxidative stress is directly involved in the pathophysiology of APS (7, 8, 177). In this context, anti-CL antibodies seem to play an important role by inducing NO and O2 − production, resulting in enhanced levels of plasma ONOO− (8). Titers of anti-CL antibodies have been found positively correlated to plasma levels of F2-isoprostanes, sensitive markers of in vivo lipid peroxidation, indicating enhanced oxidative stress (73). Moreover, the hydroperoxidation of CL has been shown to be essential for enhancing the binding of aPL antibodies to the lipids (177). β2GPI, an abundant plasma glycoprotein, is the most common target for aPL antibodies (19). Anti-β2GPI antibodies are associated with thrombotic events and are proatherogenic. These antibodies react with their target antigen in lipid rafts of the monocyte plasma membrane and trigger NF-κB activation, leading to a proinflammatory and procoagulant monocyte phenotype that is characterized by the release of TNF-α and TF, respectively (165). β2GPI can also stimulate a vigorous adaptive cellular immune response in APS patients. Oxidized β2GPI is able to induce phenotypic and functional maturation of dendritic cells, which represent the link between innate and adaptive immunity (18). In sum, the persistence of autoimmune reactions against this self-protein modified by oxidative events may contribute to local and systemic inflammation, thus sustaining endothelial dysfunction in patients with APS. Interestingly, ROS inhibition prevented the expression of an endothelial pro-adhesive phenotype, in response to aPL antibodies (110, 163). In addition, the low activity of the antioxidant enzyme paraoxonase contributes to the functional and structural arterial abnormalities observed in APS (22).
With regard to immune cells, Perez-Sanchez et al. (134) found a significant oxidative perturbation in APS leukocytes, directly related to an inflammatory and proatherothrombotic status. This perturbation has been showed to rely on the altered mitochondrial dynamics and metabolic processes, which, in turn, generate free radical species. All these events are strictly related to apoptosis induction, but the direct role of oxidative stress in modulating cell fate in APS has not yet been investigated.
Autoantibodies Play a Role in Redox Balance
As reported earlier, oxidative stress plays a key role in the induction of autoimmune responses. On the other hand, autoimmune reactions may play a role in altered redox balance. In fact, autoantibodies to antioxidant components, blocking their effects, could result in the disruption of redox balance, resulting in oxidative stress, which, in turn, leads to oxidatively modified autoantigens that serve as neo-antigens in promoting the loss of tolerance to self (126). Recently, autoantibodies specific to the C-terminus of Ral binding protein 1 (RLIP76), a protein that catalyzes the ATP-dependent transport of GSH conjugates including GSH-4-HNE, have been found in the serum of a significant percentage of patients with various diseases characterized by immune-mediated endothelial dysfunction, including SSc and SLE (107). Anti-RLIP76 antibodies lead to increased intracellular levels of 4-HNE and decreased levels of GSH, resulting in oxidative stress-mediated endothelial cell apoptosis. This evidence suggests that various autoimmune diseases related to endothelial dysfunction may arise through a common pathogenetic mechanism, possibly involving anti-RLIP76 antibodies. The female preponderance in almost all autoimmune diseases suggests that gender may be a risk factor in the development of these multi-factorial diseases (129). In general, women show higher antigen-presenting activity and mitogenic responses, and enhanced antibody production than men. The gender bias in autoimmunity has partially been explained by the role played by sex hormones, in particular estrogen, and by genetic factors (129). In this context, anti-RLIP76 antibodies are able to induce a significantly higher susceptibility to oxidative stress and to apoptosis in vascular cells from women in comparison with those from men (108). Estrogens, enhancing the cell surface expression of RLIP76, significantly increased the susceptibility of cells from women to the effects of anti-RLIP76 antibodies; whereas cells from men appeared unaffected (108). These results open a new perspective in the gender-dependent pathogenic mechanisms of autoimmune diseases that are characterized by vascular dysfunction.
A better understanding of the interactions among sex hormones, redox alterations, and immune function may provide new directions for the treatment of autoimmune diseases.
Antioxidant and Autoimmunity
The possibility of modulating cell behavior or of restoring cell homeostasis by changing the redox status is intrinsically appealing in nutrition and pharmacology. Several antioxidants have been investigated in vitro or in animal models to assess their potential therapeutic effect in conditions linked to oxidative stress. Here, we summarize results from recent studies on the therapeutic use of the main natural and non-natural antioxidants in autoimmunity.
Vitamin E is the most effective chain-breaking, peroxyl radical scavenger that protects biologic membranes against lipid peroxidation (143). In addition to its antioxidant properties, vitamin E supplementation has a variety of effects on the immune system response, including the enhancement of humoral and cellular immune reactions, subsequent regulation of cytokine balance, T-cell differentiation, proliferative responses of lymphocytes to mitogens, and reduction of anti-inflammatory reactions (99, 111). In a study aimed at determining the protective role of vitamin E, Tsai et al. (173) observed that this vitamin contributes to the integrity of cell membranes by preventing the loss of thiols. The authors suggest that this activity could probably have a role in the maintenance of intracellular calcium homeostasis. However, in SSc patients, 3 weeks of treatment with vitamin E at doses of 500 or 1000 mg/day neither decreases the basal rate of lipid peroxidation nor improves microvascular perfusion after cold exposure (30). Similarly, the results of five randomized clinical trials of vitamin E supplementation in patients with RA offered little convincing evidence of a positive effect (21). However, one of these studies suggested a superior effect of vitamin E over placebo on pain, but not on inflammatory parameters.
Dietary polyphenols are a primary source of antioxidants for humans and are derived from plants, including fruits, vegetables, spices, and herbs. Green tea polyphenols (GTP) are a group of polyphenolic compounds present in the leaves of Camellia sinensis (83). Research during the past 20 years has demonstrated that GTP are potent antioxidants which also possess antiapoptotic and anti-inflammatory activities. GTP rapidly scavenge ROS and inhibit prooxidant enzymes, such as NO synthase, lipoxygenase, cyclooxygenase (COX), and xanthine oxidase. In a mouse model of RA, oral administration of 0.2% GTP significantly reduced the protein levels of COX-2, interferon-γ, TNF-α, and IgG in the joints and was associated with a marked reduction of arthritis (59). Similarly, in a lupus mouse model, GTP feeding delayed the progression of lupus-like syndrome, including glomerulonephritis (151). In addition, epigallocatechin-3-gallate, the major bioactive polyphenol present in green tea, has been found to display antioxidant and free radical scavenging activities, to prevent lupus nephritis development through enhancing the Nrf2 antioxidant pathway, and to increase GSHPx activity (174).
Resveratrol is a polyphenolic compound found in various plants and fruits. Mechanistic studies demonstrate that resveratrol possesses anti-inflammatory, antioxidant, antiaging, and anticancer properties (122). It has been reported that resveratrol can inhibit several experimental autoimmune diseases, including diabetes, encephalomyelitis, and colitis (70, 113, 150). Although the detailed mechanisms are not fully understood, resveratrol may modulate the function of key cellular receptors, inflammatory genes or signaling transcription factors (e.g., NF-κB and signal transducer and activator of transcription [STAT] 3), and COX-2 (1). Particularly, as COX-2 inhibitor, resveratrol protects in vitro chondrocytes against oxidant injury and apoptosis (34) and ameliorates the onset of LPS-induced acute joint inflammation in rabbits (40). In a recent work, Xuzhu et al. (190) showed that prophylactic or therapeutic administration of resveratrol attenuated clinical parameters and bone erosion in collagen-induced arthritis mice. The arthritis-protective effects were associated with markedly reduced serum levels of proinflammatory cytokines and collagen-specific IgG, and with reduced IL-17 production in draining lymph nodes.
Curcumin is a natural plant polyphenol with a long tradition in folk medicine. The intricate mechanism of the action of curcumin involves various biological targets affecting the innate and adaptive arms of immunity, especially in the pathological conditions (166). Curcumin effectively modulates the function of T cells, B cells, dendritic cells, monocytes, macrophages, and neutrophils. In particular, in vitro and in vivo experiments showed that curcumin inhibited the proliferation of T cells and the production of anti-type II collagen antibodies (115). Overwhelming reports have supported the anti-inflammatory action of curcumin and its potential role in the therapy of numerous immune-mediated diseases (166).
Quercetin, which is a typical member in the flavonol subclass, is one of the most common flavonoids in the diet. Huang et al. (68) demonstrated that quercetin can suppress dendritic cell activation and, thus, may potentially be used for the prevention and treatment of inflammation, autoimmunity, and transplantation. Moreover, quercetin can induce apoptosis in RA fibroblast-like synoviocytes through mitochondrial pathway (188). Evidence for the possible clinical applications of quercetin is increasing. For instance, quercetin ameliorates experimental autoimmune encephalomyelitis by blocking IL-12 signaling and T helper 1 differentiation (118), and it improves the progression of arthritis in rat adjuvant-induced arthritis (106).
Taurine, a semi-essential amino acid naturally present in seafood and meat, is an antioxidant. It has been demonstrated that taurine inhibits the production of IL-1, IL-6, and TGF-β. For this reason, it has been proposed as a potential novel addition to the therapeutic anti-SSc weaponry (43).
Overall, despite observations in in vitro and in vivo models suggesting that the correction of antioxidant depletion by dietary means could be a viable therapeutic approach for autoimmune disorders, up to date there is little clinical evidence supporting the efficacy of antioxidant supplementation in the prevention and/or treatment of these disorders. Importantly, as many antioxidant systems interact, it remains unclear whether the supplementation with a single antioxidant can restore the activity of depleted antioxidant systems. A further level of complexity relates to the fact that ROS not only damage tissue but are also essential controllers of physiological signaling mechanisms. Inhibition of these cell-signaling roles of ROS could account for the potential adverse effects of antioxidant supplementation, such as the increased mortality revealed by review and meta-analysis of randomized trials of antioxidant supplements for primary and secondary prevention (16). Therefore, further appropriately powered studies are needed to reliably assess the effects of natural antioxidant supplements for diseases prevention and treatment.
Among the non-natural antioxidants, reduced thiols are the first reasonable candidates to accomplish the function of changing the cellular redox status and N-acetylcysteine (NAC) is the most promising of them all. NAC, the acetylated form of L-cysteine, can effectively raise intracellular GSH levels in lymphocytes (10, 63). Compared with other forms of cysteine supplementation, it has the advantages of resistance to oxidation and permeability through the cell membrane (187). In a European study of idiopathic pulmonary fibrosis patients, oral NAC (1.8 g/day) diminished disease severity and reduced the toxicity of pro-oxidant and immunosuppressant drugs (36). A recent randomized double-blinded placebo controlled study has been carried out to evaluate the metabolic, immunologic, and therapeutic impact of NAC (2.4 g/day) in SLE patients (97). NAC significantly reduced the disease activity, and this effect was associated with a markedly reduced mTOR activity in T lymphocytes. Moreover, a decreased titer of anti-DNA autoantibodies was observed. This study, although limited by the reduced number of patients investigated and by the short duration of treatment, strongly suggests a therapeutic importance of NAC for SLE based on its selectivity for mTOR modulation in T cells. Therefore, NAC may provide a safe, inexpensive, and alternative mechanism-driven as well as a potentially synergistic approach to block autoimmune reactions in SLE. A protective effect of NAC was also observed in SSc. Failli et al. (42) showed that lung macrophages obtained from SSc patients expressed a higher level of iNOS compared with cells from healthy subjects. NAC preincubation was unable to modify the release of O2 −, but it significantly reduced ONOO− production. A retrospective study involving SSc patients under long-term therapy with intravenous NAC showed that this drug was able to induce a significant improvement of pulmonary function, particularly evident in patients without radiological signs of pulmonary fibrosis (146). Moreover, a prospective observational study of SSc patients, who received NAC infusional therapy, revealed a reduction of digital ulcers and a decrease of the Raynaud's phenomenon number attacks. NAC infusion was generally well tolerated, and no patients had to discontinue the treatment (144). An amelioration of vascular renal function was also found by Rosato et al. (145) in an open-label study involving SSc patients treated with NAC intravenous infusion.
Notably, biological drugs largely used in the therapy of autoimmune disorders block in different ways and at different levels proinflammatory cytokines, also acting by modulating ROS (132) or hypoxia-induced angiogenesis (87). In particular, it has been reported that drugs which block TNF-α (infliximab, etanercept) or IL-6 (tocilizumab) reduce the oxidative stress marker levels in patients with RA, ankylosing spondylitis, and Crohn's disease (65, 84, 92). Therefore, the efficacy of these drugs in autoimmune diseases should be revaluated considering their potential antioxidant activity.
Conclusions
In the regulation of cell survival, redox molecules are a double-edge sword. ROS/RNS at low levels act as signaling molecules that promote cell proliferation and survival. However, when the increase of these molecules reaches a certain level, it may overwhelm the cellular antioxidant capacity inducing cell death by triggering not only stress-response signal-transduction pathways but also through direct oxidative modifications of the execution molecules. Studies aimed at clarifying the complex relationship between oxidative stress and both apoptosis and autophagy and its contribution to the onset and progression of autoimmune diseases are only at the beginning. A better understanding of this issue might provide new insights into the pathogenesis of autoimmune diseases and might aid in the design of novel molecular-targeted therapy.
Footnotes
Acknowledgment
This study was supported by a grant from the Italian Ministry of Health to EO (U7A).