
Abstract
Shaping the Epigenome
E
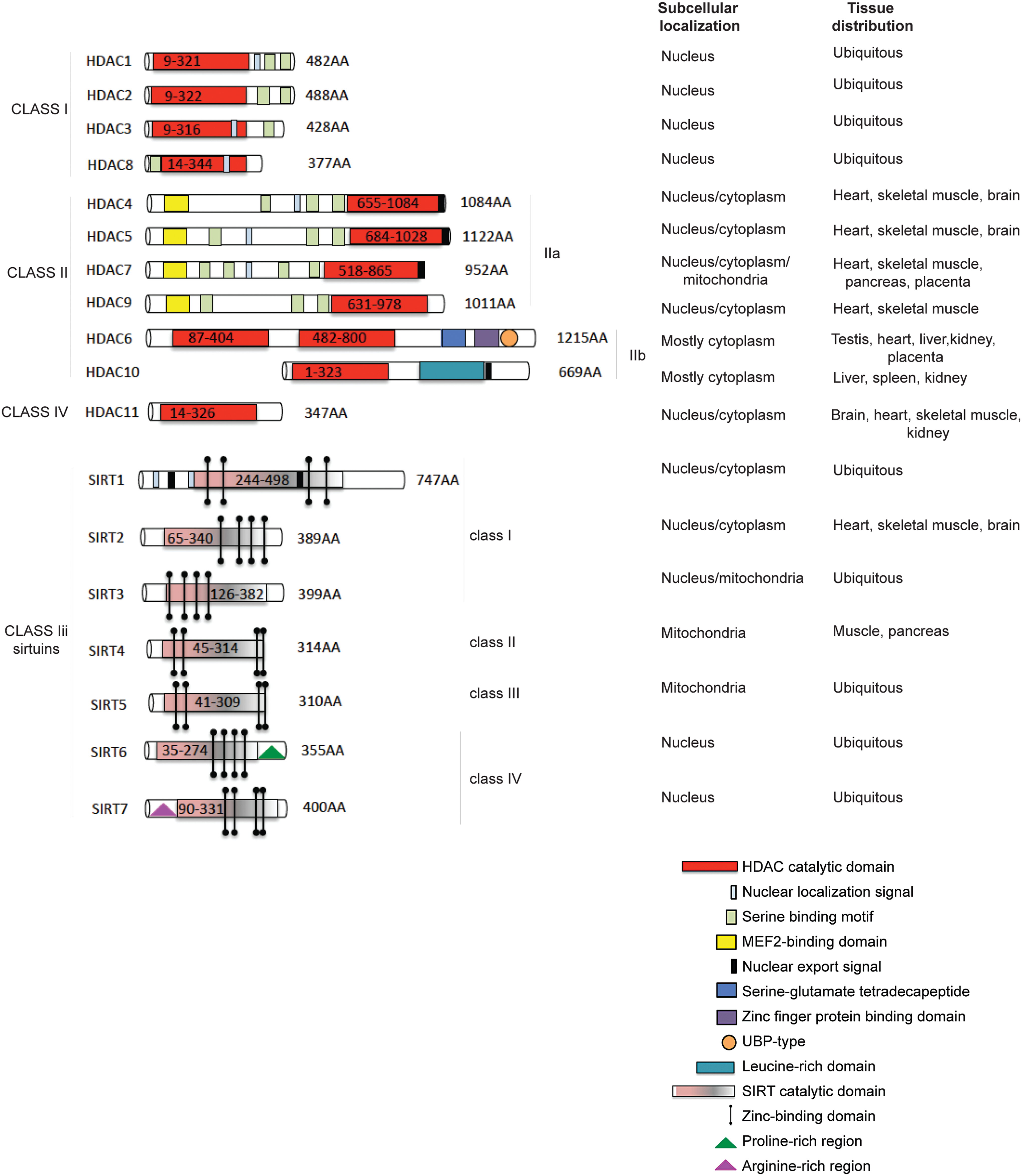
HDAC Classification
HDACs catalyse the removal of acetyl groups from the amino-terminal lysine residues of histone and non-histone proteins, such as transcription factors (TFs), hormone receptors, signaling proteins, chaperones, and DNA damage response proteins (10, 47, 68, 78). Four classes of human HDACs have been defined, based on their homology to yeast HDACs (13, 108, 228), enzymatic activities, and cellular localization (Fig. 1). HDAC1, 2, 3, and 8 belong to class I and are localized in the nucleus, presenting similarity to the yeast Rpd3 (47). Class II HDACs (229) are present in both the cytoplasm and nucleus, and they shuttle between these compartments (113, 147, 234). Class II HDACs (grouped for homology to Hda1 in yeast) are divided into two subclasses: class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6, 10). Class IV HDACs includes one member (HDAC11) (49, 71), which can be considered a “hybrid” sharing similarities to both class I and II HDACs. Sirtuins, related to Sir2 in yeast (42), have also been categorized as class III HDACs (165) (Fig. 1, lower part) and grouped as SIRT1-7 (209). These enzymes display an NAD function that is associated to a nuclear, mitochondrial, and cytoplasmic localization (31, 149). HDAC1 and 2 are highly similar with an overall identity of ∼82%. The catalytic domain on the N-terminus constitutes the main part of the proteins. Moreover, HDAC1 and 2 only act in complex with proteins required for modulating their deacetylase activity and binding to DNA (87), mediating the recruitment of HDACs to promoters (163, 190). Three protein complexes containing both HDAC1 and 2 have been characterized: Sin3, NuRD, and Co-REST (251). Both Sin3 and NuRD complexes consist of a core containing HDAC1 and 2, Rb-associated protein 48 and RbAp46 (123, 258). The core alone does not possess maximal HDAC activity, and further cofactors are needed. In addition to functioning through these complexes, HDAC1 and 2 can also bind directly to DNA-binding proteins such as Yin and Yang 1 (YY1) (249), Rb-binding protein-1 and Sp1 (51). Both activity and complex formation are regulated by phosphorylation. HDAC1 and 2 are phosphorylated (189) at a low steady-state level in resting cells, and their hyper-phosphorylation leads to a slight, but significant, increase in deacetylase activity, and, simultaneously, to disruption of the complex between HDAC1/2 and HDAC1/mSin3/YY1 (67). When hypo-phosphorylation occurs, the activity of HDAC1 and 2 decreases, although complex formation is increased (67, 173). HDAC3 (56) is closely related to HDAC8, with 34% identity, displaying the canonical domain of all class I HDACs. HDAC3 can co-precipitate with HDAC4, 5, and 7 through complex formation with SMRT and N-CoR (86). HDAC8 (69, 224, 225) consists largely of the catalytic domain with a nuclear localization signal. HDAC4, 5, and 7 represent a subgroup within class II HDACs (143). HDAC4 and 5 share an overall similarity of 70%. HDAC6 is evolutionarily most related to HDAC10. In general, however, the identity of HDAC6 with other human HDACs is low, with some resemblance to yeast HDA1, suggesting an early separation from other HDACs. HDAC6 (228) is a unique enzyme (23), in that it contains two catalytic domains in tandem, and an HDAC6-, USP3-, and Brap2-related zinc finger motif (HUB) domain at the C-terminus (264). This domain is a signal for ubiquitination, implying an involvement in degradation pathways. The catalytic domains of HDAC6 are most similar to those of HDAC9. HDAC6 and the enzyme functions as a tubulin deacetylase (98, 144, 257). Although it resides predominantly in the cytoplasm (having a nuclear export signal [NES]) to exert its function (227), HDAC6 can also be found in the nucleus along with HDAC11. HDAC9 (and its splice variants) belongs to class IIa and has a catalytic domain on the C-terminus (172, 263). HDAC10 is the most recently discovered class II HDACs (61). HDAC10 (112) is closely related (37% overall similarity) to HDAC6. HDAC10 has a catalytic domain on its N-terminus, as well as both an NES and a putative second catalytic domain on the C-terminus. HDAC10 might function as a recruiter rather than as a deacetylase (85), interacting with HDAC1, 2, 3, 4, 5, and 7, but not with HDAC6, with some contradictions present in literature. HDAC11, containing a catalytic domain at the N-terminus, is similar to HDAC3 and 8, suggesting that it might be more closely related to class I than to class II HDACs (71). In mammals, seven Sir2 homologues (SIRT1-7) have been identified, which possess primarily HDAC (SIRT1, 2, 3, and 5) or mono-ribosyltransferase activity (SIRT4 and 6) (64), targeting histone and various non-histone proteins in distinct subcellular localizations. SIRT1 has a wide range of substrates and cellular functions (171). In contrast to SIRT1, SIRT2 is predominantly cytoplasmic, deacetylating several substrates such as α-tubulin (20). For a detailed classification of SIRT3-7 (31).
HDACs and Sirtuins in Cancer: Quantitative and Functional Abnormalities
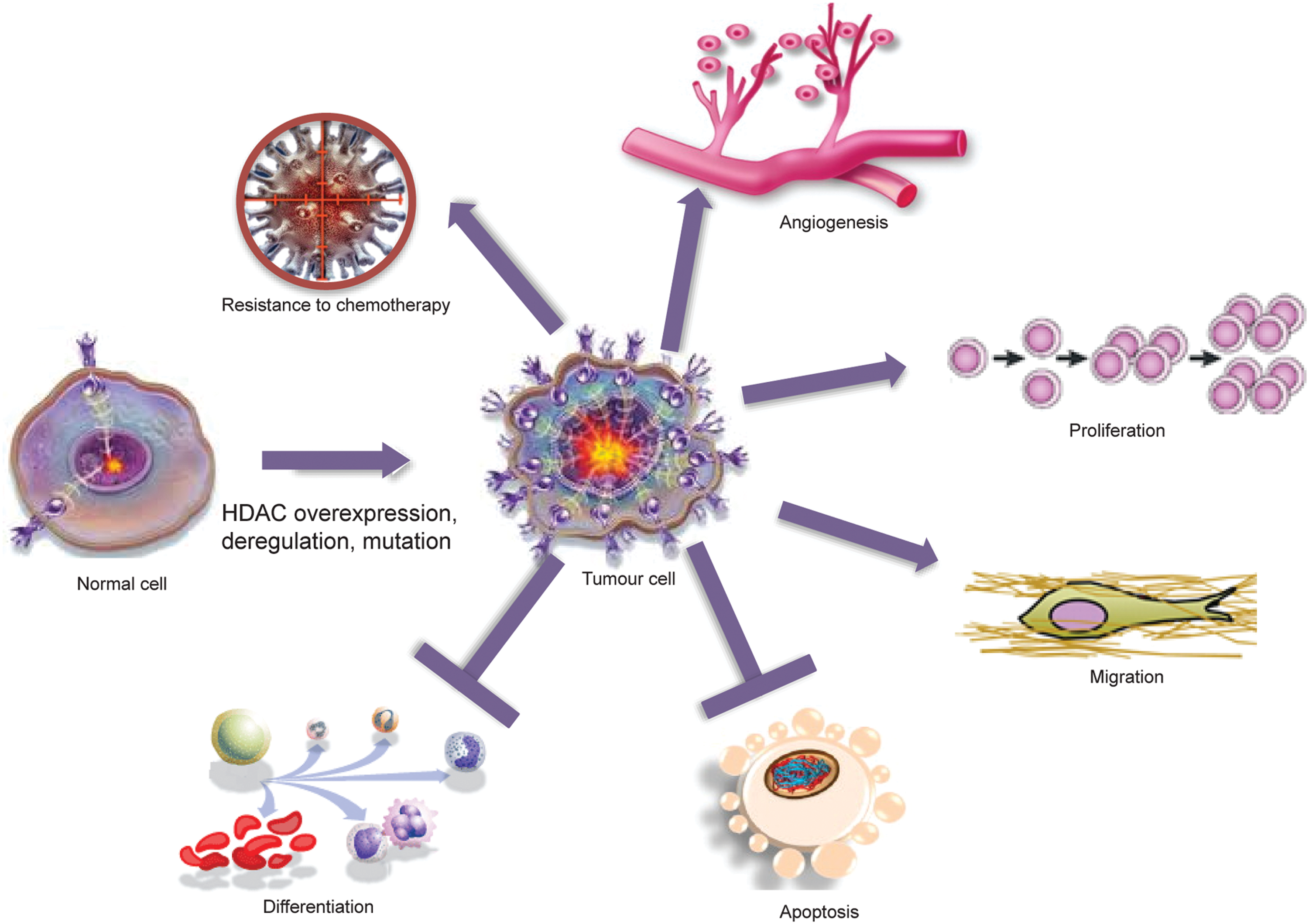
Cancer has traditionally been considered a disease of genetic defects such as gene mutations and deletions as well as chromosomal abnormalities, resulting in the loss of function of tumor-suppressor genes and/or gain of function or hyper-activation of oncogenes (121, 218). Nevertheless, the epigenetic regulation of genes and, therefore, the plasticity of the epigenome play a key role in cancer development and progression (93, 214). HDACs can act directly on chromatin, modifying histone acetylation and changing the characteristics of transcriptional factor recruitment on promoters. Consequently, HDAC deregulation frequently leads to aberrant expression of genes regulating cell proliferation, cell cycle, and apoptosis. Moreover, HDACs can promote the acetylation of structural proteins, chaperones, and TFs with a significant impact on physio-pathological pathways. Current knowledge of HDAC involvement in cancer is summarized next according to HDAC family groups.
Class I HDACs
All bonafide class I HDACs are deregulated in cancer (156). HDAC1 overexpression has been found in gastric (35, 155), breast (115), pancreatic (70, 262), hepatocellular (58, 244), lung (186), and prostate (200) malignancies, and overexpression levels correlate with prognosis and survival. High expression levels of HDAC1, 2, and 3 are associated with renal cancer (63) and Hodgkin's lymphoma (1). Interestingly, HDAC2 frame-shift mutation is the only genetic mutation identified in HDAC genes, leading to loss of HDAC2 protein and activity in human microsatellite instability (MSI)-high endometrial and MSI colon cancer cells. Unlike other class I members, HDAC8 expression appears to be cancer-type specific, as to date it has been found altered in childhood neuroblastoma (166, 167) and pancreatic ductal adenocarcinoma (48).
Class IIa HDACs
HDAC4 exerts an atypical role in cancer being overexpressed (e.g., in breast cancers) and/or down-regulated (e.g., in chondrosarcoma (204) and melanoma (252)) differently in different malignancies HDAC5 and 9 may be considered markers for medulloblastoma risk stratification (150). Their overexpression correlates with poor survival in affected patients. HDAC5 is also overexpressed in hepatocellular carcinoma (125), while HDAC7 overexpression is associated with pancreatic cancer and acute lymphoblastic leukemia (152, 169).
Class IIb HDACs
HDAC6 is implicated in breast cancer (184), diffuse large B-cell lymphomas, and peripheral T-cell lymphomas (142). Increased HDAC6 expression has been observed in more advanced—compared with early-stage cancers (239). HDAC10 expression is stronger in patients with chronic lymphocytic leukemia, and it has recently been linked to gastric cancers and reactive oxygen species (ROS) production in many cancers (130, 233).
Class III (sirtuins)
SIRT1, 4, and 7 are up-regulated in certain cancers (myeloid leukemia, prostate and non-melanoma skin cancer) (20), whereas SIRT2 is down-regulated in gliomas (94) and gastric carcinoma, as well as in melanomas (118). In melanomas, a mutation in the catalytic domain of SIRT2 (P199L) eliminates its enzymatic activity (131). Evidence suggests that SIRT2 acts as tumor suppressor and that its loss compromises the mitotic checkpoint, contributing to genomic instability and tumorigenesis. SIRT3 is, instead, up-regulated or down-regulated in different types of breast cancer (254). The quantity of the enzyme may be relevant in tumorigenesis for both loss and gain. Some studies also suggest a role for SIRT7 in breast cancer maintenance (8).
Class IV HDACs
Overexpression of HDAC11 has been found in myeloproliferative disorders as well as in Hodgkin's lymphoma (29, 198).
To date, important studies (179) have focused on aberrant HDAC recruitment to promoters through their physical association with oncogenic DNA-binding fusion proteins resulting from chromosomal translocations, or overexpression of repressive TFs that physically interact with HDACs. PML-RARα and AML1-ETO fusion proteins induce acute promyelocytic leukemia and acute myeloid leukemia (AML) by recruiting HDAC-containing repressor complexes to stably repress the expression of target genes (HDAC1 and 2) (73). HDAC6 plays a key role, by physically interacting with non-histone proteins such as HSP90, β-catenin, and epidermal growth factor (in Wnt signaling pathway) (134, 248). Altered levels of acetylation of these targets can disrupt the balance of c-Myc activation, enabling cells to escape from apoptosis. The same is true for p53 and its level of acetylation, which can induce its activity as TF. Taken together, these findings support the development of HDACi to restore the physiological pattern of gene expression and function in tumors and, possibly, other diseases.
HDAC and SIRT Modulators
HDACi represent a class of cytostatic agents that interfere with the function of HDACs and are able to modulate gene expression through indirect induction of histone acetylation (14). In particular, these compounds inhibit the proliferation of tumor cells in vivo and in vitro by inducing cell-cycle arrest, differentiation, and/or apoptosis with different kinetics and activities depending on chemical structures. Surprisingly, normal cells are often less sensitive to HDACi than are tumor cells (87). HDACi derive from natural or synthetic sources and can be classified into five main groups (16): (i) Hydroxamates, including trichostatin A (TSA), suberoylanilide hydroxamic acid (also called Vorinostat), LAQ824, LBH589 (Panabinostat), or PXD101 (Belinostat), M344, CR2408, abexinostat hydrochloride (PCI-24781) (ii) aliphatic acids, including sodium butyrate (NaB), valproic acid (VPA), and phenylbutyric acid (iii) benzamides, including MS-275 (Entinostat) (iv) tetrapeptides/depsipeptides, including Apicidin, Romidepsin, and Trapoxin B (v) sirtuin inhibitors (SIRTi), including the pan-inhibitor nicotinamide and the specific SIRT1 and 2 inhibitors sirtinol, cambinol, and EX-527.
TSA inhibits HDAC1, 4, and 6 with IC50=6, 38, and 8.6 nM, respectively [for further information, the reader is referred to ref. (39)]. The pan-inhibitor Vorinostat inhibits (177) HDAC1 and 3 with IC50=10 and 20 nM, respectively. Vorinostat also induces marked hyperacetylation of histone H4 and inhibits growth of three prostate cancer cell lines, LNCaP, PC-3, and TSU-Pr1, at 2.5–7.0 μM concentrations. Vorinostat treatment in MCF-7 breast cancer cells inhibits cell proliferation at IC50=0.75 μM, resulting in the accumulation of cells in G1 and G2-M phase. Vorinostat treatment at 1 μM for 8 h or more is sufficient to irreversibly induce the apoptosis of human multiple myeloma (MM) cells. LAQ824 (6, 80) activates the expression of p21 cell-cycle inhibitor by activating the p21 promoter with AC50=0.3 μM. LAQ824 inhibits cell growth of H1299 and HCT116 with IC50=0.15 and 0.01 μM, respectively. The antiproliferative effect of LAQ824 is selective toward tumor cell lines while inducing only growth arrest in normal fibroblasts. Furthermore, LAQ824 induces a dose-dependent increase of p21 in A549 cells and an increase in the hypo-phosphorylated state of Rb tumor suppressor. Panobinostat (5, 174) is a broad-spectrum HDACi with IC50=5 and 20 nM in MOLT-4 and Reh cells, respectively. Panobinostat induces acetylation of histones H3K9 and H4K8 as well as p21 expression while decreasing levels of c-Myc in a dose-dependent manner. Belinostat displays (77) IC50=27 nM in HeLa extracts. In vitro Belinostat inhibits the growth of tumor cells such as A2780; HCT116 induces apoptosis through PARP cleavage and acetylation of histones H3/H4, and shows enhanced tubulin acetylation in ovarian cancer cell lines. M344 (110, 178, 235) is toxic at concentrations above 10 μM, while a maximum of only 20% of the surviving cell population are induced to differentiate. In vitro, M344 shows significant antiproliferative activities against Ishikawa and SK-OV-3 cancer cell lines with EC50=2.3 and 5.1 μM, respectively, while it displays little effect on normal human endometrial epithelial cells. In addition, M344 leads to a decreased proportion of cells in the S phase and an increased proportion in G0/G1 phases of the cell cycle, induces apoptosis, and decreases the trans-membrane potential of mitochondria. PCI-24781 (28) exhibits potent antitumor activity against a variety of cell lines with 0.15 μM<IC50>3.09 μM. PCI-24781 treatment causes dose-dependent accumulation of both acetylated histones and acetylated tubulin in HCT116 or DLD-1 cells, induces p21 expression, and leads to PARP cleavage and accumulation of γH2AX. Sodium butyrate (44) induces differentiation and prevents cell proliferation. Mechanistic studies suggest that its action is often mediated through Sp1/Sp3-associated HDAC activity, leading to transcriptional activation of the p21 gene. In addition, this inhibitor down-regulates numerous genes that are associated with cytokine signaling, specifically the interferon-γ (IFN-γ) pathway. This compound has HDAC1, 7, and 8 with IC50=140 μM. VPA has IC50≈0.4 mM and exhibits anticancer, anti-inflammatory, and neuro-protective effects. MS-275 (181, 183), class I HDACi, induces the accumulation of p21 and gelsolin in K562 cells and decreases the expression of cyclin D1 and the anti-apoptotic proteins Mcl-1 and XIAP. MS-275 inhibits the proliferation of human tumor cell lines, including A2780, Calu-3, HL-60, K562, St-4, HT-29, KB-3-1, Capan-1, 4-1St, and HCT-15 with 41.5 nM<IC50>4.71 μM. MS-275 strongly inhibits human leukemia and lymphoma cells, including U937, HL-60, K562, and Jurkat. MGCD0103 (22) inhibits at nM or low μM concentrations only a subset of the nine HDACs. MGCD0103 is active against HDAC1 and 2 in vitro and in whole cells, but it does not inhibit class II HDACs. The exocyclic amino group in MGCD0103 is necessary for inhibitory activity, as HDAC-inhibitory activity against HDAC1 and 2 is completely abolished with the desamino analogue. Apicidin (88, 219), which contains an electrophilic ketone, is a potent HDACi with IC50=0.7 nM. An in vitro activity assay demonstrates Apicidin-mediated inhibition of HDAC3/NcoRat at a much higher potency than for HDAC6 (IC50=15.8 and 665.1 nM, respectively). Romidepsin (157, 161) is an HDAC1 and 2 inhibitor with IC50=36 and 47 nM, respectively. Romidepsin treatment in HeLa cells induces histone acetylation and p21 expression with EC50=3.0 nM, G2/M arrest, cyclin D1 down-regulation, and p53-independent p21 induction, leading to inhibition of CDK and dephosphorylation of Rb (resulting in early G1 phase arrest). Although SIRTi are classified along with HDACi, they act via a nicotinamide-dependent mechanism, suggesting that they should have their own class based on their chemical functionalities. To date, a number of specific SIRT inhibitors (mainly SIRT1 and 2) have been proposed for cancer therapy. Moreover, both activators and inhibitors of sirtuins might act beneficially against different types of neurodegenerations and cancers (127). In addition to nicotinamide, some other specific inhibitors have been characterized, including splitomicin and its analogues, tenovins, AGK2, sirtinol, suramin, the indole derivative EX-257, salermide, and UVI5008. Phenol derivatives, including quercetin, piceatannol, and resveratrol, possess SIRT1-activating properties. Many other compounds have subsequently been developed such as SRT1720, SRT2183, and SRT1460. For further information on SIRTi (42, 205).
Figure 2 presents a list of well-known HDACi. Many of these compounds are in clinical trials alone or in combination with other agents for the treatment of leukemias or solid tumor malignancies. Further information with regard to compounds that are currently being used in clinical evaluation is provided in the section “HDACi and SIRTi in clinical trials against cancer.” Our understanding of the mechanistic effects of HDACi will probably change significantly, establishing a new paradigm in the field of so-called “smart drugs.” New insights will have profound implications for the design of targeted therapies in cancer and other diseases.

Biology of HDACs in Cancer and Effects of HDACi
Many HDACs exist as components of multi-protein complexes, such as the transcriptional co-repressors mSin3, N-CoR, and SMRT (97, 215, 217, 245, 256, 259). These are then targeted to specific genomic regions by interactions with DNA-binding factors, including TFs, nuclear receptors, and other epigenetic modifiers, such as methyl-binding proteins, DNA methyltransferases, and histone methyltransferases. Along with their interactors, HDACs are able to modulate the expression of genes coding for signaling proteins and thus the function of key pathways, including apoptosis, Wnt pathway, cell-cycle progression and differentiation, regulation of the proteasome system, activation of kinases, and the unbalanced reaction between DNA damage repair and surveillance. At the chromatin level, therefore, HDACs work as transcriptional repressors in most, though not all, cases. Indeed, protein acetylation can have many different effects, as listed next (graphically represented in Fig. 3), underscoring how a defect in HDAC expression or function might affect biological processes. The readout of treatment with HDACi is a change in the expression level of about 10% of cellular proteins, leading to a global effect.
Protein degradation
Protein stability depends on the balance between synthesis and degradation (128). Protein degradation is mediated by ubiquitination. Both acetylation and ubiquitination occur on the same lysine residues, and a direct cross-talk exists between the two (82). Thus, HDACs can decrease the half life of several substrates by exposing the lysine residue for ubiquitination (76, 82, 104, 151). Modified lysine residues mediate many interactions, and an aberrant HDAC function could, therefore, alter the assembly of multi-complexes. HDAC6 is known to interact with Hsp90, and HDACi-mediated hyperacetylation of Hsp90 induces the activation of proteasome degradation of oncogenic client proteins such as Akt, Bcr-Abl, and c-Raf (24). One of the client proteins of HSP90 is androgen receptor, which is degraded after HDACi treatment, suggesting the use of HDACi in hormone-dependent or refractory cancers. All these findings reveal that the inhibition of HDAC6 makes cells more sensitive to induced stresses, which are useful in therapeutic strategies.
Protein–protein interactions
Acetylation/deacetylation balance regulates a growing number of non-histone proteins (as p53, nuclear factor-κB [NF-κB], p65, CBP, p300, STAT3, tubulin, PC4, GATA factors, nuclear receptors, c-Myc, hypoxia-inducible factor [HIF]-1α, FOXO1, HSP-90, HMG, E2F, MyoD, Bcr–Abl, the FLT3 kinase, c-Raf kinase, and others), affecting their interactions. HDACi modulate gene regulatory activities of TFs such as E2F1, p53, STAT1, STAT2, and NF-κB via a hyperacetylated status (75). HDACi can also regulate the activity of p53 and correlated genes to induce a p53-mediated pro-apoptotic signal in cancer treatment. Thus, many genes regulated by HDACi are likewise a target of p53, suggesting the use of HDACi in tumors due to p53 (activity or expression) loss (196, 207).
Cell-cycle progression (proliferation)
Crucial stages of cell cycle are generally controlled through the transcriptional regulation of a subset of genes, which are, in turn, regulated by acetylation/deacetylation of histone and non-histone proteins (231). HDAC and sirtuin overexpression has been observed in multiple cancers (see section “HDACs and sirtuins in cancer: quantitative and functional abnormalities”). HDACi cause cell growth arrest in the G1 phase in normal and transformed cells through the activation of and promiscuous interference with many processes, and they also induce p21, which binds to and inhibits the activity of cyclin-CDK2 or -CDK1 complexes. The expression of this gene is tightly controlled by p53, which competes with HDAC1 for a binding site (SP1) on p21 promoter region (260).
Apoptosis induction
HDACs act as regulators in intrinsic and extrinsic apoptosis processes (180). The evasion of this pathway is the principal hallmark of tumor growth and progression, as confirmed by the fact that resistance to apoptosis induction has been recognized in many cancers. In the extrinsic pathway, HDACs negatively affect the transcriptional regulation of death receptors such as tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and TNF receptor superfamily member (Fas) (38, 139, 159). In the intrinsic pathway, the overexpression or aberrant function of HDACs interferes with the expression of pro-apoptotic proteins such as Bcl-family members, with the concomitant up-regulation of anti-apoptotic proteins. HDACi are capable of inducing apoptosis in tumor cells by regulating the expression of pro- and anti-apoptotic genes through the intrinsic or extrinsic pathway. In the extrinsic mechanism, HDACi enhance the sensitivity of cancer cells to TRAIL by modulating expression of the protein and its receptors (65). Relaxed chromatin, promoted by treatment with HDACi, increases the accessibility of transcriptional machinery to the TRAIL promoter region, as well as the acetylated forms of many TFs (e.g., p53), which are strongly implicated in the up-regulation of TRAIL. In the intrinsic (mitochondrial) pathway, HDACi cause the down-regulation of anti-apoptotic proteins (Bcl-2, Bcl-xl, and Mcl-1) and a change in the expression of pro-apoptotic proteins (Bax, Bak, and Bid). Oligomerization of Bax/Bak leads to mitochondrial membrane permeabilization and, consequently, the release of cytochrome c by the activation of caspases (in particular, caspase 9) and apoptosome formation (19, 191). In the intrinsic cell pathway that is induced by HDACi, an important effect is mediated by ROS production, which is increased and regulated by Bax, Hsp90, TBP-2, and Trx (220). HDACi can also induce the cell death of transformed cells by causing mitotic defects. Extremely high histone acetylation levels can disrupt centromere structure and function with the concomitant loss of heterochromatin-binding proteins. Moreover, the level of acetylation interferes with histone phosphorylation, and, consequently, the action of proteins (many of which are spindle checkpoint proteins such as BubR1, CENP-F) recognizing phospho groups is itself inhibited. Levels of Aurora A and B are also decreased after HDACi treatment, contributing to transient arrest in promethaphase after aberrant mitosis and so, the activation of apoptosis (32).
Differentiation
The deacetylated state of histones is required for the maintenance of the undifferentiated cellular state, as genes involved in lineage progression are unexpressed. A shift to an acetylated state interferes with the proper execution of the differentiating process and contributes to cancer development. Particular attention has been focused on the role of HDACs in AML. In these systems, the presence of oncofusion proteins affects the release of HDACs from promoters (e.g., thus containing RAREs), maintaining cells in an undifferentiated state (by silencing genes required for hematopoietic differentiation) (221). HDACi can induce the differentiation in haematological malignancies by interfering not only with the HDAC(s)-oncofusion proteins complex, but also in breast cancer (154), hepatoma cells (246), endometrial stromal sarcoma (96), glioblastoma (206), and thyroid carcinoma cells (253).
Metastasis and migration
HDACs (in particular, class I HDACs) play an important role in metastasis and migration pathways, as they repress the expression level of RECK and Rho-β, and decrease the level of Kangai-1 (a metastasis suppressor factor) through β-catenin pathway (55). HDACs work in multi-complexes with metastasis-associated protein 1 and 2 (247). HDACs have also been linked to extracellular matrix formation and its remodeling, thus increasing cancer cell invasion and migration (237). In human cancer tissues, reduced histone acetylation is significantly correlated with advanced tumor stage and the depth of tumor invasion (199, 216). Treatment with HDACi up-regulates a set of metastasis-suppressor genes and down-regulates a set of metastasis-promoting genes (107), as well as represses cancer cell invasion and metastasis both in vitro (120) and in vivo (148). Many cases are reported in literature (236), though the role played by HDACi remains controversial (136).
Angiogenesis
Solid tumor metastasis and progression require the formation of new blood vessels. One of the key initiators of angiogenesis is the protein HIF-1α, which is degraded once acetylated. Deacetylation of HIF-1α is mediated by HDACs (classes I, II, III) and induces activation of the angiogenic process (by the expression of vascular endothelial growth factor [VEGF]) (74). Anti-angiogenic effects seem to be related to the down-regulation of pro-angiogenic genes, such as VEGF and endothelial nitric oxide synthase (eNOS). The mechanism by which HDACi act is still unclear, but the destabilization of mRNA coding for these genes such as protein degradation may be involved. The degradation of VEGF and eNOS seems to increase after treatment with HDACi (182) as a consequence of HSP90 hyperacetylation (25, 170). The same effect is observable on the expression level and stability of the protein HIF-1α (255).
DNA damage and oxidative stress
Hyperacetylation of histones causes chromatin relaxation and, thus, increases TFs binding to DNA. HDACi have a negative impact on biological mechanisms, ensuring genome integrity and stability, such as DNA recognition and repair pathway and regulation of ROS concentration. In open chromatin, sites usually packaged and unexposed are not protected from damage (induced by radiation, drugs, and unbalanced ROS scavengers) and, therefore, act as signals for genome integrity failure, culminating in cell death pathway activation. Thus, targeting genome integrity in rapidly cycling cells (such as tumor cells), combining HDACi and radiotherapy, could be considered a preferred strategy in cancer (discussed in section “Examples of HDACi in Combination Treatments”). ROS, such as superoxide anion, hydrogen peroxide, and hydroxyl radicals, are a major source of endogenous DNA damage. Normal cells control ROS concentration through the action of enzymatic (thioredoxin 1/thioredoxin-binding receptor-2 cross-talk) and non-enzymatic ROS scavengers (220). The activity of thioredoxin-1 is modulated by its receptor, which binds the enzyme interfering with its activity. In response to HDACi, levels of thioredoxin-binding receptor-2 increase; while thioredoxin-1 is reduced (30). In particular, the inhibition of HDAC10 causes an accumulation of ROS, increased release of cytochrome C, and activation of proapoptotic molecules such as caspase 3, caspase 9, and Bid in SNU-620 gastric cancer cells (130).
HDACs and Sirtuins in Non-Cancer Human Diseases
The therapeutic potential of HDACi has also been investigated for a wide range of other diseases. Here, we focus on cardiac, cognitive, immunological, and metabolic disorders.
Cardiac disorders
Recent evidence has suggested that different HDACs are implicated in heart diseases such as arrhythmia, heart failure, acute coronary syndromes, and hypertrophy (117). Cardiac hypertrophy is characterized by increased cell size, enhanced protein synthesis, and heightened organization of the sarcomere. In this state, fetal genes such as natriuretic peptide precursor type A (Nppa), myosin heavy polypeptide 7 (Myh7), and skeletal α-actin are reactivated; whereas cardiac contractile proteins such as myosin heavy polypeptide 6 (Myh6) and calcium-handling proteins are repressed. In addition, immediate-early genes encoding c-fos, c-jun, and heat shock proteins are up-regulated. In many of these processes, HDACs play a critical role in changing the accessibility of chromatin to transcriptional machinery and deregulating levels of acetylation in non-histone proteins (thus interfering with intracellular cross-talk). One line of evidence (103) suggests that class IIa HDACs interact with MEF2, one of the early activators of hypertrophy. HDACi-based therapy could, thus, help prevent heart failure. The mechanism by which HDACi suppress cardiac hypertrophy is still being investigated, but overexpression of Krüppel-like factor 4 (KLF4) seems to be involved (116). Conversely, much evidence suggests that treatment with HDACi causes QT prolongation and heart diseases (see section “Side effects of HDACi”) (203).
Neurodegenerative disorders
Neurodegeneration is an umbrella term for progressive loss of structure and/or function of neurons. For an in-depth description of this topic, we refer the reader to Fischer et al. (60) and the references therein. Despite the relatively recent discovery of sirtuins, the role of SIRT1 (and marginally SIRT2) in neurodegenerations seems to be clearer than that played by other HDACs (52). In Alzheimer's disease (AD) models, SIRT1 performs a protective function, as demonstrated by the fact that its overexpression or activation by small molecules (SIRT activators) reduces β-amyloid peptide content, a hallmark of AD, and ameliorates cognitive capabilities and memory. In Huntington's disease, SIRT1, a mediator of the beneficial metabolic effects of calorie restriction, protects neurons against mutant htt toxicity. The mechanisms subtending these effects are still not fully understood. A possible underlying defect in transcriptional regulation has been described for the diseases cited earlier, strongly suggesting the use of HDACi. By way of an example, VPA is able to decrease A-β production plaques in AD transgenic mice by inhibiting GSK-3β-mediated γ-secretase cleavage of APP protein. At the cellular level, transcriptional regulatory proteins such as CBP, NF-κB, p53, brain-derived neurotrophic factor, and PGC1a, a key regulator of mitochondrial gene expression, are each implicated in these diseases. These proteins are potentially amenable to modulation by HDACi and conceivably with SIRT1 activators or inhibitors (222).
Inflammation and immune disorders
Recently, several HDACi have also been recognized as promising anti-inflammatory agents (135). The most exciting field of application is the treatment of rheumatoid arthritis (26, 185, 230). The anti-inflammatory capability of HDACi seems to be due to the inhibition of transcriptional functions of important proteins, such as NF-κB, leading to a reduction in the expression of pro-inflammatory cytokines (TNF-α, interleukin-1β, and IFN-γ). HDACi treatments are currently under investigation for other immunological disorders, including inflammatory bowel diseases, systemic lupus erythematosus, and psoriasis. Growing evidence shows that HDACi exert immune-modulatory effects, causing an up-regulation of surface antigens on tumoral cells (193), leading to their enhanced recognition. HDACi up-regulate the expression of major histocompatibility complex class I and II proteins (4, 197).
Metabolic syndromes
HDACs may be considered a promising target for insulin sensitization, as demonstrated by several studies (37) in which PPAR-γ ligands are compared with canonical HDACi. HDAC3, 4, 5, and 7 work together to enhance glucose production in hepatocytes by the activation of FOXO1. This pathway leads to an increase in hepatic gluconeogenesis by the expression of G6Pase. Thus, interfering with HDACi could be beneficial for controlling obesity and diabetes (162, 238). In the context of metabolic and energetic status, the involvement of SIRT1 is more clear, because many of its deacetylation targets are key metabolic regulators. By shuttling between the nucleus and the cytosol, SIRT1 can interact with eNOS (145) and Atgs (critical components of autophagy machinery) (129), as well as with the proteins involved in mitochondrial respiration (p53 and PGC-1α) (101), gluoconeogenesis and oxidative stress (FOXOs and CRTC2), and lipid anabolism (LXRs) (133) in the nucleus.
HDACi and SIRTi in Clinical Trials Against Cancer
Based on published reports, at least 20 structurally different HDACi are currently being used in clinical trials as mono- and combination therapy for hematological and solid cancers. In this review, the focus has been limited to FDA-approved HDACi or those at an advanced stage of experimentation. All information regarding clinical trials comes from
Vorinostat
Vorinostat is at the most advanced stage of clinical development and is studied worldwide, as shown in Figure 4. A second-generation polar compound, Vorinostat binds to the catalytic domain of HDACs. This enables the hydroxamic moiety to chelate zinc ion located in the catalytic pockets of HDACs, thereby inhibiting deacetylation and leading to an accumulation of both hyperacetylated histones and TFs. Vorinostat was the first HDACi approved by FDA (2006) for clinical use in treating patients with advanced cutaneous T-cell lymphoma. Table 1 reports a list of trials (completed with and without results) having Vorinostat as a single drug. Although Vorinostat has shown strong antitumor activity against hematological malignancies, it is also currently being investigated for solid tumors. As an example, Vorinostat is most efficacious against glioblastoma, demonstrating the validity of HDACi-based therapies for brain tumor (66). Interestingly, four different trials employing Vorinostat against glioblastoma are currently being recruited (NCT00555399, NCT01378481, NCT01266031, and NCT01738646). Similar to many HDACi, Vorinostat has been used in a wide range of trials in combination with other drugs (demethylating agents and/or Bortezomib) and/or radiation (Table 2).

ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; BLCML, blastic phase of chronic myeloid leukemia; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; MM, multiple myeloma; PCN, plasma cell neoplasm; RAEB, refractory anemia with excess blasts; TCC, transitional cell carcinoma.
Table reports a list of trials started in 2009 and now closed (2013).
APL, acute promyelocytic leukemia; NSCLC, non-small cell lung cancer.
Panobinostat
Panobinostat (LBH-589), developed by Novartis, is a non-selective HDACi that is currently being used in Phase I and II clinical trials as mono- and combination therapy for hematological tumors such as non-Hodgkin's lymphoma, acute leukemia myeloblasts, AML, MM, advanced solid tumors, and breast and lung cancer (46, 54, 208) (Table 3). Table 4 presents trials in which Panobinostat is used in combination with other drugs. Panobinostat is coupled with chemicals that are able to interfere with the methylated status of DNA (NCT00946647) and with tyrosine kinase inhibitor Lapatinib (NCT00632489) (124). No results are available for any of the trials listed in Table 4, many of which are recruiting or still active, highlighting scientific interest in the development of combinatorial therapies that improve the efficacy of HDACi. In many of these interventional studies, Panobinostat is being evaluated to determine the maximum tolerated dose in different pathologies and administration regimes (NCT00686218 and NCT01496118, not present in Table 4). Panobinostat is currently used in a Phase I/II clinical trial (NCT01680094), assessing its use in HIV-affected patients on highly active antiretroviral therapy (175).
CMML, chronic myelomonocytic leukemia.
DLBCL, diffuse large-B cell lymphomas; EBV, Epstein-Barr virus.
Belinostat
Belinostat (PXD101) is a novel hydroxamic acid-type HDACi in late-stage clinical development with close to 1050 patients treated (March 2012). Belinostat is well tolerated, enabling combination with traditional chemotherapy without causing further bone marrow toxicity. Belinostat has been tested in a number of Phase I/II clinical trials in hematological cancers and solid tumors (Table 5). Data from these trials have provided evidence of the antitumor effect of Belinostat as monotherapy in peripheral T-cell lymphoma (PTCL) (95) and cutaneous T-cell lymphoma (CTCL), liver cancer (250), and thymoma. In addition, Belinostat has beneficial effects in combination with other anticancer drugs for the treatment of multiple types of cancer, including ovarian cancer (50), cancer of unknown primary, MM, AML, and bladder cancer (Table 6).
DLCL, diffuse large cell lymphoma.
Givinostat
Givinostat (originally ITF2357) was developed by Italfarmaco of Milan (Italy) patented in 1997 (WO 97/43251, US 6034096) and first described in 2005. It is in numerous Phase II clinical trials for relapsed leukemias and myelomas (NCT01761968) (79), and has been granted orphan drug designation in the European Union for the treatment of systemic juvenile idiopathic arthritis (NCT01261624, NCT01557452) and polycythaemia vera (NCT00928707). One interesting study involves assessing the use of Givinostat for Duchenne's disease (NCT01761292) (40).
PCI-24781
PCI-24781 (formerly CRA-024781) is currently being used in Phase I clinical trials for the treatment of neoplasia. The assessment of in vitro activity against tumor cell lines revealed the growth inhibition of multiple solid tumor lines, including colon, breast, lung, prostate, and ovarian cancers, as well as Hodgkin's and non-Hodgkin's lymphoma (15, 28, 210). Six different clinical interventional trials are now ongoing for PCI-24781, mainly for hematological diseases (NCT01149668, NCT00473577) and for testing the tolerability of the molecule in patients with hematological malignancies (NCT00562224, NCT00724984). A trial is now being recruited for the same drug (NCT01543763) in combination with PZP115891 (a tyrosine kinase inhibitor) (62).
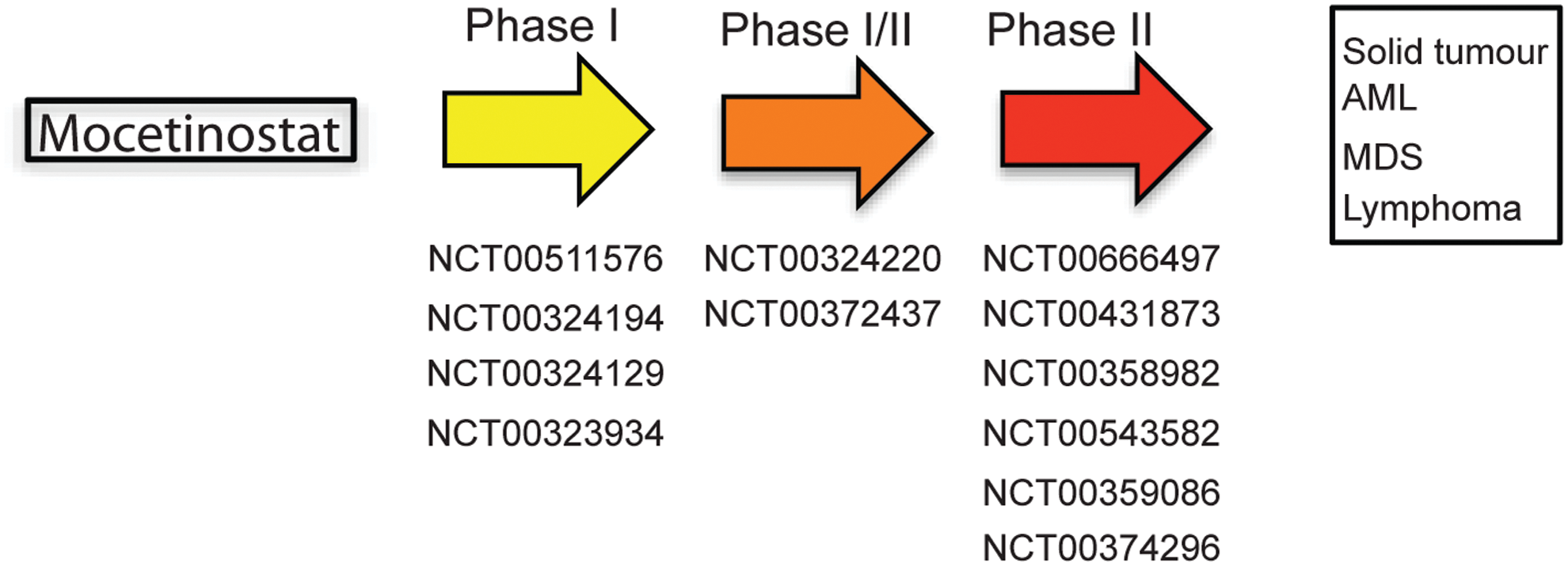
Mocetinostat
Mocetinostat (MGCD0103) has shown clinical activity in several Phase I and II clinical trials both as a single agent (18) and in combination with Azacitidine and Gemcitabine (153) (Fig. 5). It has received orphan drug designation from FDA and has been designated an orphan medicinal product by the European Medicines Agency for the treatment of Hodgkin's lymphoma (NCT00543582) and AML (NCT00323934, NCT00324129).
Entinostat
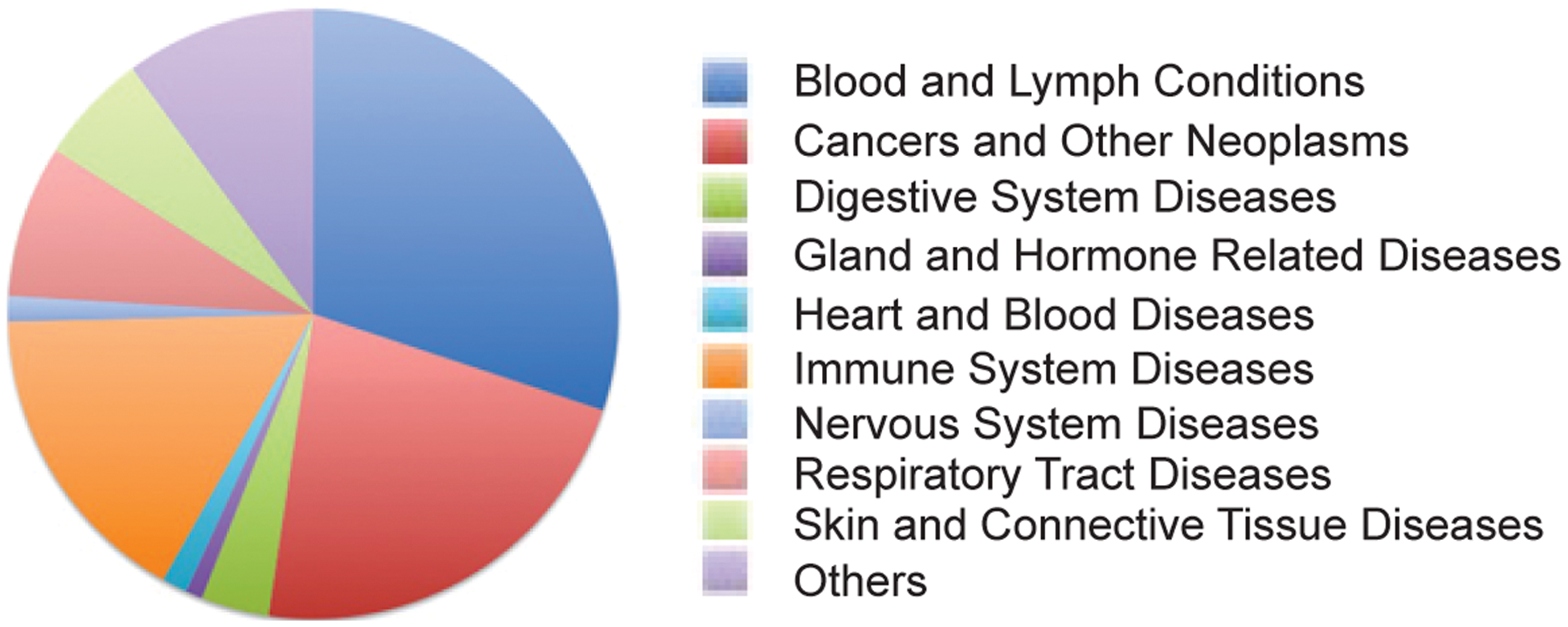
Entinostat (MS-275, SNDX-275) is a synthetic benzamide derivative shown to inhibit HDACs, and it displays antitumor activity in many preclinical models. Due to its relatively long half life, weekly and biweekly dosing schedules are explored in the clinic. A clinical trial with this agent was first performed in patients with advanced solid tumors or lymphoma in 2005. Since then, scientific interest in Entinostat has increased, and it is currently being used in Phase I/II clinical trials for recurrent advanced non-small cell lung cancer (NSCLC) combined with 5-azacytidine, or as a single drug for breast cancers (11, 109) (see Fig. 6 for disease-type classification).
Romidepsin
Romidepsin (INN, trade name Istodax) (90), a natural product obtained from the bacteria Chromobacterium violaceum, is an anticancer agent undergoing clinical trials for the treatment of CTCL (119), peripheral TCL, and other cancers (pancreatic, colorectal, renal, prostate, bladder, brain, thyroid, and ovarian) (106). In 2009, FDA granted approval for the use of Romidepsin in the treatment of CTCL in patients who had received at least one previous systemic therapy. Romidepsin is currently being used in more than 50 interventional trials, alone or in combination regimens (see
Valproic acid
VPA has found clinical use as an anticonvulsant and mood-stabilizing drug, primarily in the treatment of epilepsy, bipolar disorder, and, less commonly, major depression. In addition, it is used to treat migraine headaches and schizophrenia (around 300 interventional and/or observational trials). VPA is also an HDACi and is under investigation for the treatment of HIV and various cancers (26 studies are reported for VPA used as HDACi) (43). Phase I and II clinical trials have tested VPA, alone or in combination, for lymphocytic leukemia, AML and myelodysplastic syndrome, melanoma, HIV infection, autoimmune lymphoproliferative syndrome, and human T-lymphotropic virus type-1-associated myelopathy/tropical spastic paraparesis. Some Phase II clinical trials have involved combination therapy with hydralazine, a DNA demethylating agent, and magnesium valproate for the treatment of cervical and breast cancers, and refractory solid tumors. Other clinical trials are ongoing for HD and amyotrophic lateral sclerosis.
Clinical trials with SIRT inhibitors or activators are still in early phases, and many of these involve resveratrol. For an in-depth description of this topic, we refer the reader to Nebbioso et al. (158) and references therein, such as trials NCT01521832, NCT01485952, NCT01485965, and NCT01521585.
Limits in the Current Use of HDACi
Tumor cells are extremely plastic, and one of the key aspects of the malignant transformation process is the capability to escape normal checkpoints. Consequently, tumor cells are generally more “independent” from the environment and they are also able to modify. In many cases, tumor cells develop mechanisms of resistance to HDACi caused by epigenetic and genetic characteristics, contributing to the generation and maintenance of a neoplastic phenotype. Cellular factors implicated in resistance phenomena to HDACi include drug efflux, target status (such as overexpression, mutation, and desensitization), chromatin alteration, up-regulation of oxidative stress response mechanism, defects in pro-apoptotic pathways, and up-regulation of anti-apoptotic signals/stimuli (Fig. 7).

Effect of HDACi efflux in cells
Multidrug resistance (MDR), mediated by multiple drug efflux ATP-binding cassette (ABC) transporters, is a critical issue in the treatment of many cancers (hematological and solid), with permeability (P)-glycoprotein (P-gp), multidrug resistance-associated protein 1 (MRP1), and breast cancer resistance protein ( ABCG2), which are consistently shown to be the key effectors of MDR in cell line studies (132, 241). Pumps conferring MDR are often overexpressed in many types of cancer, requiring the use of HDACi at a higher dose or in combination with inhibitors of transporter systems (132). Overexpression of these cellular pumps is also a side effect in treatment with HDACi. Hyperacetylation of the promoter regions encoding for these genes is often observed after HDACi treatment. To date, some information is available for Romidepsin, which has been found to interact with P-gp and MRP1 (168, 242, 243). The variability of HDACi structures can help overcome problems with cell intake. Changing permeability proprieties of the molecules using a combinatorial or rational approach could, therefore, improve the efficacy of available drugs; while further studies into pharmacokinetics and pharmacodynamics could reduce required doses and aspecific interactions.
HDAC status
As previously discussed (see section “HDACs and sirtuins in cancer: quantitative and functional abnormalities”), many HDACs are overexpressed in cancer. To date, various studies have investigated the aberrant expression of HDACs in tumorigenesis as well as the progression to metastatic/refractory phenotypes. Although overexpression and mutation of HDACs contributes to inducing tumorigenesis (41), this represents a serious drawback in HDACi-based therapies. In many cases, overexpression of HDAC1 alone is able to influence the outcome of epigenetic therapy (156), without considering the compensative effects always present when a member of a specific enzymatic class is targeted. In some cases, mutation of a specific HDAC causes resistance to treatment with specific inhibitors (HDAC2 is mutated in colon cancers) (140). Resistance acquired by alterations to HDACs requires further investigation with regard to the possibility of using pan- or selective inhibitors against the specific HDAC involved in a specific cancer (see next section).
Apoptotic/pro-survival mechanisms
Altered levels of anti-apoptotic proteins in cancer cells drive resistance against HDACi-mediated apoptosis. More specifically, it has been observed that by inhibiting the transcriptional activity of the JAK/STAT pathway, resistant CTCL cells can be re-sensitized to Vorinostat-induced apoptosis (53, 240). Consistently, overexpression of anti-apoptotic proteins such as Bcl-2, Bcl-XL is sufficient to render transformed cells resistant to HDACi. This observation is common in leukemias and lymphomas, in which the expression level of Bcl-2 protein is increased (211). The same is true for NF-κB, which is able to interfere with apoptosis in NSCL cancer lines, after treatment with HDACi. A different trend is observed for p21, which is overexpressed after treatment with HDACi (in particular, specific class I HDACi), acts as a negative regulator of cell-cycle progression in a p53-dependent or -independent manner, and has been implicated in the regulation of cell death. Overexpression of p21 also correlates with the start of biochemical processes, leading to DNA repair (12, 100). Since one of the mechanisms by which HDACi induce cell death is the creation of DNA damage (and genome instability), and since these phenomena contribute to the cytotoxic effects of HDACi, p21 may act as a resistance-inducing factor.
Chromatin and epigenetic modifications
DNA methylation contributes to malignant transformation (45), such as histone acetylation/deacetylation. The balance between the acetylated status of histones and the methylated status of DNA determines expression levels of onco-suppressors and oncogenic effectors. In many reports, the treatment of cells with HDACi alone is able to restore the expression of tumor suppressor genes, while other studies claim that a hierarchy in methylation and acetylation exists, with the dominance of DNA methylation as repressive marks in cancer cells. These findings suggest combining HDACi treatment with DNA demethylating agents (see section “Examples of HDACi in Combination Treatments”).
ROS scavengers as determinants of resistance
Redox pathways play an important role in the resistance of malignant transformed cells to HDACi. Many studies hypothesize a correlation between sensitivity to HDACi and levels of ROS scavengers (36, 72). A large body of information is available for thiol reductase (Trx), which is up-regulated in half of the tumors that are resistant to HDACi treatment (141, 220). Vorinostat is able to increase the level of Trx in normal cells (an important finding that explains HDACi selectivity for cancer cells) and, conversely, in transformed cells, Vorinostat induces up-regulation of the negative regulator of thioredoxin-binding protein. Increased levels of thioredoxin-binding protein lead to ROS accumulation and so, to the activation of pro-death pathways. In many pathological systems, such as myeloid leukemias, the level of Trx is up-regulated and normal HDACi-based therapies, consequently, fail. In this scenario, not only Trx but also other proteins participating in stress response and oxidative damage recognition could act as determinants of HDACi-resistance mechanisms.
Side Effects of HDACi
As with any class of anticancer agents, some HDACi in clinical trials, such as Romidepsin (2, 146, 164), Panobinostat (46), and Vorinostat (261), have been associated with serious cardio-toxicity in the treatment of solid cancers. The most important/frequent signal in cardio-toxicity status induced after HDACi administration is the prolongation of QT interval. The biological mechanism causing this side effect is not fully understood, but it may involve the aberrant expression/function of ion channels [e.g., (hERG)K+ channel]. (hERG)K+ channel is expressed in the nervous system and heart, where it determines the timing of electrical repolarization of the action potential in ventricular myocytes. Genetic mutations in and inhibition of (hERG)K+ channel can result in long QT syndrome. The function of (hERG)K+ channel in vivo is associated with other proteins such as MinK and MiRP1, and defects in their expression levels could, therefore, cause QT prolongation syndromes. To date, it is still unclear as to whether HDACi can react directly with K+ channels or whether their effect is due to an altered gene expression pathway. The former hypothesis is not corroborated by the fact that many HDACi induce QT prolongation independently of their structures. The greatest cardio-toxicity is, however, observed after treatment with hydroxamic-derivative HDACi. Again, it is unclear as to whether hydroxamic derivatives interact preferentially with (hERG)K+ channels or whether their broad-spectrum activity against all HDACs could exacerbate cardiac side effects (195).
Examples of HDACi in Combination Treatments
Although HDACi show promise as single anticancer agents, in particular with regard to their low-molecular range of activities and minimal toxicity, many studies are investigating the use of HDACi in combination with other drugs (21, 212). In this section, we present the most frequently used combinatorial therapeutic approaches. Figure 8 shows a comparison between the number of single and multiple agents in ongoing clinical trials.
HDACi and death receptor agonists
HDACi can increase the expression and/or activity of proteins that directly transmit an apoptotic signal through death receptor pathways such as Fas and TRAIL ligands and death receptors, and downstream caspases (caspase-8 and −3), and can down-regulate proteins negatively regulating death-receptor signaling (e.g., c-FLIP, XIAP, survivin, IAP1/2). These findings provide a strong rationale for combining HDACi with death receptor stimuli. Since HDACi do not sensitize normal cells to TRAIL-mediated apoptosis, their combination with TRAIL is the most attractive option for cancer therapy (57, 99, 176, 226).
Combination of HDACi with irradiation
HDACi such as Vorinostat, TSA, VPA, and PCI-24781 enhance the radiosensitivity (
HDACi and regulators of proteasomal degradation
HDACi have recently been coupled with proteasome degradation regulators to enhance their efficacy against many cancers (92). Among all the PIs, Bortezomib is the best characterized. Proteasome inhibition results in the accumulation of mis-folded and damaged proteins, which, in turn, triggers a heat-shock protein response leading to apoptosis. The exact mechanism by which HDACi and PIs exert anticancer activity is not completely clear, but it is known to involve the regulation of Hsp90 and its client targets, such as the inhibition of NF-κB activity. ROS production and accumulation also very likely plays a role (59).
HDACi in combination with demethylating agents
Accumulating evidence has shown that the inhibition of aberrant epigenetic modifications caused by altered function/expression of HDACs could be more effective if the target strategy involving HDACi is coupled with demethylating agents. In many studies, HDACi treatment alone is insufficient to reactivate genes that are silenced by promoter hypermethylation, and is, therefore, used in combination with Decitabine, Azacitidine, and Zebularine. HDACi frequently used in combination regimens are VPA, sodium phenylbutyrate, and TSA for the treatment of AML, as well as colon, breast, endometrial, and thyroid cancers. Synergistic effects of HDACi and demethylating agents on gene expression might prolong the therapeutic time window. Although most clinical trials are still ongoing and concentrate predominantly on dosing schedules and safety of combination therapies, some studies have shown encouraging results. In patients with advanced hematological or solid malignancies, the combination of DNA-methyltransferase inhibitors and HDACi is well tolerated and induces biological effects such as DNA hypo-methylation and histone acetylation (33, 81, 102, 111, 188, 201).
HDACi with chemotherapeutic drugs
Combinatorial chemotherapeutic treatment is frontline therapy for many cancer types. Taxanes, which include paclitaxel and docetaxel, have also been used with HDACi such as TSA, Vorinostat, and PCI-24781. Since taxanes promote the stabilization of microtubules and interfere with transition from metaphase to anaphase during mitosis, the pre-treatment of cancer cells with HDACi enhances therapy by increasing tubulin acetylation. In particular, by combining class I/II HDACi with taxanes, a reduction in the proliferation of prostate, breast, ovarian, and gastric cancer cells has been observed (265). Other chemotherapeutic combinations, including DNA damage and microtubule-targeting agents, have also been used for cancer treatment. The addition of escalating doses of Belinostat in co-treatment with carboplatin and paclitaxel has been well tolerated for solid tumors (27, 50, 126). Vorinostat is also used in the carboplatin-paclitaxel chemotherapeutic regimen against NSCL. HDACi co-treatment with chemotherapeutic agents has also been employed in patients affected by anaplastic thyroid cancer. The combination of VPA and cisplatin-doxorubicin with radiotherapy decreases the volume of this tumor. However, clinical evaluation has, thus, far been limited to early-phase trials, making it difficult to draw conclusions regarding any added clinical benefit from the addition of HDACi. In conclusion, combining drugs that target different signaling pathways often lessens adverse side effects, while increasing the efficacy of treatment and reducing patient morbidity (187). In addition, multi-drug regimens using a variety of drugs and time schedules of drug administration may offer patients a more tailored therapy based on clinical status.
Open Questions: Specific or Pan-HDACi?
Unselectively inhibiting HDAC activity elicits not only therapeutic responses but also unexpected side effects. Considering the diversity of HDAC targets, selective HDACi (active against a specific HDAC class or a single isoform) may cause fewer adverse reactions and, thus, be more clinically useful. For example, class II HDACs are abundant in the heart and repress cardiac gene expression by direct or indirect association with TFs (7). As discussed in the section “Side effects of HDACi,” HDACi administration commonly causes QT interval prolongation. A selective HDACi, inactive against class II HDACs, could eliminate or reduce unwanted cardiac responses by preserving the activity of HDAC4, 5, 7 (class IIa), 6, and 10 (class IIb). To date, although considerable effort has been made toward their development, a few class-specific HDACi are available (for instance, MC1568, active on HDAC4 and 6 with IC50≈220 nM) (160). The difficulty in designing this kind of HDACi is essentially due to structural homology between HDACs and the way in which these inhibitors are developed. HDACi reported thus far are structurally diversified but, nevertheless, act as competitive inhibitors, targeting zinc ion in HDAC active sites. These compounds generally belong to the pan-inhibitor class. Sequence comparison indicates that the main differences between individual HDACs are found near the catalytic core, in the regions forming loops. Thus, inhibitors directed against this region, acting as non-competitive modulators, may display HDAC selectivity. Another way to develop non-competitive HDACi is to look at enzymatic regions that are responsible for interactions with DNA-binding proteins and/or transport proteins (for class IIa HDACs) (138). Non-competitive modulators have the advantage of being inactive (or less active) against other enzymes inside the cell requiring zinc ion as cofactor (such as RNA polymerase II, metalloprotease). The design of a new selective HDACi usually starts by taking a known active HDACi and modifying some of the structural features of the molecule without reducing its activity. Known inhibitors comprise a capping group, metal-binding moiety, and a linker region. While the capping group has been successfully modified for the design of several class-selective inhibitors (17), a combination of modifications at all three regions likely enhances their selective function. Modifications of the capping group are also useful in targeting HDACi to specific tissues. To confer tissue-selectivity properties to HDACi, they can be equipped with a surface recognition capping group that is capable of binding unique biological targets, such as expressed or overexpressed membrane receptor-binding molecules. One example of this strategy is compound CHR-2845, which is hydrolyzed by macrophage membrane receptor hCE-1 (carboxylesterase 1 receptor). The hydrolysis of CHR-2845 gives CHR-2847, the active compound accumulated only in cells expressing hCE-1 (84).
Concluding Remarks
In this review, we summarize studies on the classification, mechanisms of action, and chemistry of HDACs and their inhibitors. Several HDACi are currently being used in clinical trials for both solid and hematological malignancies. In combination with DNA-demethylating agents, chemopreventive, or classical chemotherapeutic drugs, HDACi are promising candidates for cancer therapy. Many questions regarding efficacy, tumor specificity, and gene regulation patterns still remain to be answered. A key issue is the identification of biomarkers that are predictive of positive outcome (i.e., are we aiming at the right “enemies” with the right “weapons”?). Is it possible to identify mRNAs and/or proteins that increase in response to a particular HDACi and which might also be used as indicators of drug efficacy? This is an ongoing discussion in epigenetics, and future research may help provide insights into response-based patient stratification. Due to the huge number of newly discovered epi-enzymes and their interconnection with tumorigenesis and cancer progression, a key task in future diagnostic, therapeutic, and prognostic strategies will focus on characterizing the interplay between genome and epigenome, and, thus, designing personalized epi-based single or combinatorial treatments.
Footnotes
Acknowledgments
This work was supported by EU: Blueprint (contract no. 282510), ATLAS (contract no. 221952); Epigenomics Flagship Project EPIGEN (MIUR-CNR); the Italian Association for Cancer Research (AIRC no. 11812); the Italian Ministry of University and Research (PRIN_2009PX2T2E_004); PON002782; and PON0101227. The authors apologize to the authors whose work could not be cited due to restrictions in the number of references. They are grateful to C. Fisher for linguistic editing of this article.