
Abstract
Introduction
T
Proteasomes are expressed almost ubiquitously throughout the kingdoms of life (Eubacteria generally do not contain proteasomes except some actinomycetes and mycobacteria). Although proteasomes have evolved over time, the overall layout of the inner proteolytic assemblies, called 20S core particles (CPs), has remained remarkably conserved. 20S proteasomes are C2-symmetric barrel-like structures that consist of four rings of seven protein subunits each, arranged in an αββα fashion with two outer α rings and two inner β rings. In 1995, the crystal structure of the archaeal 20S proteasome was solved (103). In prokaryotes, the α-subunits are identical and the same holds true for the β-subunits. In 1997, the crystal structure of the yeast 20S proteasome was solved (58) and in 2002, the structure for mammalian 20S was determined (164). In eukaryotes, both α-subunits and β-subunits have diverged such that, though the overall C2-symmetrical geometry is maintained, the seven α-subunits in each α ring are unique, as is the case for the seven β-subunits. In prokaryotes, each β-subunit is catalytically active. In yeast and all other eukaryotes, however, each β ring contains only three β-subunits with enzymatic activity (β1, β2 and β5). Thus, eukaryotes lack enzymatic activity of β3, β4, β6, and β7, but this loss is offset by a diverged substrate specificity of the remaining subunits. Of these, β1 is also known as “caspase-like,” because it recognizes and processes substrates having acidic residues at position 1 (P1—the amino acid occupying in the proteasome active site the position containing the scissile amide bond). The β2-subunit cuts preferentially the C-terminal of basic amino acids and is, therefore, also referred to as “trypsin-like”; whereas β5 prefers hydrophobic residues and is referred to as “chymotrypsin-like.”
Proteasome subunits are only catalytically active when they are a part of a 20S CP. The assembly of 20S particles has been subject to intensive studies, leading to detailed insights into the various consecutive steps by which these superstructures are formed (45, 163). The proteasome-assembling chaperones 1–4 form heterodimers that direct the α ring assembly. Once seven α-subunits are assembled into one ring, the β-subunits are incorporated. UMP1 is essential for correct assembly of the β-subunits. Their precursor peptides (β-propeptides) are essential for proper β ring formation. The β7-subunit is the last subunit incorporated into the ring, forming one half of a 20S proteasome. The assembly of a core 20S particle from two halves is guided by the C-terminal tail of the β7-subunits, which acts as a chaperone. Finally, CP maturation is accomplished by intramolecular cleavage of the propeptide of the inactive subunits to generate the active site (45, 160, 163). Experimental data suggest that the N-terminus of the α-subunits in the α-rings form a gate that closes the pore of the catalytic chamber, restricting the access of substrates. As a consequence, the 20S CP alone shows a basal catalytic activity, which is enhanced when bound to regulatory particles (RPs) (160).
In vertebrates, specific tissues express the interferon-gamma-inducible immunoproteasomes. In these particles, the catalytic β subunits of the constitutive proteasome are replaced by β1i, β2i, and β5i, respectively (5, 159). The immunoproteasome 20S CP are assembled de novo and cannot derive from subunit exchange starting from constitutive proteasome 20S particles, as proposed earlier (54, 56). Immunoproteasomes have a slightly different substrate preference compared with constitutive proteasomes, and this difference in cleavage correlates with MHCI peptide bonding specificity, which is a very important feature in immunology (89). Recently, the crystal structure of the mouse immunoproteasome at 2.9 Å resolution was solved; this structure revealed some differences between the immunoproteasome and constitutive proteasome active sites, thus underscoring that the substrate specificity is slightly different (79).
In 2007, Murata et al. discovered a new protein with an overall sequence highly similar to β5 and β5i, suggesting that this protein may belong to the same protein family although of a larger size (117). This protein, named β5t, is expressed specifically in thymic cortical epithelial cells, where it substitutes β5i in immunoproteasomes (162). This resulting 20S CP has been dubbed the thymoproteasome, and ensuing studies suggested a specific role for thymoproteasomes in positive T-cell selection. In 2010, our group showed by means of activity-based protein profiling (ABPP) that β5t is catalytically active. The inhibitor profile resulting from a competitive ABPP assay performed on thymoproteasomes, moreover, suggests that the β5t active site pocket is more hydrophilic than β5 and β5i (41). This altered inhibitor preference may reflect an altered substrate preference as well, which, in turn, may help explaining the role of β5t in positive T-cell selection (Table 1).
Next to the three distinct 20S proteasome CPs (constitutive proteasome, immunoproteasome, and thymoproteasome), a number of hybrid or “intermediate” 20S particles have been discovered in the past decade (30, 36, 53, 63, 88, 170). These particles may contain mixtures of constitutive proteasome and immunoproteasome active sites. Although to date only intermediate proteasomes have been identified that contain one (β5i) or two (β1i and β5i) of the three inducible catalytic subunits of the immunoproteasome, it may well be that more and more complex intermediate proteasomes exist, adding to the complexity of the 20S-CP family and its contribution to protein turnover and antigen presentation.
20S CPs are capable of degrading peptides and small or unfolded proteins, but their physiological role is limited. To become fully functional, 20S particles associate with one or two of a number of regulatory caps (50). Of these, the 19S cap is the most studied and the most important complex to associate with constitutive proteasome 20S CPs. 19S caps bind to the α-rings of a mature 20S, thus giving rise to 26S proteasomes (one 19S cap associated) or 30S proteasomes (one 19S cap at both ends of the 20S barrel, Fig. 1). 19S caps regulate 20S-mediated protein turnover in an ATP-dependent fashion by identifying and binding polyubiquitinated proteins, unfolding the substrates, and translocating these into the 20S catalytic chamber. 19S caps are assembled from 19 subunits, which can be divided in two subcomplexes: the lid and the base. The base is composed of 10 different proteins, 4 non ATPases and 6 AAA+ATPases that form a hetero-hexameric ring, which in the presence of ATP, binds the α-rings of the 20S, facilitating the opening of the gate (94). The base promotes the unfolding of the substrate, opens the pore to permit the entrance of the targeted substrates into the 20S inner chamber, and translocates these. Of the four non-ATPases proteins, two are ubiquitin receptors and the other two can bind to the ubiquitin shuttle proteins Rad23, Ddi1, and Dsk2 (94). The lid is situated on top of the base and contains nine non-ATPases proteins. Its main function is to recognize and bind polyubiquitinated substrates and deubiquitylate these. The lid subunit Rpn 11 is the only deubiquitinating enzyme (DUB) that is incorporated into the 26S proteasome. Two additional DUBs, Usp14 and Uch37, are described as proteasome-associated proteins; however, their precise binding position to the 26S is unknown (101).
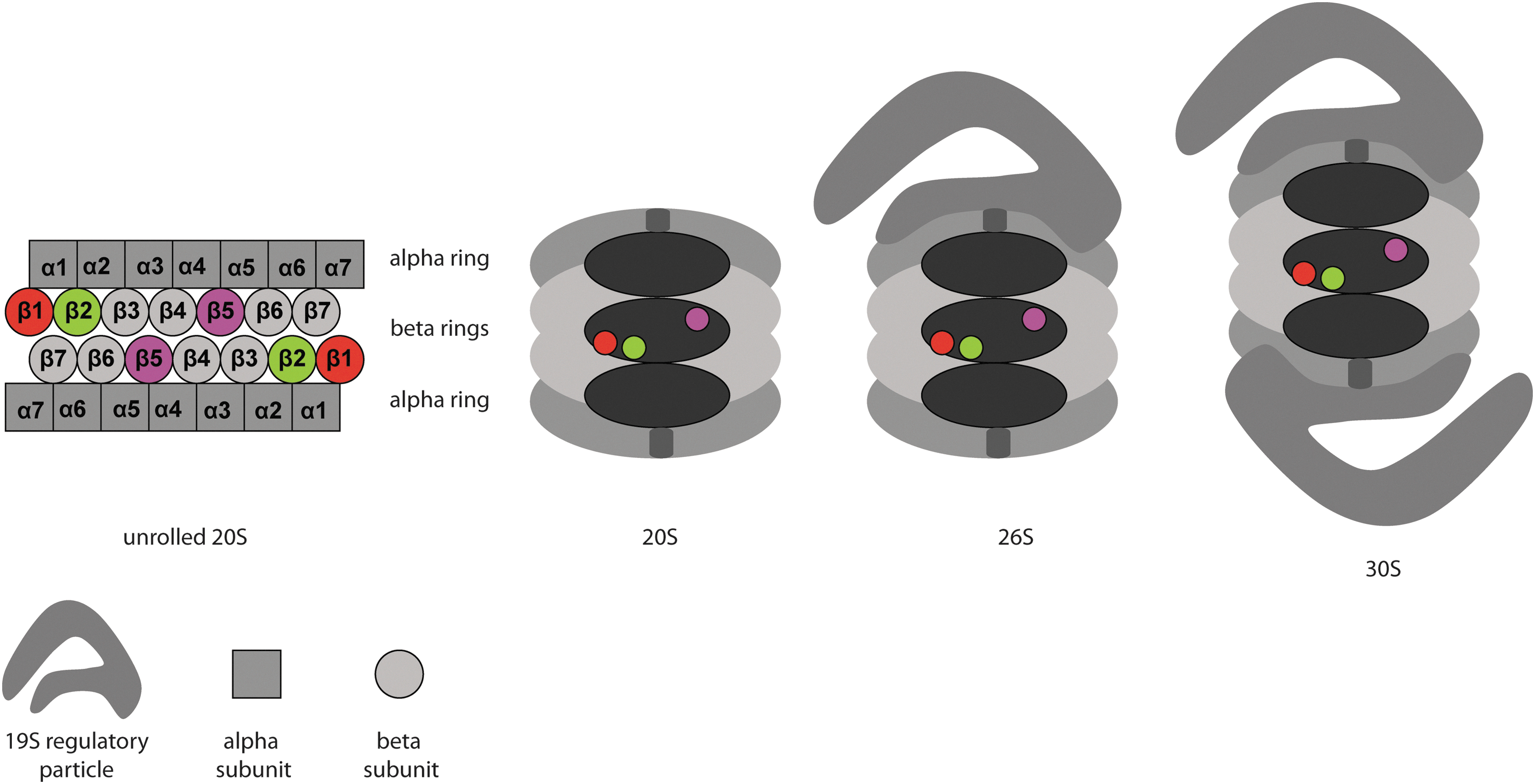
Apart from the 19S caps, other proteasome activators have been found such as the PA28 protein family and PA200. These RPs activate the proteasome in an ATP-independent manner in contrast to the 19S cap. The PA28 complex, also known as 11S, has three isoforms in higher eukaryotes, called PA28α, β, and γ. PA28α and PA28β form a heteroheptamer, while the PA28γ, which is mainly found in the nucleus, forms a homoheptamer (155). Both complexes bind to the α rings and promote gate opening. Some studies have been reported that reveal the involvement of 11S activators in the production of peptides for antigen presentation through MHC class I complexes. However, some cells and tissues that are not involved in the immune system express the 11S RPs. 11S particles may also be a part of hybrid proteasomes, with a 19S cap on one end and an 11S activator on the other (155). The monomeric activator PA200 (Blm10 in yeast) can partially open the gate of the 20S-CP, thus helping substrate entry in the proteolytic chamber. Although the 20S-CP is expressed in all eukaryotes, plants and yeasts only contain PA200/Blm10 and do not have any of the PA28 isoforms. The function of the PA200 is poorly understood, but some studies point toward its involvement in the degradation of specific substrates (160).
Proteasome Inhibitors
Many different proteasome inhibitors (PIs) have been described over the past decades. PIs are derived both from natural sources and through organic synthesis. Covalent-reversible, covalent-irreversible, and non-covalent inhibitors are known. PIs have been extensively reviewed earlier (12, 87, 140); thus, we will focus here mainly on site-selective inhibitors, for which we provide both a qualitative (different types of inhibitors) and a quantitative (potency and subunit selectivity) analysis. Figure 2 shows five classes of covalent inhibitors and their mechanisms of inhibition. The first class is represented by the peptide aldehydes, with MG-132 as its most widely used member. Aldehydes form covalent, reversible bonds within proteasome active sites, and inactivate catalytic activities by hemiacetal formation with the N-terminal threonine of the proteasome subunits. A major drawback of aldehydes is their cross-reactivity toward cysteine and serine proteases (140). A well-known class of electrophilic traps is the family of epoxyketones. Inhibitors containing the epoxyketone moiety are highly selective for the proteasome, and no off targets have been found to date (1). The structure of epoxomicin co-crystallized in yeast proteasomes reveals the molecular basis for this specificity. A morpholine ring is formed between the active site threonine and the epoxyketone, in which both the γ-hydroxyl and the free amine of the N-terminal threonine participate (60). Another class of PIs are the boronic acids, with Bortezomib as the most renowned example (2). Bortezomib (Velcade, PS-341) has been approved for the treatment of multiple myeloma (MM) patients (146). Boronates form tetrahedral adducts with active site threonines, which are stabilized by hydrogen bonding (57). Vinyl sulfones were not only initially used as cysteine protease inhibitors, but were also found to be potent PI's (19, 122). Vinyl sulfones are more readily synthesized compared with epoxyketones and have been used in many peptide inhibitors and activity-based probes (ABPs). Vinyl sulfones form a covalent adduct by conjugate addition of the hydroxyl group of the active site threonine (61). The last class of PIs discussed here are β-lactones, which form a covalent and relatively stable adduct to the proteasome by the attack of the catalytic threonine to the lactone, thereby forming a ester bond. In case of Marizomib (salinosporamide A, NPI-0052), the nucleophilic displacement of the chloride by the hydroxyl results in the formation of a tetrahydrofuran ring, which further stabilizes the adduct (59).

PIs As Drugs and Clinical Candidates
Figure 3 shows the molecular structure of several PIs that are currently used in the clinic or that are studied as clinical candidates. Bortezomib was the first PI approved by the Food and Drug administration (FDA) for the treatment of MM and refractory mantle cell lymphoma (MCL). Based on the great success of Bortezomib (52), other PIs have entered clinical trials, and Carfilzomib was recently approved for the treatment of MM. Unfortunately, patients treated with Bortezomib often develop resistance against the drug. The mechanisms behind Bortezomib resistance are poorly understood, but recent studies in cell lines indicated three main pathways by which cells can acquire resistance to PIs: (i) by up-regulation of proteasome subunits; (ii) by mutations in the β5 subunit; or (iii) by up-regulation of efflux pumps (81), although other mechanisms such as PI-resistant nuclear factor-κB (NF-κB) activity, up-regulation of chaperones such as Hsp27, Gp78- and Hsp90, overexpression of anti-apoptotic proteins such as Bcl2 and XIAP, or activation of autophagy can confer resistance to Bortezomib (26).
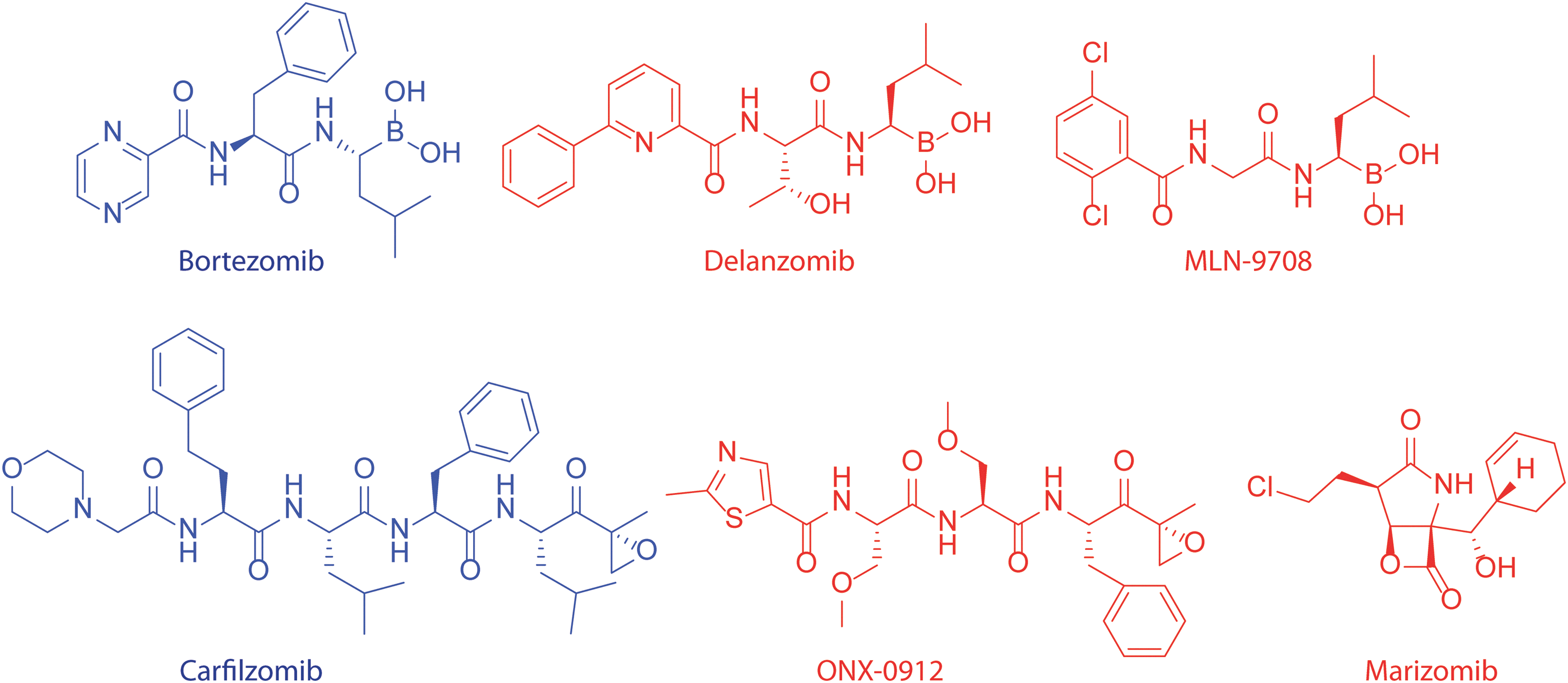
Two other boronates are currently under clinical investigation. These are Delanzomib (CEP-18770) and Ixazomib citrate (MLN-9708). Delanzomib was developed as an orally available analogue of Bortezomib [administered intravenously (34)]. Delanzomib showed promising results in toxicity studies and is currently under Phase I–II clinical investigations (135). MLN-9708, another orally bioavailable boronate currently in Phase III trials (
Current research aims at novel therapeutic applications of inhibitors/modulators of the ubiquitin proteasome system in cancer and other diseases, which emphasizes the increasing importance of these compounds for the clinic. In this Forum, some of these topics will be discussed such as the design of small-molecule noncompetitive regulators that target proteasome function by allostery and dynamics (43), the design of small-molecule noncompetitive neddylation regulators for targeted anti-cancer therapy with less anticipated cytotoxicity compared with PIs (181), how impairment of the UPS is implicated in the pathogenesis of a wide variety of neurodegenerative disorders (105), the impact of proteasome inhibition and the potential prognostic value of proteasome activities in heart diseases (35) and in atherosclerosis (175), and why the proteasome is a promising therapeutic target to combat malignant tumor growth in the lung (106).
Site-Specific Inhibitors
The chymotrypsin-like site (β5) of the proteasome is the primary target of most of the PIs. Mutagenesis studies in yeast Saccharomyces cerevisiae showed that mutation of the catalytic threonine to alanine in β1 did not cause phenotypic effects and did not lead to the accumulation of substrates. Mutation of the β2 catalytic threonine produced a slightly greater effect, namely reduced growth rate and reduced degradation of some substrates (8, 27). Genetic disabling of the catalytic threonine of β5, however, significantly retarded growth and caused accumulation of substrates (27, 68). From this, it was concluded that β5 is the most important for protein breakdown, which resulted in the development of inhibitors that (mainly) target β5. More recently however, it was observed that the inhibition of β5 alone is not sufficient to block protein degradation in HeLa cells and that co-inhibition of the caspase-like and/or trypsin-like site is necessary to completely block protein degradation (84). Bortezomib is known to inhibit β1 as well, which raised the question whether this dual inhibition of β1 and β5 is at the basis of the anti-neoplastic activity of Bortezomib. These findings stimulated the development of site-specific inhibitors (Table 2) and ABPs (Fig. 4), which will be described next.
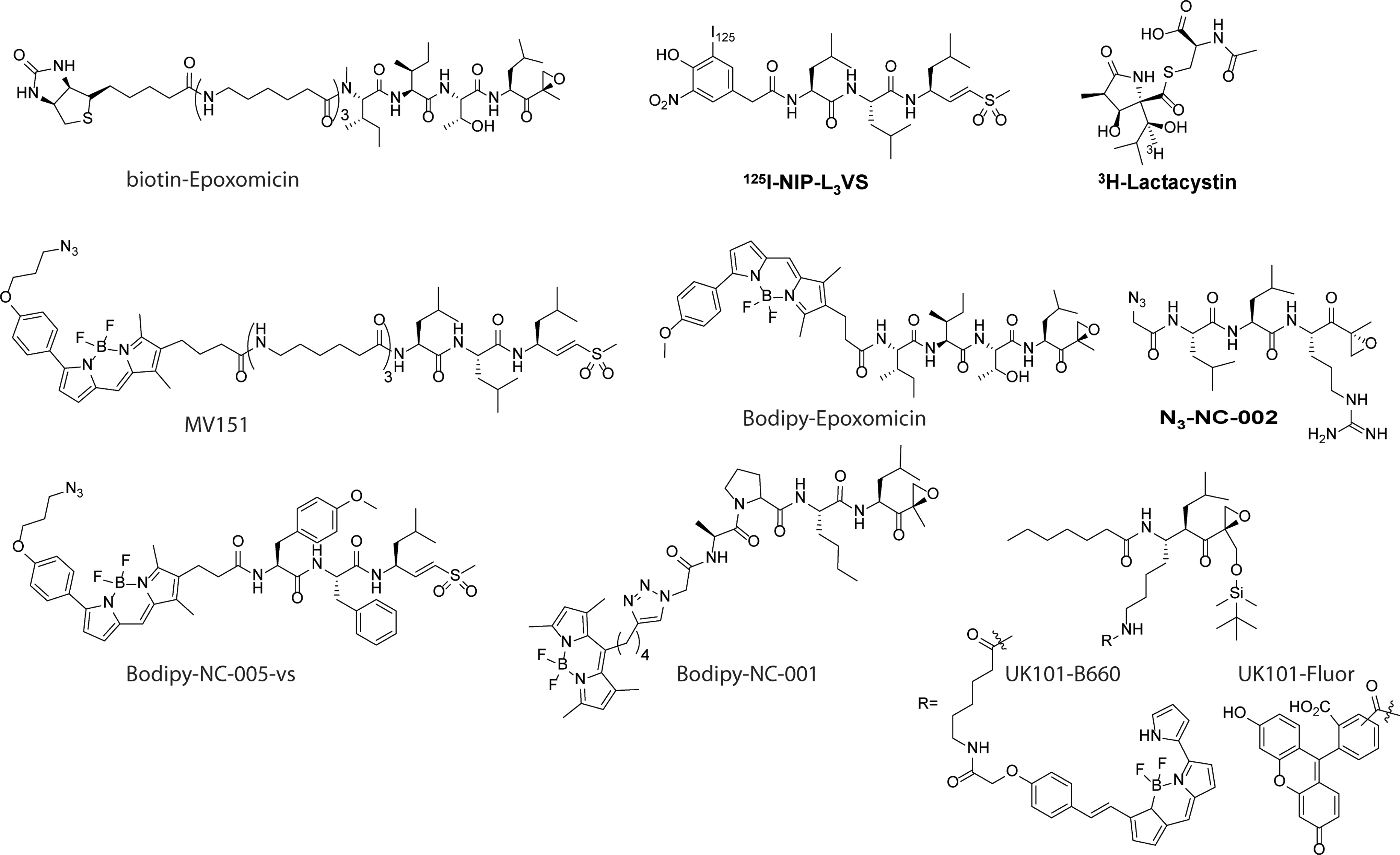
The specific subunit inhibited by the site selective inhibitor is indicated in bold.
β1 Specific Inhibitors
The caspase-like activity of the proteasome is performed by β1, which cleaves the C-terminal of acidic residues, predominantly after Glu. However, by analogy to the fluorogenic substrate Z-Leu-Leu-Glu-AMC, which is specifically cleaved by the β1 subunit (86), the incorporation of Glu α′,β′-epoxyketones in inhibitors did not provide β1 selectivity. The first β1 specific inhibitor described is epoxyketone YU-102, featuring the Pro-Phe-Leu sequence at positions P3-P2-P1 (119). More selective and potent β1 inhibitors were developed by incorporating Asp at P1, in aldehyde inhibitors (85). Based on these aldehyde inhibitors, the more potent and selective inhibitor NC-001 was synthesized that was subsequently shown to sensitize cells to β5 inhibition, thereby achieving higher cytotoxicity (21). Urea containing LU-001 was discovered by incorporation of the Val-urea-Val motif of Syringolin A in a focused library of peptide epoxyketones and peptide vinyl sulfones. LU-001 was found to be highly β1 selective and also five times more potent than NC-001 (166). β1i specific inhibitor UK-101 was identified from a series of compounds featuring different protecting groups at the primary hydroxyl of eponemycin analogues. The tert-Butyldimethyl silyl ether likely occupies the P1 prime site (P1′) of the substrate-binding channel, which is apparently beneficial for β1i inhibition. UK-101 blocks proliferation in β1i-expressing prostate cancer cells, which appeared seven times more sensitive to UK-101 than prostate cancer cells without elevated β1 levels. The peptide aldehyde, IPSI-001 is another inhibitor that is more potent toward β1i compared with β1c. However, IPSI-001 also hits β5i and, to a lesser extent, β5c. The immunoproteasome selectivity exerted by IPSI-001 can be explained by the fact that immunoproteasomes have a higher preference for more bulky, hydrophobic amino acids at P1 (norleucine in case of IPSI-001). In contrast, leucine aldehydes do not discriminate between immunoproteasomes and constitutive proteasomes. Although IPSI-001 is not potent enough for further development, it was shown that selective immunoproteasome inhibition blocks proliferation in patient blood samples and overcomes resistance to Bortezomib (92).
β2 Specific Inhibitors
The β2 site is the trypsin-like site of the proteasome that cleaves the C-terminal of basic residues and, in addition, also prefers basic residues at P3 (61, 122). The first truly β2 specific inhibitor NC-002 was designed based on Leupeptin (Ac-Leu-Leu-Arg-aldehyde), which was already shown to be β2 specific in a proteasome context (110). However, aldehyde electrophilic traps are also attacked by serine and cysteine proteases. Substituting the aldehyde by epoxyketone afforded a proteasome specific inhibitor. In analogy to the fluorogenic substrate Ac-Arg-Leu-Arg-AMC, which is specifically cleaved by β2 (86), the incorporation or Arg at both P1 and P3 (NC-012) resulted in a higher potency and specificity for β2. This increased potency appeared, however, at the expense of nearly total loss of cell permeability, likely caused by the strong cationic character. NC-002 caused higher sensitization to β5 inhibition than NC-001, and NC-022 was able to selectively sensitize MM cells to Carfilzomib and Bortezomib treatment, which is not possible using NC-001 (110). In order to overcome the possible instability of Arg epoxyketones and to improve the cell permeability of β2 selective inhibitors, a series of new inhibitors featuring para-aminomethylphenylalanine residues at P1 and/or P3 were developed
β5 Specific Inhibitors
As stated earlier, the chymotrypsin-like site of the proteasome is considered the most important for protein degradation. It is the most active site and also appears to have the broadest substrate specificity. This is also reflected by the fact that most PIs described targets at least partially β5. One of the first truly β5 specific compounds reported is YU-101, which was found to be systematically changing the P2–P3–P4 positions in epoxomicin into more hydrophobic and bulky residues. This compound showed strong anti-inflammatory effects in a mouse model (39). YU-101 is the lead from which Carfilzomib, which has been recently approved for MM by the FDA, was developed. Carfilzomib selectively inhibits β5c and β5i in purified proteasomes, although in cell lysates and more particular in living cells, β1 and β2 are also partially inhibited at higher concentrations (31, 91). Peptide aldehyde tyropeptin A, isolated from actinobacterium Kitasatospora sp., has a moderate potency and selectivity toward β5. Optimizations of tyropeptin A led to TP-110, which is much more potent and selective. Changing the N-cap to 1-naphthyl was responsible for the higher potency, and the incorporation of methyltyrosine instead of tyrosine resulted in higher selectivity (115). Highly β5 selective compounds were identified in a medicinal chemistry study in which the P4–P3–P2 positions of the peptide backbone of peptide vinyl sulfones were optimized for β5 selectivity and potency. The most promising sequences were grafted onto a new keto-oxadiazole KOD warhead, and it was found that at P3 linear side chains terminated with a bulky, hydrophobic substituent are preferential for β5 selectivity. This finding was confirmed by modeling studies along with the observation that aromatic substituents at P4 favors β5 selectivity and potency (145). In a similar way, peptide aldehydes were optimized to selectively block β5 via the incorporation of large substituents as Glu(OtBu) and Ser(OBn) at P3. In addition, the incorporation of an aromatic amino acid at P4 enhanced potency (104). Recently, a moderately potent, but highly β5 specific non-peptidic inhibitor containing a hydroxyurea moiety was identified. Crystallographic studies showed that the hydroxyurea compound did not interact with the N-terminal threonine of the subunit but it was stabilized by hydrogen bonding with β5, where the phenyl ring interacts with an S1-subpocket and the adamantane part interacts with an S3-subpocket. These new pockets are overlapping, but different from the S1 and S3 pockets that are occupied by the side chains of peptide inhibitors (44).
Based on the eminent β5 selectivity of TP-110, peptide epoxyketone NC-005 was developed, which shows a good β5 selectivity in living cells (21). Using NC-005 to inhibit β5, it was proved that the co-inhibition of β1 by NC-001 sensitizes cells to specific β5 inhibition, providing a higher cytotoxicity. During further development of this compound, it was found that changing the epoxyketone to vinyl sulfone (NC-005-mvs) produced compounds with enhanced β5 selectivity, though at the cost of slightly reduced activity (148, 169). The same observation was made with the vinyl sulfone analogues of YU-101 and PR-171, and this study led to the conclusion that not only the peptide part of inhibitors, but also the nature of the electrophilic trap influences the subunit selectivity. Interestingly, as specificity for β5 increased, cytotoxicity decreased, in agreement with the “co-inhibition theory” which states that inhibition of another β subunit next to β5 leads to higher cytotoxicity (21, 48, 110, 148). NC-005 was further optimized for β5 selectivity by the incorporation of F5Phe in P2 and by changing the naphthyl N-cap to N3Phe, leading to LU-005, which is the most potent and selective β5 inhibitor known to date (47). Recently, the sulfonyl fluoride warhead was introduced in PIs, adding a new electrophilic trap to the chemical pallet. Depending on the peptide part, selective and highly potent inhibitors for ββ5 next to more pan-reactive inhibitors were found. The molecular mechanism of action of peptide sulfonylfluorides is not yet fully understood (22). A high-throughput screening yielded capped dipeptides (CDP) as potent reversible inhibitors that are highly specific for β5 (17). CDP1 and analogues that are almost equally potent for β5c and β5i were able to inhibit NF-κB activation and proliferation of cancer cells. Moreover, CDPs with specificity for either β5c or β5i were found. CDP2 was found to be specific for β5i (most likely induced by the small P3 residue), and CDP3 was found to be specific for β5c (probably induced by the large P3 and small P1 residues) (79).
Intensive medicinal chemistry efforts led to the development of ONX-0912, which is an orally bioavailable PI that is largely selective for the chymotryptic activity of the proteasome. Interestingly, when replacing phenyl alanine epoxyketone for its leucine counterpart, some derivatives of ONX-0912 become more β5c selective (depending on the N-cap) (182). This study also led to constitutive proteasome specific inhibitor (CPSI), which is about 20×more potent against β5c compared with β5i. Some β5i selective inhibitors were also developed, among which were PR-924 and ONX-0914 (116, 133, 152). PR-924 is more selective for β5i than ONX-0914. CPSI and PR-924 have been used to determine the effect of inhibiting either β5c or β5i in hematopoietic-derived tumor cells (such as MM, which express both immuno- and constitutive proteasomes). It was found that the inhibition of only β5c or β5i was insufficient to produce an effective anti-tumor response; however, inhibiting both β5c and β5i by Carfilzomib produced an antitumor response (133). In contrast, in a different study it was shown that PR-924 inhibits growth and triggers apoptosis in MM-cell lines and primary patient MM cells, without affecting normal mononuclear blood cells (152). This paradox can be explained by the higher concentration of PR-924 used in the latter study, at which it is also likely that β5c is inhibited, resulting in apoptosis caused by complete blocking of the chymotrypsin activity of the proteasome. Selective inhibition of β5i by ONX-0914 blocks cytokine production and shows anti-inflammatory responses at only one-tenth of the maximum tolerated dose (MTD). This is in contrast to Bortezomib and carfilozomib, which induce anti-inflammatory responses at the MTD. In addition, ONX-0914 attenuated the progression of rheumatoid arthritis in a mouse model (116). The molecular basis for the selectivity of ONX-0914 for β5i was revealed recently by an elegant crystallographic study in which ONX-0914 was co-crystallized with murine constitutive and immunoproteasome. It was found that the phenyl ring at P1 causes a major rearrangement of M45 in the S1 pocket of β5c, in contrast to only a minor rearrangement in β5i. In addition, β5i has a smaller S3 pocket compared with β5c, explaining the β5i selectivity induced by Ala at P3. An analysis of the β2c/i active sites showed very spacious S1 pockets, which cannot sufficiently stabilize ONX-0914. In case of β1c, Arg45 has to be dislocated, causing a steric clash, and the S1 pocket of β1c is highly hydrophilic, which is not able to stabilize the P1 phenyl ring of ONX-0914. Although the S1 pocket of β1i is more hydrophobic, the phenyl ring of ONX-0914 does not fit optimally in the S1 pocket of β1i (79).
Activity-Based Probes
In the past decades, various ABPs for the proteasome β-subunits have been developed (Fig. 4). Generally, ABPs consist of three parts: (i) the electrophilic trap (“warhead”), which covalently modifies the active site threonine of the β-subunit; (ii) a recognition element, providing recognition by β-subunits; and (iii) a reporter group, such as a radiolabel, biotin, or fluorophore. The first activity-based proteasome probe reported is [3H]-lactacystin (28, 40), which was used to establish the binding of lactacystin to all proteasome β-subunits in a two-dimensional (2D)-gel electrophoresis experiment. The natural product epoxomicin, which was found to exhibit antitumor effects, was found to target the proteasome by using biotin-epoxomicin as a probe, followed by the detection of luminescence on treatment with avidin-horseradish peroxidase (107, 151). 125I-NIP-L3VS was used to prove that vinyl sulfones not only target cysteine proteases, but also covalently bind to proteasome β-subunits (19). All β-subunits were visualized by NLVS in a 2D-gel electrophoresis experiment using autoradiography detection of 125I. More recently, 125I-NIP-L3VS was used to screen for inhibitors of the proteasome by the incubation of cellular extracts with a potential inhibitor, followed by the labeling of residual proteasome activity by 125I-NIP-L3VS. Next, the samples were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the inhibition of a proteasome β-subunit is reflected by a decrease in the intensity of the corresponding band (18).
In the last years, various fluorescent ABPs for the proteasome subunits have been developed. The first such probe developed is the weakly fluorescent dansyl-Ahx3-L3-VS (14), which was followed by the BODIPY TMR containing MV151 (168) and Bodipy-FL-Ahx3-L3-VS (167). Using pan-reactive fluorescent probes MV151 and BODIPY-epoxomicin (169), all subunits of both the constitutive and immunoproteasome can be visualized by fluorescent scanning, directly after resolving cellular extracts that have been incubated with the probes on SDS-PAGE. Figure 5 shows a schematic overview of the workflow used for activity-based proteasome profiling. Visualization of proteasome activity by fluorescent ABP is straight forward, time efficient and provides a higher resolution compared with either biotinylated or radiolabeled probes. MV151 and BODIPY-epoxomicin can be used both in living cells and in cell extracts (168, 169). Next to ABPs that target all subunits, β-subunit-specific ABPs have been developed. Based on NC-001, BODIPY-NC-001 shows highly specific labeling of both β1c and β1i, without labeling of the other subunits. The same applies to BODIPY-NC-005-vs, which is based on NC-005-mvs, which only labels β5c and β5i subunits (169). However, generating a fluorescent ABP for β2 proved to be more difficult: The attachment of a BODIPY fluorophore to LU112 yielded a compound that also labels β5 (48). Therefore, a two-step labeling using N3-NC-002 should be used to specifically label β2 subunits (110). UK101-B660 and UK101-Fluor, both based on the β1i selective inhibitor UK101, are used to selectively label β1i, both in cell extracts and in living cells (23). Interestingly, the fluorophore is not attached to the N-terminus of the inhibitor, but to the P2 substituent, as the S2 pocket is rather large and solvent exposed, enabling the introduction of bulky substituents (Fig. 4). In general, the proteasome ABPs presented in Figure 4 can be used to quantify relative proteasome activity, to perform competitive ABPP, to test the inhibition profile of potentially new inhibitors, and as imaging probes according to the scheme in Figure 5 (97).

Molecular Mechanisms of PI-Induced Apoptosis
A review of clinical, preclinical, and biochemical literature on the use of PIs in organisms, tissues, and cells shows several corroborative observations: PIs induce cell cycle arrest and caspase-mediated apoptosis that somehow affects oncogenically transformed cells more than healthy tissues (6). This suggests that proteasome inhibition has a stronger impact on fast proliferating tissues (37) and that PIs are remarkably “clean” in their mode of action by specifically targeting only the active subunits of the proteasome (1, 4). Having said so, a plethora of exceptions to this dogma have become known. For instance, PIs exhibit adverse effects in the clinic, indicating that healthy tissues are affected; the anti-neoplastic effects are limited to the treatment of several fast proliferative myeloma types of cancers but they are less effective against quiescent cells or solid tumors and transformed cells evolve resistance to PI treatments (129). Work in cell cultures showed that prerequisite of PIs for apoptosis induction is that they should deactivate at least two out of the three active proteasome subunits with potencies which eliminate >50% of the subunits activity (84, 109). It has been observed that MM cells showing increased proteasome stress (balance between poly- and free ubiquitin) with the proteasome workload exceeding the proteasome capacity to process substrates are more sensitive to PI-induced apoptosis (15). Next to this, tremendous scientific efforts have been undertaken in the past decade to unravel the molecular mechanisms of PI-induced apoptosis. It is remarkable that proteasomes—major factors in protein homeostasis in every cell type—are, in fact, valid therapeutic targets, and a detailed understanding of the mode of action of clinical proteasome drugs in affecting apoptosis may provide invaluable information for designing future drugs. What makes understanding the mechanisms of apoptosis induction by PIs even more complicated is that the knowledge of the cell biological basis evolved alongside the development of more specific PIs, leading to several controversies in the literature. On top of this, the cellular effects of PIs mentioned earlier may induce both cell-protective and cell death pathways simultaneously, stressing the need to understand the kinetics and the cross-talk between the different effects.
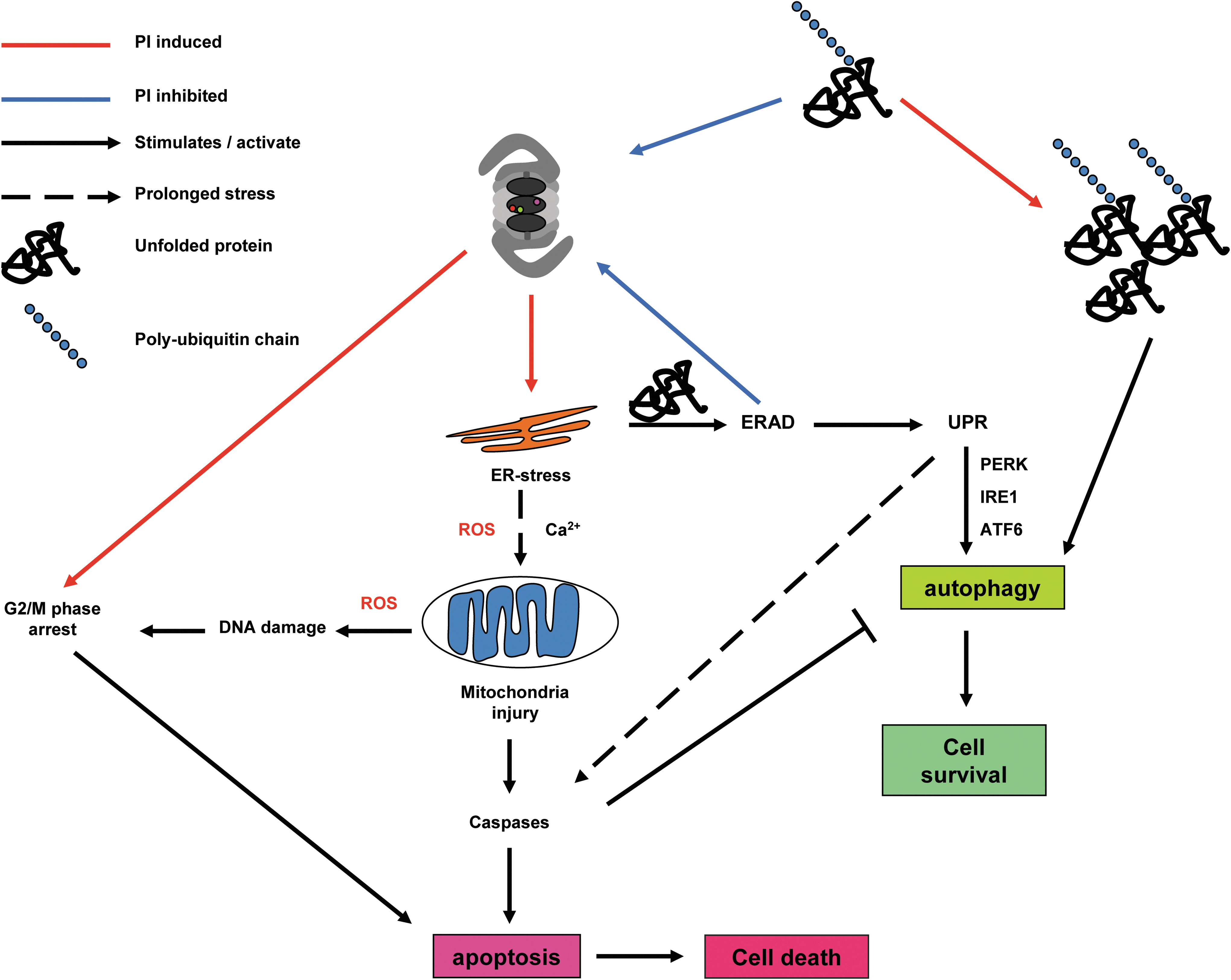
Besides its role of guarding the cellular amino-acid homeostasis by degrading damaged or misfolded proteins, the UPS is instrumental for defining the repertoire of peptides used for antigen presentation of every cell and is vital for regulating signal transduction molecules that decide between cell survival and cell death in both the cytoplasm and the nucleus (1, 123). Proteasome inhibition has been reported to have numerous effects on cells (32, 123), including the following: (i) cell cycle arrest by activation of G2/M checkpoints; (ii) perturbation of cyto-protective and pro-death signaling transduction pathways; (iii) ER stress and Ca2+ release; (iv) oxidative stress by reactive oxygen species (ROS) production; and (v) depolarization of mitochondrial potential and, as a consequence of these effects, apoptosis. In addition, in patients, cell death is caused by non-cell autonomous mechanisms such as inhibition of interleukin-6 (IL-6) secretion, inhibition of vascular endothelial growth factor secretion, and angiogenesis (4, 129).
In the mid 1990s, the discovery that the UPS regulates cell cycle progression (83) and NF-κB signaling (131) combined with early observations that PIs induce apoptosis (37) sparked the idea that the proteasome might be a suitable anti-neoplastic target which should be targeted with specific inhibitors (3). Cell cycle progression is driven by oscillations in the activity of cyclin-dependent kinase (CDK) complexes with cyclins. CDK is activated by cyclins (short-lived regulatory proteins that undergo fast degradation at exit from cell cycle) and inhibited by p21 (WAF1/CIP1) or p27 (KIP1) proteins at the G2/M and G1/S transitions, respectively (83). Cell cycle-dependent phosphatases (CDC25A-C) antagonize the CDK complex kinase activity to ensure strict control of the cell cycle and fidelity of proliferation. Levels of all proteins engaged in this pathway are tightly controlled by the UPS, and intervention via PIs disrupts the cell cycle accompanied by observations of p53 stabilization, a decrease in NF-κB level, and accumulation of CDK complex activators and inhibitors, in different cell types, both in cell culture and in animal models (1, 3, 75, 99). It is not clear why cells arrest at the G2/M and not at the G1/S checkpoint, but this might be explained by cell cycle-dependent lifetime of the p27 protein (130) or by the inability of the UPS to degrade cohesion complexes that hold together sister chromatids before mitosis (138). It is also not clear how cells induce apoptosis under prolonged G2/M arrest. Investigations focused on the elevated levels of the tumor suppressor protein p53 as a signaling molecule in this. The p53 protein is a short-lived sensor of DNA damage and oncogene activation, and in non-stressed p53, levels are maintained at a low concentration via ubiquitination by the specific E3 ligase MDM2 (67). Stress and DNA damage sensing kinase pathway (mitogen-activated protein kinase [MAPK] and ATM)- mediated p53 phosphorylation (177) prevents its degradation and activates the transcription factor function of p53 that drives the expression of pro-apoptotic genes such as Bax (113). Earlier investigations showed a controversial role for the tumor suppressor gene p53 on PI exposure, where apoptosis was p53 dependent (102), p53 independent (69, 134) or showed mixed effects (75), indicating that forced accumulation of p53 might not be a universal pathway for PI-induced apoptosis.
The NF-κB family is an ubiquitously expressed group of transcription factors which are essential for leukocyte differentiation that drive a strong pro-survival program encompassing the synthesis of growth factors such as IL-6, cell adhesion molecules (E-selectin), detoxifiers (cyclooxygenase-2, nitric oxide synthase), and anti-apoptotic factors (Bcl-2) in response to noxious stimuli, including (oxidative) stress, bacterial/viral antigens, inflammation, and UV radiation (1). NF-κB is sequestered in the cytoplasm by its inhibitory binding partner (IκB), which after receptor activation is phosphorylated, poly-ubiquitinated, and degraded via the UPS, enabling the free NF-κB to translocate to the nucleus and activate transcription (49, 131). Reports from the Anderson lab (75, 111, 112) revealed the elevated NF-κB activity in hematopoietic cancers, which justifies the rationale of using PIs to inhibit this pathway for malignant cell survival. Of particular interest is their analysis of the gene expression profile in an MM cell line exposed to Bortezomib at concentrations that induce cell cycle arrest and apoptosis (111). Data showed the expected down-regulation of survival pathways and anti-apoptotic proteins as well as the up-regulation of cell death signals not only via the canonical mitochondrial release of cytochrome c and caspase 9 activation but also via the Jun kinase and death receptor/caspase 8-dependent apoptotic pathway. Expression of the 26S proteasome complex genes was found elevated, and surprisingly, protein folding chaperones such as heat shock protein 70 went up, indicating the activation of a stress response. It was previously reported that NF-κB inhibition might not be enough to induce cell death in MM cells (73), a finding supported by studies on carcinoma cells (42) and myeloma cells (126).
In 2003, three studies, with partially overlapping observations, of PI-induced apoptosis appeared. The combined results suggest that (i) disruption of the unfolded protein response (UPR) leads to apoptosis (95); (ii) generation of ROS and mitochondrial dysfunction triggers apoptosis; (100) and (iii) induction of the pro-apoptotic Jun kinase pathway along with ROS kills leukemic cells (179). The accumulation of polyubiquitinated and improperly folded proteins is an undisputed result of proteasome activity inhibition that imposes an unfolded protein burden on the ER (126, 174). The ER is the cell organelle that serves functions in lipid metabolism, regulated Ca2+ storage, and chiefly, the assembly, folding, and post-translational modification of newly synthesized proteins (38). Misfolded proteins are retained in the ER and retrotranslocated into the cytosol for proteasome-based degradation, a process called ER-associated degradation (ERAD) (153). PIs can block ERAD, leading to protein accumulation in the ER, which not only activates the cytoprotective phase of the UPR, but also causes cytosolic accumulation of misfolded proteins in the nucleus and cytosol and the heat-shock response in the cytosol. The UPR consist of three branches that are activated by distinct sensors: the rapidly induced double-stranded RNA-activated protein kinase-like ER kinase (PERK); the evolutionarily conserved inositol requiring enzyme 1 (IRE-1); and the activating transcription factor 6 (ATF6), recently reviewed by Hetz (72). The three sensors are transmembrane proteins that contain a luminal ER domain which interacts and senses unfolded proteins and a cytosolic part that conveys the signal to the nucleus in order to modulate gene expression programs. The initial signals from the UPR as conveyed by PERK result in a general slowdown in protein synthesis for immediate alleviation of the ER protein burden. IRE-1, in turn, induces the synthesis of lipids, ERAD proteins, and chaperones to increase the ER protein-processing capacity and ATF6 induces the synthesis of ER-resident protein folding chaperones such as BiP (member of the heat shock protein HSP70 family) (172). Interestingly, the UPR seems to be activated in two waves: a first acute signaling through PERK, IRE-1, and an autophagy signal mainly aimed at repressing protein synthesis, followed by a second wave of IRE-1, ATF6, and PERK signaling to accommodate and equip the ER for facing a stress situation (72). However, in the case of sustained ERAD block and protein burden, the IRE-1 and AFT6 signals decline; while the PERK signaling persists and eventually leads to apoptosis induction via the eIF2α/activating transcription factor 4 (ATF4)/C/EBP-homologous protein (CHOP) pathway (158).
The question of how a basically cytoprotective pathway such as the UPR can also drive a cell's commitment to apoptosis has just recently been elucidated (64), and it might be physiologically relevant for host defence against the intracellular organisms Mycobacterium tuberculosis (149). PERK-mediated phosphorylation of the ubiquitous translation initiating factor eIF2α leads to its inactivation and, therefore, to global inhibition of mRNA translation but specifically induces ATF4 translation (65). ATF4 drives the expression of another transcription factor, CHOP, which has pro-apoptotic effects by repressing transcription of the anti-apoptotic Bcl-2 protein, induction of TRAIL-R2 death receptors that activate caspase 8-mediated apoptosis, cytotoxic Jun kinase activation, and elevation in ROS by up-regulating the ERO1α (ER oxidase 1α) that promotes disulfide bond formation in newly translated proteins. (158) The recent mechanism proposed by the Kaufman lab (64) states that immediately after an insult eIF2αα phosphorylation slows down translation, and subsequent induction of ATF4/CHOP and their downstream gene targets function to restore protein synthesis. In case protein synthesis increases before proteostasis equilibrium is achieved, ERO1α activity continues to increase the ROS levels driving the cell in a pro-apoptotic state that will lead to cell death.
A dissection of the timing of activation, the sequence of events, and the magnitude of the signal induced by the three UPR branches discussed earlier has been performed both in cell lines (98) and in tissues of UPR gene-defective mice (65) using ER specific inhibitors of protein folding and trafficking such as thapsigargin (Tg) and tunicamycin (Tm). Although ER stress induction by PIs is undisputed in the literature, the nature of a PI effect on the three UPR branches is less clear. At one end of the spectrum Bortezomib induced apoptosis by disrupting the IRE1 signalling in myeloma cells (95) or by inhibiting PERK and eIF2α phosphorylation but activating the ER resident caspase 4-mediated apoptosis in pancreatic cells (121). These observations can be explained from the mechanism detailed earlier: Although IRE1 signaling seemed disrupted (95), the paper showed a clear accumulation of CHOP that can drive apoptosis. In pancreatic cells, CHOP and eIF2α activities were found (121), which according to the Kauffmann model (64) of ER-stress-induced transcription regulation increases protein synthesis, leading to apoptosis. At the other side of the spectrum, PIs were found to induce apoptosis via the PERK/ATF4/CHOP terminal UPR in MM (126) and head and neck squamous cell carcinoma cells (42). Interestingly, PI-induced UPR via PERK can also activate the expression of cytoprotective elements such as the anti-apoptotic Mcl-1 protein (78) and the nuclear factor-erythroid 2-related factor 2 (Nrf2) transcription factor Moreover, accumulating evidence points toward the existence of an ER-mediated apoptotic cascade proceeding via the ER-resident caspase 4 activation (16, 96, 178) besides the two canonical apoptosis pathways regulated by death receptors via caspase 8 and mitochondrial damage in conjunction with capsase 9.
A second mechanism of PI-induced apoptosis which has been the subject of intense scrutiny comes from the observation that PIs cause intracellular ROS levels to steadily rise, inducing an oxidative state that pushes the cell toward cell death. Anti-oxidants such as vitamin C, N-acetylcysteine, and gluthathione are able to quench the ROS molecules and prevent apoptotic death (24) except in one study which found that vitamin C can complex to Bortezomib, preventing it from inhibiting the proteasome (183). Although studies with PIs equipped with structurally unrelated warheads to Bortezomib (108, 132) show cytoprotective effects on antioxidant treatment, the results of antioxidant studies should be interpreted carefully.
Cells robustly maintain their reduction/oxidation (redox) homeostasis in a reducing state to prevent oxidative damage or degradation of vital bio-molecules (51). This “reductive field” regulates levels of ROS molecules, providing them with a physiological function as signaling molecules for differentiation, cell cycle progression, growth arrest, and apoptosis. ROS molecules such as the superoxide anion (O2 •−) hydroxyl radical (OH•), hydrogen peroxide (H2O2), and several other organic radicals are side products of electron transport chains in the mitochondrial respiration cycle, of enzymatic metabolism (for instance, p450 cytochrome), or function as signaling molecules produced by the NADPH oxidases (NOXs) family (20). From the atoms necessary for life, sulfur is easily oxidized and sulfur-containing amino acids such as methionine and cysteine are prone to react with ROS. Cysteine is the main nucleophile in the active site of most phosphatises (82), ubiquitin chain E1, E2, E3 ubiquitin ligases, and their antagonists DUBs (13), which are essential enzymes for the post-translational control of vital pathways in the cell. Evidence accumulates that ROS can exert both physiological and pathological effects by oxidizing active cysteines and that a plethora of regulatory proteins (NF-κB, p53, pyruvate kinase, and ATM, among others) have evolved ROS-sensing propensities by strategically incorporating cysteine residues which on reacting with ROS influence the activity of the protein (120).
ROS are continuously produced by the leakage of electrons in the mitochondrial respiratory chain, in the ER by the activity of the ERO1α flavoenzyme needed for disulfide bond formation of newly translated proteins, in phagosomes for host defense against microorganisms, and by NOXs at the cytosol side of the plasma membrane on recruitment by major signaling receptors to participate via ROS production to the amplification of their signaling cascades in growth and proliferation (e.g., neuronal growth factor), immune response (e.g., toll-like receptors), and apoptosis (e.g., tumor necrosis factor α,TNFα) (10). Although the first evaluations that PI-induced ROS is necessary for apoptosis were performed in solid-tumor model systems (42, 100), the PI effects in hematopoietic malignancies such as MCL (134), leukemia cells (108), and MM (25, 124) were shown to be more robust. Mitochondria and ER-stress-induced ROS (42) were indicated as a source of ROS generation, because all studies found a decrease of the mitochondrial membrane potential (Δψm) that is indicative of mitochondrial damage, leading to the leakage of ROS in the cytoplasm. Mitochondria received most attention, because they are both an ROS producer and a convergence point for ROS-induced apoptosis, which on damage release cytochrome c that along with Apaf-1 and pro-caspase 9 form the canonical apoptosome system that activates executioner caspases to induce cell death (24). Treatment with anti-oxidants reduced the ROS levels and the apoptotic events, indicating that ROS play an essential role in PI-induced apoptosis. However, the use of organelle-specific ROS reporters would be advisable for the future for a more precise determination of the ROS source.
Interestingly, differences in apoptosis induction pathways were found between different PIs with Bortezomib mainly functioning through the mitochondria/caspase 9 pathway (25, 42, 100) and NPI-0052 mainly through the Fas associated death domain (FADD)/caspase 8 pathway (25, 108). Bortezomib has been shown to repress the cyto-protective Bcl-2 protein (25), leading to release of the pro-apoptotic Bax protein, which along with Bak injures the mitochondria (154). Activation of the FADD is more difficult to explain, but it might proceed via JNK signaling or terminal UPR response to ER stress (158). It should be mentioned that the pro apoptotic Bak and Bax proteins also reside in the ER and are suggested to regulate Ca2+ storage and apoptotic events (147). The release of Ca2+ in the cytoplasm can trigger apoptosis by activating the Ca2+-dependent CaMKII that signals to downstream apoptotic pathways (161). Alternative mechanisms of Bortezomib-induced apoptosis are the stabilization of the pro-apoptotic protein NOXA in medulloblastoma independently of p53 activity (128) or p53 dependent in MCL (134). Both were ROS dependent and function, because NOXA binds to and displaces the anti-apoptotic Mcl-1 from a complex with Bak (176), which on release binds to Bax, promoting mitochondrial injury. As an exception, PI-induced but ROS-independent apoptosis was found in colon cancer models to proceed by p53 stabilization, driving the expression of pro-apoptotic p53 up-regulated modulator of apoptosis that, in turn, promoted Bax-activated apoptosis (32). Evidence is accumulating that the ER plays a central role in PI-mediated apoptosis by the release of Ca2+ in the cytoplasm, UPR signaling, and via an ER resident caspase 4 pathway (16, 96, 178) that gets activated on ER stress. Recent work shows that caspase 4 is recruited to the ER by transmembrane protein 214, which was essential for ER-stress-induced pro-caspase-4 activation and apoptosis (96). Taken together, a logic interpretation is that ROS report on the stress state and integrity of an organelle such as the ER or the mitochondrium, and, in some cases, excessive ROS production might be a symptom of their injury.
The onset of apoptosis by PIs has been linked to the activation of intracellular stress-sensing kinase cascades such as the MAPK pathways, which physiologically govern cell proliferation, stress response, and survival (180). Of the three MAPK modules, the Jun-N-terminal kinase (JNK) and p38 MAPK branches are associated with the induction of apoptosis; while the extracellular signal-regulating kinase (ERK) is cytoprotective (80). PIs appear to induce apoptosis in MM cells, in part, by suppressing ERK and activating JNK (74, 179, 180), which was accompanied by an increase in ROS production. It remains unclear whether ROS production led to JNK activation and ERK repression or merely a symptom of damaged mitochondria after action of Bax-Bak membrane destabilizing complexes. Interestingly, caspase 8 activation was found to take place (179, 180), indicating that the extracellular death receptor pathway was activated. Studies of ER stress and JNK activation showed that the IRE1 branch of UPR binds to TNF receptor-associated factor 2, an adaptor protein of the TNFα receptor and via a kinase signaling cascade can activate JNK (165). Recent studies, reviewed by Tabas and Ron (158), show that both IRE1 and the PERK branches of the UPR can activate the pro-apoptotic JNK pathway or directly interact with mitochondrial membrane-permeabilizing factors, which link the ER stress effects of PIs with the four known pathways of apoptosis induction via the extrinsic caspase 8, the mitochondrial caspase 9, the ER resident caspase 4, and the Ca2+-dependent CaMKII. Lately, Bortezomib has been used in clinical experiments with organ transplantation as an agent to deplete healthy plasma cells that produce donor-specific anti-human leukocyte antigen antibodies, which are responsible for graft rejection [reviewed by the Woodle and colleagues (171)]. Although the mechanism of cell death is not known, this work suggests that some of the mechanisms discussed earlier are also at play in healthy tissues.
Resistance to PIs
PIs ability to overcome the resistance to classic anticancer therapies brought about a wave of initial enthusiasm (75). However, PI-resistant tumor clones appeared that employ various mechanisms of protection, including up-regulating proteasome synthesis (79), drug efflux pumps such as Pgp (143), and PI metabolizing enzymes (129). Interestingly, PI-induced UPR via PERK can also activate the expression of cytoprotective elements such as the anti-apoptotic Mcl-1 protein (78) and the Nrf2 transcription factor. Nrf2 phosphorylation liberates it from its inhibitor Kelch-like ECH-associated protein 1, driving the expression of some 200 genes involved in oxidative stress/redox signaling (29) that can ensure resistance to PI treatment.
Constitutive activation of Nrf2 is emerging as a prominent molecular feature in many tumor types (66), and Nrf2 phosphorylation likely restores the redox balance in oxidatively stressed tumors and clears electrophilic xenobiotics. Elevated Nrf2 levels in acute myeloma leukemia (144) correlated with reduced ROS levels and sensitivity for Bortezomib treatment, and it has been shown that Nrf2 and Nrf1 (137, 156) also up-regulate the expression of proteasome genes that increases the capacity to remove damaged proteins and scavenge PIs (157). Other mechanisms of PI resistance in human myelomonocytic THP1 cells were an Ala49Thr mutation in the highly conserved binding pocket of the β5 subunit accompanied by overexpression of the PSMB5 gene (127). Another study of induced Bortezomib adaptation in leukemia and myeloma cells showed increased expression of functional β5, β2, and β1 subunits, 11S activator caps, alongside reduced protein biosynthesis and transcription of chaperones (142).
Aggresome formation (121), up-regulation of chaperones HSP27 and HSP70 (121), and autophagy are also pathways that convey resistance to PIs. Autophagy meditates the breakdown of insoluble protein aggregates and aggresomes in the cytosol through encasing it in a double-layered membrane that is lately fused with the lysosome for degradation into its constitutive components (139). Autophagy can take over the processing of proteasome substrates, mitigating cellular stress and, ultimately, apoptosis and cell death. The link between autophagy and the proteasome is still uncertain, but evidence is pointing toward ER-stress mediated autophagy (9, 141). However, the role of autophagy in PI-induced apoptosis is controversial, as some studies suggest that autophagy is a mechanism of resistance to PIs (9, 141); while others suggest that autophagy might enhance PI lethality, perhaps depending on the cell type and the cell state being either normal or oncogenically transformed (33, 77, 118). An intriguing study in yeast proteasomes showed that S-glutathiolation, a post-translational modification, controls the 20S gate opening. The 20S CP itself might be under redox control, as the activity of S-glutathiolation on two discrete cysteine residues of the α5 subunits that control 20S gate opening proved to open the gate, increasing the protein processing power of the proteasome (150).
It should be noted that the mechanistic knowledge discussed here comes from studies in different cell culture models, primary cell cultures, and animals. Immortal cell cultures often have disturbed genetic patterns that might not reflect the defects encountered during oncogenesis in vivo, so they might react differently to PI stress. The un-physiologically high glucose concentrations in cell culture media might be taken into account as well; they affect the cell metabolism and may influence the PI response. There are clear differences in PI response between tissues, as hematopoietic cells show fast activation of terminal UPR, intensified ROS production, mitochondrial damage, and onset of apoptosis. On the contrary, adherent growing neuronal, epithelial, or fibroblast cell models or the ones closer to normal tissues such as mouse embryonic fibroblasts show a higher tolerance to PI requiring higher PI dosing, more delayed apoptotic responses, less ROS induction, and apoptosis onset via other pathways than the mitochondria. This might be, in part, explained by the composition of the proteasome in the cell, either constitutive or immune proteasome, that is often overlooked in studies; however, it is not unimportant, because PIs have different affinities for constitutive and immune proteasome. Physiological details such as the total proteasome concentration in the cell, the presence of efflux pumps, and perhaps, the shape of the cell might matter. In a spherical cell, mitochondria might be closer to the ER; thus, ROS species emanating from the stressed ER (172) might reach other organelles faster by diffusion and affect a larger area than in the case of a polarized and elongated cell where the signaling gradient might be more diffuse, giving the cell more time to take counteractive actions by activating defence mechanisms such as anti-oxidant or anti-apoptotic factor production. It has been postulated that secretory cells such as the B-cell which produce large quantities of antibodies or β-cells from pancreatic islets endowed with the production of insulin poses an intrinsically stressed ER that activates a terminal UPR earlier than other cell types (129). Poor oxygen transport into solid tumors might render the cells hypoxic, which activates the hypoxia-induced factor-1α that, in turn, can activate Nrf2 to drive ROS quenching genes (120), making these cells less vulnerable to PI-induced ROS.
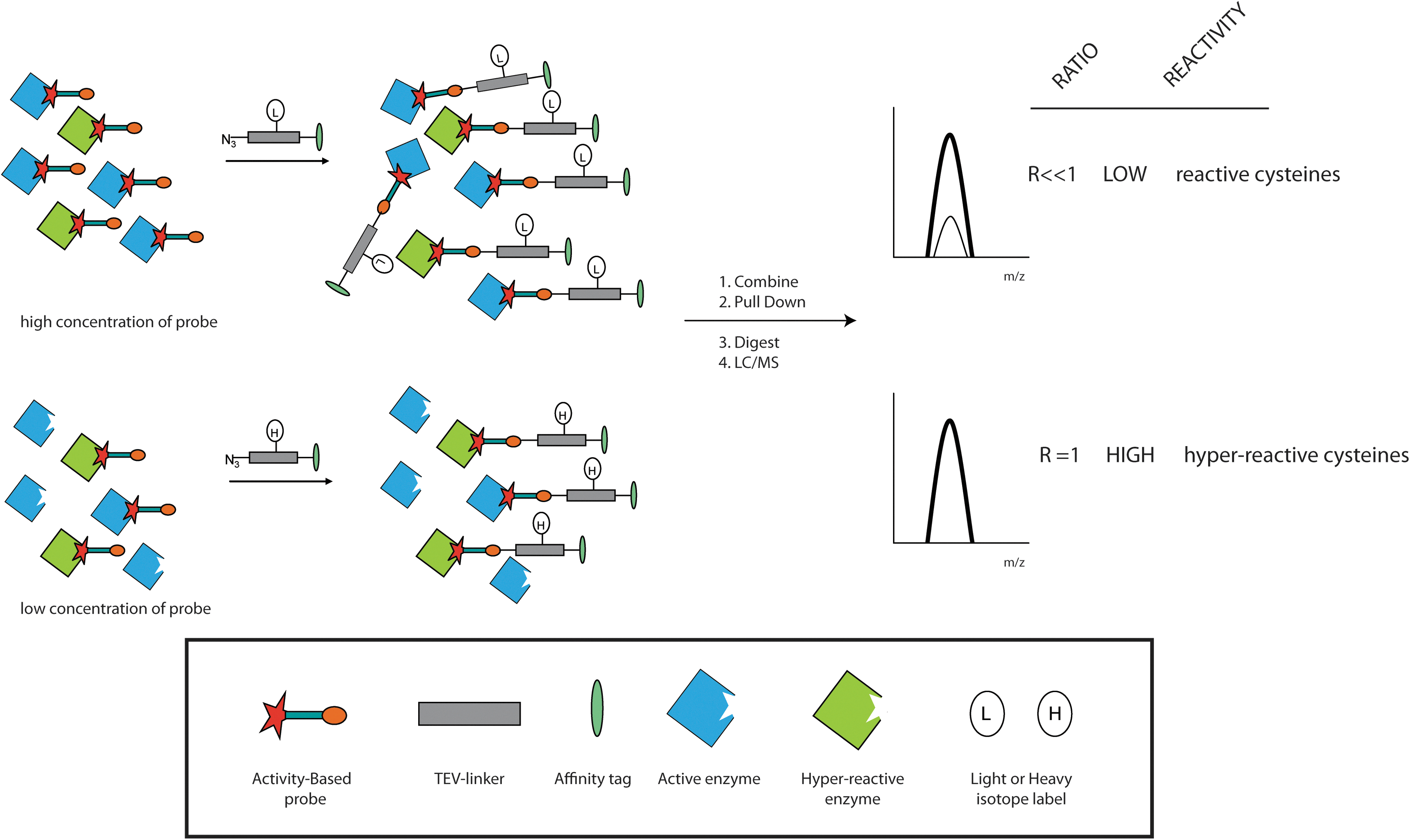
In conclusion, this review illustrates that many genes and cellular events are involved in PI-induced apoptosis. Global systems biology approaches may be used to identify the gene partners, their regulation, post-translational modification, and kinetics in order to establish which pathway is chiefly responsible for the induction of apoptosis. A cell's decision to commit to apoptosis might be a convergence of signals from several pathways underscoring the need for system-wide analysis. Moreover, ROS and Ca2+ levels should also be determined, because these factors have important regulatory and signaling function. First of all, the concentration, constitution, and activity of the proteasome in a given cell population should be determined (97). Second, global mass spectrometry-driven proteomics studies can be used to determine the protein concentrations of as much as possible proteins in order to see which pathways are up- or down-regulated. Third, an analysis of the transcriptome is necessary to determine the response at the level of gene transcription. Fourth, these measurements should be performed at several time points after PI treatment to determine the kinetics of different pathway responses. With the advent of superior mass spectrometry methods and machines, the determination of post translational modification (PTM) status of proteins has become increasingly feasible as in the case of kinases (46) and phosphatases (82) activities. An interesting method to probe the reactivity of cysteine side chains has been recently launched (173), which might be instrumental for determination of the oxidation state of proteins, an important PTM to be scored when dealing with ROS-induced phenomena. This technique uses a smart adaptation of the general alkylation principle of cysteines by iodoacetamide combined with global activity-based profiling, to determine the hyper reactive cysteines in the proteome suggested to play a role in the catalytic site of enzymes or function as ROS sensors (Fig. 6). A combination of these techniques might provide us with a clearer picture of the course of events during PI-induced apoptosis and will surely afford novel start points for therapy.
The global picture emerges that under physiological conditions, the cell is kept in a reductive state in order to prevent oxidative damage of essential bio-molecules such as DNA, RNA, proteins, and lipids. Blocking the proteasome in the nucleus, cytoplasm, and the ERAD leads to the arrest of NF-κB signaling, increased p53 levels, cell cycle arrest, ER stress inducing some form of UPR signaling, possible ER-resident caspase 4 activation, and elevated production of ROS. The release of Ca2+ from the ER, depletion of glutathione pools, and signaling via the JNK pathway injure the mitochondria, impairing the oxidative phosphorylation energy pathway that further increases ROS production, pushing the cell into an oxidative phase. If protein synthesis continues, ROS production will further damage the mitochondria and induce cytochrome c release and activation of caspase 9 that, in conjunction with caspase 8 activation via up-regulation of death receptor signaling, mediate the onset of cell death by apoptosis (Fig. 7).
Footnotes
Acknowledgments
The authors are grateful to the Netherlands Genomics Initiative, the Netherlands Proteomics Center, and the Netherlands Organization for Scientific Research (NWO) for funding this work.