
Abstract
Introduction
C
During the last decades, intensive research has led to the accumulation of comprehensive knowledge of the cellular and molecular pathology of atherosclerosis. Formerly thought to be the result of simple vascular lipid accumulation and calcification, we now recognize atherosclerosis as a complex inflammatory disease (Fig. 1) (55, 157).
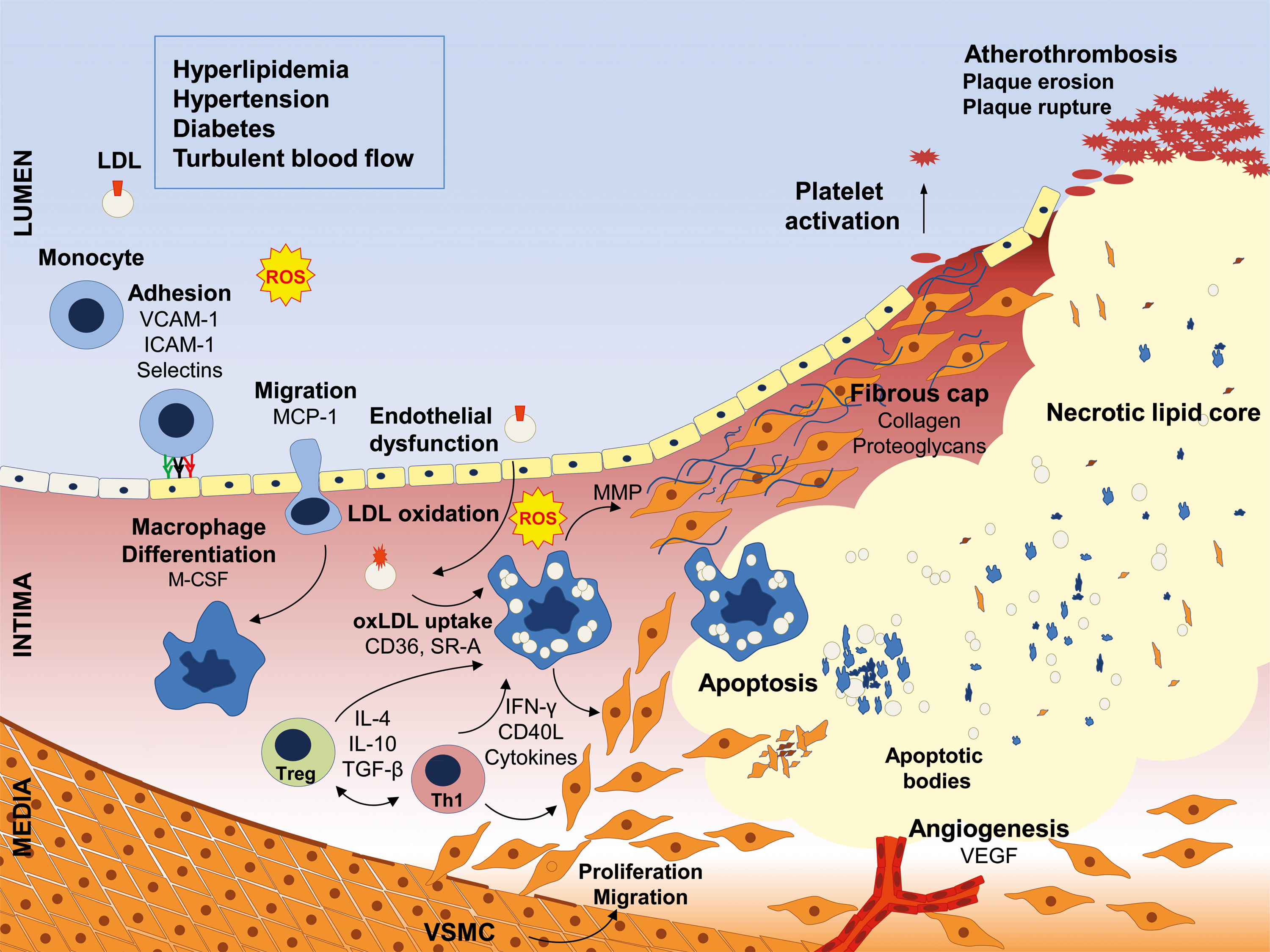
The inner layer of the arterial wall (the endothelium) regulates vascular tone and inhibits leukocyte and thrombocyte adhesion. The key mediator of endothelial function is nitric oxide (NO), synthesized by the endothelial NO synthase (eNOS), whose expression and function is stringently regulated (97). Impairment of endothelial function by pro-atherogenic stimuli is the first step in a cascade of disease-promoting events. Pro-inflammatory cytokines increased amounts of low-density lipoprotein (LDL) and reactive oxygen species (ROS), as well as nonlaminar turbulent blood flow at sites of arterial branching, all of which can lead to endothelial dysfunction. This is characterized by endothelial activation, reduced NO bioavailability, and increased permeability. Loss of barrier function of the endothelium facilitates subendothelial retention of LDL particles. Activated endothelial cells express adhesion molecules such as vascular adhesion molecule 1 (VCAM-1) and intracellular adhesion molecule 1 (ICAM-1), and they secrete chemotactic substances such as monocyte chemoattractant protein 1 (MCP-1), thereby enabling the attraction, adhesion, and subsequent transmigration of leukocytes into the intimal space (51, 52). Monocyte-derived macrophages internalize modified (e.g., oxidized) subendothelial LDL particles via scavenger receptors, which leads to the formation of lipid-laden foam cells. Migrated leukocytes produce a plethora of growth factors, cytokines, proteinases, and ROS, which further boost plaque development. The interaction of macrophages and T lymphocytes affects the lesion environment by the induction of pro-inflammatory cytokines; for example, tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ) (64), which enhance the attraction of even more leukocytes. In addition, this promotes the dedifferentiation of vascular smooth muscle cells (VSMC) from a quiescent contractile to a proliferating synthetic phenotype, which is characterized by the enhanced capacity for production of extracellular matrix proteins (45). Proliferating VSMC subsequently migrate into the intima and participate in the formation of the fibrous cap of the atheroma. Further leukocyte attraction, VSMC proliferation, and collagen synthesis lead to lesion growth. Simultaneously, apoptosis and necrosis of resident vascular cells and inflammatory cells occurs, leading to the formation of a necrotic core covered by a fibrous cap (126, 127). As inflammation persists, leukocytes release matrix metalloproteinases and inhibitors of collagen synthesis, resulting in destabilization of the fibrous cap and, ultimately, in plaque rupture (Fig. 1) (140).
The ubiquitin-proteasome system (UPS) is the major pathway for intracellular protein degradation in eukaryotic cells (57). Beyond its function to degrade dysfunctional proteins, the UPS is involved in various regulatory processes, including transcription, apoptosis, and cell cycle (68). The UPS has been recognized as a key regulatory pathway in cardiovascular diseases, especially due to its role in inflammation and management of oxidative stress. Central to its role in inflammation is the UPS-dependent regulation of the inflammatory transcription factor nuclear factor kappa-light-chain-enhancer of activated B-cell (NF-κB) (149). Under conditions of oxidative stress, an intact UPS is important for maintaining cellular protein homeostasis and function, as it removes damaged proteins and prevents aggregation and cross-linking of proteins (57).
The UPS comprises two parts: one responsible for the recruitment of proteins, the other for the degradation of recruited proteins. Proteins are recruited for degradation by ubiquitination, a process by which ubiquitin is covalently attached to target proteins by a series of three enzymes. First, the polypeptide ubiquitin is activated ATP dependently by an E1 activating enzyme; then, it is transferred to one of several different E2-conjugating enzymes; and then, finally, covalently linked to the substrate protein by a specific ubiquitin E3 ligase (56). The existence of hundreds of E3 ligases that are specific for different protein substrates accounts for the selectivity of ubiquitination (192). Repetition of this process results in a polyubiquitinated protein substrate that is recognized by the 26S proteasome.
Ubiquitinated proteins are degraded into short peptides by the 26S proteasome, which is composed of the barrel-shaped 20S core particle and one or two 19S regulatory particles (192). Proteolysis takes place in the central chamber of the 20S core particle, where the subunits β1, β2, and β5 of the two inner β rings expose their proteolytically active sites, exhibiting caspase-like, trypsin-like, and chymotrypsin-like activities, respectively (38). The two outer α rings of the 20S particle control substrate entry (61) bind the 19S regulatory particles, which themselves recognize and bind ubiquitinated substrates (192), as well as facilitate unfolding and transfer into the proteolytic core of the 20S particle (138, 170). Alternatively, the 11S proteasome activator (PA28) can associate with the 20S proteasome (109, 119). After pro-inflammatory stimulation, the β subunits of the 20S particle are replaced by the IFN-γ-inducible subunits β1i, β2i, and β5i, thereby leading to the formation of immunoproteasomes that exhibit altered cleavage specificities as compared with the constitutive 20S proteasome (66, 107). The replacement of only one or two of the β subunits results in the formation of intermediate-type proteasomes (34).
Selective proteasome inhibitors are not only valuable research tools for the investigation of UPS function, but meanwhile also represent an efficient treatment strategy for malignant diseases. In the last few decades, a plethora of structurally diverse peptide inhibitors have been designed [for overview (9, 105)]. Most of them target the active sites of the 20S core particle. The peptide boronate bortezomib (2) was the first proteasome inhibitor to be approved by the U.S. Food and Drug Administration (FDA) for the treatment of multiple myeloma (MM) and mantle cell lymphoma. Other applications are currently being studied; for example, the treatment of antibody mediated rejection in kidney transplantation (159). More recently, the second generation proteasome inhibitor carfilzomib (36), an epoxyketone-based irreversible proteasome inhibitor, was approved for the treatment of MM. New, structurally diverse, inhibitors have reached phases of preclinical and clinical testing for anticancer treatment [for overview (105)]. Although proteasome inhibitors are predominantly used as anticancer drugs, their anti-inflammatory and immunomodulatory effects are of substantial therapeutic interest.
The current medical therapy of atherosclerosis is restricted to the treatment of traditional risk factors (e.g., hyperlipidemia, hypertension, and diabetes). Although intensive efforts are being made in the field, no targeted anti-inflammatory therapy is available to date (27). Based on the discovery that the proteasome is involved in activation of the pro-inflammatory transcription factor NF-κB (149), the therapeutic potential of proteasome inhibitors in inflammation was recognized early.
This review describes our current knowledge about the involvement of the UPS in the regulation of key processes in atherosclerosis and how the disease influences the activity and function of the UPS. We will summarize the results of in vitro and in vivo experiments that have investigated the effect of proteasome inhibition and discuss the potential of proteasome inhibitors for anti-inflammatory therapy of atherosclerosis.
Key Processes of Atherosclerosis Are Regulated by the UPS
The UPS is essential for the survival and homeostasis of all eukaryotic cells. The fundamental role in many cellular processes also implies an involvement of the UPS in the onset and progression of many diseases. Atherosclerosis is a vascular disease that is characterized by inflammation, oxidative stress, proliferation, and apoptosis. All of these processes are regulated by the UPS, suggesting a role for the UPS in atherosclerosis (Fig. 2). One of the major functions of the UPS is the degradation of misfolded and damaged proteins. Protein misfolding accompanies normal protein synthesis, but it can be dramatically enhanced by conditions such as oxidative stress (57). Protein misfolding results in the exposure of hydrophobic regions, which are recognized by molecular chaperones of the heat shock protein (Hsp) family (57). These chaperones promote protein refolding, and they also interact with ubiquitin E3 ligases, such as Parkin and carboxyl terminus of Hsc70-interacting protein (CHIP), which promote the polyubiquitination of irreversibly misfolded proteins, leading to their degradation by the proteasome (78). If the amount of irreversibly misfolded proteins exceeds the capacity of UPS, protein aggregation occurs. Experimental evidence suggests that free 20 S proteasomes without the regulatory 19S particle are crucial for the efficient degradation of oxidatively damaged nonubiquitinated proteins (167), although it is debatable whether this mechanism has relevance in vivo. Furthermore it was suggested that immunoproteasomes play a particular role in the maintenance of protein homeostasis under cytokine-induced oxidative stress, as immunoproteasomes are more efficient at degrading ubiquitinated proteins (163). However, others found that immuno- and constitutive proteasomes degrade ubiquitin conjugates at similar rates (137).
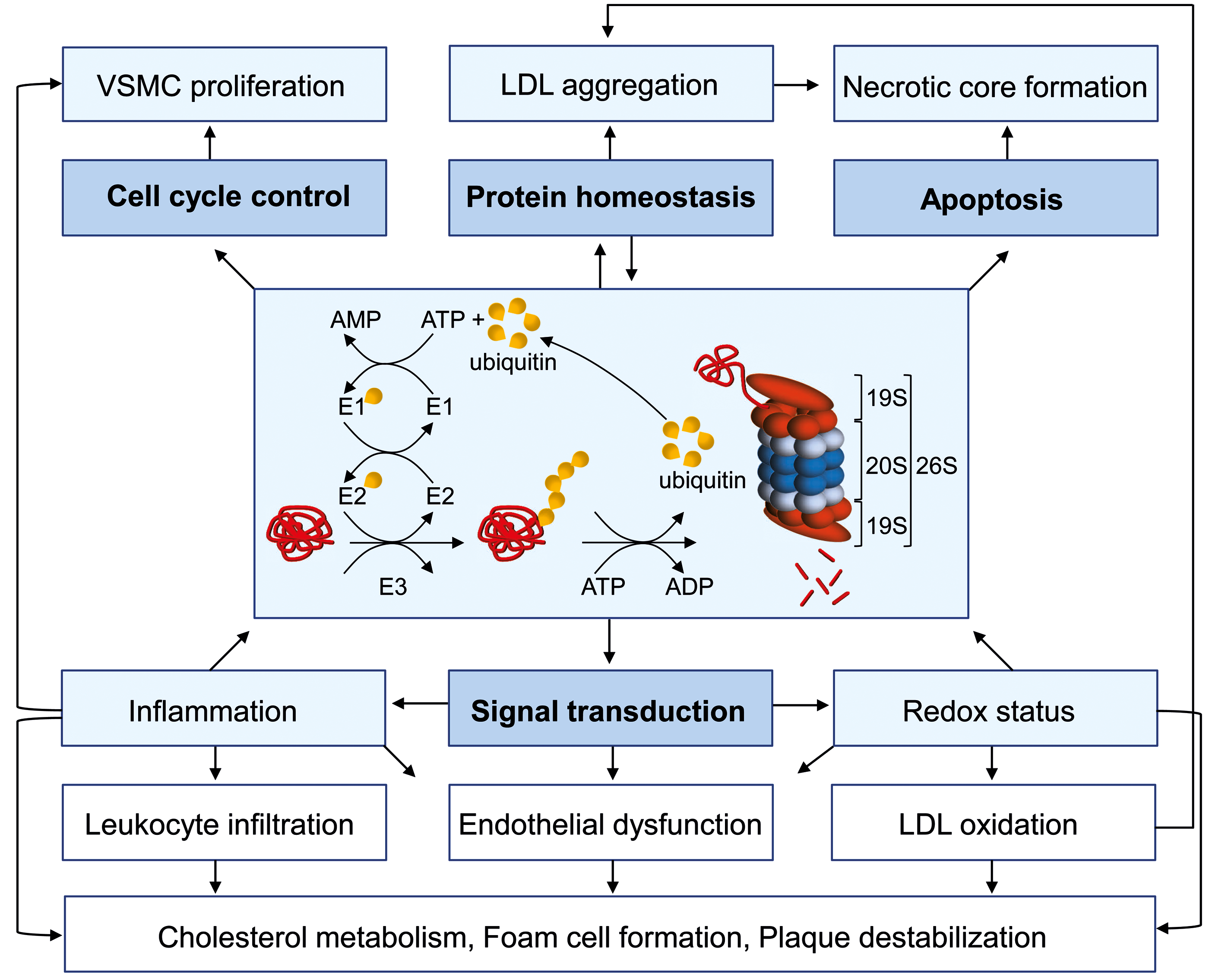
In atherosclerosis, the UPS is proposed to be dysregulated (see The UPS is (Dys)regulated in atherosclerosis section), which in the face of elevated expression of Hsp60 (106) and Hsp70 (11), the activation of the unfolded protein response (206), and the presence of protein aggregates with a characteristic structure known as amyloid in atherosclerotic tissue (156) raises the question as to whether atherosclerosis bears characteristics of a protein quality disease (70). If this hypothesis holds true for certain stages of atherosclerotic disease, this would have consequences for the therapeutic targeting of UPS activity.
Autophagy, a degrading process by which aggregated proteins, damaged cell structures including up to whole organelles, are enclosed in membrane vesicles and degraded on fusion with lysosomes, has been linked to the UPS [for overview (103, 113)]. On proteasome inhibition, autophagy becomes activated, providing an important compensatory mechanism in the event of impaired proteasomal activity (40). Even though the role of autophagy in atherosclerosis is poorly understood, evidence suggests that autophagy is stimulated in plaques by oxidized lipids, inflammation, and hypoxia, and it influences the composition and stability of the advanced atherosclerotic plaque (120, 161).
As emphasized earlier, inflammation has been recognized as a driving force in lesion formation, progression, and destabilization. A central role in vessel inflammation was assigned to the activation of the transcription factor NF-κB. Transcription factors of the NF-κB family comprise five members: relA (p65), relB, c-Rel, p105/p50, and p100/p52, all containing the Rel homology domain, which facilitates dimerization and translocation to the nucleus. Healthy human vessels contain p50 and p65 that are distributed in the cytosol; whereas in intimal and medial cells of atherosclerotic lesions, p50 and p65 are predominantly localized in the nucleus (16). Further studies have implicated a particular role of the so-called canonical activation pathway of NF-κB activation in vascular inflammation (134). In the canonical pathway (Fig. 3), the proteasome mediates the degradation of the inhibitor molecule of NF-κB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκBα). IκBα is a protein that masks the nuclear localization signal of NF-κB, thereby preventing the nuclear import of the p50/p65 complex under normal conditions. Pro-inflammatory stimuli such as TNF-α and interleukin-1 (IL-1), two cytokines shown to promote atherogenesis (3), activate IκB kinase (IKK)-2, which phosphorylates IκBα, leading to the ubiquitination and proteasomal degradation of IκBα (95), subsequently enabling nuclear translocation of NF-κB. NF-κB nuclear translocation facilitates the transcription of a plethora of genes shown to be involved in atherogenesis, such as endothelial adhesion molecules (E-selectin, VCAM-1, and ICAM-1) (154) and the chemokine MCP-1 (99).

The UPS also regulates the termination of canonical activation by prompt degradation of promoter-bound p50/p65 (158). In this same manner, the proteasome also terminates p50/p65 transcription in the lesser-known atypical pathway (30), which is activated by a phosphotyrosine-dependent but proteasome-independent mechanism (79). This pathway has been shown to be activated by oxidative stress in vascular cells (22). In addition, the UPS is also required for the proteolytic processing of the p100 precursor of p52 in the noncanonical NF-κB activation pathway (Fig. 3), which can be activated by CD40 ligation (176).
The complex involvement of the proteasome in the activation and termination of NF-κB-mediated transcription in three distinct pathways could have important implications for proteasome inhibition in atherosclerosis. Depending on the prevailing activation pathway, impaired proteasomal function could either inhibit or promote vascular inflammation.
The disruption of oxygen homeostasis contributes to the pathogenesis of atherosclerosis from the initial to the complicated stage. Two major regulators of oxygen homeostasis are regulated by the UPS: the transcription factors nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) (Fig. 4A, B).

Oxidative stress in the vascular system arises from the increased production and/or the insufficient scavenging of ROS (60). Major risk factors for atherosclerosis such as hypertension, diabetes, adipositas, and aging are associated with increased oxidative stress. The relationship between oxidative stress and vascular disease has attracted a great deal of attention (21, 46, 65, 112). However, despite strong experimental evidence linking oxidative stress to vascular disease, clinical trials with oral supplementation of antioxidants were discouraging (14, 164, 179, 180). Alternative therapeutics that instead strengthen the endogenous anti-oxidative defense system may prove to be more effective at restoring redox homeostasis in cardiovascular disease.
The expression of important enzymes of the cellular anti-oxidant defense system, such as hemeoxygenase-1 (HO-1) and superoxide dismutase 1 (SOD1), is regulated by the transcription factor Nrf2 (Fig. 4A). Under homeostatic conditions, Nrf2 is bound to its inhibitor Kelch-like ECH-associated protein 1 (KEAP1). KEAP1 is an adaptor that associates Nrf2 with a Cullin3-dependent ubiquitin ligase, thereby mediating the rapid ubiquitination and the subsequent degradation of Nrf2 by the 26S proteasome. Conditions of oxidative stress result in the dissociation of Nrf2 and KEAP1, thereby attenuating the degradation of Nrf2 and enabling an enhanced translocation of the transcription factor into the nucleus. Consequently, this leads to the transcriptional activation of numerous genes of the endogenous anti-oxidant system via binding to anti-oxidant response element sites. The atheroprotective response of endothelial cells to laminar shear stress depends on Nrf2 activation (73). Nrf2 target genes SOD1 and HO-1 have been implicated in anti-atherogenic protection (82, 114, 184). Consequently, Nrf2 and the UPS are being recognized as an interface with therapeutic potential in vascular disease (26).
Seventy years ago, the anoxemia theory of atherosclerosis suggested that hypoxia is a key factor for the progression of atherosclerotic lesions (76). Decades later, the existence of hypoxic areas was verified in atherosclerotic lesions in animal models of atherosclerosis (15). A major transcription factor mediating cellular adaptation to hypoxia is HIF-1 (Fig. 4B). HIF-1 acts as a heterodimer that is composed of two subunits: HIF-1α and HIF-1β. Although HIF-1β subunits are constitutively located in the nucleus, the abundance of HIF-1α subunits is inducible by hypoxia (75, 86, 193). Hydroxylation at two proline residues of HIF-1α, mediated by oxygen-dependent prolyl hydroxylases (18, 83, 84), links the protein to the von Hippel–Lindau tumor suppressor protein (pVHL) (28, 128), a ubiquitin ligase (E3), which ubiquitinates HIF-1α, thereby mediating the extremely rapid proteasomal degradation of HIF-1α in cells under normoxic conditions (28, 92, 143, 160, 178). Under hypoxic conditions, oxygen-dependent prolyl hydroxylases are inhibited and the hydroxylation of HIF-1α is prevented, which stabilizes HIF-1α and promotes its translocation into the nucleus and dimerization with HIF-1β. HIF-1 dimers bind to the hypoxia response element and induce the transcription of genes that are responsible for the cellular response to hypoxia.HIF-1 affects several processes in atherosclerosis (53): foam cell formation (87), smooth muscle cell proliferation (39), hypoxia-induced cell death (59), angiogenesis, and plaque neovascularization (169). Besides hypoxia, pro-inflammatory cytokines (89, 90) as well as ROS (25) are activators of HIF-1. Importantly, growing experimental evidence implicates an intimate link between NF-κB-mediated inflammatory signaling and HIF-mediated hypoxic signaling pathways (146). Considering the complex interaction of the two important transcription factors, it becomes obvious that the outcome of proteasome inhibition can hardly be explained by a simple monocausal view (see Targeting key processes of atherosclerosis with proteasome inhibitors section).
Hypercholesterinemia is a major risk factor for atherosclerosis. The UPS is involved in the regulation of cellular cholesterol synthesis as well as in cellular cholesterol efflux. The enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase catalyzes the conversion of HMG-CoA to mevalonic acid, the precursor for the biosynthesis of cholesterol and many other important nonsterol isoprenoids (58). When cholesterol levels are low, HMG-CoA-reductase is stable. However, when cholesterol levels are increased, HMG-CoA-reductase is rapidly degraded by the UPS (153). E3 ligases gp78 and TRC8 are reported to be involved in the sterol-accelerated degradation of HMG-CoA-reductase (88), despite others not being able to confirm this (185).
ATP-binding cassette transporter A1 (ABCA1) promotes the efflux of free cholesterol and phospholipids from nonhepatic cells by loading to the extracellular acceptor apolipoprotein A-I (apoA-I), thereby forming high-density lipoprotein (HDL). This subsequently enables plasma cholesterol transport back to the liver—a process known as reverse cholesterol transport (RCT). Experiments demonstrated that the UPS influences RCT by ubiqitination and proteasomal degradation of ABCA1 (7, 47, 142).
The switch of VSMC from the quiescent (contractile) to the proliferating (synthetic) phenotype is a major factor contributing to atherosclerotic plaque growth. The UPS plays an important role in the re-entry of VSMC into and progression through the cell cycle as well as in the impairment of the contractile gene expression (200). The sequential degradation by the 26S proteasome of cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors (CKIs) regulates the cell cycle (155). It has been shown that the S-phase kinase-associated protein-2 (Skp2), an F-box component of the SCFSkp2 E3 ligase complex, promotes VSMC proliferation and neointima formation in vivo (199). SCFSkp2 confers the polyubiquitination of CKIs such as p27 and p21, thereby enabling the release of CDKs and promoting cell cycle progression (136). Thus, Skp2 may be considered a potential therapeutic target for preventing VSMC proliferation.
The UPS controls cell death via apoptosis and related pathways, including transcriptional regulation of apoptotic gene expression, outer mitochondrial membrane permeabilization, and caspase activation (12, 17, 139). Apoptosis in atherosclerotic lesions can be triggered by inflammatory cytokines and modified and aggregated lipoproteins. The impact of apoptosis on atherosclerosis depends on the disease stage, the affected cell type, and the localization. To simplify, in early disease stages, the apoptotic death of VSMC and macrophages can delay lesion progression; whereas in advanced plaques, cell death promotes growth of the necrotic core and destabilizes the lesion (54, 189). In view of the ambivalent role of apoptosis in atherosclerosis along with the fact that the cellular response to proteasome inhibition ranges from the induction of apoptosis to the protection from cell death (131), it becomes obvious that a multitude of factors determine the final outcome of targeting the proteasome in atherosclerosis.
As outlined in this section, the UPS is important for the regulation of the function of all cell types involved in the pathogenesis of atherosclerosis, and in the changes they undergo during disease progression. Processes regulated by the UPS are not confined to a specific stage of the atherosclerotic disease, but they play a role throughout disease progression, as described elsewhere (150, 171).
The UPS Is (Dys)regulated in Atherosclerosis
Considerable evidence from in vitro and in vivo studies suggests that the UPS regulates vascular cell homeostasis and that it is dysregulated in certain pathological conditions. Oxidative stress is considered as having different effects on proteasome degradation ability depending on the extent of substrate oxidation: Mildly oxidized protein substrates are degraded effectively, whereas the degradation susceptibility of extensively oxidized proteins decreases due to the formation of protein aggregates (62). Lipid peroxidation products such as 4-hydroxy-2-nonenal (HNE) (145), HNE cross-linked oxidized proteins (49), and age-related lipofuscin/ceroid (168) are capable of inhibiting the proteolytic activity of the proteasome. Moreover, all components of the proteasomal system, including de-ubiquitinating enzymes, are impaired by extensive or chronic oxidative stress (85, 141, 204). Oxidative stress has been shown to induce the dissociation of the 20S core particle from the 19S regulatory particle of the 26S proteasome in eukaryotic cells, leading to impaired 26S proteasome activity and the accumulation of ubiquitinated proteins (195). Consequently, sustained oxidative stress and severe conditions such as ischemia-reperfusion injuries are shown to cause decreased proteasomal activity (19, 98, 198).
Macrophages are central to atherosclerotic lesion progression and are capable of producing ROS. The proteasome has been shown to be essential for the induction of several defense proteins to protect macrophages from oxidative stress (81). However, oxidized LDL (oxLDL) is able to inhibit proteasomal activity in macrophages, thereby leading to protein accumulation and macrophage apoptosis (191). On the contrary, aggregated LDL was shown to suppress the apoptosis of foam cells by inducing an E2 ubiquitin-conjugating enzyme mediating proteasomal degradation of pro-apoptotic p53 (100).
Several studies highlight the influence of risk factors of atherosclerosis on vascular cell functions. Hyperglycemia in diabetes mellitus can modulate proteasomal activity in endothelial cells. Hyperglycemia-induced formation of dicarbonyl methylglyoxal covalently modifies the 20S proteasome, thereby decreasing its activity (151). On the contrary, it has been shown that high glucose can also enhance proteasomal activity in endothelial cells, which leads to tetrahydrobiopterin (BH4) deficiency and endothelial dysfunction due to the enhanced proteasomal degradation of guanosine 5′-triphosphate cyclohydrolase I (GTPCH), an enzyme that is essential for BH4 synthesis (203). Enhanced 26S proteasomal activity has been attributed to tyrosine nitration of the proteasomal regulatory subunit PA700 (19S) in diabetic (115), hypertensive (201), and hyperlipidemic mice (202), suggesting that this is a common mechanism for endothelial dysfunction in cardiovascular diseases.
NO, whose importance for vascular function goes far beyond its vasodilating properties, was shown to provide cytoprotection from oxidative stress in endothelial cells by stimulating proteasomal activities and up-regulation of the immunoproteasomal subunits β5i and β1i (110, 111, 182). In VSMC, NO is known to inhibit proliferation by modulating the expression of cell cycle regulatory proteins (63, 80). This could be due to NO-mediated inhibition of 26S proteasomal activity through S-nitrosylation, differential regulation of 20S proteasomal subunit expression (94), and a decrease in the activity and expression of the proteasome activator PA28 in the vasculature (187). Another mechanism explaining the decrease in VSMC proteasomal activity was demonstrated to be the NO-induced inhibition of isopeptidase T, a protease that is responsible for hydrolyzing unanchored polyubiquitin chains to free monoubiquitin (186). Taken together, the studies highlight the importance of NO to regulate UPS function in vascular cells. A reduced NO bioavailability leads to the dysregulation of the UPS in endothelial and VSMC, thereby contributing to endothelial dysfunction and VSMC proliferation.
An immunohistochemical analysis of human atherosclerotic plaques is used to describe the overall status of the UPS in advanced atherosclerosis. A role for the UPS in fatal coronary plaque progression was demonstrated in a postmortem study showing higher ubiquitin immunoreactivity in infarct-related compared with noninfarct-related coronary arteries (67). Despite the absence of mechanistic explanations, the enrichment of ubiquitin in the lipid core and shoulder regions, co-localization with neointimal smooth muscle cells, T cells, macrophages, and apoptotic cells suggests disturbance of the UPS in these cells (67). The majority of data from human tissues comes from analyses of carotid thrombendarterectomy (TEA) specimens. TEA specimens also show highly advanced atherosclerosis, the result of decades of plaque development, with complications such as rupture and atherothrombosis leading to symptomatic cerebral ischemia. According to studies by Versari et al. and Marfella et al., TEA plaques from symptomatic patients with a more unstable morphology display significantly more ubiquitin conjugates. Interestingly, the outcomes from these studies differ in terms of proteasomal activity. Versari et al. reported a decreased proteasomal activity in the presence of increased oxidative stress and apoptosis. Macrophages and smooth muscle cells were shown to co-localize with ubiquitin conjugates (190). The authors connected the impairment of proteasome activity in the face of increased oxidative stress to disease progression in advanced atherosclerosis. It was hypothesized that persistent overload of the proteasomal capacity leads to the accumulation and aggregation of dysfunctional proteins and, ultimately, to cell death. This mechanism is also evident in neurodegenerative diseases, and it shows that aspects of atherosclerosis can be considered a protein quality disease (70). In support of this theory, amyloid can be detected in atherosclerotic tissue (74, 156, 181). In contrast, Marfella et al. detected enhanced proteasomal activity with increased inflammatory cell infiltration, higher levels of NF-κB, and lower levels of IκB in symptomatic compared with asymptomatic patients (121). Similar results were also shown in TEA specimens of diabetic patients (122), postmenopausal women (124), and hypertensive patients (125). In an autopsy-based study, unstable plaques from the middle cerebral artery showed a higher expression of ubiquitin conjugates and NF-κB than stable plaques and normal vascular tissue (175).
Based on the conflicting observations in these studies, an ongoing discussion has evolved as to whether over-reactivity or insufficiency of the proteasome system is the prevailing influence that drives plaque progression and complication (37, 68). Both hypotheses would have opposite implications for the treatment of atherosclerosis with proteasome inhibitors. However, it was soon realized that both processes might play an important role, depending on the stage of the atherosclerotic disease. While proteasomal activity could be preserved or even enhanced in early atherogenesis, thereby driving the inflammatory response via NF-κB, late-stage atherosclerosis might be characterized by impaired proteasomal activity and defective elimination of dysfunctional proteins (68). Currently a definite answer to this question is still missing. We assume it will remain an unanswered question unless efforts are made to investigate the vascular UPS in much earlier stages, preceding that of TEA specimens. With regard to human specimens, this effort is, of course, hampered by low tissue availability and ethical issues. Thus, a close-meshed investigation of proteasomal capacity in animal models of atherosclerosis could be considered a reliable alternative (Fig. 5).
It is worth a look into the field of aging research at this point, as aging is a strong independent risk factor for atherosclerosis. Nonatherosclerotic aged vessels exhibit characteristics of atherosclerostic vessels, including endothelial dysfunction and intimal thickening (194). Along with an age-related decrease in the rate of protein synthesis, aging leads to a gradual decrease of proteasomal activity. This has been observed in various tissues (33) and it is characterized by the accumulation of ubiquitinated proteins in aged tissue (20). In addition, there is an age-related decrease of the anti-oxidative defense system, resulting in the accumulation of oxidatively modified proteins (20). In addition, ubiquitinated aggregated proteins can impair proteasomal activity (10), leading to proteasomal dysfunction. Atherosclerotic TEA specimens from elderly patients also exhibit higher ubiquitin levels and lower proteasomal activity compared with specimens from a younger cohort (123). Thus, the overlap of these two processes (vascular aging and pathologic atherosclerosis) will certainly complicate the discrimination of the physiological from pathologic response of the UPS in atherosclerosis.
To summarize, in vitro and in vivo evidence suggests that pro-atherogenic stimuli (oxidative stress, modified LDL, and reduced NO bioavailability) influence the proteasomal activity of vascular cells. It is conceivable that a multitude of atherosclerotic risk factors lead to a dysregulation of the UPS in the diseased vasculature, yet uncertainty remains as to whether proteasomal activity is impaired or enhanced in the course of atherogenesis.
Targeting Key Processes of Atherosclerosis with Proteasome Inhibitors
The increasing recognition of atherosclerosis as an inflammatory disease has naturally raised the question as to whether the targeting of NF-κB activation with proteasome inhibitors in vascular cells would be an effective anti-inflammatory treatment. Table 1 summarizes several studies investigating the effect of proteasome inhibition on endothelial cells, VSMC, leukocytes, and platelets. For a number of potential reasons (as outlined next), the treatment of these cell types with various proteasome inhibitors resulted in complex, partly opposing outcomes—potentially atheroprotective and potentially adverse effects (Table 1).
eNOS, endothelial nitric oxide synthase; IL, interleukin.
A considerable number of in vitro studies delineated the anti-inflammatory, vasculoprotective potential of proteasome inhibitors. Especially, it is clear that the reduction of cytokine-induced expression of adhesion molecules on endothelial cells, and the decrease in the stimulated release of inflammatory mediators from leukocytes is attributed to impaired NF-κB activation in most studies. However, the number of processes modulated by the UPS in atherosclerosis (see Key processes of atherosclerosis are regulated by the UPS section) illustrates that proteasome inhibition could potentially affect multiple cellular pathways. In fact, we observed anti-inflammatory effects of proteasome inhibition in vitro and in vivo that were not caused by impaired NF-κB activation but rather by the reduction of oxidative stress (117).
Endothelial dysfunction, characterized by the loss of NO bioavailability and subsequent impaired endothelium-dependent vasodilation, is involved in atherosclerosis from the initiation to the complication phase and can even precede the existence of macroscopically detectable atherosclerotic lesions (35). The key determinants of NO bioavailability are NO production and NO consumption by ROS (21). These processes can be influenced beneficially by using proteasome inhibitors; however, effects have been shown to vary with increasing doses. The treatment of isolated rat aortic rings with low proteasome inhibitor doses improved endothelium-dependent vasodilation, which could be attributed to the induction of an anti-oxidative response in endothelial cells as well as to a suppression of vasoconstrictor endothelin-1 (116, 130, 172). In addition, proteasome inhibition was shown to rescue endothelial dysfunction in diabetic, hypercholesterinemic, and hypertensive mice by enhancing the synthesis of the essential eNOS cofactor BH4 (201 –203). However, opposing results have been shown in hypercholesterinemic pigs, in which proteasome inhibitor treatment impaired endothelium-dependent vasodilation in coronary arteries (69) while improving blood flow in the renal circulation (24), suggesting organ-specific effects of proteasome inhibition. Despite these conflicting outcomes (see Table 1), its interaction with several essential regulatory pathways qualifies the UPS as a potential target for beneficial modification of endothelial (dys)function (171).
Recently, it was reported that the treatment with PR-957, a selective inhibitor of the immunoproteasomal subunit β5i (large multifunctional peptidase 7 [LMP7]), caused a potentially favorable regulation of T-cell differentiation. In an experimental colitis model, this inhibitor suppressed the expansion of T-cell subsets Th1 and Th17 with no effect on Th2 differentiation, but it enhanced the development of regulatory T cells (91). Among lesion infiltrating immune cells, the balance of T-cell subsets influences plaque progression and plaque stability [for overview (64)]. CD4+ T-helper cells (subdivided into Th1, Th2, Treg, and Th17) are detectable in human atherosclerotic plaques. Th1 cells secrete pro-atherogenic cytokines such as IFN-γ and TNF-α; whereas cytokines released by Treg, such as IL-4 and IL-10, are regarded as anti-inflammatory and anti-atherogenic. The impact of Th2 and Th17 helper T cells in atherosclerotic plaques is, however, ambiguous. Upcoming inhibitors of the immunoproteasome could soon enable a more targeted proteasome inhibition, as immune cells constitutively express higher levels of immunoproteasomes. Though not tested to date in atherosclerosis, selective inhibitors of the immunoproteasome provide an interesting perspective.
Another potentially protective effect of proteasome inhibition was demonstrated in the context of lipid metabolism. Here, proteasome inhibitors increased the expression of ATP-binding cassette transporters A1 and ATP-binding cassette transporters G1, thereby enhancing the cholesterol efflux from macrophages and promoting reverse cholesterol transport in vivo. Short-term treatment of BL6 mice with the proteasome inhibitor bortezomib resulted in the enhancement of ATP-binding cassette transporters A1 and a decrease of scavenger receptor class B, member 1, albeit this was not accompanied by a measurable increase in HDL cholesterol serum level (142).
Atherothrombosis occurs, after the erosion or disruption of an atherosclerotic lesion, and it can result in fatal complications such as myocardial infarction and stroke. A few studies suggest anti-thrombotic effects of proteasome inhibitors. In vitro bortezomib has been shown to have an inhibitory effect on platelet aggregation induced by ADP (6). In renovascular hypertensive rats, treatment with the proteasome inhibitor PSI prevented experimental arterial thrombosis (148). The rate of venous thromboembolism is considerably lowered in MM patients treated with bortezomib compared with other chemotherapeutic regimens (29, 166, 205). Without doubt, the anti-thrombotic effect of proteasome inhibitors is worth gaining more attention by cardiovascular researchers, particularly as clinical data in MM patients treated with bortezomib strongly support the experimental observations.
However, the considerable number of studies showing beneficial effects are counterbalanced by reports describing opposite outcomes: namely the induction of oxidative stress, proapoptotic effects, impairment of endothelial function, and enhanced inflammation caused by proteasome inhibition (see Table 1). This controversy led to the following questions: Is proteasome inhibition in atherosclerosis beneficial or detrimental (50, 118, 150)? What are the reasons for different outcomes?
As a matter of course, proteasome inhibition does not affect single proteins or single pathways, but it causes an extensive modulation of cellular function. Thus, among other issues, the degree of proteasome inhibition decides whether a proteasome inhibitor acts as poison, inducing cell death, or as remedy, modulating cellular function (131). This consideration gave rise to studies that have performed comprehensive transcriptomic and proteomic analysis in primary human umbilical vein endothelial cells (HUVEC) treated with differing doses of proteasome inhibitors (13, 130). High-dose proteasome inhibition induced apoptosis in HUVEC, whereas low doses did not affect cell viability. Instead, this low-dose treatment resulted in vasculoprotective expressional patterns in endothelial cells, marked by the up-regulation of several enzymes of the antioxidant defense system, eNOS, and proteins involved in stress response, as well as by the down-regulation of nicotinamide adenine dinucleotide phosphate oxidase subunit Nox-4, vasoconstrictor endothelin-1, and the chemokine MCP-1. These expressional changes are apparently an adaptive cellular response of endothelial cells to nontoxic proteasome inhibition, resulting in an improved protection against oxidative stress (42, 117, 130) and a reduction of the inflammatory response (117), and, subsequently, in improved endothelial function (116). The following experiments revealed that an underlying mechanism mediating the anti-oxidative effects of low-dose proteasome inhibition in cardiovascular cells is the stabilization of Nrf2 (41, 42).
Moreover, divergent effects of proteasome inhibition may be explained by cell type-specific responsiveness. In general, proliferating cells seem to be more sensitive to proteasome inhibitor-induced cytotoxicity than quiescent cells (43). The susceptibility to apoptotic effects of proteasome inhibitors seems to correlate with the rate of protein synthesis (23, 132, 188), accounting for the higher sensitivity of proliferating VSMC and activated macrophages to bortezomib-induced apoptosis compared with resting macrophages (188).
Furthermore, different cell types have divergent compositions of proteasome subtypes. For example, noninflamed endothelial and VSMC predominantly express constitutive proteasomes, whereas vessel infiltrating immune cells constitutively express high amounts of immunoproteasomes. Considering that IFN-γ, an important pro-inflammatory mediator in atherosclerosis, is a strong inducer of the expression of immunoproteasomal subunits, it seems obvious that during the process of atherogenesis a shift in proteasome subunit composition can occur. For cardiac proteasomes, it has been shown that different proteasome inhibitors exert differential inhibitory effects on the various cardiac proteasome subtypes and that different proteasome subtypes are inhibited by the same dose of proteasome inhibitor to a different extent (108). Proteasome subtype composition in the course of atherosclerosis has not yet been investigated, but it is conceivable that proteasome subtype composition influences the response to treatment with proteasome inhibitors.
So far, post-translational modifications such as phosphorylation, nitrosylation, ubiquitination, and acetylation of proteasomal subunits (162) are not systematically investigated in atherosclerotic tissue. However, it is likely that such modification affects not only proteasomal activity (115) but also the susceptibility to proteasome inhibition in cells involved in atherosclerosis.
In conclusion, multiple variables influence the effect of proteasome inhibitors on vascular cells. The cellular context in which a proteasome inhibitor is used substantially influences the outcome. In vitro evidence has elucidated proteasome inhibitor dosage, duration of the treatment, the subunit composition of the targeted proteasomes, and the proliferative status of a cell (proliferating or quiescent) as important variables. These variables represent a challenge for in vivo studies with proteasome inhibitors in atherosclerosis.
Proteasome Inhibition in Atherosclerotic Animal Models
The fact that several key processes of atherosclerosis can be efficiently targeted by proteasome inhibitors in vitro has raised interest in the effect of proteasome inhibition on atherosclerosis development in vivo. At present, the number of animal studies investigating the effect of proteasome inhibitors on atherosclerosis is rather low. With one exception (48), these available animal studies have qualified the effect of systemic treatment with proteasome inhibitors in hypercholesterolemia-driven animal models of atherosclerosis, in which pigs or transgenic mice (low-density lipoprotein receptor-deficient mice LDLR−/−, Apolipoprotein E-deficient knockout mice ApoE−/−) are fed a high cholesterol diet to induce atherosclerotic lesions (69, 188, 197).
Using boronate-type proteasome inhibitor MLN-273 in a 12-week treatment (s.c. twice weekly) of female hypercholesterinemic pigs, Herrmann et al. showed aggravation of early coronary atherosclerosis by proteasome inhibition, displayed as intimal thickening of porcine coronary arteries (69). This led to a 70% reduction of chymotrypsin-like activity in coronary arteries at 24–48 h post MLN-273 injection. MLN-273 treatment also led to increased expression of adhesion molecules (VCAM-1, E-selectin) and macrophage content in coronary arteries, as well as to increased oxidative stress and impaired endothelium-dependent vasodilation (69). Interestingly, in a preceding study of the same group, with the same treatment, endothelial function was improved in the renal circulation (24), suggesting organ-specific effects of this inhibitor in pigs.
A more recent study investigated the effect of proteasome inhibition on atherosclerosis in a widely used hypercholesterolemic mouse model. Employing a carotid artery injury model in female ApoE−/− mice, van Herck et al. comprehensively investigated the effect of low (100 μg/kg bodyweight) and high (500 μg/kg bodyweight) doses of the proteasome inhibitor bortezomib on early and advanced carotid plaques. An intraperitoneal injection of bortezomib resulted in a 49%±9% and 77%±2%, respectively, decrease of chymotrypsin-like activity in the liver measured at 4 h postinjection (188). The four-week bortezomib treatment did not affect collar-induced carotid plaque size in either stage, but both doses changed plaque composition in advanced atherosclerotic lesions toward an unstable plaque phenotype, showing necrotic core enlargement, increased apoptosis, and a decrease in α-SMC-actin and collagen content. Accompanying in vitro experiments confined apoptosis to VSMC and macrophages, of which both cell types showed signs of endoplasmatic reticulum stress due to the accumulation of ubiquitinated proteins (188). The authors concluded that bortezomib treatment promotes a rupture-prone plaque phenotype in the advanced plaques of ApoE−/− mice.
The two studies mentioned earlier suggest a contributing role of proteasome inhibition in atherosclerosis, whereas two more recent studies show the beneficial effects of proteasome inhibition in early atherosclerosis. Uremia-induced accelerated atherosclerosis in rabbits was shown to be reversed by treatment with the proteasome inhibitor MG132 (20 μg/kg bodyweight daily, intramuscular injection) (48). These MG132-treated uremic rabbits had lower aortic NF-κB expression, higher IκBα expression, and lower TNF-α levels as compared with the nontreated uremic control group (48).
In order to test whether the potent anti-inflammatory and anti-oxidative effects observed by low-dose proteasome inhibition in vascular cells in vitro (42, 117, 130) can be translated into anti-atherogenic properties in vivo, the effect of low-dose bortezomib treatment on primary early lesion formation in the aortas of Western diet-fed male LDLR−/− mice was investigated (197). In preceding dose establishing experiments, an inhibitor dose as low as 50 μg/kg bodyweight (intraperitoneal injection twice weekly for 6 weeks) was identified as an effective, nontoxic treatment, which resulted in the inhibition of liver chymotrypsin-like activity by 32.8% measured at 24 h postinjection. Subsequently, bortezomib treatment attenuated early atherosclerotic lesion formation, which could be attributed to the reduction of oxidative stress and vascular inflammation in the absence of pro-apoptotic effects. Gene expression microarray analysis showed that gene expression changes induced by atherosclerosis were attenuated by bortezomib treatment toward an expression pattern similar to healthy animals. This attenuation held especially true for genes associated with the gene ontology term “oxidation reduction.” A favorable regulation of genes by bortezomib was also reflected in the reduced expression of atherosclerosis-relevant genes (e.g., ICAM-1, VCAM-1, IL-6, Nox4, and MCP-1) (197). These results suggest that the protective adaptive response to low-dose proteasome inhibition seen in vitro (130) is reproducible in early atherosclerosis in vivo.
While two of the studies mentioned earlier show aggravation of atherosclerosis, the other two show the beneficial effects of proteasome inhibition in atherosclerosis. Despite these differences, overall, the involvement of the UPS in the pathophysiology of atherosclerosis is highlighted, despite the varying results. Study outcomes were found to differ in terms of oxidative stress, inflammation, and apoptosis. The question arises as to whether this could be, besides the employment of different animal models, due to differences in dosage as well as due to different stages of atherosclerosis being investigated.
The effect of proteasome inhibition on atherosclerosis in these studies clearly varies with the resulting degree of proteasome inhibition. A low degree of proteasomal inhibition showed beneficial effects in early atherosclerosis (197). This effect could be lost with higher inhibitor doses (Fig. 6) (69, 188).

In terms of the different stages under investigation: In the study by van Herck et al., while investigating both early and advanced lesions, bortezomib had no effect on early lesion composition; whereas even a low bortezomib dose promoted features of instability in advanced lesions (Fig. 6) (188). This is in support of the notion that late-stage atherosclerosis itself might be characterized by impaired proteasomal activity, leading to insufficient elimination of dysfunctional proteins as discussed earlier in this review. As a matter of fact, proteasomal dysfunction and insufficiency at this stage would be further deteriorated by additional proteasome inhibition, leading to increased apoptosis and destabilization of plaque architecture. Conversely, preserved or even enhanced proteasomal activity in early-disease stages enables proteasome inhibition as a treatment option to effectively tackle inflammation and ROS production.
Despite partly opposing study results, available animal studies emphasize the influence of the UPS on atherosclerosis development. Future animal experiments will be needed to elucidate the stage-specific function of the UPS. Studies should take into account the strong dose dependency of proteasome inhibitor-mediated effects as well as disease stage-specific effects to validate the potential and the limitations of utilization of proteasome inhibitors in atherosclerosis.
Challenges and Perspectives
Since key processes of atherosclerosis are regulated by the UPS, we are convinced that the UPS is an attractive therapeutic target. To our knowledge, only inhibitors of the 20S proteasome have been investigated in the context of atherosclerosis; while other UPS-related targets, such as proteasomal regulatory particles, ubiquitin ligases, and deubiquitinating enzymes, have not been addressed, leaving an open field for future research and drug development.
Conflicting results of in vitro and in vivo studies has resulted in questions with regard to the feasibility of proteasome inhibition in atherosclerosis. In our view, inconsistent results of the studies mentioned earlier should not discourage future studies with proteasome inhibitors. Even though the therapeutic potential of proteasome inhibitors is obvious, inhibitors such as bortezomib are far from being used as systemic treatment in atherosclerosis. In this context, it should be considered that bortezomib was primarily developed as an anti-cancer agent which does not necessarily fulfill all requirements for potential application in vascular disease. Future proteasome inhibitors targeting inflammation in atherosclerosis need to have suitable characteristics for exactly this application. A prerequisite enabling the formulation of these requirements is comprehensive knowledge on the role of the UPS in atherosclerosis. This knowledge should be extended by systematic investigation of the UPS in all stages, from initiation to complication, and in all cell types involved. To achieve this, approaches using both appropriate animal models and human atherosclerotic tissue are needed.
A future successful proteasome inhibitor first and foremost should have anti-oxidative and anti-inflammatory effects and exhibit minimal toxicity. However, how can this be achieved?
At this point, it should be noted that adverse side effects rule out long-term application of bortezomib. Bortezomib-induced peripheral neuropathy and gastro-intestinal irritation are likely to be attributed to off-target effects against various serine proteases such as cathepsin A, cathepsin G, chymase c, and dipeptidylpeptidase II (5). The newly developed inhibitor carfilzomib, already underway in clinical trials, is obviously more specific; consequently, has fewer side effects; and therefore, might better qualify for an application outside tumor therapy.
Another promising alternative could be proteasome inhibition using reversible, noncovalently binding inhibitors, such as TMC-95 A. This mode of inhibition is suggested to exhibit increased target selectivity and induce less cytotoxic effects in nontransformed cells (9).
Experimental data indicate that sustained inhibition of at least two proteasomal active sites efficiently inhibits protein breakdown and is cytotoxic, whereas partial inhibition of only the chymotrypsin-like site is not sufficient to block protein degradation (104, 165), but results in a protective response of vascular cells (130). Therefore, site-specific inhibitors, which specifically target one catalytic subunit (105), could have potential for noncytotoxic treatment.
The specific targeting of the immunoproteasome is another promising approach. β5i-selective inhibitor of PR-957 (ONX0914) has been proved to be effective in treating experimental rheumatoid arthritis (135), lupus (77), and colitis (91). PR-957 not only caused a reduction in autoimmune antibody production but also effectively decreased the concentration of a number of inflammatory cytokines (135) and resulted in a favorable shift in T-cell differentiation, as described earlier (91). Potentially, proteasome inhibitors specifically targeting the immunoproteasome could be considered a successful strategy in atherosclerosis. However, the function of immunoproteasomes in the initiation, progression, and complication of atherosclerosis should be investigated in detail.
Atherosclerosis is recognized as a chronic inflammatory disease, but long-term systemic anti-inflammatory treatment is expected to be associated with an inacceptable risk-to-benefit ratio (27, 177). Therefore, chronic systemic treatment with even well-suited proteasome inhibitors may not be feasible. Alternatively, innovative techniques enabling a lesion-directed delivery of therapeutics to atherosclerotic vessels on a controllable time scale (96) could expand the application spectrum for anti-inflammatory agents, including proteasome inhibitors.
For vessel restenosis, an undesired renarrowing of arteries after angioplasty in patients with occlusive vascular diseases, the local application of proteasome inhibitors could be considered a feasible approach. A proof-of-concept experiment showed that short-term local application of a single high dose of proteasome inhibitor MG132 in a rat carotid balloon injury model effectively reduced neointima formation corresponding to a strong anti-proliferative effect (129).
Concluding Remarks
Exploring opportunities that target the UPS in atherosclerosis is challenging. However, a substantial gain of knowledge in both the understanding of atherosclerosis and the UPS can be expected and will further stimulate this important area of research (Fig. 7). Therefore, it will be essential to reinforce basic research using animal models and human specimens to delineate the role of the UPS in the initiation and progression of atherosclerosis and to identify new molecular targets for treatment, such as disease-associated ubiquitin ligases, deubiquitinating enzymes, and regulatory subunits of the proteasome. The clinical use of second-generation inhibitors, such as carfilzomib, will provide information about their therapeutic potential in and outside the field of oncology. New proteasome inhibitors, in particular site-specific inhibitors, will be developed and should be tested in atherosclerosis. In the future, techniques for a lesion-targeted application could support this translation from basic science into clinical application, thereby avoiding disadvantages of chronic, systemic application. Moreover, the establishment of appropriate biomarkers and novel noninvasive imaging techniques will enable a monitoring of the effects of therapeutic interventions. We are convinced that targeting UPS will remain a promising therapeutic approach as this knowledge on atherosclerosis widens and new inhibitors emerge.

Footnotes
Acknowledgments
The authors would like to apologize to researchers in the field whose work could not be cited. They are grateful to Katrina Binger for critically reading this article. This work was supported by a grant from the foundation Deutsche Stiftung für Herzforschung [F/02/05] and a grant from the Charité-Universitätsmedizin Berlin to AL.