
Abstract
Introduction
O
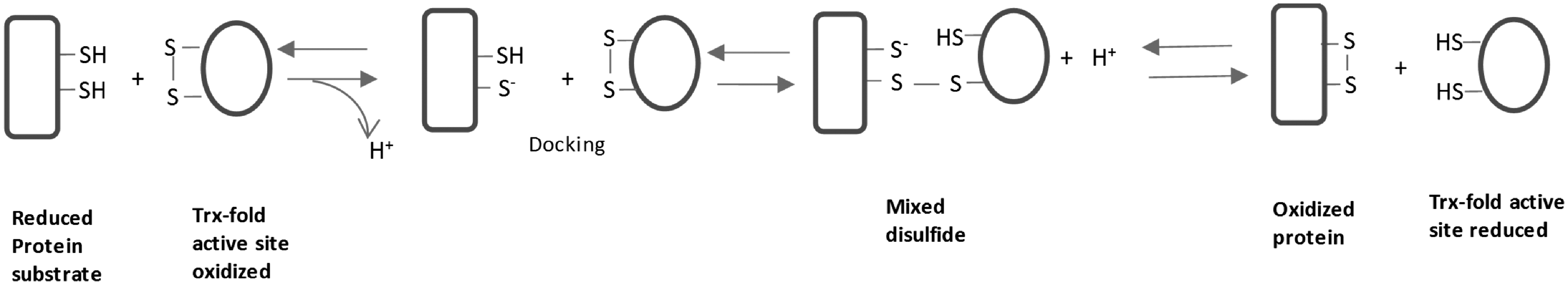
In this equation, A-(SH)2 is a protein substrate with a dithiol, and B-S2 is an enzyme catalyzing the disulfide bond formation. After the reaction, the dithiol in the protein substrate A has been changed to a disulfide bond. Most of the enzymes catalyzing the disulfide bond formation belong to the thioredoxin (Trx) superfamily, containing Trx folds in their structure (Fig. 2A) (Table 1). Trx is a ubiquitous small protein and plays critical roles in many cellular functions such as redox regulation and DNA synthesis via its disulfide reductase activity. Trx receives electrons from thioredoxin reductase (TrxR) and nicotinamide adenine dinucleotide phosphate (NADPH) and catalyzes substrate reduction, donating electrons to an oxidized substrate, while disulfide bond formation catalyzed by protein disulfide isomerase (PDI) and similar oxidases involves the transfer of electrons from the reduced substrate to the active site of PDI (Fig. 1). In this review, we focus on how Trx superfamily proteins assist the disulfide bond formation during the oxidative protein folding process.

ER, endoplasmic reticulum.
Trx Superfamily Proteins
Trx was originally discovered as a hydrogen donor for ribonucleotide reductase (74, 83) and can catalyze the reduction of various protein disulfides by different reductants (45, 76). The primary structure determination revealed that Escherichia coli Trx contains an active site with -CGPC- motif (42), which is now known to be conserved in nature. The crystal structure of Trx showed that it contains a distinct structural motif consisting of a four-stranded β-sheet and flanking α-helices (Fig. 2A) (48, 55), which is named the Trx fold (84). The structure was first identified in five classes of proteins: Trxs, glutaredoxins (Grx), glutathione S-transferases, DsbA, and glutathione peroxidases (84). Now, more and more proteins, including peroxiredoxin (Prx), arsenate reductase (ArsC), quiescin sulfhydryl oxidases (QSOX), and vitamin K epoxide reductase (VKOR), have been found to possess this structure. The Trx superfamily proteins illustrate diversity in function and domain structure (Fig. 2) (8), but most these proteins are involved in redox-related reactions.
General Physical Chemistry Basis for Thiol–Disulfide Exchange Reaction
Disulfide bond generation involves thiol-disulfide exchange, and thus, the pKa value of the active site Cys residue and the redox potential are two key factors that determine the thermodynamics and kinetics of this redox reaction (56, 104). Redox potential represents the reduction capacity in redox reactions and pKa determines the existence of thiolate in an active site Cys. The thiol-disulfide reaction involves the nucleophilic attack of a thiolate on a disulfide bond (Fig. 1) (99, 130), a deprotonated form is much more active than a thiol (60). To determine whether a chemical reaction such as the thiol-disulfide exchange reaction is thermodynamically favorable, change in Gibbs free energy (ΔG) of the reaction should be considered. The ΔG for a reaction like (a) can be calculated as follows:
where, R is the gas constant, T is the absolute temperature, ΔG0 is change in standard Gibbs free energy of reaction, n is number of electrons per mole product, F is the Faraday constant, and
If reaction (a) is at equilibrium, ΔG=0,
To determine the standard redox potential of a protein, a reaction with oxidized glutathione (GSSG) (b) is normally used. Thus, the protein
where, P-(SH)2 is a protein involved in the redox reaction with GSSG; K′ is the apparent equilibrium constant.
Since reduced glutathione (GSH) is the most abundant small thiol in the cells and provides major thiol buffer systems to maintain the cellular redox environment, generally, the half-cell redox potential (E
h) of the GSH buffer system is used as a reference of the cellular redox state (64). The high value of E
h represents a weak reducing capacity. The E
h can be calculated from the ratio of GSH to GSSG and total amount of glutathione according to Equation (b3) (Fig. 3A).
where in the equation,

Oxidative Protein Folding in ER
Protein folding is a fast process, the local structure such as α-helices can occur less than 100 ns, and β-sheet can form in 1 μs (26, 27). Thus, it is not surprising that in a cell, some proteins can fold cotranslationally when the nascent chain is still attached to the ribosome. However, other proteins need to translocate to specific compartments, such as ER and mitochondrial IMS, to complete their folding into three-dimensional structures with the help of chaperones, thiol-disulfide oxidoreductases, and other catalysts (26). After getting the correct structure, including the correct disulfide bonds, the proteins with high stability can be then transported from the ER to the Golgi complex and secreted to extracellular environments (29).
Structure and function of PDI
The best known enzyme to catalyze the oxidation and isomerization of disulfide bonds in ER is PDI (28, 40). Human PDI is a 55-kDa enzyme and consists of 491 amino acid residues. Yeast PDI is essential for yeast cell survival (109) and the indispensability is due to its disulfide oxidase activity (135). PDI is multifunctional in the protein folding process. Besides the thiol-disulfide oxidoreductase activity, PDI is known to have the chaperone and antichaperone activity (40, 101, 102). Moreover, PDI is a substrate for TrxR and can act as a Trx-like protein (80).
The early amino acid sequence analysis showed that the structure of PDI contains multidomains (28). Now, it is known that PDI have four



The active site containing domains
Recently, the crystal structure of human PDI showed that PDI is a flexible molecule (117) and that the four Trx domains are arranged as a horseshoe shape (118, 127) (Fig. 2D). The PDI has a closed reduced conformation and an open oxidized form. The oxidized PDI binds the unfolded/reduced substrate with its large accommodating cleft and extensive hydrophobic area to initiate the oxidative folding process. After the transfer of the disulfide bond to its substrate, PDI gets reduced and switches to a closed conformation, which causes the release of the oxidized substrates, including the native protein with correct disulfide folding and the misfolded protein. The later can bind reduced PDI and convert to native protein through isomerization (Fig. 4) (127).
PDI-mediated oxidizing equivalent transfer pathway
Since the ratio of [GSSG]/[GSH] is much higher in ER than that in cytosol and PDI can pass the oxidizing equivalent to the substrate thiols in vitro, GSSG was originally assumed to be the main oxidant of PDI in ER (50). However, the mutation of the gshA gene resulted in no detectable GSH in the yeast, but did not affect the oxidative protein folding (30). In contrast, endoplasmic reticulum oxidoreductin 1 (Ero1p), a 57-kDa protein, is required for disulfide bond formation in the ER (30, 100). Moreover, the protein oxidation can be recovered in ero1 mutant yeast by the decrease of cellular GSH caused by the gshA gene deletion, indicating that GSH contributes to a reducing equivalent in yeast ER (19). There are two Ero1p-like proteins in mammalian cells, Ero1-Lα and Ero1-Lβ. These human proteins can be used to complement the deficiency of Ero1p in the yeast. The ero1 yeast mutant was shown to be sensitive to temperature and DTT, while expression of the two human proteins in the yeast mutant alleviated their DTT and temperature sensitivity (13, 97). Moreover, in the ero1 mutant, protein thiol oxidase Erv2p is screened to be the protein to restore the viability and disulfide bond formation to complement the deficiency of Ero1p (36). Both Ero1p and Erv2p are flavoproteins (36, 121) and the oxidizing equivalent is from flavin adenine dinucleotide (FAD) and oxygen (122). The oxidation of PDI by Ero1 is a thiol-disulfide exchange process, while also producing H2O2 (Fig. 5A) (36). Ero1p contains multicysteine pairs. The active site cysteine pair C352–C355 is adjacent to the FAD cofactor and the C100–C105 pair on a flexible loop. The oxidizing equivalents pass from FAD to C352–C255, then shuttle to C100–C105, and finally to PDI (35, 111). The property of the two pairs of cysteines consisting of the active site for the transfer of oxidizing equivalent has also been found in Erv2p. The first C121–C124 pair is adjacent to the isoalloxazine of the FAD, and the second C176–C178 pair is located in a flexible C-terminal segment. The mutation of any of these Cys residues will disrupt the electron transfer activity of Erv2p (37). Very interestingly, besides these active site cysteine pairs, several noncatalytic Cys pairs participate in the regulation of Ero1 activity and prevent the ER from getting into further oxidizing conditions (6, 112). The
On the other hand, the role of GSH in the oxidative folding has been re-evaluated (5, 16, 40). GSH was shown to be required to maintain the redox state for native disulfide bond formation in the ER (Fig. 5A). Depletion of GSH resulted in the acceleration of disulfide bond formation, but this disulfide bond formation is non-native, indicating that GSH may play a critical role in isomerization (16). A DTT wash-out experiment showed that oxidation of PDIs and glutathione levels is rapidly and efficiently regulated, indicating that there may be Ero1α-driven and GSSG-driven oxidation in ER (7). In yeast cells overexpressing Hgt1, an extensive GSH and GSSG transporter, supplementation with GSH can convert the fully oxidized Ero1p to reduced form, indicating that GSH may diffuse into the ER and reduce regulatory disulfides in Ero1p (72). More recently, PDI1p was shown to be a key regulator for Ero1p activity. PDI1p responses to free thiol and glutathione redox levels in the ER to mediate Ero1p activity (65). Moreover, several other disulfide formation pathways involving Trx superfamily proteins have been proposed in the ER, which will be described below.
PDI homologues
Many PDI-like proteins have been found in eukaryotic cells. In Saccharomyces cerevisiae, there are five putative PDI genes, but only the PDI1 gene is essential. The other nonessential PDI homologues in yeast are Eug1p, Mpd1p, Mpd2p, and a transmembrane protein Eps1p (135). These proteins can be oxidized by Ero1p in vitro and the N-terminal domain in PDI1p has the highest reaction rate (125). In human cells, about 20 proteins have been defined as PDI-like proteins in the ER, and the structure of all the proteins contains at least one Trx-fold domain (40). ERp57, pancreas-specific PDI (PDIp), ERp72, P5, ERp44, ERp29, ERdj5, ERp27, and many transmembrane PDI family proteins have been well studied and many of them have showed the catalyzing activity on the disulfide bond formation in vitro (40, 68). The PDI family members show distinct substrate specificity, which may explain why there are so many PDI homologues present in the ER (57), but the physiological functions of most proteins are not clear (40).
Thiol-dependent peroxidases
The H2O2 generated in the metabolism mediated by Ero1 and PDI, together with H2O2 produced by NADPH oxidase and mitochondrial respiration, can be used as an oxidizing source for oxidative protein folding. Several ER-resident thiol-dependent peroxidases, including Prx4 and two glutathione peroxidase homologues, GPx7 and GPx8, participate in transferring the oxidizing equivalent to substrate thiols (Fig. 5B) (59, 137). These proteins belong to the Trx superfamily, but they do not have the classic CXXC active site motif. Prx4 is a typical 2-Cys Prx. Like other typical 2-Cys Prxs (Prx1–3), the reaction mechanism of Prx4 involves in an initial reaction of peroxidatic Cys124 with H2O2 to produce water and a Cys sulfenic acid intermediate. Then, the sulfenic acid can react with a resolving Cys residue (Cys245) in the adjacent Prx4 molecule, leading to the formation of homodimeric Prx4 with intermolecular disulfide bonds. Different from other typical 2-Cys Prxs, which can receive electrons from the Trx system to run the catalysis cycle (77), Prx4 has both Trx- and glutathione-dependent peroxidase activity (59). The structures of reduced and oxidized Prx4 are stable decamers consisting of five dimers, forming a ring donut structure (Fig. 2F) (15, 128). The Prx4 reduction can be efficiently and specifically catalyzed by the PDI family proteins, including PDI and ERp46 (57, 116); GSH enhances this process (116). Moreover, input of mammalian Prx4 can complement the temperature sensitivity of ero1 yeast mutant. Ero1 deficiency in mammalian cells has only modest effects in disulfide bond formation, but Ero1 knockout mouse embryo fibroblast cells were intolerant of Prx4 knockdown (138). These results show that Prx4 plays a critical role in oxidative protein folding in the ER.
GPx7 and GPx8 are selenium-independent glutathione peroxidases, most of these type of GPxs do not receive electrons from the GSH system, but from the Trx system. The structure of human GPx8 exhibits a typical Trx fold (120). Interestingly, the H2O2 scavenging activity of GPx7 and GPx8 can be catalyzed by many PDI family members and Ero1α has been shown to interact with GPx7 and GPx8 in vivo (95), indicating that the formation of two GPx-mediated disulfide bonds may be another pathway to utilize the H2O2 produced by Ero1α.
Quiescin sulfhydryl oxidase
The QSOX family of proteins catalyzes disulfide bond formation, reducing molecular oxygen to produce H2O2 (Fig. 5C). In humans, QSOX can be secreted into extracellular fluids and also was reported to be located intracellularly in the ER, Golgi, and secretory granules (69). QSOX contains one or two Trx domains and an Erv domain (Figs. 2G and 6) (3). One Cys pair is located in the Trx1 domain and two Cys pairs are in the Erv domain, which is also a FAD-binding domain. The proposed mechanism involves the reduction of O2 by FAD to generate the disulfide bond in the FAD proximal Cys pair, and the transfer of the disulfide bond to another Cys pair in the Erv domain, then to the Cys pair in the Trx domain, and finally to the substrate by a thiol-disulfide exchange reaction, a similar oxidizing equivalent transfer process of the yeast Erv2-PDIp system (69).
Vitamin K epoxide reductase
Mammalian VKOR is a membrane protein that catalyzes the reduction of vitamin K 2,3 epoxide and vitamin K to vitamin K hydroquinone, which is required to sustain blood coagulation (119). A bacterial homologue of VKOR shows that the enzyme contains four transmembrane helix bundles that surround a quinone, an additional transmembrane segment, and a Trx domain (Fig. 2H). Three pairs of Cys-Cys are located in the Trx domain and two are in the transmembrane bundle, which are critical for the electron transfer. The oxidizing equivalent may be transferred from quinone to the adjacent Cys pair and then to the Cys pair in the Trx domain and finally to substrate dithiols (Fig. 5D) (75). VKOR strongly reacts with the ER membrane anchored PDI homologue TMX, an integral membrane protein of the ER with an unusual CPAC active site sequence (110). Recently, Ero1, Prx4, and VKOR were found to be of importance for cell viability, secretion, and recovery after reductive challenge. No involvement of QSOX1 in the ER oxidative process was detected in human hepatoma cells (108).
S-Nitrosylation of PDI and ER Stress in Metabolic and Neurodegenerative Diseases
A delicate balance is maintained in the redox states controlling the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and the antioxidant systems that detoxify them. Increased ROS/RNS production or a decreased antioxidant capacity will lead to oxidative/nitrosative stress (1, 92, 123). A particular striking example is the accumulation of misfolded proteins through severe ER stress in neurodegenerative diseases like Parkinson's diseases and Alzheimer's diseases (92, 93). Lipton et al. identified S-nitrosylated PDI (123) as an inhibited protein, which lost its function to prevent ER stress and protein aggregation and loss of proteasome function (1, 92, 93). In addition, S-glutathionylation of PDI has been shown following nitrosative stress (124). A consequence of this is to regulate estrogen receptor α stability and function (136). ER stress is known to dramatically affect protein folding homeostasis and be associated with many metabolic diseases through the unfolded protein response (14). Recently, some small molecule chaperones have yielded promising results. For example, chemical chaperones tauroursodeoxycholate and 4-phenylbutyrate, which reduce ER stress and prevent protein misfolding, have been used in small clinical trials, demonstrating therapeutic potential in multiple metabolic diseases (14).
Oxidative Protein Folding in Bacterial Periplasm
The study on oxidative protein folding in bacteria started from the identification of DsbA as a protein required for the disulfide bond formation in 1991 (11). E. coli has been used as the model to investigate bacterial oxidative protein folding mechanisms. By screening for the sensitivity/resistance to sublethal levels of DTT, the other periplasmic enzymes, involving oxidative protein folding, DsbB, DsbC, and DsbD, have been identified and cloned (88 –90). Now, the DsbA-mediated thiol-disulfide pathway gets more and more clear and DsbA becomes a potential drug target because of its role as a virulence-associated factor in some pathogenic bacteria (41, 113).
Dsb family proteins
E. coli DsbA is a monomeric 21 kDa periplasmic oxidase that catalyzes disulfide bond formation with a C30PHC33 active site, a classic Trx active site motif (11). The structure of DsbA consists of a Trx domain and a helical domain (Fig. 2B) (85). DsbA is a highly active oxidizing enzyme with a redox potential of −124 mV and a very low pKa (about 3.5) for the N-terminal Cys30 in active site motif (34, 94, 132). The special properties make the oxidized enzyme able to accept electrons from reducing substrates easily via a thiol-disulfide exchange reaction to become reduced. The dithiol in the active site of DsbA then can be oxidized to disulfide by DsbB, a 20-kDa protein (88) that is located in the bacterial inner membrane (Fig. 7). DsbB contains four membrane-spanning structures and two periplasmic loops, in which four essential Cys are located, the Cys41–Cys44, and the Cys104–Cys130 pairs (38, 54). DsbB has similar structural features to Ero1p and Erv2p, all have the four helix bundles and other two domains containing active site Cys pairs (35, 37, 53). The redox cycling process involves the electron transfer from Cys30 of DsbA to Cys104 of DsbB, then to Cys130, and Cys130 attacking the Cys41–Cys44 pair and finally transferring electrons to ubiquinone under aerobic conditions and menaquinone under anaerobic conditions (Fig. 7) (9, 67). In the case of electron transfer from PDI to Ero1p or Erv2p, cofactor FAD serves as the electron acceptor.

Different from PDI, which has oxidase and isomerase activity, DsbA functions almost entirely as an oxidase. The misfolded protein disulfide bond isomerization in E. coli is performed by the DsbC-DsbD system (89, 90). DsbC is a homodimeric periplasmic protein, with subunit molecular weights about 23 kDa. DsbC is a V-shaped structure consisting of a Trx domain and an N-terminal dimerization domain, which are joined via a helix linker (Fig. 2C) (86). The location of active site Cys98–Cys101 and adjacent residues resembles that of the other Trx superfamily proteins, including DsbA (86). A DsbC homologue DsbG, also located in the E. coli periplasm, functions as a disulfide isomerase (4, 12). In wild-type cells, DsbC and DsbG are in a reduced form and electrons are obtained from DsbD (103). DsbD consists of 546 amino acid residues and contains two periplasmic domains, an N-terminal domain (DsbDα), and a C-terminal domain (DsbDγ), which are connected by a central transmembrane domain (DsbDβ) (Fig. 6). The N-terminal domain has an immunoglobulin-like fold, the central domain is predicted to have eight α-helices, and the C-terminal domain possesses a Trx fold. Each of these domains contains a pair of Cys, which contributes to its function (33, 61). The electrons may flow from the Cys pair in the central transmembrane domain of DsbD to the C-terminal Cys pair, then to the N-terminal Cys pair, and finally to the DsbC and DsbG active site. The DsbD central domain receives electrons from the cytoplasmic Trx system (Fig. 2C) (18, 107).
Trx and Grx in Oxidative Protein Folding
Trx and Grx systems are the major cellular disulfide reductases in cells (46). Trx is the electron donor for many critical enzymes, such as ribonucleotide reductase, Prx, and methionine sulfoxide reductase, and thus is involved in a wide range of cellular functions (46). Grx has been shown to be a good oxidase and can accelerate protein-dependent folding together with PDI (79, 134). It is known that many ER-resident Trx-fold-containing proteins, including PDI, CaBP1, and CaBP2, are substrates for TrxR (78, 80) and disulfides in secreted proteins like insulin can be reduced by Trx (43, 44, 47). Moreover, the Trx system can provide the electrons for the vitamin K cycle in vitro (114), but the exact roles of the Trx and Grx systems for oxidative protein folding in the ER need further investigation.
For oxidative protein folding of E. coli, Trx can provide the electrons to E. coli DsbD and contribute to the isomerization of incorrectly paired disulfide bonds (Fig. 7) (107). Detailed studies show that the transmembrane domain of DsbDβ is symmetrical; the Cys163–Cys265 pair in DsbDβ is water exposed in the central of the membrane, and accessible to both partners of Trx, the cytosolic Trx and DsbD Trx domain DsbDγ (18). The
However, not all thiol-disulfide exchange reactions are thermodynamically favorable, judging from
Concluding Remarks
In summary, all cells consist of various compartments with different specific functions. The oxidative compartments, including the ER, mitochondrial IMS, and bacterial periplasm, are responsible for the secretion of the correctly folded proteins. The Trx superfamily proteins serve as key bridges to facilitate correct oxidative protein folding, involving thiol-disulfide exchange reactions with their active Cys pairs in the Trx structure. The thiol-disulfide exchange processes occur not only in one compartment, but also involve the electron transfer between different compartments via Cys pairs in membrane proteins.
Footnotes
Acknowledgments
The authors acknowledge the support from the Swedish Research Council Medicine (3529), the Swedish Cancer Society (961), the K.A. Wallenberg Foundation, Åke Wiberg Stiftelse, and the Karolinska Institutet.