
Abstract
Introduction
W
A number of NMR pulse sequences have emerged in recent years, which greatly reduce experimental acquisition times (e.g., selective optimized flip-angle short transient and fast heteronuclear single quantum coherence) (22). These techniques are a promising approach for studying short-lived protein structures, but they invariably cause a substantial loss of sensitivity and are (presently) limited to two-dimensional spectral acquisition rates of around 1 Hz (22, 66). An alternative approach for studying protein folding “intermediates” is to perturb the native structure via the addition of mildly denaturing solvents under equilibrium conditions (53, 81). This can allow for the acquisition of high-resolution structural data (by NMR and other methods), but it is questionable whether intermediates populated under nonnative conditions are the same as those that are transiently populated as the protein folds under native conditions (20, 47). Biophysical NMR has proven to be an invaluable tool for studying protein dynamics, particularly with the advent of Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments, which allow quantitative measurements of the rate of transition between two (or more) conformational states (39, 67). However, CPMG and most other NMR-based methods for studying protein dynamics are subject to the inherent limitations of NMR, which puts restrictions on analyte size, sensitivity, and the accessible timescale.
Mass spectrometry has developed rapidly over the past three decades as a robust and sensitive technique for studying protein folding, structure, and dynamics (17, 37, 38, 44). Of the two “soft” ionization techniques most often used for studying proteins—matrix-assisted laser desorption ionization and electrospray ionization (ESI)—ESI has proven to be more appropriate for direct structural analyses because of the link between protein structure in solution and the extent to which the protein acquires charge upon ionization (14, 54). However, direct ESI measurements alone provide an exceedingly coarse picture of structure, offering no information beyond overall compactness. To achieve true spatial resolution with “site-specific” measurements of structure and dynamics, ESI mass spectrometry can be combined with structure-dependent labeling techniques, the two most common being hydrogen/deuterium exchange (HDX) and oxidative labeling. This review focuses on experimental methods and applications of oxidative labeling, which provide a facile and comparatively “artifact-free” measurement of protein structure and dynamics through the lens of solvent accessibility.
Structure-dependent labeling techniques
HDX is likely the most widespread mass spectrometry (MS)-based labeling technique for studying conformational dynamics and protein structure (3, 31, 65, 104). Briefly, in “forward” exchange, proteins in H2O are diluted into D2O, resulting in deuterium uptake on the protein where protons are solvent exchangeable (i.e., NH, OH, and SH groups). By MS, this process is detected as a mass increase in the protein, usually in a time-dependent manner. Side chain exchange is ultra-rapid and largely independent of structure and is thus usually neglected; however, backbone amide proton exchange occurs on a manageable timescale and is highly structure dependent. One of the major technical challenges of HDX-MS is the occurrence of hydrogen back-exchange, which has the effect of reducing the sensitivity of the measurement to structure (120). A number of creative approaches have been devised to circumvent this problem, most involving cooling and minimization of the duration of the proteolysis step (85).
Chemical cross-linking is another MS-oriented labeling technique used for structural analysis (and to a lesser degree, for dynamics). It involves the formation of intramolecular and intermolecular covalent bonds between amino acid side chains, ultimately yielding distance constraints that give information on tertiary and quaternary protein structures (105). This approach does provide low-resolution structural information, and the development of higher-resolution cross-linking probes is ongoing, but interpretation of fragmentation patterns from cross-linked peptides can be exceedingly complicated, and this problem worsens with increasing resolution and analyte size (42, 64, 106).
Covalent labeling techniques relying on reactive oxygen species (ROS), such as hydroxyl radical (•OH) footprinting, have emerged as a highly complementary approach to conventional structural analyses. •OH react with amino acid side chains to form unreactive covalent bonds, resulting in a “permanent” label that is not subject to scrambling effects or back-exchange (64). In addition, oxidative labels are relatively easy to retain through conventional collisionally activated dissociation (CAD)-based peptide fragmentation. Care must still be taken, however, as dissociation during MS/MS can still occur via a number of pathways, including neutral loss of methane sulfenic acid (CH3SOH) via methionine oxidation (82), C-terminal cleavage of the amide bond from cysteine oxidation to cysteine sulfinic acid (107), and histidine oxidation (7). Although HDX is sensitive mainly to backbone amide hydrogen bonding, oxidative labeling is more sensitive to side chain solvent accessibility, a property that is thought to arise from the fact that many ROS, particularly •OH, have similar size and polarity to water (96). Although HDX and oxidative labeling are complementary techniques, their experimental workflows and data interpretation considerations are dissimilar. For a thorough comparison of these approaches, see reference (46).
It should be noted that in all nonisotopic labeling regimens, there is a chance that the incorporation of label, which is after all a chemical modification of the protein, can perturb the structure (45, 64). The degree of structural perturbation in oxidative labeling depends on two main factors: the timescale of ROS exposure (30) and the nature of the label (100, 111). Radical exposure times in the range of 10 μs to minutes may induce nonnative conformational changes, resulting in a labeling profile that reflects multiple nonnative structures (45). The “natural” lifetimes of ROS in solution depend on the nature of the reactive species and can range from nanoseconds to hours (93). ROS lifetimes are typically controlled by introducing a radical scavenger, such as ascorbate, the concentration of which is critical to balancing sufficient labeling with structural perturbation (112). In the case of •OH, several strategies such as peptide internal standards (100) and fluorescent dyes (111) have been used to normalize exposure. High protein concentrations, which are often required when looking at weak protein–protein interactions, can complicate oxidative labeling due to the monomeric proteins themselves acting as ROS scavengers (2, 26, 28).
Of the ROS suitable for oxidative labeling experiments, •OH are the most commonly used, mainly because they are highly reactive and can be readily generated using a broad range of approaches and under a wide range of conditions (98).
Spatial resolution
Spatial resolution in MS-based structure-dependent labeling experiments can be achieved by fragmentation of the protein after labeling, localizing the label to specific peptides that can be mapped back onto the structure (if one is available). Fragmentation is achieved either before ionization using a protease-like trypsin or pepsin (bottom-up) or postionization using a gas-phase fragmentation technique, such as CAD, electron capture dissociation, or electron transfer dissociation (top-down) (79, 80, 97). Top-down experiments are particularly useful as they can allow site-specific measurements (72). In the case of top-down HDX experiments, however, extreme care must be taken to avoid hydrogen “scrambling,” which randomizes the labeling profile (35). In addition, nonergodic fragmentation methods, which are required to avoid scrambling are generally inefficient, resulting in sensitivity issues, and they impose a size limit (36, 118, 119). As mentioned previously, oxidative labeling is generally thought to be free of scrambling artifacts, although the possibility cannot be entirely ruled out in the case of rare CAD-based fragmentation mechanisms, such b-ion cyclization, which can cause sequence scrambling (87).
Generating ROS
At elevated levels of oxidative stress, •OH are the most destructive ROS in biology: they have been implicated in damaging carbohydrates, altering protein function, cleaving DNA and other nucleic acids, and collapsing lipid membranes (91). The short half-life of •OH (10−9 s) ensures that oxidative damage from this species is restricted to very near the site of generation. Unfortunately, in a biological context, the majority of •OH is generated by homolytic cleavage of the much longer-lived and free-diffusing precursor hydrogen peroxide (H2O2) (23). Homolysis or degenerative reduction of H2O2 can occur in a number of ways, many of which can be harnessed to generate •OH in oxidative labeling experiments in vitro.
The Fenton chemistry has been established for almost 120 years and is one of the earliest methods known to produce •OH (21). The reaction, initially discovered by Fenton himself (21), and later described mechanistically by Haber, Willstätter, and Weiss (29), includes a means to generate •OH radicals from H2O2 using reduced transition metals M(n) as part of a larger catalytic cycle, as shown in Scheme 1.

Fe(II) was established as the original “Fenton reagent”; however, other redox active metals have shown a propensity for “Fenton-like” chemistry—some producing •OH faster than the original reaction (108, 111). Many of these Fenton-like reactions occur naturally in cellular environments and have been implicated in aging as well as a variety of diseases (78, 89). The second-order rate for the production of •OH from aqueous Fe(II) is 60 M −1 s−1; too slow to be of practical use in oxidative labeling experiments. However, Fe(II) chelation, for instance by ethylenediaminetetraacetic acid, results in a 100-fold rate enhancement (108). This type of chelated Fenton chemistry was first used in oxidative labeling as a footprinting method for the identification of protein–DNA binding surfaces (16, 102). It has since found use in identifying protein–protein interactions (33), probing protein solvent-accessible surface areas (88), and more recently to probe the differences in conformation of oxidized versus unmodified proteins by ion mobility-MS (90).
In Fenton-based oxidative labeling experiments where the reduced transition metal is limiting, for instance in experiments that probe metal binding sites using the bound metal itself as the Fenton-like reagent, oxidation can be enhanced by closing the catalytic cycle using a reducing agent, as shown in Scheme 2 (49):
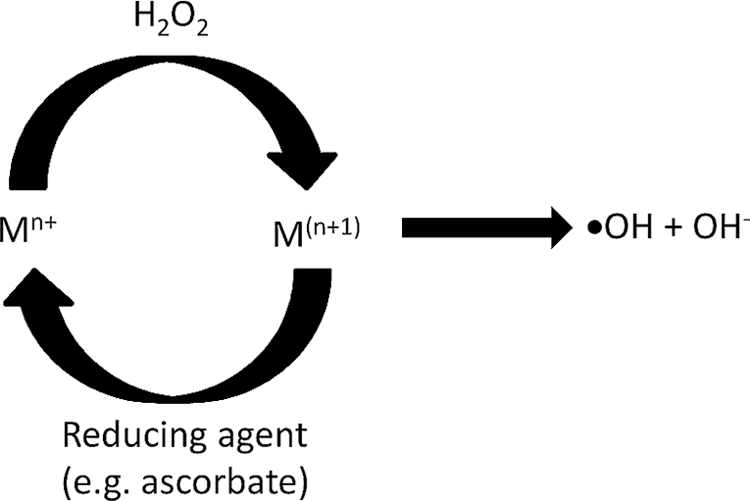
Where in this case, ascorbate plays the dual role of reductant, which ensures the continuous production of •OH, and scavenger of oxidation products, which helps localize oxidation to the immediate vicinity of radical production (8). The Fenton-like reactions are a slow way of generating ROS, with some processes taking 30 min or more for oxidation to go to completion. The addition of sodium persulfate (S2O8 2−) and use of microwaves can increase reaction times to as short as 2 min (9).
One of the most effective ways to produce •OH is by exposing water to high-energy sources like X-rays, γ-rays, and β particles (98, 111). The pioneering work of Chance and coworkers (57) showed that modifying amino acid side chains with •OH produced from X-ray sources can be combined with mass spectrometry for characterizing protein structure. Synchrotron X-rays generate •OH from water as shown in Scheme 3:

And under aerobic conditions, the following reaction occurs, producing superoxide anions and hydroperoxyl radicals, as shown in Scheme 4 (57).
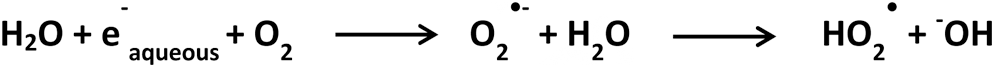
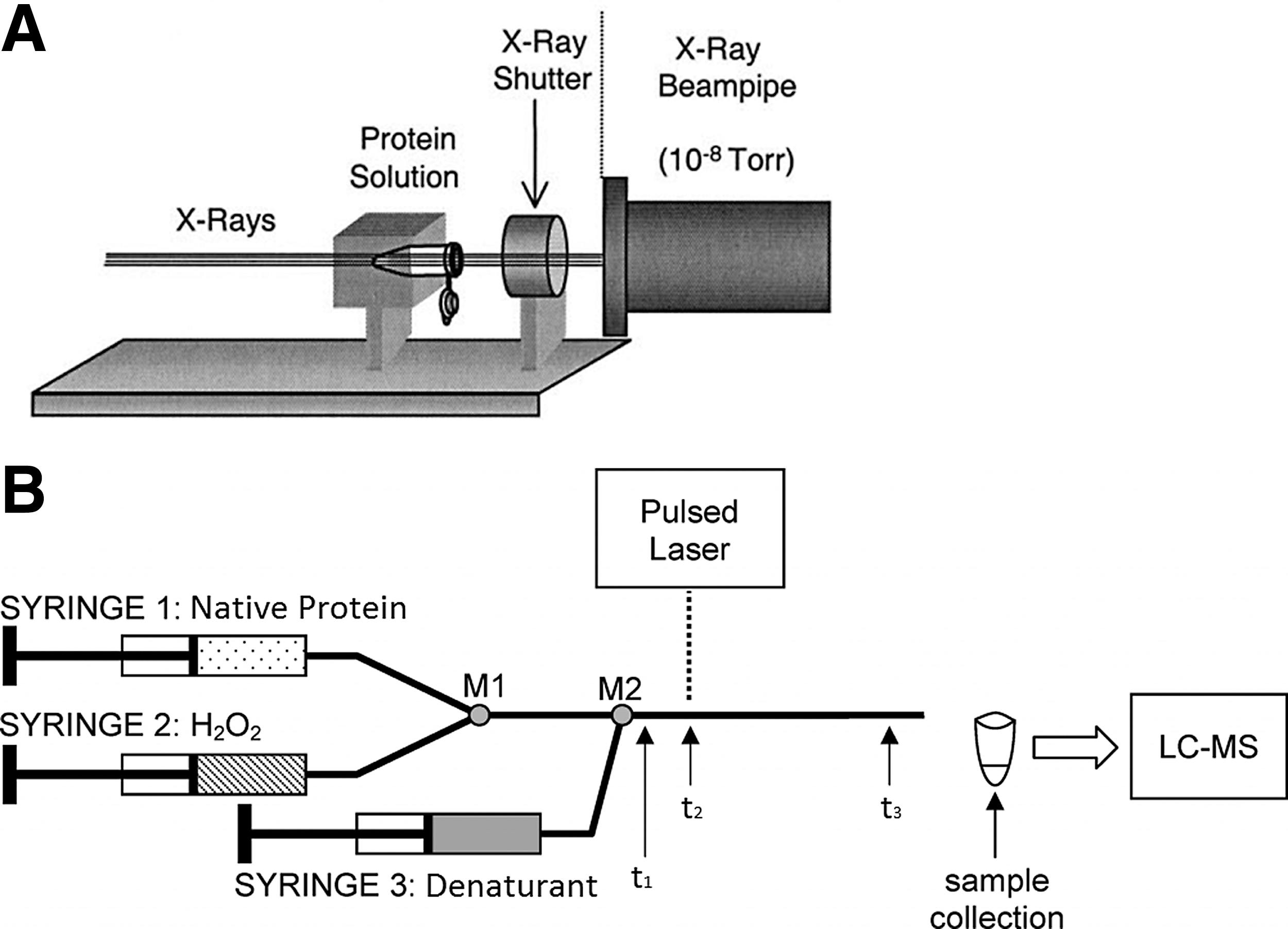
These radiative techniques represent the fastest way to produce ROS without the need for chemical additives (i.e., H2O2) but have a number of disadvantages, such as working with harmful radiation sources and limited access to synchrotron beam lines. A schematic depiction of a synchrotron X-ray experimental setup is provided in Figure 1A.
UV photochemical dissociation and laser photolysis can induce the homolytic cleavage of H2O2, providing an exceedingly straightforward and efficient way to produce •OH, as shown in Scheme 5 (103).
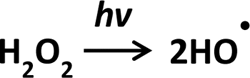
This was first attempted with a continuous UV light source and 15% (5 M) H2O2 solution to identify the solvent accessible surface area of lysozyme and β-lactoglobulin (89). Lengthy exposure of proteins to •OH have been shown to perturb the native structures, thus oxidation must be fast relative to conformational rearrangement, which is possible in principle given the diffusion controlled nature of the reaction. Nanosecond pulsed UV lasers can generate •OH over a period of 3–5 ns, an approach now commonly known as fast photochemical oxidation of proteins (FPOP) (30).
Although •OH from FPOP are generated quickly, the duration of their presence in solution must be mediated to avoid ROS exposures in excess of 100 μs (25). This can be achieved through the use of radical scavengers such as glutamine, which can drastically reduce the lifetime of •OH, ensuring that protein oxidation occurs over a period of a few microseconds (25). The removal of excess H2O2 from solutions post-FPOP with catalase is critical to preventing any further unwanted oxidation of the protein. In addition, methionine is often added with catalase to quench stable thiol radicals (112). While most pulsed laser studies characterize proteins under equilibrium conditions (30), Stocks and Konermann devised a continuous flow method for kinetic studies, requiring only a UV laser and UV transparent glass capillaries, as shown in Figure 1B (94).
One additional means of producing ROS in oxidative labeling experiments bears mention because it often occurs unintentionally in ESI mass spectrometry experiments. Electric discharge oxidation (EDO) occurs when there is a high-energy discharge during ESI, particularly in an oxygen-rich environment (when EDO is desired, oxygen is used as a nebulizing gas) (58, 59). The process is initiated near the electrospray tip, where intense electric field results in the stripping of an electron from O2, generating an •O2 + radical, as shown in Scheme 6:
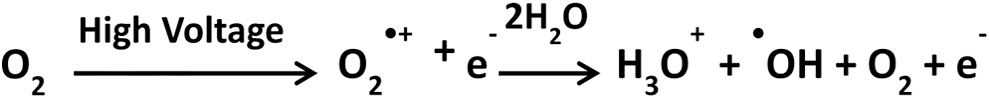
The target protein is subsequently oxidized during the electrospray process, before release into the gas phase (or collected for subsequent analysis). It should be noted that oxidation of proteins by EDO under normal electrospray conditions (where direct discharge is avoided) is usually negligible and thus does not generally cause artifactual oxidation in non-EDO oxidative labeling experiments. Where EDO is intended, there is some question as to whether the labeling pattern remains reflective of the native structure under the harsh ionization conditions used; however, it was recently demonstrated using ion mobility MS that, at least in the case of limited oxidation, the oxidized conformers of egg lysozyme and bovine ubiquitin were identical in size to those of their unmodified counterparts (18). Applications using electrical discharge are not discussed extensively in the following sections; however, examples of its use include characterization of the interaction between ribonuclease (RNase) S-protein and its S-peptide target (110).
There are many additional means of generating •OH; a few examples include photosensitization (62), electron radiolysis (71), sonolysis of water (83), and production through use of fast neutrons (92). Of all of the available options, however, the use of UV photolysis of H2O2 seems to be growing the fastest, likely due to the widespread availability of “benchtop” UV lasers.
Intrinsic Reactivity of Amino Acids to •OH
•OH react with side chains at rates ranging between 2×107 and 3×1010 M −1 s−1 (23, 57). The relative reactivity of the amino acid side chains with respect to •OH under aerobic conditions, established from γ-ray and X-ray radiolysis of water combined with mass spectrometry, is as follows: Cys>Met>Trp>Tyr>Phe>Cystine>His>Leu∼Ile>Arg∼Lys∼Val>Ser∼Thr∼Pro>Gln∼Glu>Asp∼Asn>Ala>Gly in an environment that is completely solvent accessible (Fig. 2) (42, 57). Where multiple ROS are produced (as is the case in X-ray-mediated radiolysis of water under aerobic conditions), the majority of amino acids react predominantly with •OH; however, sulfur-containing side chains can also be modified by molecular oxygen from air, and proline is exclusively modified by molecular oxygen (57, 99). This complexity can in some cases preclude a quantitative analysis of the labeling rates but does not prevent the semiquantitative or qualitative analysis of largescale protein conformational changes, such as large-scale folding and unfolding events (100).

Upon covalent modification of side chains by •OH, the labeling patterns can be probed by top-down or, more commonly by bottom-up liquid chromatography MS/MS, giving detailed structural information (Fig. 3) (45, 57, 99). The most common consequence of oxidation via •OH is a +16 Da mass shift due to the addition of a carbonyl oxygen or hydroxyl group (with concomitant removal of hydrogen). However, many other mass shifts are possible as shown in Figure 2.

Applications
Activity-linked conformational changes in proteins and large protein complexes
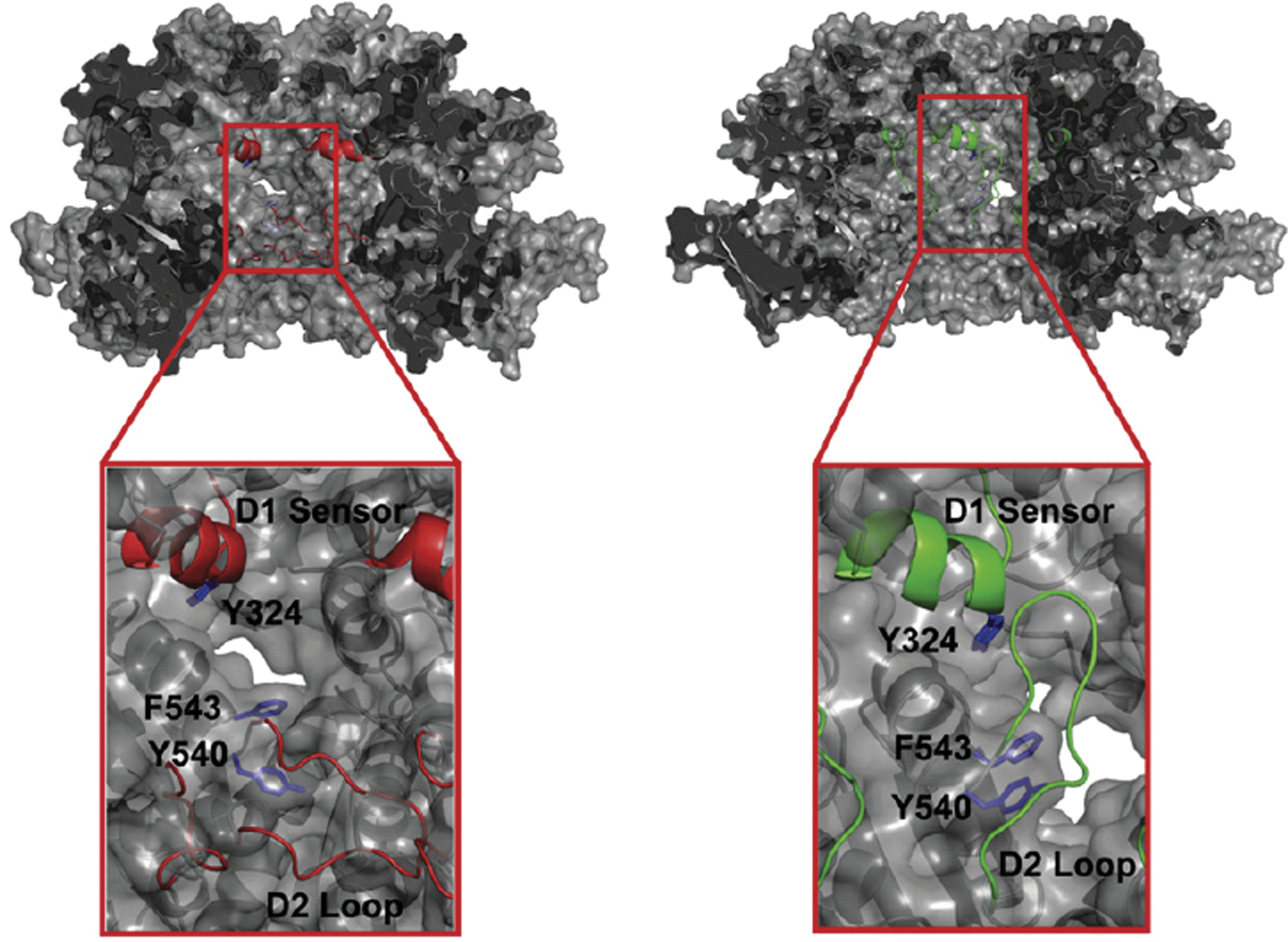
One of the primary advantages of oxidative labeling is the ability to investigate conformational changes in large protein complexes. A classic example of this is the work of Bohon et al. on the ClpA component of the cylindrical Escherichia coli ATP-dependent protease complex (ClpAP) (4). ClpA is an∼500 kDa hexamer responsible for substrate recognition and translocation to the ClpAP proteolytic core (40). It has been suggested that ClpAs D2 loop binds target protein substrates tightly in “prehydrolytic” conformation (27). Upon ATP or ATPγS hydrolysis, the D2 loop then “drags” the unfolded protein substrate down to the proteolytic subunit (34).
Given the size of the complex, and the substantial nature of the conformational change in the D2 loop, this system is ideal for the analysis by oxidative labeling (4). Models based on the crystal structure had predicted that the D2 loop should be buried in the ClpA hexamer core and thus solvent inaccessible in the oligomeric ATP-bound form of the protein (Fig. 4, left). However, oxidative labeling revealed that D2 loop was at least partially solvent accessible, which was in better agreement with a model in which the D2 loop was positioned “up,”, close to the site of substrate binding and in contact with the D1 “sensor” region (Fig. 4, right). These results are easily rationalized with respect to ClpAs function within the ClpAP complex: Upon ATP-binding and hexamerization, the D2 loop is initially positioned up to facilitate substrate recognition and binding. Upon hydrolysis of the nucleotide, the D2 loop is drawn into the hexamer pore, dragging the substrate toward the ClpAP proteolytic domain and ultimately facilitating substrate release. This study serves as an example of the potential pitfalls in developing functional models from methods that provide only the ground-state structure of the protein.
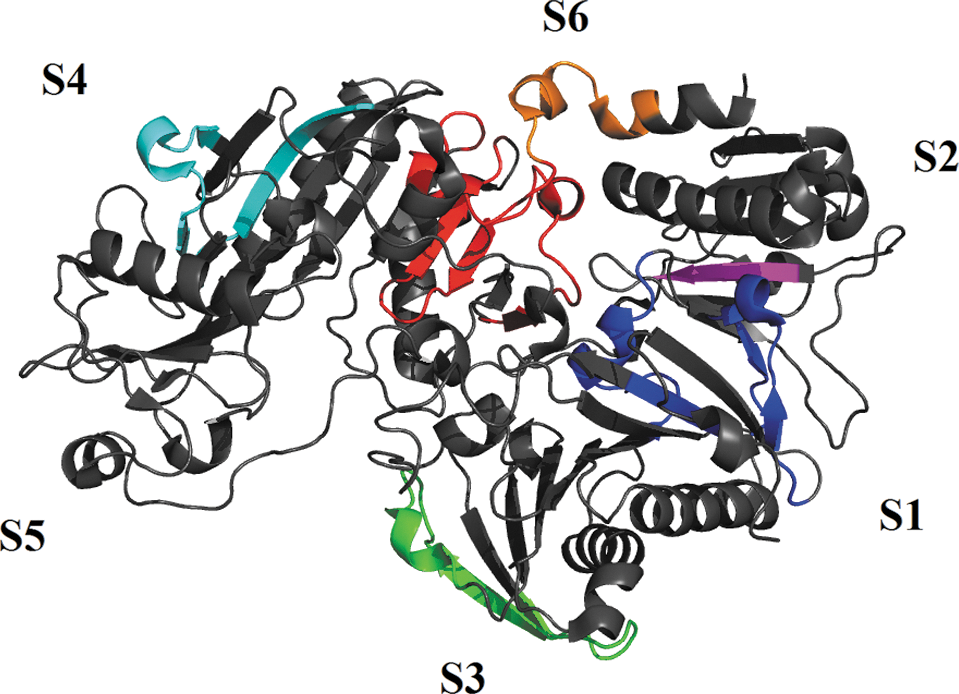
The ability to probe activity-linked conformational changes by oxidative labeling is not limited to large protein complexes. A relatively early example of this is a 2003 study by Kiselar et al., who probed Ca2+-binding-dependent conformational changes in gelsolin (43). Gelsolin is a Ca2+-dependent protein that regulates actin filament assembly/disassembly. Structurally, it is composed of six homologous subdomains termed S1–S6 (10, 115). Binding of Ca2+ to gelsolin had been extensively studied by conventional biophysical techniques (10, 43, 51, 77); however, very little was known about structural rearrangements induced by binding, an ideal application for •OH footprinting (43).
Seven peptides were found to be Ca2+ sensitive (Fig. 5): residues 49–72 (blue), 121–135 (blue), 431–454 (cyan), and 722–748 (red and orange) showed increase in oxidation, whereas residues 276–300 (green) and 652–686 (red) showed a significant decrease. Residues 162–166 (magenta) are buried in the absence of Ca2+; however upon binding, a significant increase in oxidation occurred. By measuring a full concentration dependence of Ca2+ on labeling efficiency in each of the observed peptides, the authors were able to unravel a three-state binding mechanism with a highly cooperative binding process at low (sub-μM) Ca2+ concentrations followed by a noncooperative (or at most moderately cooperative) transition associated with lower affinity Ca2+ binding. Of course, in oxidative labeling experiments, information about binding stoichiometry is often inferred from structural transitions, thus the data also provide insights into the structural consequences of Ca2+ binding. In this case, the high-affinity cooperative binding step was linked to the “unmasking” of the actin binding site, whereas the low-affinity uncooperative step was associated with a general extension of the protein structure through the loss of contact between subdomains 4 and 6, corresponding to the fully activated protein (43).
Identifying intermolecular interfaces in large protein complexes
One of the first uses of protein footprinting by oxidative labeling was the identification of protein–protein interaction surfaces in large protein complexes. In the following example, Gau et al. used FPOP-based oxidative labeling to study the tetramerization interface of the three most common isoforms of apolipoprotein E (ApoE) (24). ApoE is essential in regulating lipid metabolism and redistribution in tissues and cells but is extremely challenging to study structurally (56). In lipid-free solution, its three most common isoforms, E2, E3, and E4, predominantly form tetramers (116). Only the lipid-free N-terminus has been structurally characterized, mainly because the C-terminus has a tendency to undergo uncontrolled oligomerization (109). The FPOP data provided good evidence that ApoE isoforms share a common tetrameric structure, with important contacts in the linker (183–205) and C-terminal (232–251) region. This work represented one of the first applications of FPOP and illustrated the ability of oxidative labeling to identify protein interaction interfaces in challenging systems. Additional early examples of protein–protein interaction surface footprinting include studies on the transferrin–transferrin receptor interaction (52) and the interaction between cofilin and G-actin (26).
Quantitative analyses of secondary structure stability
Oxidative labeling is typically applied in a semiquantitative or qualitative manner, as in the examples above. However, provided that artifactual changes in structural stability can be controlled, oxidative labeling is suitable in principle for quantitative measurements of the thermodynamic stabilities of secondary structures. Among the first such studies was a straightforward analysis of myoglobin helix stabilities using equilibrium denaturation and synchrotron radiolysis by Maleknia and Downard (60). The level of oxidation as a function of urea concentration was measured on peptides spanning three α-helices: residues 1–16 (helix A), 17–42 (helix B/C), and 103–118 (helix G). In Figure 6, helices A and B/C showed two-state cooperative unfolding (m values 1.7±0.2 kcal mol−1 M −1 and 2.5±0.5 kcal mol−1 M −1, respectively) coupled to global unfolding of the protein, with ΔG0 F-U values of 5.4±0.7 kcal mol−1 and 7.6±1.6 kcal mol−1, respectively. However, helix G exhibited a unique, noncooperative unfolding profile (m=0.6±0.1 kcal mol−1 M −1, ΔG0 F-U=2.0±0.2 kcal mol−1). This was consistent with previous reports, which had suggested the presence of “residual structure” in helix G in the “unfolded” protein (13, 53). To further understand the mechanism of helix G's unique behavior, a substructural profile was obtained via MS/MS after exposure to 3.5 M of urea. The results showed that the C-terminal portion of helix G was more solvent accessible and thus likely less thermodynamically stable than the N-terminal portion (60).

As a pioneering study, this work demonstrated a number of key features of oxidative labeling, namely the ability of fast labeling techniques to modify proteins without significant structural perturbation, the ability to make accurate quantitative measurements of secondary structure stability, and the ability to make qualitative assessments of substructural stability.
Ultra-rapid kinetic protein folding experiments
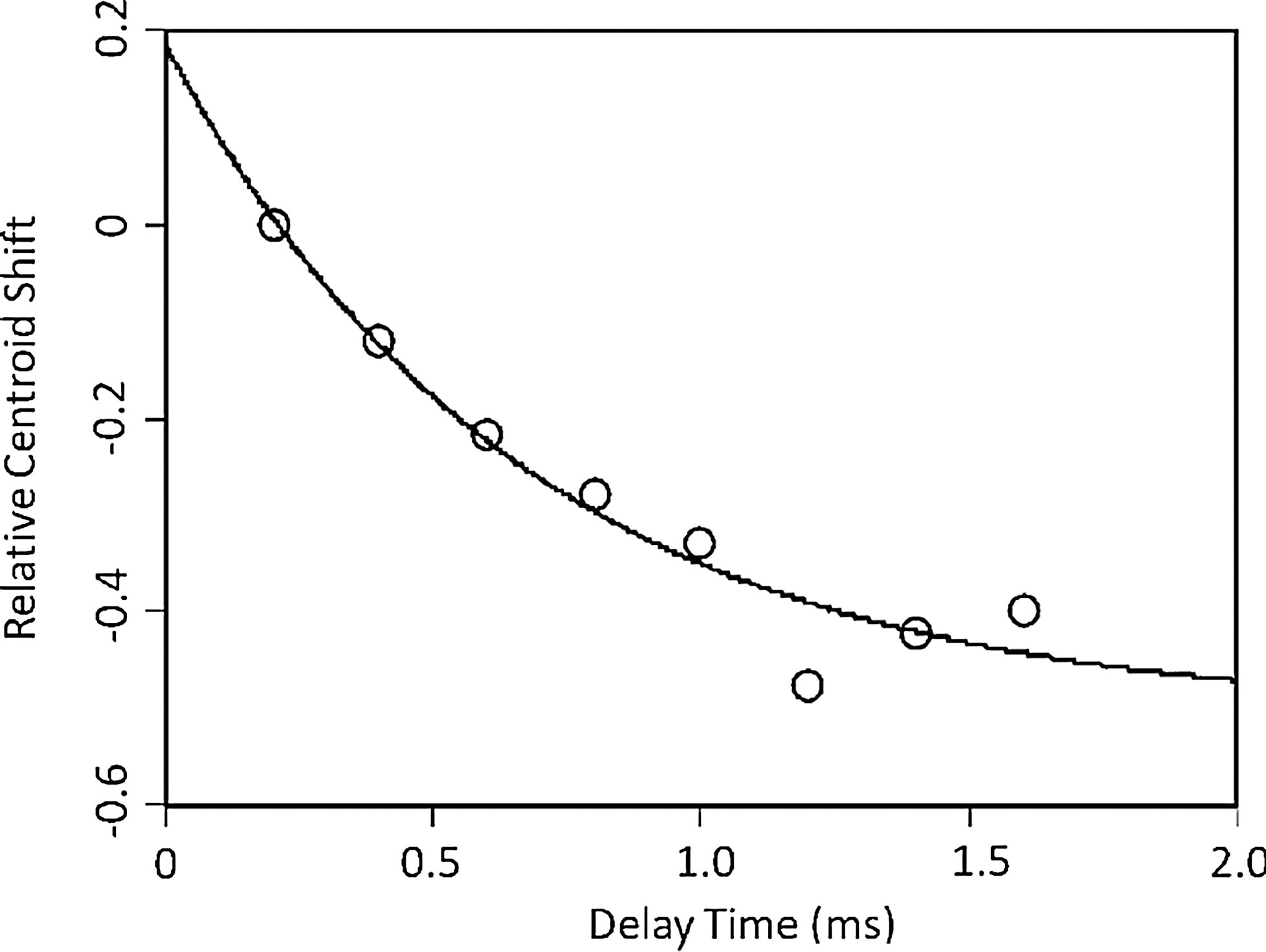
The early steps of protein folding are often exceedingly fast, occurring in nanoseconds to microseconds range, and are thus a challenging target for kinetic experiments (114). Chen et al. have combined FPOP and T-jump structure perturbation to monitor rapid refolding in the protein Barstar, a common T-jump model protein because it is unfolded at 0°C and refolds in response to heating (12). The Barstar folding mechanism includes two intermediates. The first (I1) is established within ∼1 ms, and the second (I2) between 10 and 100 ms with full transition to the native state taking ∼100 s. However, the first transition has not been structurally characterized using conventional methods because of its short lifetime. To apply FPOP to this problem, Chen et al. used a two-laser setup, one to initiate the T-jump and the other to generate •OH from H2O2. Time courses for folding were obtained by varying the delay time between the onset of folding (T-jump laser pulse) and labeling (FPOP laser pulse).
By monitoring the time-dependent shift in the peak centroid of the most populated charge state, the rate constant for equilibration of the first folding step was measured to be 1.5 ms−1 (Fig. 8). This was in a good agreement with previous fluorescence-based measurements, which had yielded a rate 3.1 ms−1 (69). One distinct advantage of using an oxidative probe over fluorescence detection is the nonreliance on a fluorophore (12). Oxidative labeling experiments could also in principle provide structurally resolved information on this ultra-rapid process; however, in this recent proof-of-principle study, fragmentation was not applied. Hybrid T-jump/FPOP experiments are a new and promising approach with a broad range of applications that remain to be fleshed out, not only in folding but also in ligand binding or any protein system that is amenable to perturbation/relaxation.
Simultaneous study of folding and dimerization
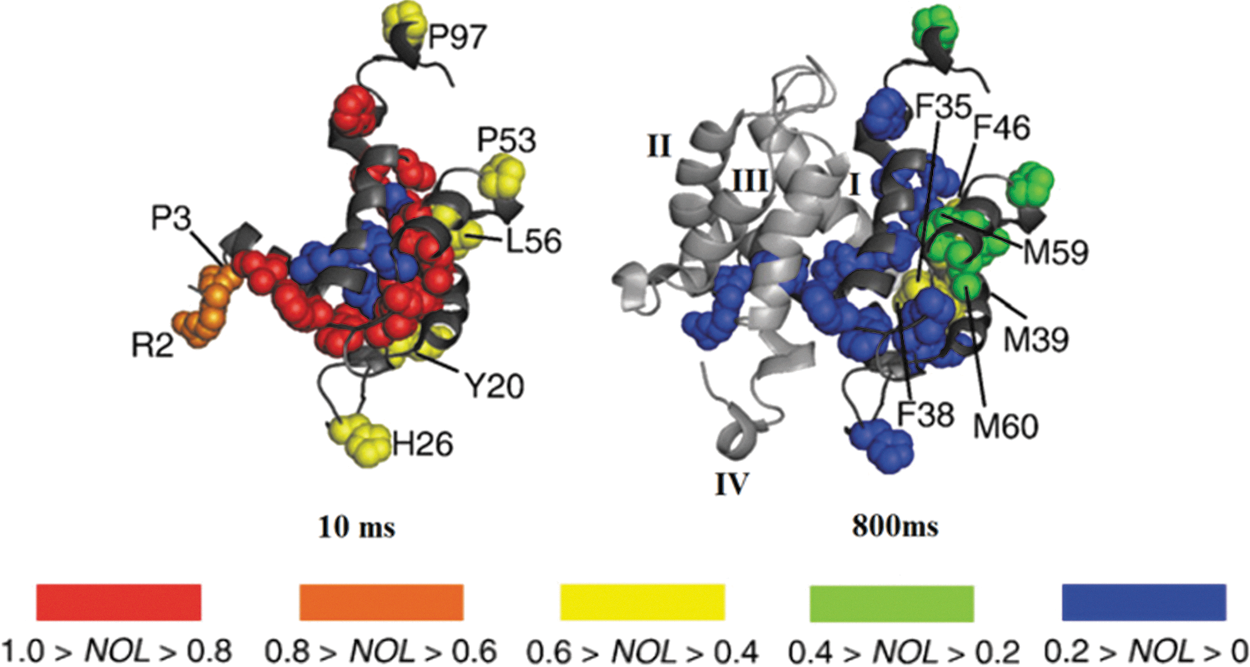
Oxidative labeling is of course an equally viable approach for monitoring slower processes in protein folding, including the interplay between folding and native oligomerization. Recent work from the Konermann group simultaneously investigated S100A11-folding and dimerization using FPOP coupled to a continuous-flow reactor (95). S100A11 belongs to the S100 family of proteins that is involved in tumor suppression and DNA repair (68, 70). The monomeric form of the protein folds into four helices (I–IV) and can form compact dimers with hydrophobic interface comprised of regions from helices I and IV of both subunits (15). NMR, circular dichroism (CD), and time-resolved ESI-MS suggested that the transition pathway from acid denatured monomer to native dimer involves a monomeric “burst phase” that still retains some of its “native-like helicity” (73).
Refolding and dimerization was triggered in the flow reactor by pH jump from 2 to 7, and the protein was labeled at different times by generating •OH at different points along the flow tube (Fig. 1B). Globally, the protein was labeled the most at 10 ms, and by 800 ms, folding and dimerization were complete. From spatially resolved data, it was determined that the S100A11 monomer retained some residual structure at pH 2, forming intramolecular contacts between elements of helices I and IV at the expense intermolecular hydrophobic interactions (associated with dimerization). At 10 ms, the burst phase monomeric species has formed, and the normalized oxidation level for residues that are distant from the dimerization interface (Y20, H26, P53, L56, and P97) is lowered to 0.5 (Fig. 7, left). At 200 ms, residues that are involved in dimerization (F35, F38, M39, F46, M59, and M60) acquire additional protection, indicating the completion of dimerization. However, elements of helices II and III (Fig. 7, right) still retain some exposure to the solvent, which is lost over a period of 800 ms (well after dimerization is complete). This implies the presence of the transient, native-like intermediate that is fully dimerized but not fully folded. Overall, these data provide an exquisite example of the link between folding and oligomerization and the power of oxidative labeling to structurally characterize folding intermediates.
Identification of metal binding domains
Vachet and coworkers have applied the Fenton-like metal-catalyzed oxidation (MCO) to study copper binding in Human beta-2-microglobulin (β2m), a 12 kDa component of major histocompatibility complex class I (50, 101). Previous work had demonstrated that unfolded β2m binds to Cu2+ more strongly than its native state and displays weaker conformational stability in the presence of Cu2+ (9, 19). Misfolding of this protein leads to dialysis-linked amyloidosis, hence it is crucial to obtain insights to copper binding sites under native and nonnative conditions (19).
Under native conditions, β2m was reacted with MCO for 30 min. Upon subsequent proteolytic digestion, four peptides were generated: I1-R3, S20-K41, I92-M99, and W95-M99. Three of them (I1-R3, S20-K41, and I92-M99) had 16 Da increase, whereas I1-R3 had a 1 Da decrease resulting from hydrogen abstraction (loss of NH2 and H as result of oxygen addition) attributable to Cu binding at the N-terminus. In the peptide S20-K41, a number of potential oxidation sites were present; however, H31 was exclusively oxidized indicating the presence of Cu2+ in close proximity to that site. A number of artifactual oxidations were identified, particularly in peptides I92-M99 and W95-M99, which both show modification on M99 including in negative controls with no H2O2. This artifact could have been due to a nonspecific oxidation reaction (i.e., with molecular oxygen) or due to second weaker Cu2+ binding site sensitive to high Cu2+ concentrations (19, 64).
Under nonnative conditions (8 M urea), nonartifactual oxidations were detected at I1, H31, H51, H84, revealing the locations of the additional Cu2+ binding sites. These results provided for the first time a clear structurally resolved picture of copper binding in β2m, with the native protein binding through the N-terminus and H31, with different (yet also highly specific) Cu2+ binding in the denatured protein. The data also suggest that amyloidogenic aggregation of β2m is initiated by destabilization of the N-terminus (50).
Structural characterization of integral membrane proteins
Studies on membrane proteins have always been challenging due to their propensity to precipitate when deprived of their natural lipid environment. This challenge and others has resulted in a dearth of high-resolution X-ray and NMR structures for this important class of proteins. Oxidative labeling provides an alternative (albeit lower resolution) approach, enabled by the fact that many membrane proteins tend to be methionine-rich, making them highly susceptible to oxidation at exposed sites (117).
Pan et al. have applied time-resolved •OH labeling for folding studies on the membrane protein bacteriorhodopsin (BR) in the presence of the retinal cofactor (75). BR forms seven transmembrane helices (A–G) connected by short loops, which pocket prosthetic retinal chromophore (Fig. 9) (32). Kinetic folding mechanisms of BR have been proposed by using various techniques, such as stopped-flow fluorescence, CD, UV-Vis spectroscopy, and mutational analyses (5, 6, 55). However, due to the low resolution of these techniques, an in-depth structural characterization of various intermediates in BR refolding could not be achieved.
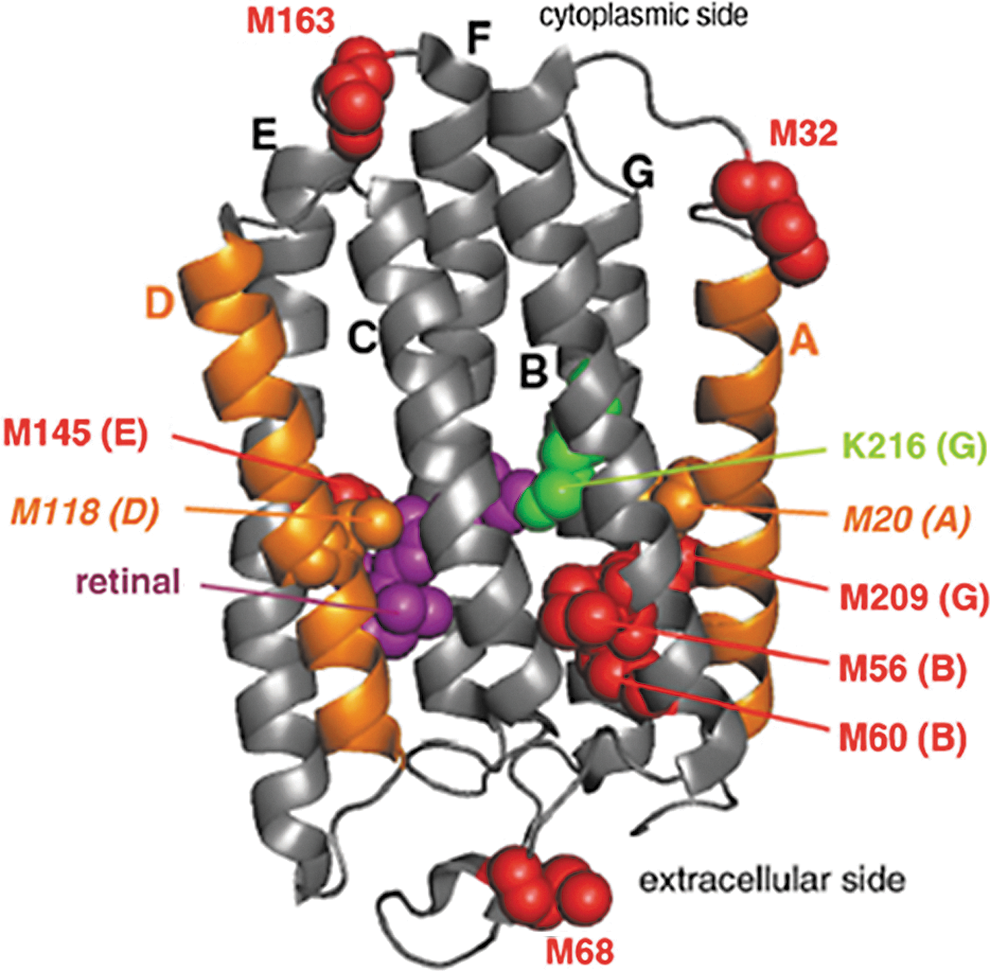
Based on the footprinting data, M20 (helix A) and M118 (helix B) undergo different processes through which they become solvent inaccessible during BR refolding and reconstitution into the membrane. M20 became protected independent of cofactor binding. M118 remained exposed in the absence of retinol; however, it slowly became protected (over∼4 s) after retinol binding. In separate mutagenesis experiments, helix C (L93M) or helix F (V179M) were engineered to confirm that the central portion of helices C and F remain shielded from solvent in sodium dodeyl sulfate denatured BR (74).
These data were ultimately assembled into a detailed stepwise mechanism for BR folding: Formation I1 (cofactor-free intermediate 1) occurs within 20 ms; helix A is formed first whereas helix D remains disordered. Formation of I2 (cofactor free intermediate 2) occurs after 4 s, this transition involves consolidating the protein structure. Once I2 is formed, noncovalent retinal binding induces folding of helix D, generating the “early” IR intermediate (noncovalent protein–cofactor complex). The “late” IR intermediate is formed as the cofactor is further adjusted into the binding pocket of the protein, which occurs over a period of roughly 10 s. Establishment of the covalent linkage to the chromophore ring then results in native BR. This mechanism represents a minimal model for the BR refolding and reconstitution process but is nonetheless the most detailed mechanism to date for an integral membrane protein.
Conclusions and Outlook
Since its introduction nearly 15 years ago, oxidative labeling with analysis by mass spectrometry has matured from the early phases of technological development to an established approach for probing the solvent-accessible surfaces of proteins as they undergo biologically relevant conformational changes and binding. While new methods for generating ROS continue to be introduced, current facile techniques for rapidly generating ROS, particularly photo-induced dissociation of H2O2, are likely to drive a substantial increase in the adoption of oxidative labeling as a general structural technique. Another important driving force behind widespread adoption is the broad applicability of the method, with future applications extending well beyond those reviewed in the present work. In particular, structurally resolved studies on rapid folding processes and conformational dynamics represent an underinvestigated area to which oxidative labeling is uniquely well suited. The same can be said of studies on the structure and dynamics of membrane-bound proteins and protein complexes. In addition, there is also much room for growth in areas that are already somewhat more established, particularly in the study of conformational transitions in large protein complexes in response to ligand binding.
The concept of quantitative analysis by oxidative labeling, while it has shown to be possible in principle, remains an underexplored area that merits attention as a future direction. However, fully realizing the potential of oxidative labeling as a quantitative tool will require additional fundamental studies on the intrinsic reactivities of solvent-exposed amino acids in the context of folded proteins (i.e., considerations of side chain interactions) and even some remaining questions about the influence of the primary sequence (i.e., will a residue appear to react at the same rate if it is flanked by two more reactive residues?). Another tantalizing possibility is the incorporation of the oxidative labeling workflow onto a microfluidic device, such as the one developed by Rob et al. for HDX (84 –86). Combining oxidative labeling and HDX into a single device could provide a convenient way to probe the impact of oxidative label incorporation on dynamics (which is somewhat different than the issue of the impact of the incorporation of label on structure) and, if the effect were small, might be used as a simultaneous probe of solvent accessibility and hydrogen-bond strength.
In summary, oxidative labeling represents a powerful, widely applicable tool in structural biology, with recent methodological advances that promise to enable increasingly widespread adoption in the next decade and beyond.
Footnotes
Acknowledgments
The authors thank Dr. Michael Gross and Dr. Lars Konermann for helpful discussions.