
Abstract
Introduction
P
Much as the recent understanding of PD as a clinical syndrome broadened from one focused narrowly on motor symptoms, so has the understanding of PD as a neuropathological syndrome broadened from one focused on dopamine neurons in the midbrain substantia nigra to one encompassing widespread regions of the central nervous systems (CNS) and peripheral nervous systems (PNS) (16, 17, 35, 159). In addition to the substantia nigra, neuronal loss has been convincingly described for noradrenergic neurons in the pontine locus ceruleus, cholinergic and noradrenergic neurons in sympathetic centers in the spinal cord and periphery, and cholinergic neurons in the dorsal motor nucleus of the vagus nerve (DMV) in the medulla, among other areas (19, 86, 95, 96, 103, 104, 119, 131, 209). Furthermore, the pathological hallmark of PD, Lewy bodies as detected by abnormal α-synuclein (AS) immunoreactivity, is spread through an even wider variety of neurochemical subtypes, including cells in the olfactory bulb, cerebral cortex, pontine raphe, spinal cord, and enteric nervous system (ENS) (16, 35, 77, 139, 165). Thus far, data indicate that many of these areas are more vulnerable than the midbrain dopamine neurons to Parkinsonian pathology and death. In fact, the extreme focus on pathology in dopamine neurons over the past century may have limited our understanding of PD as a more systemic neurological disorder (159, 169, 261).
This view of PD is more a rediscovery than a new one. When James Parkinson first described the disorder in 1817, he described a multitude of nonmotor symptoms and hypothesized that the “proximate cause” was a “diseased state of the medulla spinalis…extending, as the disease proceeds, to the medulla oblongata (216).” Frederic Lewy described the pathological hallmark of PD, the Lewy body, in the early 1900s in the basal forebrain and medulla before noting them in the midbrain (172, 173). As the field has returned to considering PD as a systemic disease, many theories concerning its pathophysiology focused solely on dopamine neurons have required reevaluation. Although the dopaminergic phenotype is still an important component related to selective neuronal vulnerability in PD, it is not the only one. In particular, anatomical and pathophysiological links between the PNS and CNS and the specific subtypes of affected neurons within each are prerequisites to any comprehensive theory of PD pathogenesis.
The DMV and the vagus nerve provide such a link. Located in the lower brainstem, the DMV contains widely ramified neurons projecting to nearly every part of the periphery. In addition, the vagal complex has extensive interconnections to the rest of the brain. Stimulation of the nerve has profound effects in both central and peripheral directions with the vagal nerve stimulation widely used as a treatment for epilepsy and depression refractory to medications and experimentally used for its cardioprotective, antiasthmatic, anti-inflammatory, and GI promotility effects (32, 59, 116, 154, 181, 245, 271).
In this review, I will review the basic anatomy of the DMV, discuss the impact of PD on the nucleus, and review potential consequences of and causes for DMV degeneration in PD.
Functional Anatomy of the Vagus Nerve and DMV
The vagus nerve is a mixed cranial nerve (the 10th) that contains (in descending order of abundance) (i) afferent visceral sensory (predominantly autonomic) fibers from the heart, lungs, GI tract and accessory organs (spleen, liver, gallbladder), sweat glands, and other areas; (ii) efferent visceral motor (autonomic) fibers to a roughly similar distribution of tissues; (iii) efferent somatic motor fibers to several skeletal muscles of the pharynx and larynx; (iv) afferent special sensory fibers mediating taste in portions of the pharynx; and (v) afferent general sensory fibers from parts of the ear and surrounding skin (Fig. 1) (234).
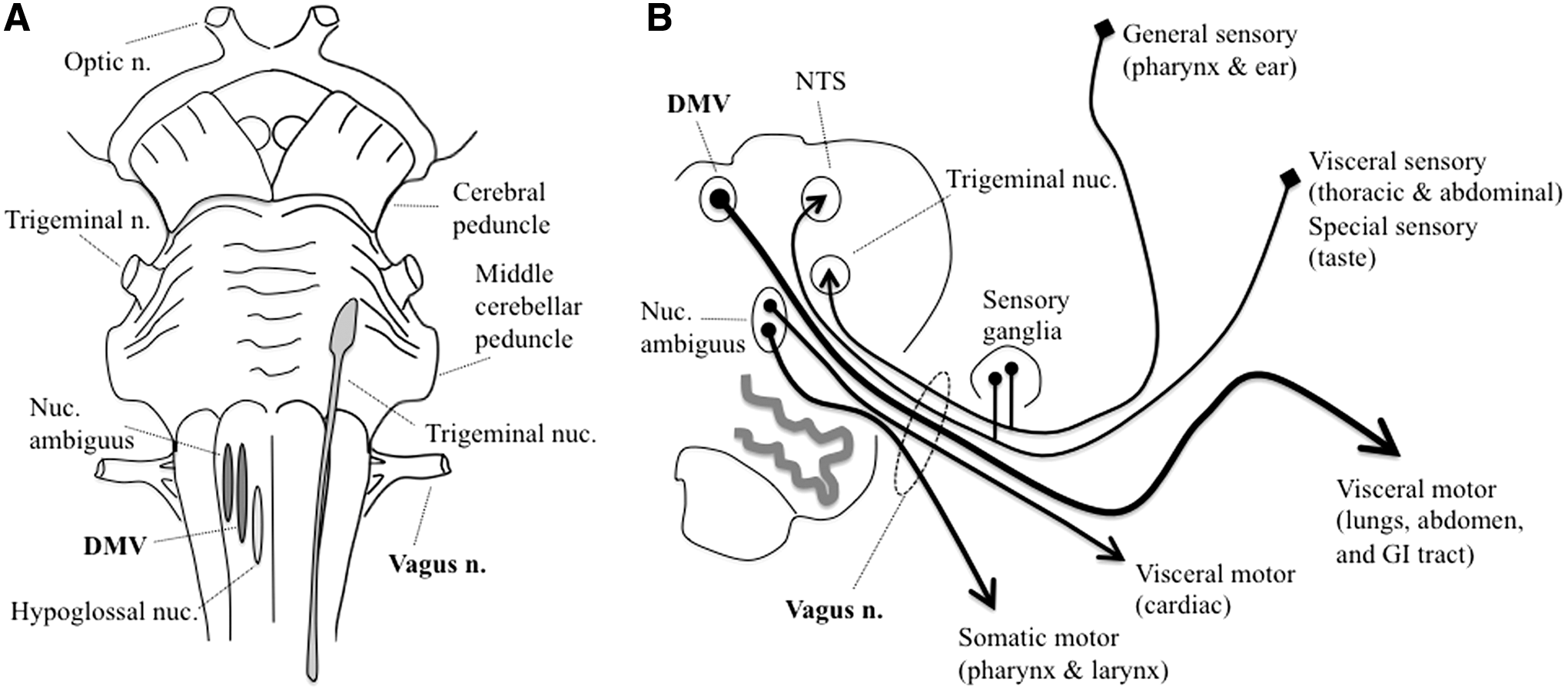
Vagus nerve functions are regulated by a central autonomic network that integrates visceroceptive, humoral, and environmental information and modulates autonomic, endocrine, behavioral motor, emotional, attentional, and antinociceptive output (18, 45, 182). The structures forming the central autonomic network are widely distributed at all levels of the nervous system, including the cerebral cortex, basal forebrain, basal ganglia, midbrain, thalamus, hypothalamus, pons, and medulla (45, 46, 180, 202). Although less well-studied than for motor function, the basal ganglia appear to play a similar comparator role in autonomic function modulating the iterative processing of the central network; both anatomical and neurophysiological connections have been described (5, 54, 67, 90, 92, 105, 112, 118, 138, 158, 176, 178, 183, 223 –225, 259, 264).
The DMV is a paired nucleus on each side of the caudal medulla that runs longitudinally along the dorsomedial aspect of the brainstem (Fig. 1). It gives rise to preganglionic parasympathetic autonomic fibers that innervate the GI tract and accessory organs from the distal third of the esophagus to approximately the splenic flexure of the colon, although there is some evidence for more distal projections (22). Cardiovascular preganglionic parasympathetics arise predominantly from the nucleus ambiguus rather than the DMV (206).
DMV neurons have thin, widely ramified axons with minimal myelination that are among the longest in the body (33, 169). This is in contradistinction to other fibers in the vagus nerve (afferent and somatic motor efferent) with adjacent locations in the medulla that have more robustly myelinated neurites with simpler branching structures. Acetylcholine is the major neurotransmitter for DMV neurons, although others are thought to function as cotransmitters, including substance P and catecholamines.
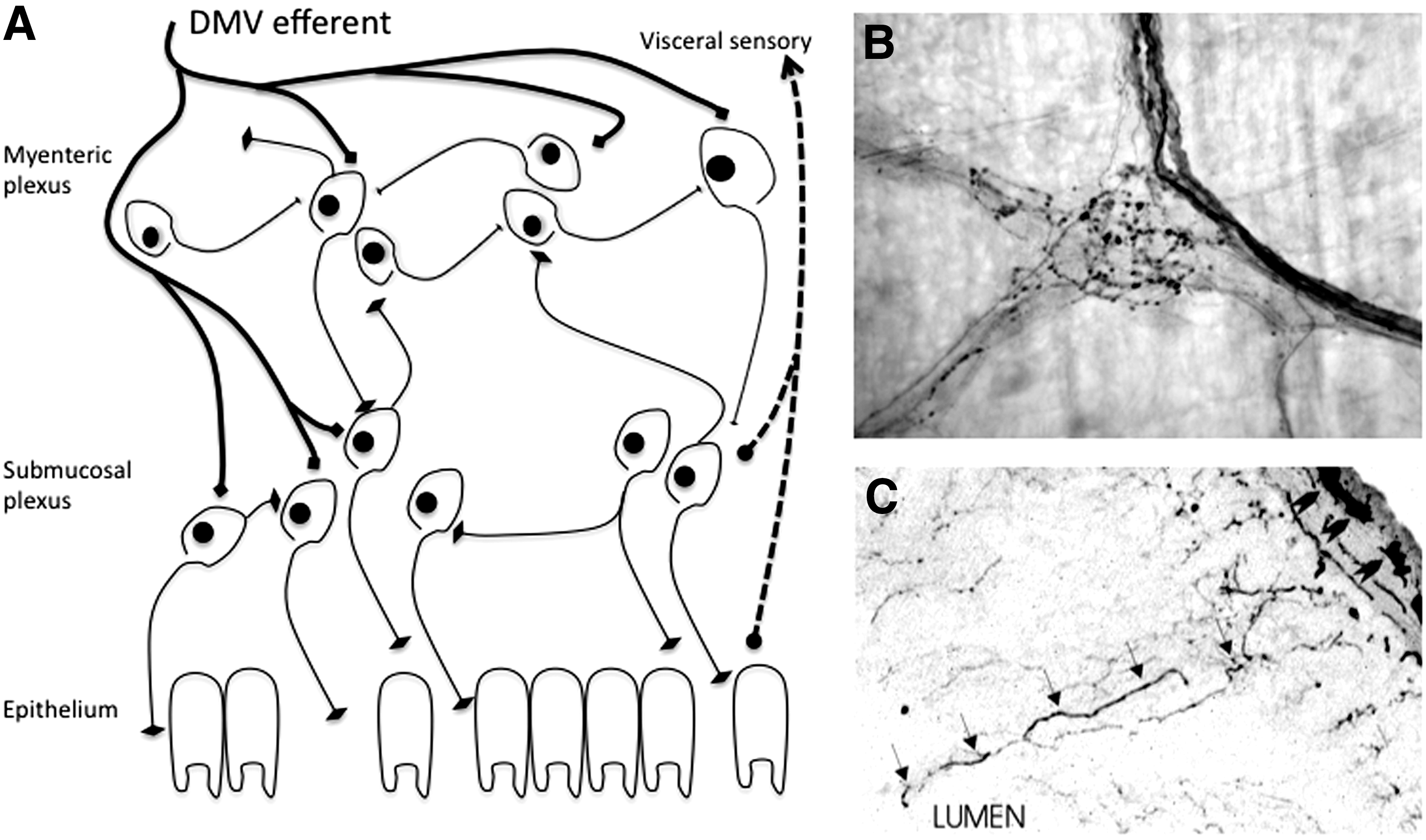
Parasympathetic innervation from the DMV is important for control of GI motility and reflexes, especially in the stomach (219, 237, 267, 268). About 25%–30% of fibers in the abdominal vagus nerve are efferent fibers that modulate function of the ENS, a semi-autonomous network of neurons lining the entire alimentary canal to control GI function (142). Individual axons from DMV neurons ramify widely into an extensive network contacting nearly every enteric neuron, especially in the proximal GI tract (Fig. 2) (22, 205). Vagotomy has long been known to result in GI dysfunction; less well known are GI complications due to pathological lesions of the DMV itself (240, 241, 254).
In addition to its classical function as a modulator of GI function, the DMV has recently been described as a potent inhibitor of inflammation via the cholinergic anti-inflammatory pathway (32, 248, 279). Studies have shown that activation of vagal parasympathetics attenuates the systemic inflammatory response to a variety of insults, including endotoxin administration (32). This effect is mediated via the α7 subunit of the nicotinic acetylcholine receptor, which is expressed on macrophages (279). In addition to the anti-inflammatory effect of vagus nerve stimulation, physical or pharmacological vagotomy has a proinflammatory effect, in which the branch of the vagus nerve that innervates the spleen has been shown to be particularly important (137, 222, 246). The afferent signaling for this iterative inflammatory/anti-inflammatory loop has both neural and humoral components and is modulated by multiple central and systemic signaling mechanisms (76, 208, 220, 221, 247, 248, 288, 289).
Pathophysiology of the DMV in PD
There are two main ways that PD may affect the vagal system. One is the direct impact of neuronal damage to vagus neurons. For example, loss of DMV innervation to the gut causes GI problems by cutting a hard-wired pathway. The other is the downstream deleterious effect of central neurodegeneration on iterative processing of autonomic regulation. For example, from a motor standpoint, it is now accepted that motor parkinsonism is a circuit disorder, rather than a straight line from dopamine depletion to motor dysfunction. Given the evidence accumulated thus far, autonomic symptoms in PD may be caused by both mechanisms. Whereas this review will primarily focus on the direct damage accumulated by DMV neurons in PD, considering the DMV as one node in a distributed network is helpful to place these findings in context.
Disrupted central regulation of DMV output
Loss of substantia nigra neurons and subsequent striatal dopamine depletion is a primary driver of PD motor symptoms by disrupting basal ganglia circuit function (39, 49, 61, 101, 121, 171, 188, 189, 207, 249, 255). Loss of integrity in the circuitry of the striatum increases the oscillatory activity in corticocortical loops by propagating to basal ganglia output nuclei (63, 69, 102, 187, 239). Experimental studies have shown that pharmacological blockade of dopamine receptors (61, 87) or lesion of the nigrostriatal pathway (284, 285) increases the neuronal synchronization and oscillatory activity in the basal ganglia (40, 101, 121, 170, 280). In the parkinsonian brain, dopaminergic drugs (127, 274) and deep brain stimulation have been found to reduce synchronization in conjunction with their therapeutic effect (70, 274). While this abnormal circuit activity has been intensively investigated in regard to motor function, its contribution to other symptoms is less clear.
The relationship between dopamine depletion and autonomic dysfunction has been examined in a few previous studies. In rats, the salivary response is dependent upon the activity of dopamine receptors in the striatum and decreased by striatal lesions (223 –225). Baroreflex regulation of blood pressure has been reported to be dependent on the nigrostriatal pathway in rodents (5, 67, 176, 178, 183). In addition, GI motility can be modulated by basal ganglia activity, especially in the caudate (112, 158, 264). In some PD patients, deep brain stimulation in the subthalamic nucleus has been reported to improve cardiovascular and GI dysfunction (134, 161, 292).
More specific to the vagal system, animals with lesions of the nigrostriatal tract show reduced choline acetyltransferase expression in the DMV and decreased levels of acetylcholine in the gastric wall (291). Although dopamine receptors are expressed by both cholinergic and catecholaminergic neurons in the DMV (43), a direct anatomical link between the two structures has not yet been demonstrated.
It is enticing to hypothesize that there are loop circuits regulating the autonomic function analogous to those demonstrated for somatic motor control and that these loops may not only link a distributed network of brain nuclei but also the peripheral and ENS. This is likely to be a fruitful area of research in the future; however, at present, the existence and dysfunction of such loops are largely hypothetical, while direct pathological damage to the DMV has been clearly and repeatedly demonstrated in PD patients.
Neuropathology of the DMV in PD
Although PD has predominantly been considered a disease of dopaminergic neurons in the substantia nigra, it has long been known that there is substantial neuropathology in other areas. For example, extensive cell loss on the order of >50% in the DMV has been described consistently in PD since 1925 (82, 88, 126). This loss is age dependent in PD in that, older age correlates with fewer DMV neurons; however, there is no age-dependent loss of DMV neurons in individuals without PD (95). Preganglionic parasympathetic neurons in the DMV are preferentially vulnerable to degeneration (82, 119, 120).
Lewy bodies, eosinophilic proteinaceous intracellular inclusions that are a pathological hallmark of PD, were described in the DMV before their recognition in the substantia nigra (172, 173, 226). Additionally, the DMV has been noted to have the highest number and density of ubiquitin-positive degenerating neurites of any brain nucleus in PD. Intriguingly, the density of degenerating neurites in the DMV correlates with the duration of PD, while the density in the substantia nigra does not (94).
In 1997, mutations in α-synuclein (AS) were described as an autosomal dominant cause of parkinsonism (231). Soon after, it was described that AS was a major component of Lewy bodies in all cases of idiopathic PD (260). These findings led to a renewed appreciation for Lewy pathology and careful examination of its extent and character in PD. Using immunostaining techniques, AS aggregation, particularly in neurites, has been shown to be very prominent in the DMV (35, 145, 217).
Lewy neurites have been described in the ENS in nearly 100% of PD patients at autopsy (Fig. 3) (6, 155, 236, 275 –277). The vast majority (>97%) of ENS synuclein pathology in PD is not cytoplasmic Lewy bodies in enteric neuron cell bodies, but neuritic Lewy pathology around the ENS, a finding that is recapitulated in AS transgenic mice (34, 276, 277). The distribution of pathology in both humans and transgenic mice mirrors that of efferent DMV innervation to the ENS nearly exactly, where neuritic AS aggregation and Lewy pathology are common proximally (stomach) and rare distally (rectum) (6, 17, 205, 276, 277).
It has been suggested that AS neuritic pathology affects the DMV very early in the course of PD, and furthermore, neuritic AS pathology in the GI tract occurs at least as early, if not earlier (33 –35). In fact, Braak has proposed a pathological staging system of PD based on AS pathology derived from characterization of AS neuritic pathology in brains from PD patients as well as aged patients where incidental AS neuritic pathology was found on neuropathological examination (33, 35). The scheme proposes that PD pathology begins in the DMV, with subsequent spread of AS pathology rostrally from the DMV to pontine nuclei (locus ceruleus, raphe), to midbrain (substantia nigra), and last, to the cortical areas. This model is quite controversial, with some authors in agreement and others less so (41, 144, 145, 177, 217). In particular, a theory of progressive pathogenesis based on cross-sectional postmortem data and the assumption that patients with incidental Lewy pathology would ultimately develop PD is problematic. However, there is uniform agreement that the DMV is consistently and severely affected by PD pathology early in the disease course.
Consequences of DMV Degeneration in PD
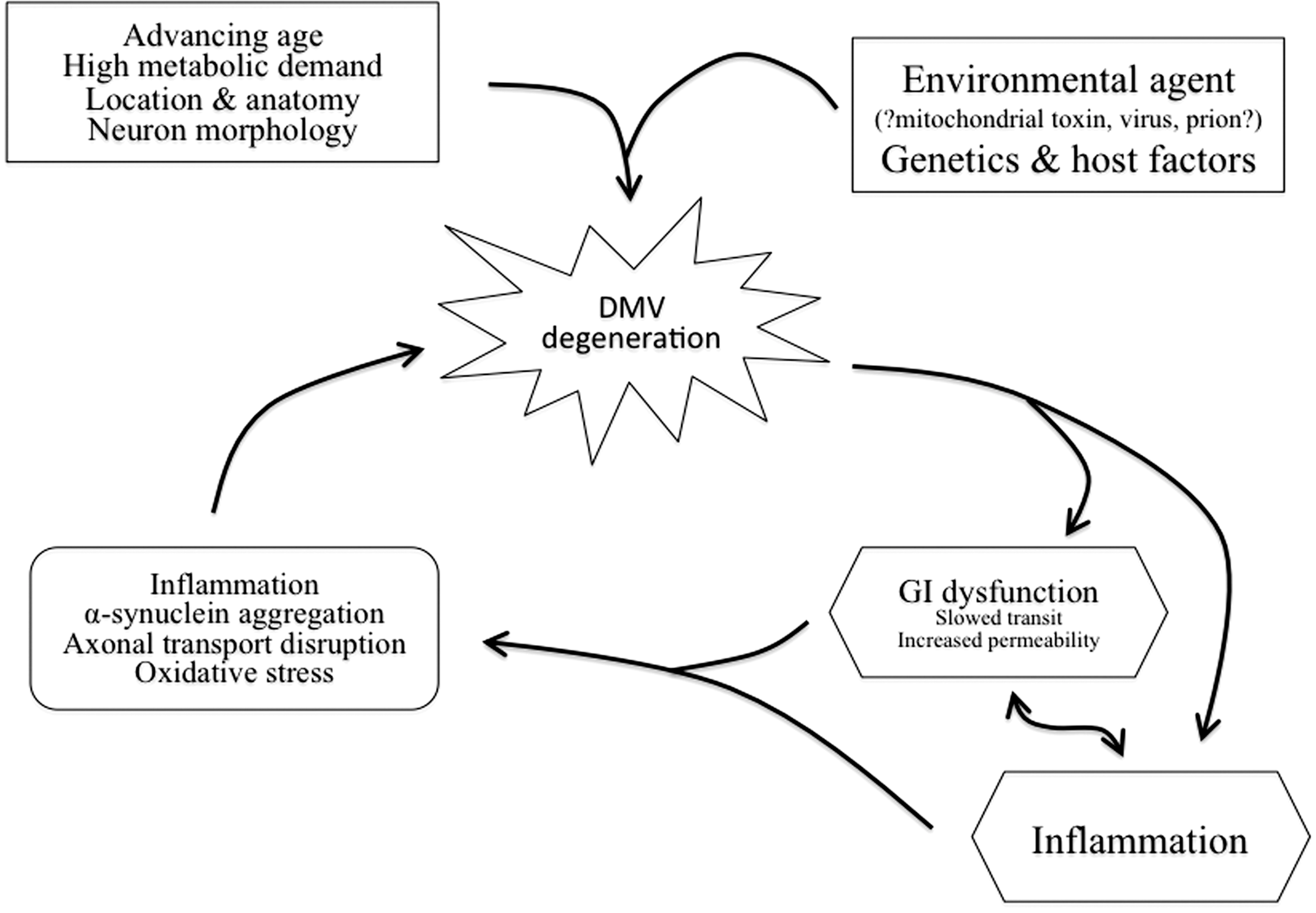
Given the functions of the vagus nerve, there are multiple potential consequences to dysfunction and degeneration of the DMV that may impact PD patients, including GI dysfunction and unchecked inflammation. In addition to direct impact on patient symptoms and quality of life, damage to DMV neurons could contribute to a feed-forward cycle in the pathophysiology of PD whereby loss of DMV function worsens neuropathology (Fig. 4).
GI dysfunction
GI dysfunction is a prominent nonmotor symptom in PD that occurs in nearly every patient at some point in his or her illness (85, 227, 228). Symptoms span the entire alimentary tract and include early satiety and nausea from delayed gastric emptying, bloating from poor small bowel coordination, and constipation and defecatory dysfunction from impaired colonic transit (84, 85, 196, 227).
A comprehensive survey of PD patients in the late 1980's found that problems with saliva occurred in 70% of PD patients, dysphagia (trouble swallowing) in 52%, nausea in 24%, and constipation in 29%, whereas the same symptoms were reported by less than 10% of age-matched control individuals. In addition, defecatory dysfunction was reported by two-thirds of PD patients—twice the control prevalence (84).
Subsequent studies have confirmed the high frequency of GI abnormalities in PD. Abnormal gastric emptying has been described in 43%–88% of PD patients and can worsen as PD progresses (79, 106, 107, 125, 153, 201). Interestingly, objective gastric motility abnormalities are frequently detected even in PD patients without subjective GI complaints (50). The incidence rate of constipation in PD has been estimated to be anywhere from 29% to 81%, and problems with defecation are also much more common in PD (∼60%) than in age-matched controls (∼25%) (83, 250, 256, 257).
GI dysfunction has many important consequences for PD patients. First, GI symptoms negatively impact the quality of life. They have significant psychosocial impact by causing alterations in eating habits, contributing to feelings of embarrassment, and invoking the need for social and caregiver adjustment (196, 250). Second, GI dysfunction in PD is associated with serious, possibly life-threatening complications, including pulmonary aspiration, weight loss, malnutrition, megacolon, and intestinal pseudo-obstruction (1, 12, 85, 147, 190, 204, 251). Third, GI dysfunction dramatically impacts motor PD symptoms by causing erratic absorption of oral medications, motor fluctuation, and medication side effects (10, 79, 80, 106, 107, 156, 201). Finally, it has recently been hypothesized that GI disturbance may be a sentinel event in the manifestation of PD. Constipation can appear decades before motor symptoms in PD, and recent data from a large-scale study suggest that constipation is associated with an increased risk of future PD (2, 269).
The majority of GI symptoms have objective correlates related to abnormal motility of the GI tract. For example, not only do patients describe symptoms of early satiety and bloating but also they have evidence that slowed stomach motility and delayed gastric emptying are responsible for those symptoms (50, 79, 107). Similarly, patients experiencing constipation have objective evidence for slowed colonic transit, and patients with defecatory dysfunction have electrophysiologic sphincter abnormalities (8, 9, 84, 143). The total GI tract transit time is also significantly prolonged in PD (66).
In addition to dysmotility, it has recently been suggested that PD patients have increased GI permeability and gut leakiness. Two small studies have indicated using noninvasive testing that absorption of nonmetabolizable sugars is greater in PD patients, implying that permeability across the GI mucosa is higher (89, 252). One has shown an increase in Escherichia coli density in the epithelium and lamina propria and an indication that PD patients have greater exposure to bacterial endotoxin (89). These findings correlated with abnormal AS immunoreactivity and markers of neuroinflammation; however, data are scarce, and the extent and causes of increased gut permeability are unknown.
Despite the frequency of GI dysfunction and GI neuropathology in PD, it is not clear whether or not there is a causal relationship between the two findings, and clinicopathological correlation studies have only begun to be performed (6, 72, 89, 164, 165). Recent evidence from colon biopsies indicates both synuclein aggregation and neuronal loss in the submucosal plexus of the ENS possibly correlated with GI symptoms in some way (163, 165, 232). This is in contradistinction to the myenteric plexus of the ENS which, although it contains a greater burden of synuclein pathology, does not exhibit loss of neurons (6). As far as the potential role of vagal pathology, there is a case report of motor fluctuations first developing in a PD patient immediately after vagotomy (78). In addition, transgenic animals expressing abnormal AS in the DMV exhibit age-dependent GI dysfunction similar to that seen in PD in the absence of synuclein pathology in enteric neurons themselves (205). Experience from the midbrain suggests that clinical symptoms in PD are driven primarily by neuronal loss rather than aggregation of α-synuclein, so quantitative evaluation of neuron populations that control GI motility is an important step toward determining the pathological underpinnings of GI symptoms in PD. In this regard, exploration of DMV pathology is more advanced than that for other neuronal populations that modulate GI motility since neuronal loss has been convincingly described in PD; however, no direct correlations between vagal pathology and GI symptoms have been completed (82, 88, 126). Obviously, these would be difficult studies to perform, since pathological evaluation of the DMV necessarily occurs postmortem, but organizations such as the Arizona Parkinson's Disease Consortium and the Banner Sun Health Research Institute Brain and Body Donation Program have recently begun to address this need (6). Equally intriguing is the potential to use advanced imaging techniques such as functional magnetic resonance imaging to monitor DMV activity in real time in PD patients or animal models with GI symptoms (150, 186, 268, 270, 282, 283).
Unchecked inflammatory responses
Activated microglia, accumulation of proinflammatory cytokines, nuclear factor kappa B pathway activation, and oxidative damage to proteins have all been described in the brains of individuals with PD (132, 262, 263). Intriguingly, the midbrain from patients or primates exposed to the dopamine neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) years earlier (supposedly a one-time event) displayed similar microglia activation and signs of active neurodegeneration at autopsy suggesting that common mechanisms may underlie degeneration induced by divergent triggers (14, 160, 195). In addition, the chronic use of nonsteroidal anti-inflammatory drugs, in particular, ibuprofen, has been associated with a lower incidence of PD (51, 52, 253).
Experimental evidence from rodent models also supports a role for both innate and adaptive immune system modulation of dopamine neuron survival. Exposure of dopamine neurons to the gram-negative bacterial endotoxin lipopolysaccharide, after either direct infusion into the midbrain or intraperitoneal injection, results in delayed loss of dopamine neurons associated with persistent neuroinflammation in the midbrain (91, 98, 99, 235). Even dopaminergic damage caused by noninflammatory insults such as MPTP or 6-hydroxydopamine is prevented by blockade of proinflammatory cytokines such as tumor necrosis factor α, inhibition of cyclooxygenase, or alteration of adaptive immunity by transfer of T lymphocytes (20, 37, 91, 136, 168, 194, 242, 243, 265). Importantly, transient initiation factors (i.e., toxins, bacterial or viral infections, particulate matter, and pesticides) may trigger an active, self-perpetuating cycle of chronic neuroinflammation (i.e., increased production of chemokines, cytokines, reactive oxygen species/reactive nitrogen species, and adhesion molecules by activated microglia), which serves to further promote clustering of activated microglia around dopamine neurons and may contribute to irreversible neuronal dysfunction and cell death (13, 38).
Although selective loss of midbrain dopamine neurons from the substantia nigra pars compacta is the typically recognized pathological hallmark of PD, as discussed above, many levels of the nervous system are pathologically affected by PD, with the DMV, one of the most severely and earliest affected spots (82, 88, 126). The usual interpretation of these findings has been that the same pathological processes that impact dopamine neurons affect DMV neurons. That may certainly be correct; however, the early involvement of the DMV and a broader vagal system may have additional implications for PD pathogenesis. In particular, the possibility that early loss of DMV neurons and a resultant deficiency of vagal anti-inflammatory signaling exacerbate damage to dopamine neurons is intriguing because if true, it would open a window of opportunity to protect dopamine neurons in PD. Whereas not explored in any neurodegenerative diseases to date, the hypothesis that anti-inflammatory actions mediated by the vagus nerve could be neuroprotective is gaining traction in other brain diseases such as stroke and traumatic brain injury (53, 133, 154). It has been known for years that people who smoke have a lower incidence of PD and that nicotine is protective in animal model systems (30, 117, 149, 233, 238). Given that the anti-inflammatory effects of the vagus are dependent on stimulation of nicotinic acetylcholine receptors, it is possible that the protective effect of nicotine against PD is explained by the anti-inflammatory effects of nicotinic receptor activation (148, 214).
Causes of DMV Degeneration in PD
There are myriad proposed mechanisms of neurodegeneration in PD with the precise one likely a combination of environmental and host factors, including genetics. This section will review proposed causes of neurodegeneration with a focus on the unique qualities of DMV neurons that may make them particularly susceptible to damage in PD. In addition, the consequences of damage to DMV neurons described above are likely contributors to a feed-forward cycle in the pathophysiology of PD (Fig. 4). A slower GI transit increases the exposure to potentially toxic environmental agents, increases inflammation by altering the luminal microbiotic environment, and disrupts brain-gut feedback loops; increased epithelial permeability has similar consequences. For example, although runaway inflammation may be a consequence of DMV damage, it may also be a contributing cause.
Inflammation and neuroinflammation
Although it has not yet been studied, DMV neurons are theoretically susceptible to many of the same inflammatory insults as dopamine neurons. Perhaps, more prescient with regard to the role of inflammation in PD is the unique position bridging the central and PNS. In fact, DMV neurons are potentially exposed to a myriad of inflammatory insults via axon terminals in proximity to the GI lumen and, as such, the outside world (Fig. 2). This anatomy has led to the provocative idea that PD may be caused by an unidentified environmental agent that gains access to the CNS via the GI tract and vagus nerve (36).
The anatomy (or highway) to support such a theory is fairly well known (193, 198). More intriguingly, mice exposed to the influenza virus not only show viral infection in serial progression from the ENS to the DMV to substantia nigra dopamine neurons but evidence of AS aggregation, microglial activation, and neurodegeneration in the same regions (140). This raises the possibility that a localized, transient event could propagate in time and space to become a widespread self-sustaining neurodegenerative cycle.
A clinical association between the GI tract and inflammation in PD is just starting to be investigated, but recent epidemiologic and clinical evidence suggests a link. Pro-inflammatory cytokines and markers of glial activation are upregulated in the colon from PD patients (75). In addition, eradication of Helicobacter pylori (a common cause of gastric inflammation and ulcers) modifies PD progression and clinical deterioration (28, 81). Moreover, serum Helicobacter antibody profile may predict the presence, severity, and progression of PD (281). Recently, a polymorphism in the CARD15 gene previously associated with inflammatory bowel disease (IBD) has been associated with PD (26); vice versa, LRRK2, a causative PD mutation, is a susceptibility locus for IBD (15). Another putative PD gene, NR4A2, has also been linked to GI inflammation (122, 162). In rats, IBD exacerbates in vivo damage to midbrain dopamine neurons by an inflammation-mediated process (273).
As discussed in the next section, recent evidence suggests that one potential trigger agent could be aggregated synuclein itself acting via a prion-like mechanism (124). In addition to propagation of pathology by direct transfer, synuclein can trigger neuroinflammation, causing activation of macrophages, microglia, and astrocytes (3, 167). In fact, neuroinflammation and AS dysfunction can potentiate each other in animal model systems and drive chronic neurodegeneration (98, 100).
AS aggregation
Genetic abnormalities in AS cause parkinsonism clinically indistinguishable from idiopathic PD; pathological data are more limited but indicate a similar distribution of Lewy pathology and neuronal loss (231, 258). As described above, AS is a major component of Lewy bodies in all cases of PD, and all types of neurons lost in PD display AS-positive Lewy pathology (260). The converse is not necessarily true in that it is not clear every neuron containing aggregated AS invariably dies in PD. In addition, the toxic nature of AS is still being debated, and it is possible that aggregation is an attempt at a cytoprotective response rather than a cause of neuronal death (7, 60). Regardless of those caveats, hypotheses about mechanisms of neurodegeneration in PD must address AS.
The distribution of pathological AS aggregation has been discussed above, but AS in its native form is also expressed in DMV neurons. In rodents, AS is expressed at high levels in DMV neurons as compared with other neurons affected in PD (175). In addition, AS is very highly expressed in all efferent axons from the DMV to the ENS, and there is also expression of native AS in a proportion of enteric neurons (229). Given the widely ramified nature of each DMV axon, the burden of native synuclein expression in the GI tract is substantial, particularly in proximal segments like the stomach, which correlates with the distribution of AS neuritic pathology observed in PD (6).
The exact pathophysiological mechanisms of neuronal death related to AS are unknown, but hypotheses abound. One involves oligomerization, fibrillation, and aggregation, in which native or mutant AS forms toxic multimeric constructs that disrupt cellular processes (130). Another is that overexpression or mutation of AS causes abnormalities in mitochondrial morphology and function (135, 191). Direct mitochondrial accumulation of AS with resultant complex I inhibition has been reported in cellular models, rat brain, and PD brains (74, 179, 212, 213). A third suggests that overexpression of AS disrupts axonal transport and causes morphological changes in nerve terminals before cell body loss (56, 166). Another implicates increased generation of toxic oxygen or nitrogen free radical species causing indiscriminate cellular damage or specific activation of downstream apoptotic pathways (31, 135). Finally, impaired protein degradation may facilitate all the possible mechanisms listed above by accelerating accumulation of abnormal, misfolded, or aggregated AS (24). At present, it seems most likely that all of the above mechanisms interact to form a complicated interconnecting pathophysiological network either caused or exacerbated by AS.
Particularly interesting for DMV neurons, whose terminal fields nearly abut the GI lumen, has been the recent finding that AS and its associated pathology can propagate from cell to cell (124). The question was initially raised after a postmortem neuropathological evaluation of neuronal transplants placed into the striatum of PD patients in the 1980's revealed AS pathology and neurodegeneration in the transplanted cells (151, 152, 174). Subsequently, it was discovered that AS can transfer from neuron to neuron and seed pathology in the receiving neuron by a prion-like mechanism both in vitro and in vivo (73, 123, 192, 199, 200). Pathological forms of AS have been shown to induce Lewy-like pathology in normal mice and even more remarkably, to cause selective degeneration of dopamine neurons after injection into the striatum (184, 185, 192). This process apparently involves extracellular release of AS, perhaps transynaptically, although the precise mechanisms and location of transfer have not yet been conclusively determined (4, 62).
Defective axonal transport
A common theme in the neuropathology of PD that has emerged over the last few years is the prominence of pathology in neurites. The earliest dopaminergic pathology in PD is thought to be a loss of axons projecting to the posterior putamen (21, 64, 157, 244). Similar pathology to dopamine terminal fields in the striatum has been described in animal models of PD, including MPTP treatment in primates and mice and rotenone intoxication in rats, and the damage is apparent before loss of dopamine cell bodies in the substantia nigra (42, 58, 129). Axonal damage has also been described in other sites of PD pathology, such as the hippocampus, where it is thought to correlate with cognitive dysfunction (23, 97). This has led to a theory suggesting that PD neuropathology may be a dying-back phenomenon progressing from loss of synaptic function to distal axonopathy and eventually to neuronal death (Fig. 5) (68, 110, 197, 286).
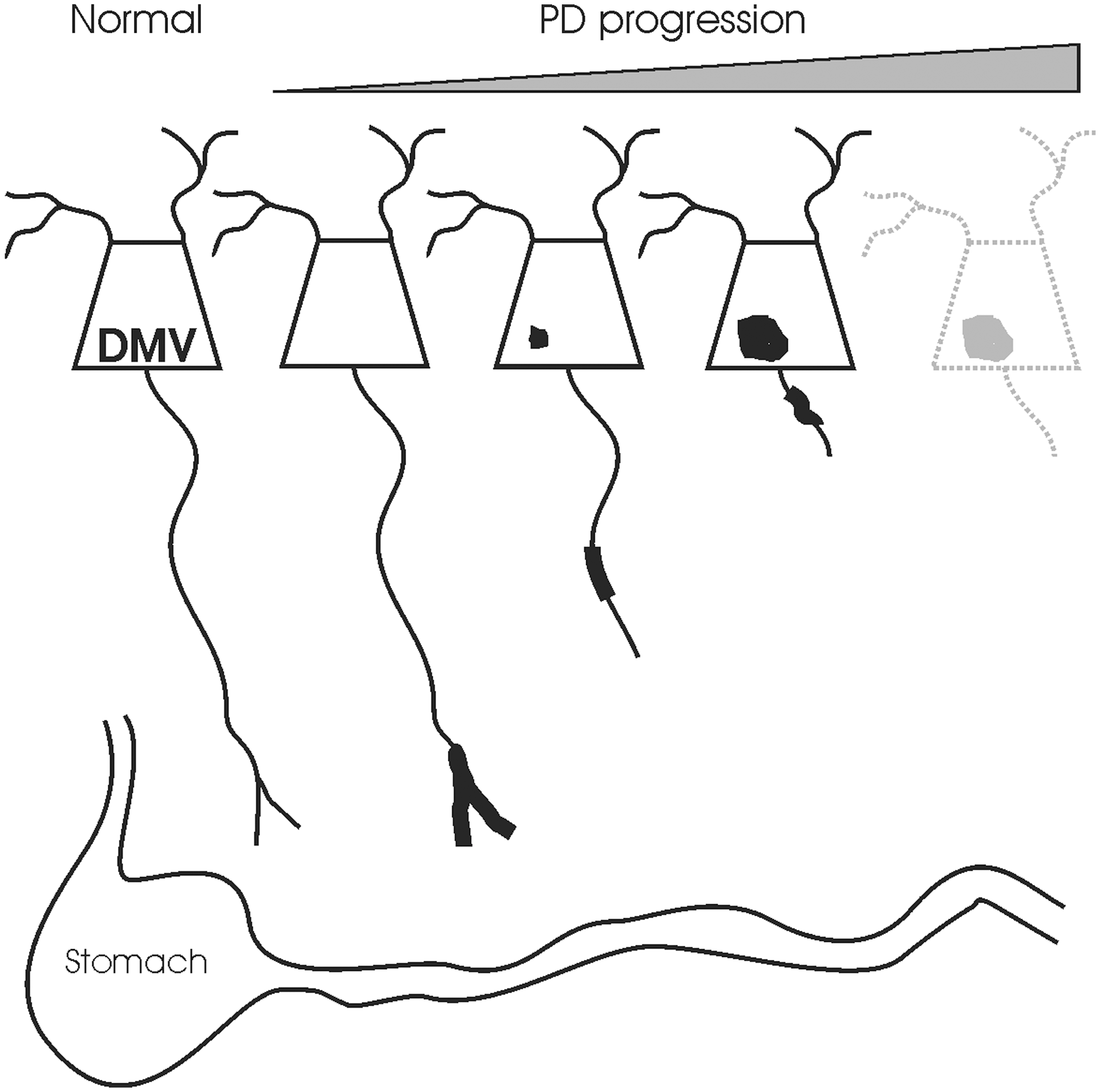
This has also been a burgeoning theme in the study of the autonomic nervous system in PD. AS neuritic pathology is often the most prominent, only, or perhaps the earliest pathology observable in areas relevant to GI motility (6, 34, 276). As discussed above, the DMV is not only a prominent site of Lewy pathology in PD but also one of the most common brain areas to be affected by incidental Lewy pathology, which is typically manifest as neuritic AS aggregation (35, 278).
Notwithstanding the general belief that axonal damage is the earliest manifestation of PD pathology, there have been few direct investigations into the involvement of axon terminals in PD symptomatology and pathogenesis and none in noncatecholaminergic neurons, such as those in the DMV. However, investigations of the cardiac peripheral sympathetic nervous system in PD provide some interesting parallels. Sympathetic terminal loss is the neuropathological correlate to cardiovascular symptoms associated with parkinsonism (110, 111). In the heart, cross-sectional postmortem studies suggest that damage to cardiac sympathetic nerve terminals precedes axon loss in the sympathetic nervous system and functions as a herald of neuron cell body pathology and loss (77, 93, 210, 211). DMV axons are very long and lightly myelinated meaning they may be particularly vulnerable to axon-mediated injury, since their terminals are far from the cell body at the end of a poorly protected and metabolically demanding axon.
Mechanisms of nerve terminal and axon damage in PD have not been fully elucidated (Fig. 6). Dopaminergic toxins like MPTP are concentrated in terminals by uptake via the dopamine transporter whereupon they inhibit mitochondria causing degeneration; this mechanism is active to a lesser extent in norepinephrine neurons, like those in the pontine locus ceruleus (141, 203). Abnormal catecholamine metabolism, such as that seen in mice with genetic hypoactivity of the vesicular monoamine transporter 2, has also been shown to preferentially damage catecholaminergic nerve terminals before causing selective degeneration of catecholaminergic cell bodies (44, 264). MPTP disrupts axonal transport, which can lead to terminal degeneration by interfering with the provision of necessary cellular components, such as mitochondria (29, 31, 68, 109, 197). As a result, mitochondria in axons and terminals are under particular stress, and studies demonstrate that synaptic mitochondria are highly vulnerable to mitochondrial complex I inhibition (65). Overexpression of AS has recently been shown to disrupt axonal transport and cause morphological changes in dopamine terminals before cell body loss in cellular models and in mice (56, 166). When viewed in the context of the central role that mitochondria may play in PD pathogenesis, current results suggest that the combination of disrupted axonal transport and mitochondrial dysfunction may be particularly damaging (27, 47, 68, 215, 218, 272, 290).
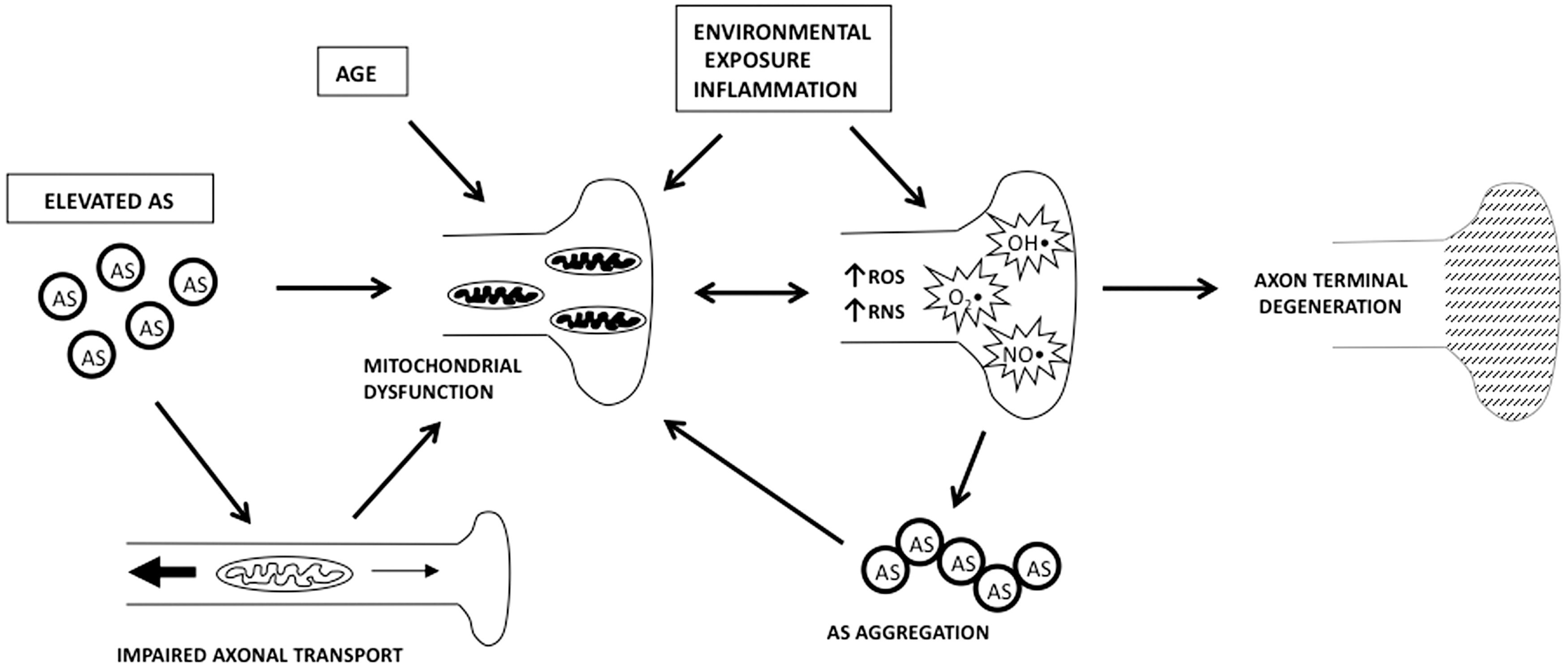
High metabolic demand and oxidative stress
High cellular energy demand has been described as a robust marker for selectively vulnerable neurons in a variety of neurodegenerative diseases (57, 113, 114). In PD, neurons vulnerable to parkinsonian neuropathology, including those in the DMV, have a high rate of metabolic demand. One factor in that demand is axonal transport through long, thin, highly branched, poorly myelinated axons as discussed above (33, 169), but synaptic activity is the main driver of the neuronal metabolic demand and consumes the majority of mitochondrial energy produced (11, 146). Axon morphology plays a role in this demand as well, since neurons of this shape tend to be highly metabolically active, at least partially as a result of the greater energy requirement for neurotransmission in such cells. Transmission is inherently slower in small-caliber fibers and poor myelination means there is limited energy benefit from saltatory conduction. Additionally, the energy cost of terminal maintenance is high due their long axon length and resultant axonal transport demands.
Adding to these factors is the recent discovery that DMV neurons, in addition to other neurons susceptible to PD, are vulnerable to metabolic stress due to their intrinsic activity patterns and the mechanisms by which those patterns are generated and maintained (108). Cholinergic DMV neurons exhibit an autonomous pacemaker activity accompanied by a robust extracellular calcium entry that is only weakly buffered by endogenous proteins. Neuronal activity and calcium entry both factor into elevating mitochondrial oxidant stress in DMV neurons. These characteristics are not unique to DMV neurons, but present in several neuronal subtypes vulnerable to degeneration in PD, including dopamine neurons in the substantia nigra and noradrenergic neurons in the locus ceruleus, thereby suggesting a common phenotype that defines neurons at risk in PD (47, 48, 261).
This hypothesis is yet to be fully tested in vivo, but oxidative metabolic stress related to mitochondrial dysfunction has been hypothesized to be intimately involved in PD pathogenesis (272, 290). A deficiency in complex I of the electron transport chain has been consistently described in PD, and the complex I inhibitors, MPTP and rotenone, mimic behavioral and neuropathological features of PD in animal models (25, 27, 64, 215, 272, 290). As might be expected, cells more dependent on mitochondrial respiration are more susceptible to mitochondrial inhibitors such as rotenone. The earliest behavioral abnormality associated with systemic administration of rotenone to rats is delayed gastric emptying, a result suggesting that neurons regulating GI motility, such as those in the DMV, may be particularly vulnerable to mitochondrial inhibition as is suggested by the recent physiological data (115).
Synthesis and Future Directions
The DMV is uniquely vulnerable to damage from PD. This selective vulnerability appears to be based both on its anatomy and intrinsic properties of its neurons. From an anatomical standpoint, efferent fibers in the vagus nerve from the DMV course throughout the GI tract forming a close link between the peripheral and CNS and a point of proximal contact between the environment (in the GI tract lumen) and brainstem areas where PD pathology is believed to be set in motion. In addition, since the GI tract is the largest immune organ in the body, vagal terminals are particularly exposed to damaging inflammatory insults. Intrinsically, DMV neurons are under high levels of oxidative stress due to their expression level of AS, fragile axons, and specific neuronal physiology. Moreover, several consequences of DMV damage, namely, GI dysfunction and unrestrained inflammation, may be the contributing causes of neurodegeneration and propagate a vicious cycle of injury that consumes the DMV and spreads to other vulnerable brain regions.
Although perhaps two among many, the DMV and vagus nerve may be crucial nodes in the network of pathophysiology that ultimately leads to PD. Their accessible anatomy may prove beneficial for PD therapeutics in the future if it can be harnessed, via neural stimulation, gene therapy, or targeted pharmacological intervention, to provide a system in which to study PD pathogenesis and a highway with direct access to neurons particularly vulnerable to the disease process.
Footnotes
Author Disclosure Statement
Dr. Greene has no conflicts of interest.