
Abstract
Introduction
H
The goals of the current article are to provide a summary of selected biological effects of H2S that are relevant in cancer cell biology, to review the experimental evidence on the role of endogenous cancer cell-derived H2S in cancer biology, and to overview the therapeutic potential of CBS inhibition for cancer therapy.
Biological Effects of H2S with Relevance for Cancer Biology
H2S, as a vasodilator and pro-angiogenic mediator
Vasorelaxation is one of the first recognized biological effects of H2S. Often compared with NO, H2S exerts a concentration-dependent vasodilatory effect in blood vessels. The mechanisms of H2S-mediated vasodilation include the activation of KATP channels, a variety of other channels, inhibition of phosphodiesterases, and a synergy with NO (132). The physiological vasodilatory effect, which appears to be more prominent in the microvasculature than in large resistance vessels, is evidenced by the development of progressive hypertension in mice deficient in CSE (142), although it should be also noted that the hypertension was not observed in another strain of CSE-deficient mice (49).
In the late 2000s, the pro-angiogenic effect of H2S was recognized. This effect involves all prototypical hallmarks of angiogenesis, such as endothelial cell proliferation, migration, and stimulation of the formation of tube-like structures. The pathways involved in this effect include KATP channel activation, Akt activation, and a synergistic interaction with NO through a cooperative inhibition of phosphodiesterases and the consequent activation of the cGMP/protein kinase G pathway (14, 20, 90, 119, 131). Endogenous production of H2S by endothelial CSE is also involved in the angiogenic effects of vascular endothelial growth factor (VEGF), a key endogenous growth factor and tumor-derived angiogenic hormone (90, 94).
H2S, as a bioenergetic stimulator
Although initially H2S was solely considered a mitochondrial “poison” via the inhibition of cytochrome c oxidase (mitochondrial complex IV), more recent studies unveiled a more complex, concentration-dependent modulation of mitochondrial function and cellular bioenergetics by H2S. In various cell types (including intestinal epithelial cells and hepatocytes), low concentrations of H2S act as mitochondrial electron donor, resulting in bioenergetic stimulation (36, 67, 120). Endogenous H2S produced by 3-MST or by CSE can also serve a role as a bioenergetic stimulator (31, 80, 81). The stimulatory effects of H2S on mitochondrial electron transport occur at the level of mitochondrial complex II through the interaction of H2S with the redox-sensitive protein sulfur-quinone-oxidoreductase. At higher concentrations, the stimulatory effects of H2S are superceded by an inhibitory effect at complex IV (87, 120).
H2S, as promoter of proliferation and cell survival pathways
H2S stimulates the proliferation of endothelial cells, fibroblasts, hepatocytes, and various cancer cells (8). The signaling mechanisms involved include activation of specific kinase pathways (e.g., MAPK and PI3K/Akt) and inhibition of selective phosphatases such as PTEN and PTP1B (8, 14, 47, 75, 144). The underlying molecular mechanisms include post-transcriptional protein modification by H2S (sulfhydration) (e.g., in the inhibition of PTEN and PTP1B) (65, 75) and intracellular formation of polysulfides from H2S followed by oxidative inactivation of the proteins (38, 61). Of potential relevance to cancer cell biology, the sulfhydration of nuclear factor kappa B (NF-κB) by H2S has also been shown to inhibit apoptosis (103).
Enzymology, expression, and transcriptional regulation of CBS
For detailed information on the enzymology, tissue expression levels, and transcriptional and post-transcriptional regulation of CBS, we refer the reader to multiple specialized reviews (7, 51, 76, 77, 107). CBS (EC 4.2.1.22) is traditionally introduced as the first enzyme of the transsulfuration pathway. Its physiological function is the elimination of homocysteine by conversion to cysteine. CBS deficiency is the most common cause of homocystinuria, an inherited metabolic disease, manifested from infancy, where many inactivating CBS mutations have been identified (76, 141). The catalytic activity and crystal structure of human CBS, a homotetramer consisting of 63-kDa subunits, has been characterized in substantial detail. The enzyme binds two cofactors, pyridoxal-phosphate (PLP) and heme. Each CBS monomer binds two substrates (homocysteine and serine), and its activity is allosterically modulated by S-adenosyl-L-methionine (SAM), which binds to and induces a rotation of the C-terminal CBS motifs and relaxation of the loops delineating the entrance to the catalytic site of the enzyme (Fig. 1). A truncated form of CBS (a 45 kDa “active core”) that exhibits increased catalytic activity due to a loss of the C-terminal CBS motifs has also been described (28, 58, 148).

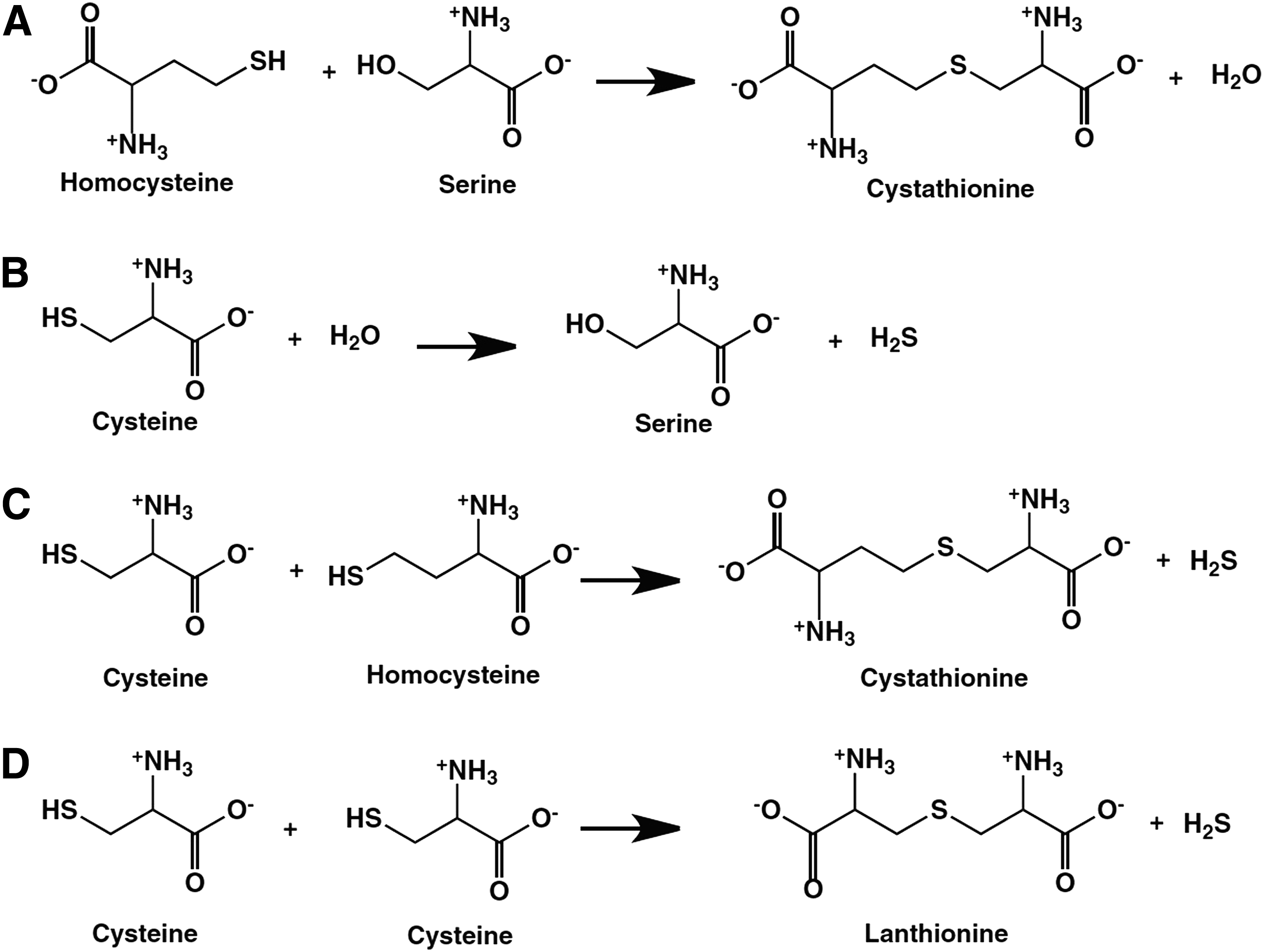
The reactions catalyzed by CBS are shown in Figure 2. In addition to the “canonical” pathway that is considered the major elimination route of homocysteine (Fig. 2A), CBS can produce H2S via at least three pathways, by the conversion of cysteine+H2O to serine+H2S (Fig. 2B), by the condensation of cysteine+homocysteine to yield Cystathionine+H2S (Fig. 2C), and by the condensation of two cysteine molecules to lanthionine+H2S (Fig. 2D). The catalysis involves two separate pockets of CBS, one, where the PLP external aldimine is formed and a separate one, where the nucleophilic amino acid is bound (107, 108).
CBS is physiologically a cytosolic enzyme, with highest expression in the brain [where H2S has been implicated as a neurotransmitter (1, 60, 100)], the liver, where the majority of physiological homocysteine “detoxification” takes place (29, 99) (via the reaction shown in Fig. 2A), and the kidney (29, 127). The cellular levels of CBS are regulated by transcriptional (29, 32, 73, 132), epigenetic (96), and post-translational (41, 107, 123) mechanisms. CBS expression is also under the control of hormonal regulation. For example, glucocorticoids and insulin regulate its expression in the liver and testosterone regulates its expression in the kidney (99, 127). Although normally cytosolic, CBS can also translocate into the nuclear and mitochondrial compartments under certain conditions (55, 123).
Role of the CBS/H2S Axis in Colorectal Cancer
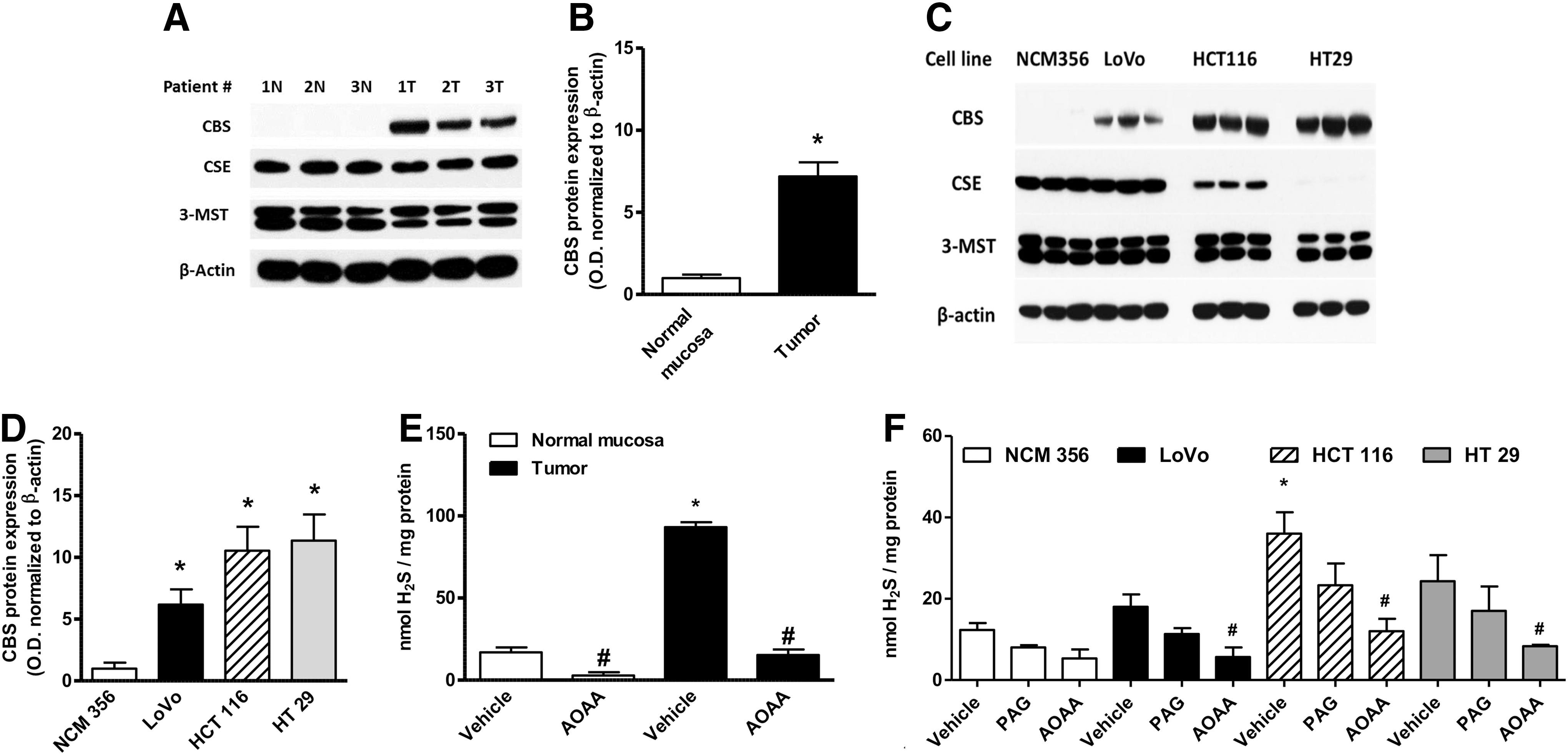
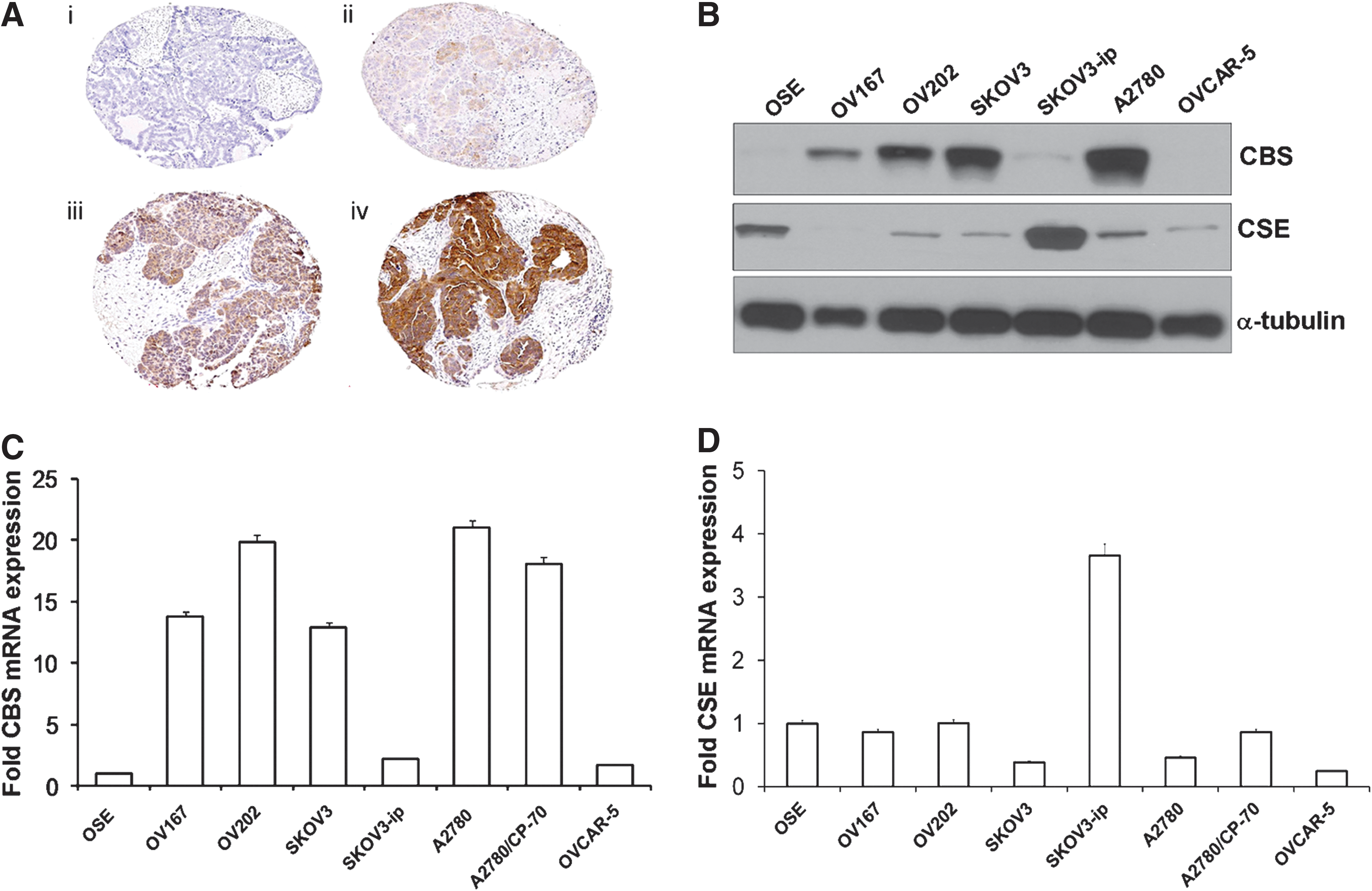
Colorectal cancer cells have a substantially higher expression of CBS than the surrounding normal mucosal margin (Fig. 3A, B) (117). Similarly, various human colon cancer-derived cell lines exhibit high expression of CBS relative to a normal colon cancer cell line (NCM356), whereas the expression levels of CSE are variable (Fig. 3C, D). The increased CBS expression correlates with increased capacity for H2S production, as measured in homogenates from human colon tumor tissue and colon cancer cell lines in vitro (Fig. 3E, F). The expression of the other H2S-producing enzymes (CSE and 3-MST) is not up-regulated in the colorectal cancer cells or the tumor tissue; the expression of 3-MST was fairly uniform in all cells and tissues studied, while CSE expression levels show more variability (Fig. 3A, C).
To address the function of CBS in colon cancer cell biology, we use the shRNA-mediated gene silencing to achieve >50% reduction in CBS protein expression in the human colonic epithelial cancer cell line HCT116 (Fig. 4A). For comparison, cells expressing a non-targeting lentiviral vector (shNT) were used. We also compared a separate HCT116 cell line with silencing of CSE, another H2S-producing enzyme, in order to verify the specificity of CBS in colorectal cancer.

CBS silencing resulted in a significant inhibition of HCT116 cell proliferation; whereas CSE silencing did not inhibit it, and, in fact, it tended to enhance it at later time points (Fig. 4A). Conversely, adenoviral overexpression of CBS into non-tumorigenic colonic epithelial cells (NCM356) resulted in the stimulation of cell proliferation (Fig. 4B) (117). These findings indicated a proliferation-stimulating role for endogenously produced H2S in colon cancer cells.
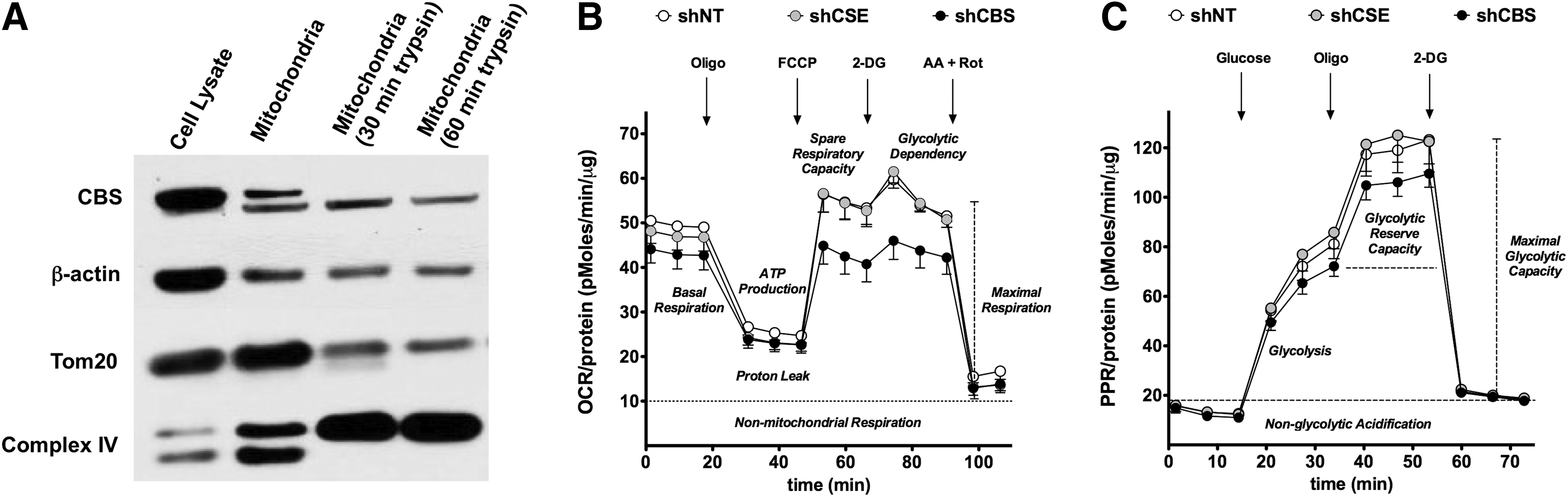
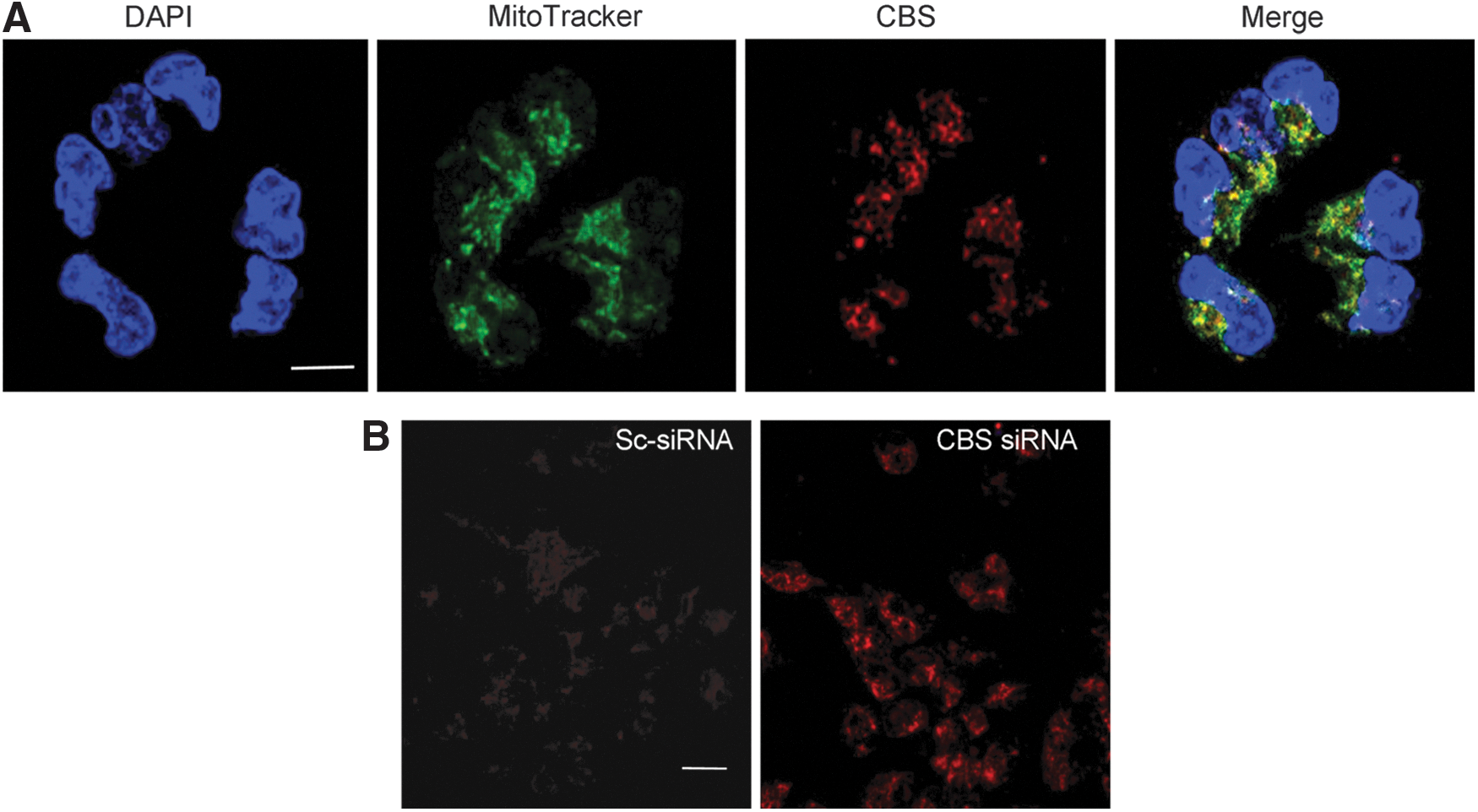
We next investigated the potential role of the CBS/H2S in the regulation of the bioenergetic status of the colorectal cancer cell. Although CBS is physiologically a cytosolic enzyme, we were surprised to find that a substantial proportion of CBS is localized to the mitochondria of HCT116 cells (Fig. 5A) (117). We speculated that translocation of CBS to the mitochondria places the enzyme to close proximity of one of its cellular targets (mitochondrial electron chain proteins), perhaps enabling a more effective transmission of H2S. Consistent with this hypothesis, CBS silencing suppressed both basal oxygen consumption and stimulated oxygen consumption induced by the administration of a mitochondrial-uncoupling agent (Fig. 5B) and also attenuated the glycolytic activity of the cancer cell (Fig. 5C) (117). We hypothesize that this is related to the known stimulatory effect of H2S on GAPDH (84). In contrast to CBS silencing, shRNA-mediated suppression of CSE failed to significantly affect either the proliferation or bioenergetics of HCT116 cells (Fig. 5B, C) (117).
We also explored the potential role of L-cystathionine (the other product of CBS). HCT116 cells contained high levels of L-cystathionine, which was further stimulated by the addition of L-cysteine/homocysteine. However, the addition of 1 mM L-cystathionine to HCT116 cells failed to increase their proliferation rate or bioenergetic function (117). Since the various biological responses (proliferation, mitochondrial electron transport) that were inhibited by CBS silencing and can be recapitulated by the administration of H2S donors (but not of L-cystathionine), we conclude that H2S is an independent product of CBS which is primarily responsible for the proliferative and bioenergetic actions observed.
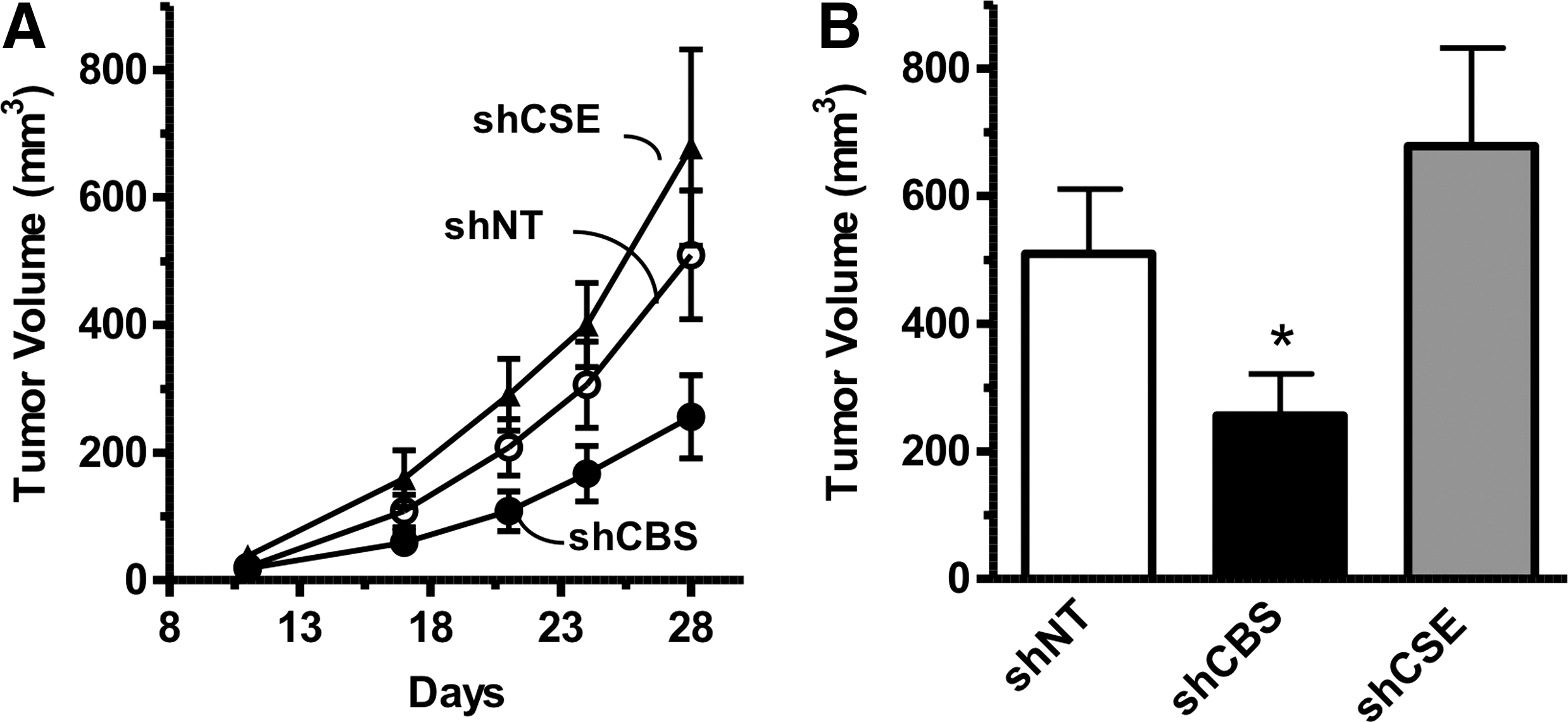
Consistent with the in vitro findings, shRNA-mediated knockdown of CBS reduced the growth rate of HCT116 tumor xenografts, while CSE silencing had no effect (Fig. 6A, B) (117). CBS suppression significantly reduced the density of CD31-positive blood vessels within the tumor tissue, and reduced both the prevalence of larger blood vessels and vessel branching (117). These data are consistent with the hypothesis that CBS-derived H2S, in addition to acting as an autocrine mediator, also acts locally in the tumor microenvironment in a paracrine manner to stimulate tumor angiogenesis. Finally, direct intra-tumor administration of the CBS substrate L-cysteine increased peritumoral blood flow, while pharmacological inhibition of CBS decreased it (117).
The autocrine and paracrine functions of colon cancer-derived H2S are also supported by an independent study of Yamagishi et al. (140). These investigators used HT29 cells [another human colon cancer line in which we have found high expression of CBS (117)]. When implanted into nude mice, Yamagishi et al. observed that the proliferation of HT29 cells was inhibited by a sodium hyaluronate-based physical barrier, which was designed to prevent the effusion of gaseous mediators (such as H2S) from the tumor. They also detected significant amounts of H2S inside the colon cancer tissue, although its enzymatic sources have not been identified. Based on their in vitro experiments in cell-free systems, the authors hypothesized that H2S is produced by the “Maillard reaction” (L-cysteine+D-glucose) (140), although it is currently unclear whether this reaction, in fact, takes place in tumor cells.
We have studied the proliferative and bioenergetic effects of the CBS activator, SAM, in HCT116 cells and the non-tumorigenic colonic epithelial cell line NCM356 (79). As expected, SAM exerted only minor stimulatory effects on the proliferation and energetics of NCM356 cells, which expressed low levels of CBS and produced low levels of H2S, which was increased by SAM to a slight degree. On the other hand, SAM induced a pronounced, concentration-dependent stimulation of H2S production in HCT116 cells, which was converted into an inhibition at the highest SAM concentration tested. The stimulatory effects of SAM-induced were absent in HCT116 cells with CBS silencing, indicating that these effects, indeed, are related to the allosteric stimulation of CBS, and the consequent enhancement of intracellular H2S levels. With longer SAM exposure times, however, the inhibitory effects of SAM became more prominent (on both proliferation and energetics/cell survival). These effects were comparable in HCT116 and NCM356 cells, and were not attenuated by CBS silencing. Taken together, these data indicate that the stimulatory effects of SAM are mainly related to the stimulation of the CBS pathway, while the inhibitory effects seen with higher concentrations/longer exposures of SAM are due to a significant CBS/H2S-independent cytotoxic component. Based on these data, inhibition of cell proliferation by SAM does not appear to represent a tumor cell-selective approach (79).
Role of the CBS/H2S Axis in Ovarian Cancer
Our findings of colorectal cancers have recently been confirmed in ovarian cancer (10). These investigators assessed the expression level of CBS in primary ovarian cancer specimens, and found high expression of CBS in the cytosol of primary ovarian tumors, especially in serous carcinoma, as well as in various ovarian cancer cell lines (Fig. 7). CBS was already present in early stages, but its levels further increased in more advanced forms of ovarian cancer (10).
Similar to our studies in HCT116 cells, Bhattacharyya et al. determined the functional effects of CBS silencing on ovarian cancer cell biology using CBS-specific siRNA-mediated gene knockdown. To exclude nonspecific targeting, two different siRNAs were used. The efficiency of the silencing was determined as 80% using Western blotting. CBS silencing resulted in a significant decrease in proliferation in all ovarian cancer cell lines studied (Fig. 8) (10). Similar to HCT116 cells, CBS silencing in A2870 ovarian cancer cells markedly suppressed cellular metabolism (mitochondrial electron transport); both basal oxygen consumption responses and the enhanced respiratory responses induced by a mitochondrial uncoupler were inhibited (10). The effects of CBS silencing were also associated with a decrease in cellular NAD/NADH ratio, a partial suppression of cellular ATP levels, and a significant decrease in cell viability (10). Similar to our earlier findings in colon cancer cells, a large proportion of CBS in ovarian cancer cells was localized to the mitochondrial compartment (Fig. 9A) (10). As expected, silencing of CBS significantly decreased total cellular glutathione levels in the ovarian cancer cells, while exogenous administration of GSH improved cell viability (10). Moreover, CBS silencing resulted in an increase in cellular reactive oxygen species (ROS) levels (Fig. 9B) (10), consistent with the role of H2S as an endogenous mitochondrial antioxidant. Since H2S exerts a protective/antioxidant effect on mitochondria (93, 113, 135), we speculate that this effect may be especially relevant for cancer cells, which often contain partially uncoupled mitochondrial electron transport and produce high levels of mitochondrial ROS (102, 134).

The effect of CBS silencing was also evaluated with regard to the activity of two critical redox-sensitive signaling pathways, p53 and NF-κB. CBS silencing in A2780 cells increased the expression of p53, while the expression of the RelA/p65 subunit of NF-κB decreased and the activation of NF-κB was attenuated (10). Although the exact connection of these responses to H2S and to cell proliferation and cell viability remains to be determined, these data confirm earlier suggestions (117, 118, 120) on the existence of an intricate interplay between H2S production, cellular bioenergetics, cellular redox status, and intracellular signaling pathways.
Consistent with our findings in colon cancer cells, the other product of CBS, L-homocysteine, failed to affect cell viability or proliferation in ovarian cancer cells, while “rescue” experiments with an authentic H2S donor (Na2S) restored cell proliferation in CBS-silenced ovarian cancer cells (10).
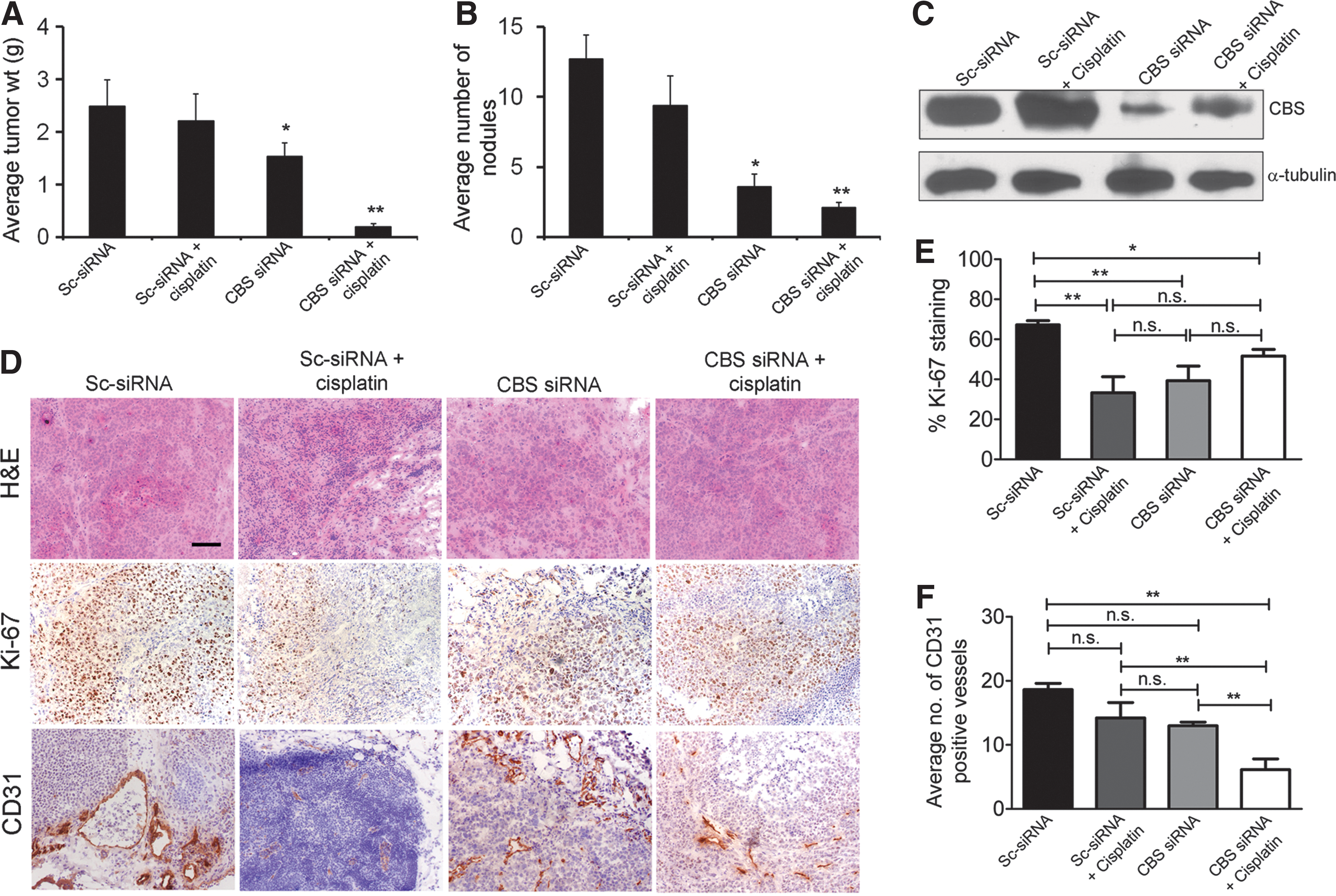
Nanoliposomal siRNA silencing of CBS in ovarian cancer xenografts significantly reduced tumor growth, with a 40% reduction in tumor burden and a 70% reduction in the number of nodules (Fig. 10A, B) (10). CBS silencing resulted in a significant growth inhibition in vivo, as assessed by Ki-67 staining, a nuclear proliferation marker (Fig. 10D). Finally, the number of CD31-positive blood vessels in the tumor was decreased, consistent with a paracrine angiogenic effect of tumor-derived H2S (10), similar to our earlier observations (117) in colon cancer cells. In the ovarian cancer model, the potential additive or synergistic effect of CBS silencing with cisplatin was also explored. In vitro, CBS silencing produced a slight potentiation of cisplatin cytotoxicity in A2780 cells; while in the in vivo experiments, the combination of cisplatin and CBS silencing produced a near-complete inhibition of ovarian cancer cell volume and nodule number (Fig. 10A, B) (10). Based on in vitro data, it is conceivable (although it remains to be directly tested) that the depletion of glutathione plays a role in the CBS-silencing mediated enhancement of cisplatin cytotoxicity.
Taken together, the studies in colon and ovarian cancer cells clearly establish CBS and CBS-produced H2S as a novel, endogenous, regulator of tumor progression.
Increased Expression of CBS and/or Increased Production of H2S in Other Forms of Cancer
Previous studies suggested increased expression of CBS and/or the increased production of H2S in various cancers. The earliest published report dates back to 1987 when Klein et al. detected elevated CBS activity in neuroblastoma surgical resection homogenates; these increases were also associated with elevated urinary cystathionine levels in the patients (62). In fact, the observation that cancer patients present with cystathionuria dates back as far as the early 1960s (34, 35); detection of cystathionine in the urine of patients was considered of diagnostic potential (33, 128). Stabler et al. showed markedly elevated urinary levels of cystathionine in the urine of prostate cancer patients; the levels correlated with the progression of the disease (111). While there are multiple pathways that produce cystathionine, an intriguing possibility may be the contribution of CBS up-regulation in cancer cells to this response. However, this hypothesis remains to be experimentally tested.
In myeloma samples, oligonucleotide microarray analysis showed a 16-fold increase in CBS gene expression (23) and a 7-fold up-regulation of CBS transcripts in biliary cancers (43). In an expression profiling survey, conducted in 2005, most cell lines of the NCI60 collection of human cancer cell exhibited increased CBS expression (assessed by Western blot analysis), with the highest expression levels noted in breast, ovarian, and renal cancer lines (146). In a more recent study, the prostate cancer cell lines LNCaP, PBH-1, and DU145 showed a marked increase in CBS expression, which correlated with increased H2S production (39).
Several lines of evidence from recent studies demonstrate significant increases in exhaled H2S levels in cancer patients (4, 48, 66, 140); however, the identification of its biological source was not investigated. In addition, elevated urinary thiosulfate (the stable breakdown product of H2S) levels were found in prostate cancer patients; the mean thiosulfate levels in prostate cancer patients were almost 50 times higher than in the control groups and 5 times higher than in patients with benign prostatic hyperplasia (19). Again, the cellular sources of thiosulfate were not defined in this study.
We speculate that there may be three potential mechanisms of the increased H2S levels in cancer patients. (i) Tumor cell-derived H2S may be absorbed into systemic circulation and exhaled through the lungs and/or excreted into the urine via its oxidative degradation to thiosulfate. (ii) CBS may be up-regulated as a consequence of local or systemic inflammation [as reviewed in Refs. (29, 132)]; thus, cancers develop in this pre-inflammatory microenvironment). (iii) Normal intestinal microbiota produces significant amounts of H2S; however, in the presence of cancer, additional tumor-derived H2S may contribute to elevate basal systemic H2S levels [as reviewed in Ref. (120)].
Effect of CBS Inhibition in Preclinical Models of Cancer
Aminooxyacetic acid (AOAA) is a prototypical pharmacological inhibitor of CBS that is widely used in H2S biology. Although the selectivity of AOAA is limited, it is currently the best pharmacological tool that is used to inhibit CBS-derived H2S production in vitro or in vivo. Using HCT116 cells, we and others have shown that AOAA treatment recapitulates all of the effects of CBS gene silencing, including the inhibition of cellular bioenergetics, proliferation, and H2S production (117). AOAA treatment also suppressed the migration and invasion of HCT116 cells toward fibroblast conditioned media and reduced endothelial cell migration toward HCT116 colon cancer cells in a multi-chamber co-cultures assay (Fig. 11). The efficacy of AOAA was comparable to that of a VEGF neutralizing antibody in this experimental system (Fig. 11). Furthermore, studies conducted in ovarian cancer cell lines (10), the breast adenocarcinoma cell line MDA-MB-231 (124), and pancreatic ductal adenocarcinoma cells (109) showed that AOAA treatment attenuated cellular proliferation, viability, and bioenergetic functions in vitro.

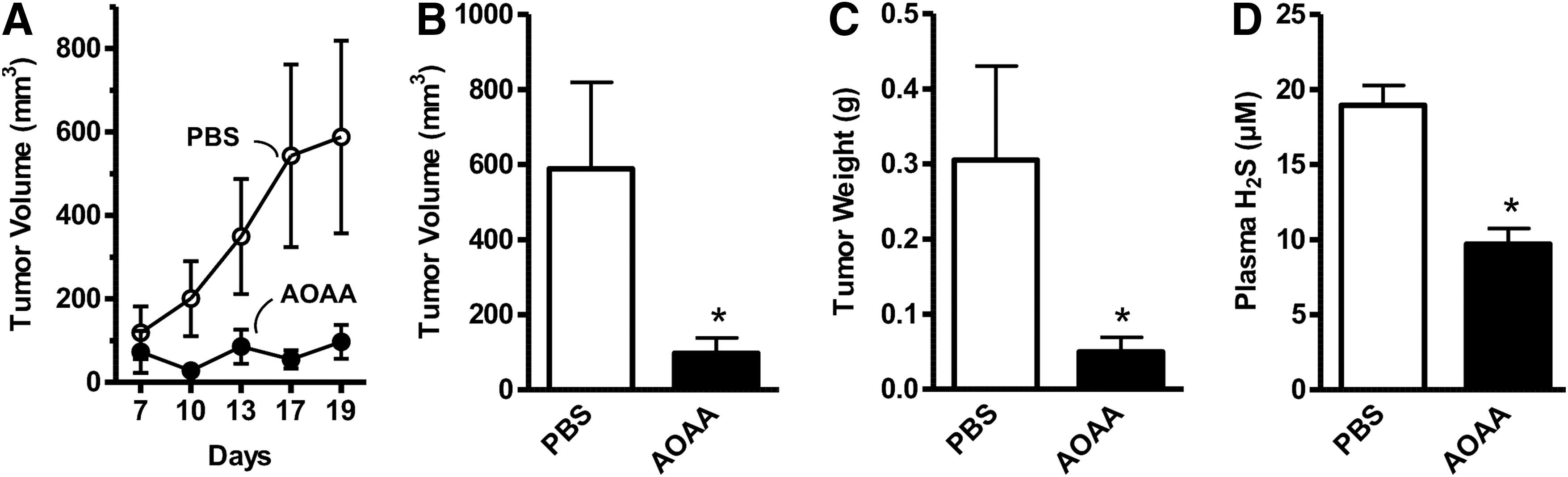
In vivo, AOAA exerted a marked inhibition of subcutaneous HCT116 cell tumor xenografts in athymic-nude mice (Fig. 12) (117), and the growth of patient-derived tumor xenografts (PDTXs) from three different colon cancer patients (Fig. 13). Moreover, in vivo treatment of mice with AOAA also attenuated the growth of MDA-MB-231 cell xenografts in nude mice (Fig. 14) (124).

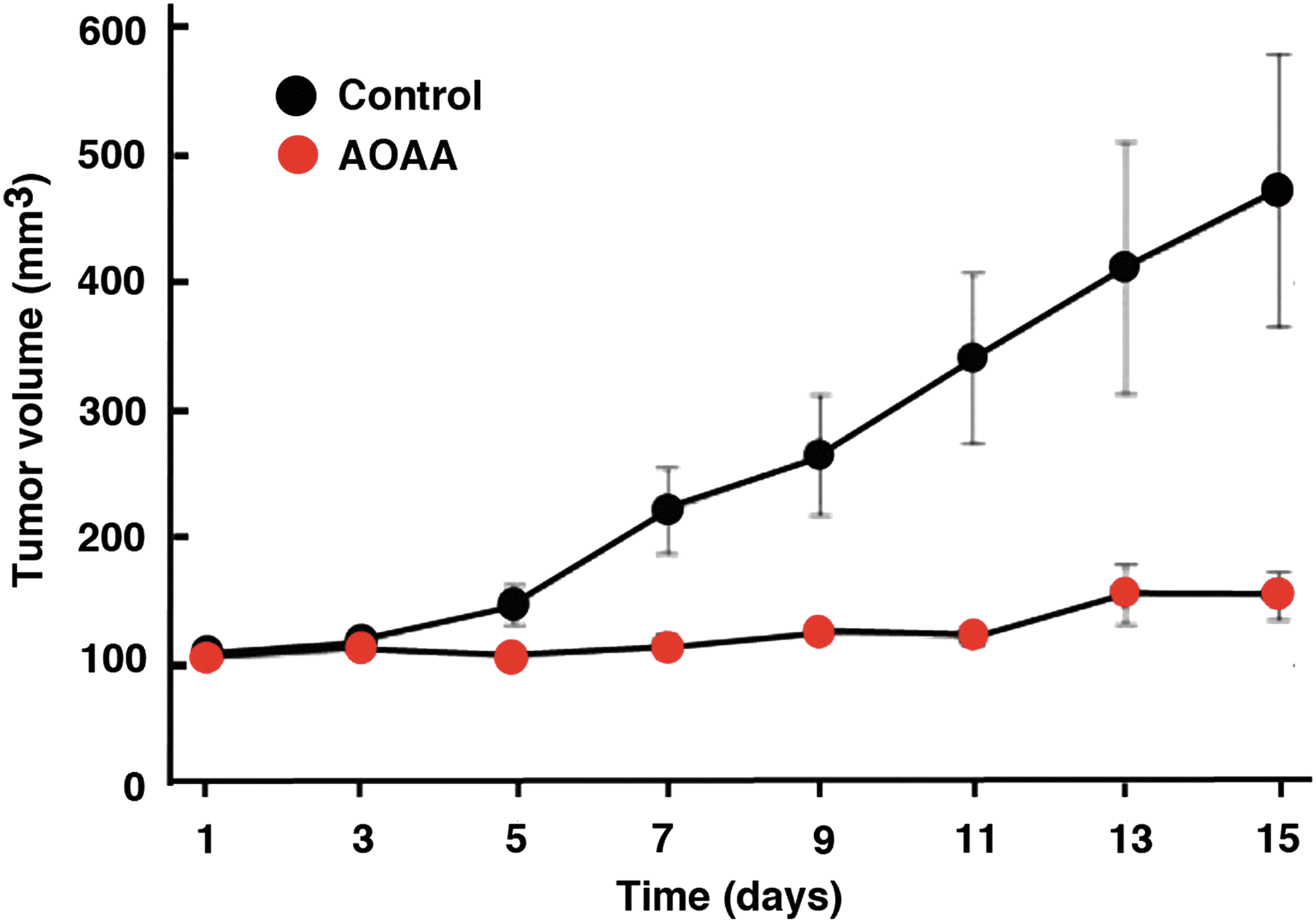
CBS inhibition also attenuated metastasis formation, as demonstrated in our recent studies, and enhanced the effect of oxaliplatin therapy (16a). Taken together with the studies demonstrating the synergy between cisplatin and CBS silencing in the ovarian cancer cells (10), CBS inhibition may enhance the efficacy of standard-of-care chemotherapeutic agents to reduce both recurrent and metastatic disease.
Pharmacological Inhibitors of CBS
The field of CBS inhibitors is in its infancy, because before the discovery of the role of CBS in cancer, there was no biomedical reason to synthesize or develop CBS inhibitors. In a recent study conducted in a collaborative effort between the Papapetropoulos group and our own group (5), a number of previously identified H2S biosynthesis inhibitors were tested with regard to the enzymatic activity of human recombinant GST-tagged CBS and CSE enzymes. For CBS inhibition, the potency was the highest for AOAA (IC50=9 μM), while trifluoroalanine and hydroxylamine showed weaker inhibition (IC50=66 and 278 μM, respectively) (Fig. 15A). Propargylglycine (PAG) and L-aminoethoxyvinylglycine were confirmed as fairly selective CSE inhibitors without any inhibitory effect on CBS for approximately 1 mM. Beta-cyanoalanine was also a preferential CSE inhibitor, which only exerted a slight inhibitory effect on CBS at 1 mM and above. The known PLP-dependent enzyme inhibitors D-cycloserine, isoniazid, parsalmide, pargyline, and methylseleno-cysteine exhibited a 6%–26% inhibition of CBS activity at 100 μM and a 10%–21% inhibition at 1 mM. None of the compounds tested showed selectivity to CBS compared with CSE (5).
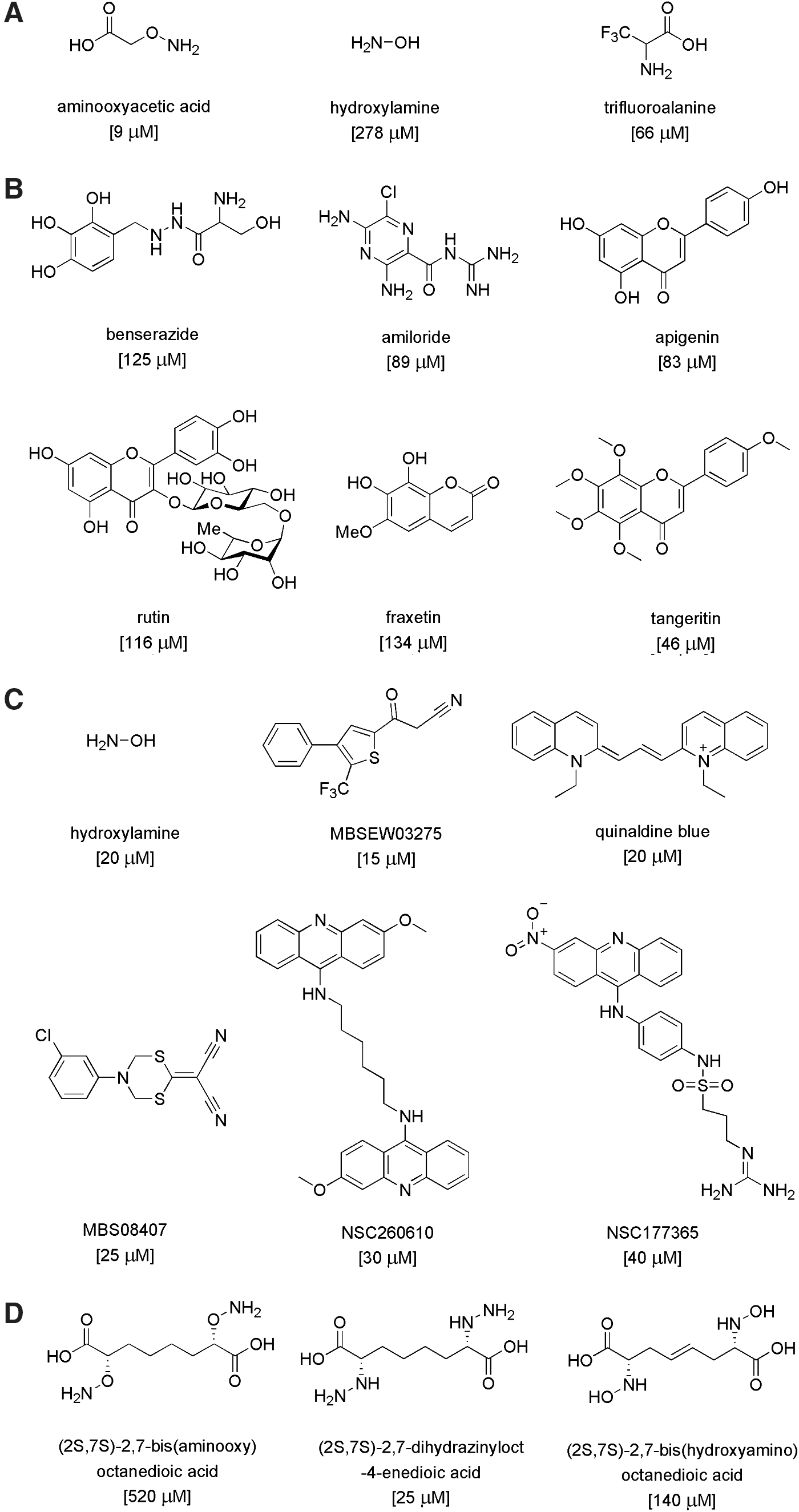
In late 2013, two articles were published on small-molecule screening for CBS inhibitors. Both projects used cell-free assays utilizing recombinant CBS enzymes and employed fluorescent H2S readouts (125, 147). Thorson et al., using a truncated version of human CBS (D414-551), screened a composite library of 1900 compounds (including compounds with known biological activity, clinically used drugs, and natural products) from the Microsource Spectrum Collection. An initial screen showed that twelve compounds at a concentration of 150 μM exhibited significant inhibitory activity in their fluorescence assay. Subsequent evaluations confirmed, the activity of several inhibitors included benserazide, a clinically used compound and known inhibitor of L-3,4-dihydroxyphenylalanine decarboxylase (a PLP-dependent enzyme), amiloride (a potassium-sparing diuretic and clinically used drug), and several flavonoids, including apigenin, tangeritin, and rutin. The structures of the compounds mentioned earlier and their reported IC50 are shown in Figure 15B. However, the relative potency of these compounds, compared with prototypical CBS inhibitors (e.g., AOAA or hydroxylamine) in their assay, is unknown.
Zhou et al. used recombinant human full-length CBS in a tandem microplate assay (147) to screen a chemical library consisting of ∼20,000 compounds assembled from NCI, FDA, and the Johns Hopkins Clinical Compound Library, Maybridge, ChemBridge, and other sources. Hit confirmation assays included selectivity testing against CSE, as well as the confirmation of direct binding by the hit molecules using a surface plasmon resonance assay, followed by in silico docking analysis. Controls included hydroxylamine (which, as expected, inhibited both CBS and CSE) and PAG, which, [in accordance with earlier studies (5)] was selective for CSE).
The screening yielded eleven CBS inhibitor compounds with IC50 values lower than 50 μM. Almost all of these inhibitors belonged to different structural classes, with NSC111041 and NSC67078 being the most potent inhibitors (IC50: 4 and 12 μM, respectively). Five compounds: MBSEW03275, quinaldine blue (also known as pinacyanol; code name in the screen: JHU-8555), MBS08407, NSC260610, and NSC177365 showed some selectivity for CBS compared with CSE, with MBS08407 showing the highest (>16-fold) selectivity, and an IC50 of 25 μM (147) (Fig. 15C). The potency of AOAA was not reported; however, another CBS inhibitor (hydroxylamine) had an IC50 of 20 μM under the assay conditions employed for the screening.
Others have used a structure-function paradigm to develop potential CBS inhibitors (105). Their efforts focused on three series of putative CBS inhibitors, featuring oxyamino, hydrazino, and hydroxylamino functionalities, designed to engage the active site cofactor PLP in stable oxime, hydrazone, and nitrone adducts, respectively. Since these molecules are functionalized analogues of CBS product L-cystathionine, the assays were designed such that the compounds competed with endogenous (L,L)-cystathionine at the active site of CBS via CBS “reverse reaction” involving the hydrolysis of (L,L)-cystathionine to L-serine and L-homocysteine in the presence of CBS. The L-homocysteine formed was trapped and measured with Ellman's reagent. The most potent inhibition (25–30 μM) was observed with some of the hydroxyamino compounds. The oxamino and hydrazino derivatives exhibited low potency (IC50 between 500 and 3000 μM), although the inhibition became more potent (140–1200 μM) when the compounds were pre-incubated with the enzyme, indicating a slow binding kinetic. The most potent compounds from each class were (2S,7S)-2,7-bis(aminooxy)octanedioic acid, (2S,7S)-2,7-bis(aminooxy)octanedioic acid, and (2S,7S)-2,7-bis(hydroxyamino)octanedioic acid (Fig. 15D). Direct measurements demonstrated that these compounds form complexes with the CBS prosthetic group PLP as expected.
In the same work (105), an additional approach was also described, aimed at synthesizing and testing quartenary α-branched amino acids as a new class of potential electrophile-releasing triggers for CBS (as well as other PLP-dependent enzymes). For this series of compounds, CBS inhibitory effects were assessed using a radioactive assay. The inhibitors were preincubated with hCBS dimer (D,L)-homocysteine followed by the addition of [14C]L-serine. The radioactive product of the reaction was chromatographically separated and quantified by scintillation counting. These compounds were characterized as weak inhibitors; 30 mM O-difluromethyl L-serine or S-difluromethyl L-cysteine produced an ∼50% inhibition of catalytic activity after 60 min of pre-incubation with the enzyme.
To be complete, the gaseous transmitters CO and NO should also be mentioned, as they represent a special “class” of endogenous CBS inhibitors. These molecules bind to the heme group of CBS. The biochemical characteristics of these interactions have been explored in significant detail (16, 95, 112).
Different experimental conditions yield different relative IC50 values for CBS inhibition. For instance, CBS activity and the inhibitory potency of various compounds are influenced by the source and structure of the recombinant CBS used (i.e., species, full length vs. truncated etc.), as well as by the assay conditions (e.g., pH, buffer, time of pre-incubation of test compounds with the enzyme, read-out of the assay, plate format, etc.). As an example, a slight modification in the composition of the assay buffer produced a shift in the IC50 of hydroxylamine from 20 μM to above 200 μM (147). Therefore, inclusion of reference compounds/standards (e.g., hydroxylamine, AOAA) in the screening assays is essential to enable comparisons between different screening campaigns conducted in different laboratories. In the absence of such reference standards, the IC50 values reported in Figure 15A–D only can be compared within each screen, but not between different screens. For example, in our previous screen in human CBS, AOAA and hydroxylamine were determined to have respective IC50 values of 9 and 278 μM (5). In contrast, Zhou et al. established the IC50 of hydroxylamine as 20 μM under the conditions applied for screening (147); in other words, all of the identified hits in this screening campaign were comparable, in terms of potency, to hydroxylamine (Fig. 15C). Thus, there is a high likelihood that according to the present state of the art, all currently known CBS inhibitors lay in the AOAA-hydroxylamine range, perhaps with AOAA remaining the most potent molecule. However, this remains to be directly tested in future studies.
With regard to therapeutic repurposing of existing FDA-approved drugs, it appears that those identified as CBS inhibitors to date lack the necessary potency and/or selectivity for CBS-targeted therapy. For example, inhibition of CBS by amiloride or benserazide requires concentrations of 100 μM or higher, which would be difficult to achieve therapeutically, as the achievable plasma concentration of these drugs in humans is in the low nM range (52, 53). Based on the published database of clinically used compounds that are candidates for repurposing (18), Zhou et al. referred to quinalidine blue/pinacyanol chloride as an FDA-approved anticancer compound (147). However, other than the fact that the compound used to be a part of in vitro staining techniques for chromosome banding or beta-amyloid staining (86, 101), we were unable to find any in vivo studies or clinically relevant information on this compound in the literature. Therefore, the status of this compound as a potential human therapeutic agent remains unclear.
Taken together, recent work in the field of CBS inhibitors may have identified potentially useful chemical scaffolds for future medicinal chemistry optimization. It is likely that some of these compounds target the PLP site of CBS, increasing the potential for cross-reactivity with other PLP-dependent enzymes.
The Pharmacological Profile of AOAA: Implications for Cancer Therapy
Although referred to as the prototypical CBS inhibitor, the pharmacology of AOAA is complex (78) and not entirely specific. Its mode of action entails an irreversible binding to the prosthetic group PLP in CBS and therefore other PLP-dependent enzymes. For example, AOAA inhibits CSE (5). However, when compared under the same assay conditions, AOAA is a much weaker inhibitor of CSE (IC50=1 μM) when compared with CBS (IC50=9 μM) (5). In isolated rat aortic rings assay (a tissue with low levels of CBS), pretreatment with AOAA inhibits L-cysteine-induced smooth muscle relaxations to the same extent as the CSE inhibitor PAG; while the combined treatment of PAG and AOAA exerted no greater inhibition of relaxation, suggesting that AOAA can inhibit CSE in vascular tissue (5) (Fig. 16). Thus, the use of AOAA as an anticancer therapy may potentially cause hemodynamic side effects due to the physiological actions of CSE/H2S on vasodilator tone.
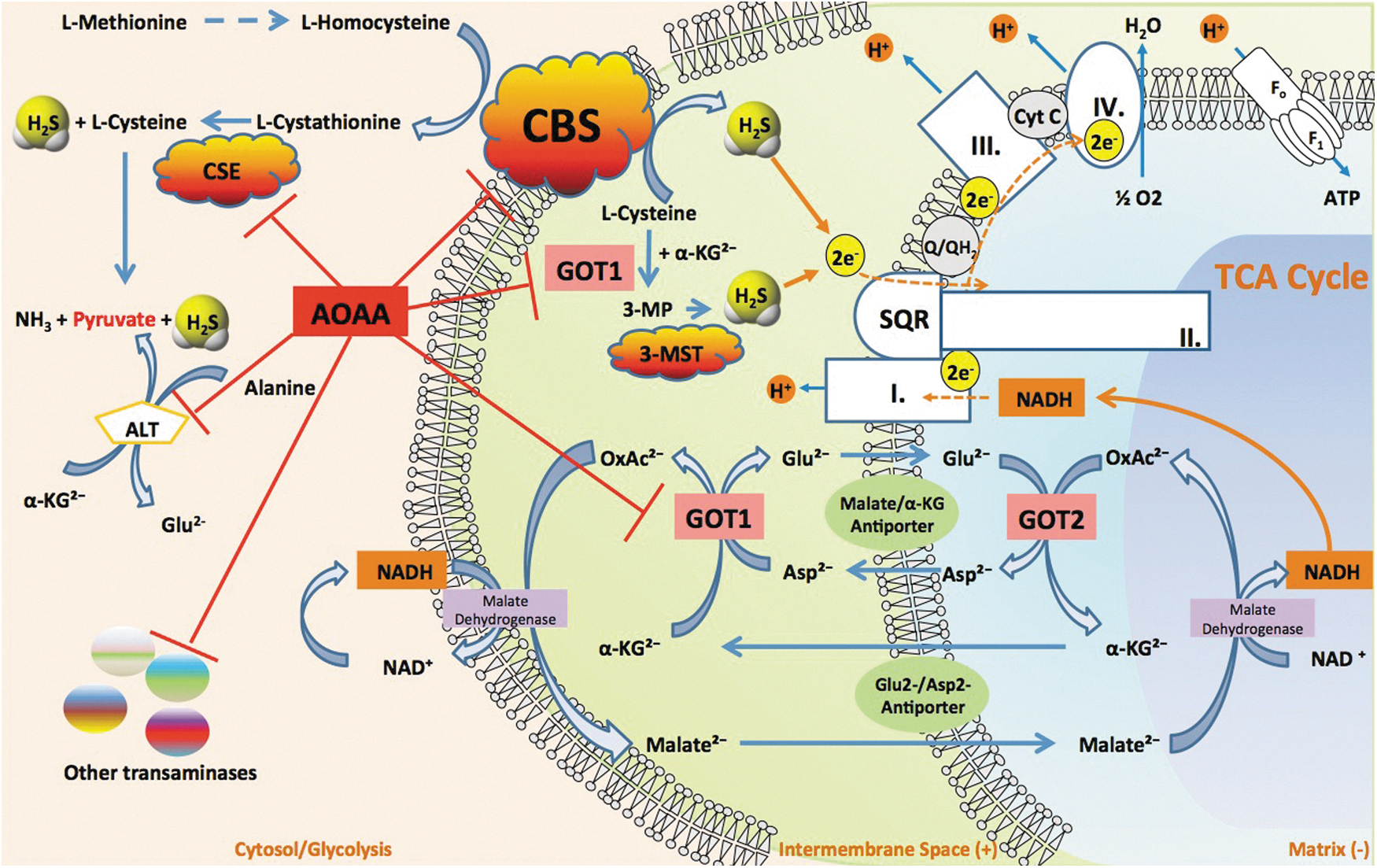
It is possible that AOAA exerts additional inhibitory effects on H2S production through the indirect inhibition of 3-MST. 3-MST generates H2S from 3-mercaptopyruvate (3-MP), which is produced by the cysteine aminotransferase (CAT, EC 2.6.1.3)-dependent condensation of cysteine and α-ketoglutarate through the following reaction: L-cysteine+2-oxoglutarate→mercaptopyruvate+L-glutamate. CAT is identical to cytosolic aspartate aminotransferase (EC 2.6.1.1, also known as glutamate oxaloacetate transaminase 1 [GOT1], aspartate aminotransferase, or aspartate transaminase [AST]). In its latter (GOT or AST) function, the enzyme catalyzes the following reaction: L-aspartate+2-oxoglutarate→oxaloacetate+L-glutamate. Inhibition of CAT by AOAA would be expected to inhibit the formation of 3-MP and therefore, it would be expected to inhibit 3-MST-induced H2S production (Fig. 16).
To further increase the complexity of the subject, we also have to mention that GOT1 is an essential component of the malate/aspartate shuttle. In cancer cells, this mechanism has been shown to link glycolysis to the transfer of electron donors into the mitochondria and this transfer is inhibited by AOAA (22, 37, 124, 133). Moreover, the function of GOT1 is linked to glutaminolysis, another partially cancer-selective metabolic pathway and anticancer target (109). Knockdown of GOT1 and other essential enzymes in the glutaminolysis pathway have recently been shown to suppress cancer cell metabolism (109). Therefore, we have recently suggested (78) that AOAA may exert its anticancer effects through the simultaneous suppression of the malate/aspartate shuttle, glutaminolysis, and the CBS/H2S axis. In effect, through these diverse, but interrelated pharmacological actions in the cancer cell, AOAA can be viewed as an inducer of “synthetic lethality” (Fig. 16).
Although not an FDA-approved drug, AOAA was given to patients with Huntington's disease and tinnitus in several non-randomized clinical trials in the 1970s and 1980s (42, 91, 92). In these disorders, AOAA was used to inhibit an enzyme in neurons, glutamic acid decarboxylase or GABA transaminase (GABA-T), a PLP-dependent transaminase. These clinical studies established dose-limiting toxicities in human subjects and suggested that AOAA can be reasonably well tolerated. However, the study design and drug formulation would not be acceptable if we applied today's regulatory standards. Nevertheless, with appropriate chemical and regulatory preparations, AOAA or closely related compounds may be amenable for re-introduction for cancer therapy in the future.
H2S Donation Versus H2S Biosynthesis Inhibition in Cancer
Literature searches focusing on “hydrogen sulfide” and “cancer” keywords will identify a significant number of articles which support the concept that cancer cells can be killed via H2S donation. On the surface, these studies appear to contradict key concepts outlined earlier. However, many of the contradictory findings regarding the effects of H2S on cancer cells may be explained by a careful consideration of a bell-shaped dose-response curve that characterizes H2S activity in biological systems.
Some of the published reports use complex molecules, where the H2S donor group is linked with a larger molecule, which also has its own independent pharmacological action. Prime examples of such parent molecules include non-steroidal anti-inflammatory drugs (NSAIDs) such as diclofenac, naproxen, or sulindac (17, 30, 57, 63). In these studies, typically no information is presented on the relative contribution of H2S versus the parent molecule to the anticancer effects, even though it is well known that NSAIDs exert anticancer effects in many preclinical models of cancer through inhibition of the pro-inflammatory cyclo-oxygenase signaling pathways. In fact, the beneficial property of the H2S donating group is likely related, at least in part due to its vasodilatory/cytoprotective effects on the gastric mucosa, which permits higher doses of the parent NSAIDs without gastric irritation. Moreover, simple calculations of the H2S “content” of these molecules, and a comparison of these numbers with the effects of authentic H2S donors makes it unlikely that the H2S contained in these structures can reach sufficiently high levels to induce cytotoxicity in the cancer cell. For these reasons, we have excluded the combined molecules mentioned earlier from our present analysis. In another study, Ma et al. report on the anticancer effect of the garlic extract component S-propargyl-cysteine (72). Although this compound is often discussed as a H2S pathway modulator, its pharmacological profile is extremely complex, as it includes kinase activation, up-regulation of CSE, and many other actions (9, 72). Thus, while the compound can induce apoptosis in vitro and may reduce tumors in vivo, we believe that its actions are too complex to be evaluated in the current review.
We will focus solely on studies where the effect of H2S donors was directly tested on the viability and/or proliferation of various cancer cell lines in vitro (6, 8, 13, 85, 139). For instance, Wu et al. using colon cancer cells (includig HT29 and HCT116) reported that H2S (400–1000 μM) decreased cell proliferation and induced autophagy in a concentration-dependent manner (139). Moore and colleagues used the slow-release H2S donor GYY4137 in multiple human cancer cell lines (HeLa, HCT116, Hep G2, HL-60, MCF-7, MV4-11, and U2OS) and found that the H2S donor decreased cancer cell survival at 400–800 μM (68). Similarly, Attene-Ramos et al. used CHO cells and HT29 cells, and showed a concentration-dependent decrease in cell viability at concentrations above 100 μM (6) and also when used in combination with caustic agents such as hydroxylurea (6). Finally, Cai et al. evaluated a wide range of H2S concentrations in HCT116 and SW480 cells and reported a concentration-dependent stimulation of cell proliferation between 10 and 50 μM H2S, a plateauing of the effect at 200 μM, and an inhibition of proliferation at 1000 μM (13).
Other studies reported diverse effects of H2S and cell growth/cell death in various transformed and non-transformed cell lines in vitro (6, 8, 13, 83, 85, 121, 139). Many of these reports are summarized in a review article by Baskar and Bian (8). A critical evaluation of the conditions and the concentrations of the H2S donors suggests that the effects of H2S are bi-phasic, as in a bell-shaped curve, where lower concentrations of H2S are pro-survival and proliferative, while higher concentrations of H2S become cytostatic, cytotoxic, and antiproliferative. We also hypothesize that the net effects seen in cell proliferation are the result of the net balance of endogenously produced and exogenously administered H2S. We assessed the proliferation rates of HCT116 colon cancer cells in the presence of different concentrations (0.03–3 mM) of either NaHS (fast-release) or GYY4137 (slow-release) H2S donors, with or without pretreatment with the CBS inhibitor AOAA. In the absence of AOAA, low concentrations (0.03–0.3 mM) of NaHS stimulated proliferation whereas higher concentrations (1–3 mM) inhibited growth (Fig. 17A). CBS inhibition with AOAA shifted the dose response to the left, not only decreasing basal proliferation but also limiting toxicity only to cells exposed to 3 mM NaHS (Fig. 17B). In the absence of AOAA, all concentrations of GYY4137 (0.03–3 mM) stimulated HCT116 cell proliferation over the basal growth rate (Fig. 18). However, only the highest concentration of GY4137 (3 mM) stimulated HCT116 cells when CBS was inhibited (Fig. 18). These data demonstrate that colon cancer cell proliferation exhibits a biphasic (bell-shaped) dose response to H2S. The precise nature of the cellular response (increased or decreased growth) is determined by the rate of H2S production (fast- vs. slow-release H2S donors) as well as by the concentration of donor relative to the endogenous (basal) level of CBS-dependent H2S production. Our model provides a useful framework to reconcile some of the controversies in the literature regarding the role of H2S in the regulation of cancer cell proliferation.
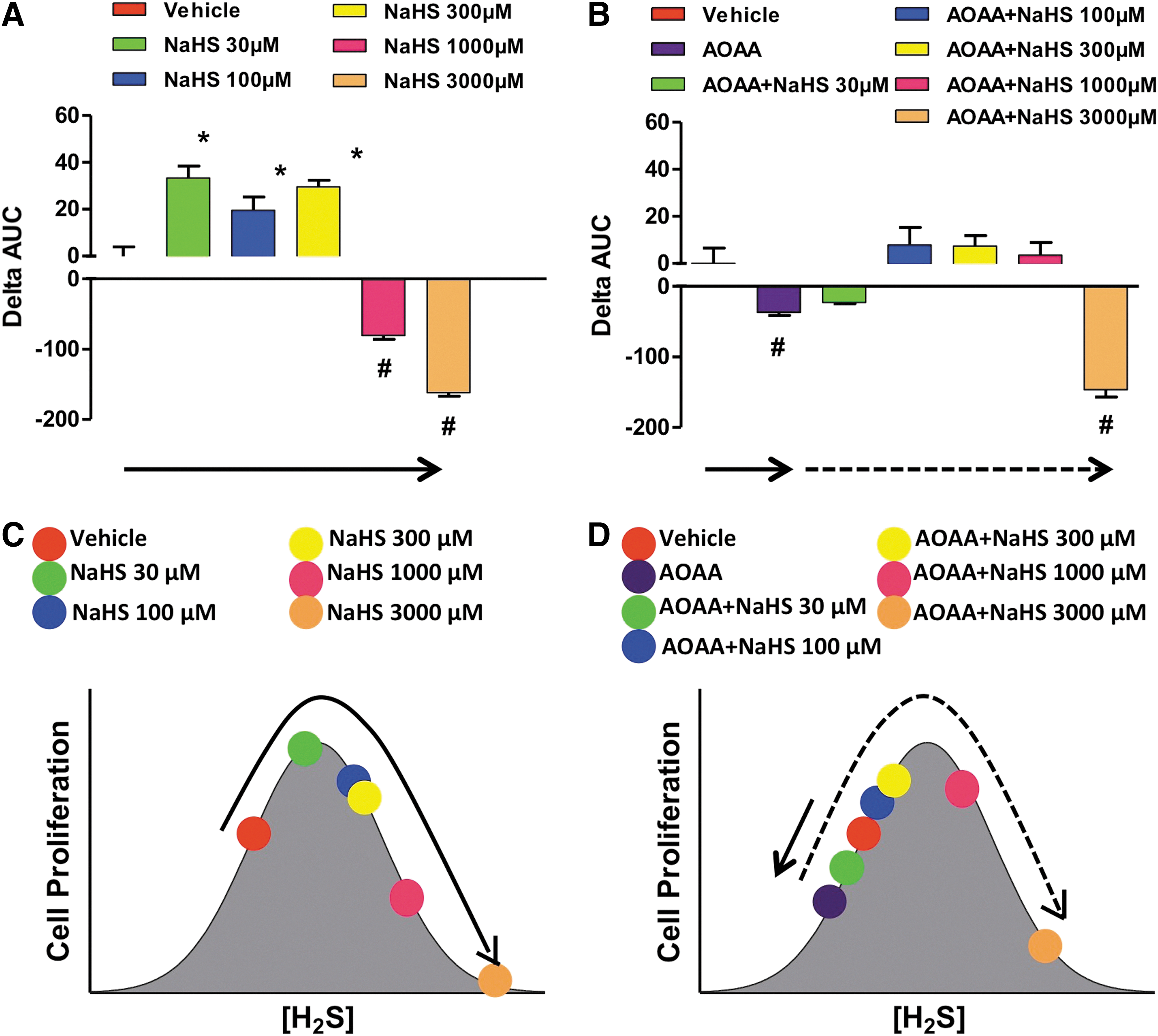
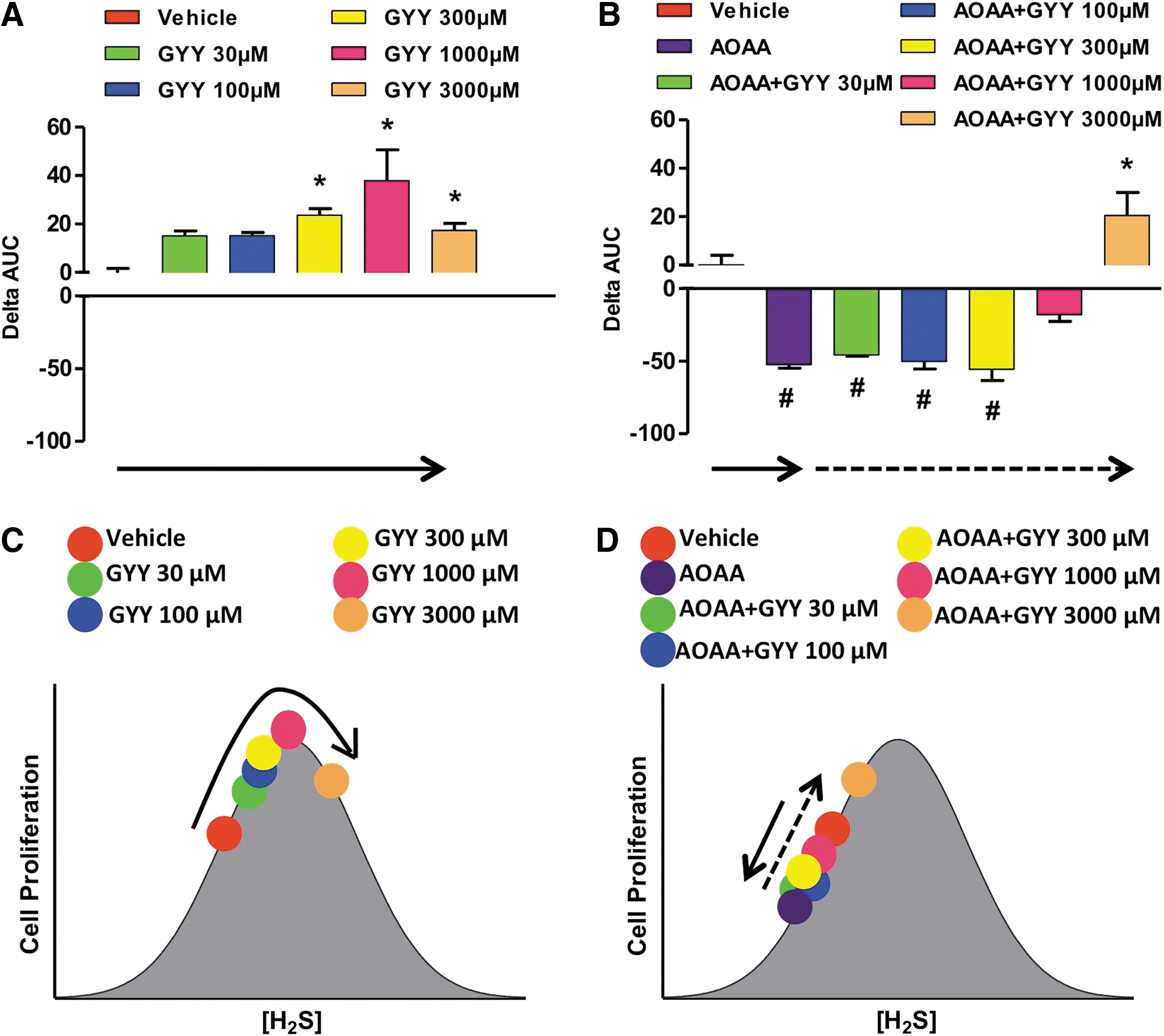
Similar to the bell-shaped pharmacological effects of the H2S donors, lower concentrations of the allosteric CBS activator, SAM increase proliferation; whereas higher concentrations (3 mM) inhibit it (Fig. 19A). When CBS was catalytically inhibited or physically absent, the allosteric activator was unable to modulate cellular H2S production or cancer cell proliferation (Fig. 19B, C, bottom cartoons).
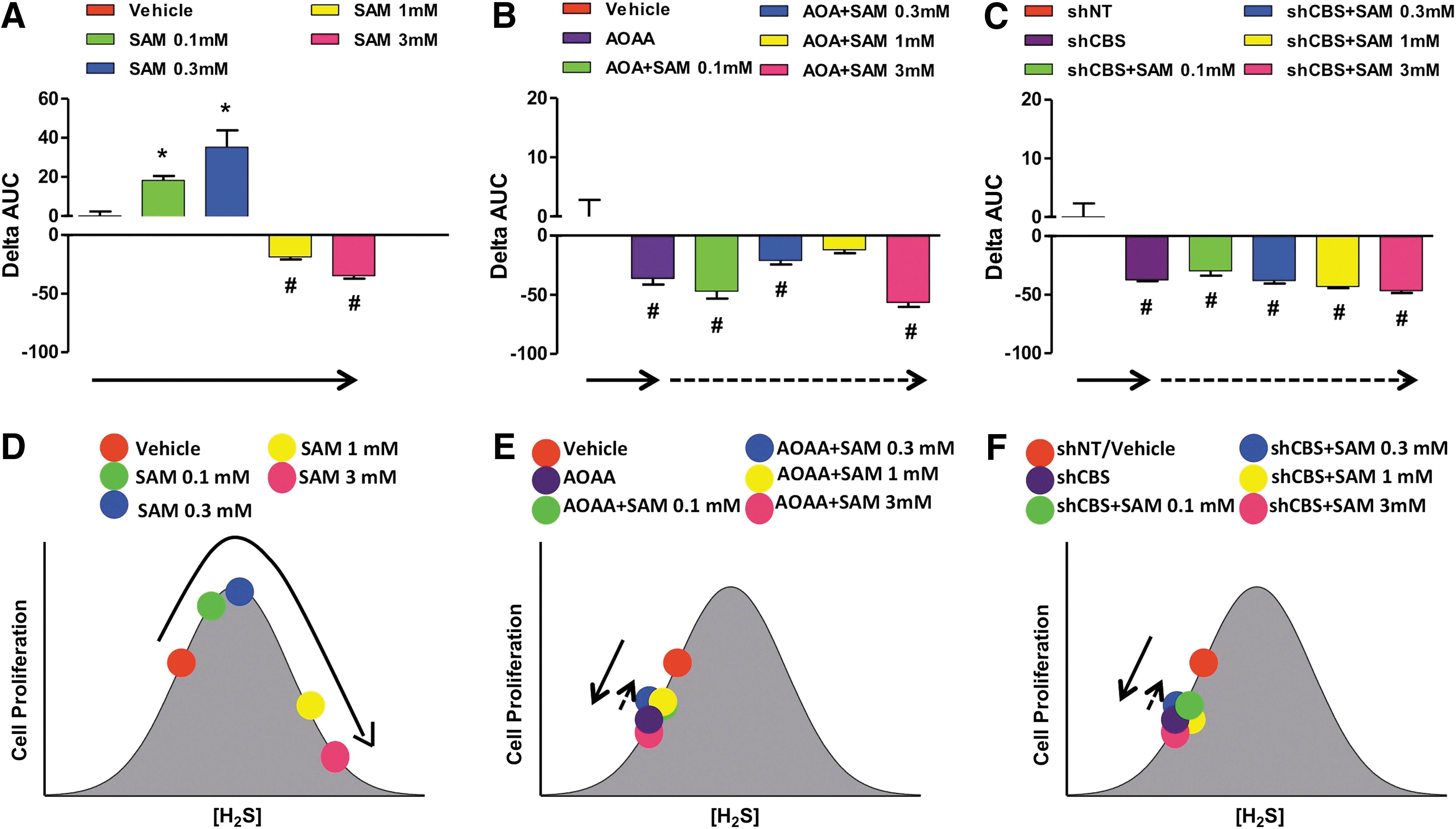
We should emphasize that the earlier schemes only depict a reductionist model, as the model only considers ambient (endogenous/exogenous) H2S concentrations and simple outcome measures (such as survival or proliferation). The terms “local concentration of H2S” and the “rate of release of H2S from H2S donors” are often used interchangeablely. Thus, in most cases, donors with higher H2S release rates produce higher ambient H2S concentrations. In other situations, H2S donors with different rates can produce not only quantitative but also qualitative differences in cellular responses (68, 136). This aspect of H2S biology remains to be further explored and integrated into our model. In addition, the complexities of the temporal relationship between H2S donation and its effect should be considered. For example, pretreatment of cells or animals with H2S can induce the expression of protective genes and pathways (e.g., ones governed by the antioxidant master switch Nrf2), resulting in altered cellular phenotypes at later time points (i.e., where ambient H2S levels are no longer detectable, the classical phenomenon of “preconditioning”) (15, 143). While these mechanisms are primarily studied in the context of cardiovascular biology, Koike et al. have shown that pretreatment of cancer cells with polysulfides (which have H2S-donating properties, as well as many other pharmacological actions) can induce Nrf2-dependent protective phenotypes (64).
In summary, the simple bell-shaped model outlined in the current section is consistent with the majority of published data and provides a useful framework to reconcile some of the controversies regarding H2S donation versus H2S biosynthesis inhibition.
Unanswered Questions and Future Directions
The field of H2S and cancer is a new and expanding research area. The later sections represent a partial list of open questions and briefly outline potential future research directions.
Why have the cancer cells chosen such an unusual route to support themselves?
We speculate that H2S-mediated metabolism/signaling may represent a “regressive response” by the tumor cell, similar to the Warburg effect, another cancer-specific metabolic alteration, where the tumor cells can switch to anaerobic metabolism. As previously discussed (120), billions of years ago, before the presence of atmospheric oxygen on Earth, H2S was a common metabolic “fuel” for bacteria and other simple organisms. In fact, even present-day organisms living in the oxygen-poor but H2S-rich environment of deep-sea vents utilize H2S to support their metabolic functions. By synthesizing large amounts of intracellular H2S, tumor cells may regress back to such primordial metabolic pathways. One important difference between the H2S-dependent metabolism of the tumor cells and the Warburg effect is that based on what we know about H2S-dependent mitochondrial mechanisms, we do not believe that H2S-dependent metabolism will be maintained in severe hypoxia, because the H2S-fueled electron transport requires oxygen as a terminal electron acceptor at complex IV.
Is CBS-derived H2S the reason that dogs can smell cancer?
Two decades of literature show that dogs can be trained to detect cancer patients, in some cases with astonishing precision (46, 110, 137). The dog's nose can detect H2S down to parts per billion concentrations. When coupled with multiple lines of recent data showing that exhaled H2S and urinary H2S is increased in cancer patients (as discussed earlier), it may possible that dogs sense cancer patients by detecting H2S. However, we believe that dogs detect a complex “signature” of various volatile molecules (including H2S). Such volatile signatures may be useful for the future detection of cancer, either by dogs or by various “artificial nose” systems currently under development (21, 70).
What is the mechanism of up-regulation of CBS in cancer?
Currently, it is not known whether the mechanism involves transcriptional, post-transcriptional mechanisms, or both; further, the trigger/signals that up-regulate CBS are unknown. When CBS expression was studied in the NCI60 panel of tumor cells, the increased CBS protein expression statistically correlated with increased CBS mRNA levels (146), suggesting that transcriptional regulation is a distinct possibility. In addition, both Wang and Kruger have recently published on post-transcriptional regulatory mechanisms of CBS expression, and discovered that CBS is subject to intracellular degradation by various proteases (e.g., the Lon protease) (41, 106, 123), suggesting that perhaps CBS protein stability may be mediated by decreased CBS-degrading proteases in the cancer cell. Bhattacharyya et al. found that the up-regulation of CBS appears to be a relatively early event in ovarian cancer progression (10). However, for colorectal cancer, the temporal sequence of CBS up-regulation, in relation to the progression of colorectal cancer (expression levels in premalignant intestinal polyps vs. early-stage cancers), remains to be further studied.
What are the local and systemic levels of H2S in biological fluids in normal biological conditions and in cancer patients?
A significant controversy in the field of H2S relates to quantification methods for biological levels of H2S. The absolute value of the H2S level in extracellular fluids or in plasma depends on the experimental method used (e.g., derivatization of H2S with monobromobimane, or other methods, or measurement using the “methylene blue” assay, or free H2S gas measurements using headspace analysis, or readings taken by H2S electrode), as well as the tissue type studied, with vascular tissues exhibiting substantially high levels than many other tissues (29, 89, 120, 138). Further work is needed to address this question. No reports exist on circulating H2S levels in cancer patients. In our nude mice studies xenografted with human HCT116 colon cancer cells, we were unable to detect significant alterations in plasma H2S levels using the “methylene blue” method. Tissue levels of H2S are determined by both H2S production and H2S degradation (26). In cancer cells, partially uncoupled mitochondria may produce increased levels of ROS, and the increased cancer cell-derived H2S may be accompanied by increased H2S degradation due to increased oxidative stress, resulting in the primary accumulation of H2S metabolites, rather than H2S. A full analysis of the levels of H2S and its metabolites (e.g., thiosulfate) in cancer remains to be conducted in future studies.
How does the CBS/H2S pathway in cancer cells relate to various known pathways of cellular metabolism or cell proliferation?
It is not yet known whether up-regulation of CBS in cancer cells is a part of a global reprogramming of the trans-sulfuration pathway, which subsequently affects cellular metabolism and signaling. Future studies need to characterize these complex alterations. Ramasamy et al. characterized the expression of thiosulfate sulfurtransferase (TST, an isoform of the H2S-detoxifying enzyme family rhodanese) in HT29 colon cancer cells and in biopsies of colon cancer and showed that TST expression increases in proliferating HT29 cells can be induced by low-to-intermediate concentrations of authentic H2S; while TST is down-regulated by higher H2S concentrations (98). In addition, the same authors reported that there is a reduction in TST expression in sections of human colon cancer (98). Although the exact pathophysiological role of these alterations remains to be explored, the cellular effect is the net result of H2S production and its degradation and so, it is expected to be regulated by both changes in the expression/activity of enzymes that produce it (such as CBS) and enzymes that degrade it (such as TST).
Deplancke and Gaskins conducted a microarray analysis of gene expression changes in response to H2S exposure in the colonic epithelial cell line IEC-18 (25) and found that H2S increased the expression of cytochrome c oxidase subunit V, perhaps as a compensatory response to alterations in mitochondrial electron transport. Importantly, VEGF expression was also found to be up-regulated. There are several studies in the literature showing that VEGF up-regulates H2S production and/or that H2S up-regulates VEGF expression (11, 45, 71, 90, 94, 97, 103, 122, 130, 131). Thus, the possibility of a positive feedback cycle (with increased tumor angiogenesis, as a possible outcome) needs to be investigated in future studies.
What are the interactions of the CBS/H2S system with other gaseous mediators in cancer?
There are three gaseous mediators, NO, CO, and H2S, that utilize partially overlapping, and partially synergistic pathways (56, 93, 114). Many known interactions exist between the three transmitters and the respective enzyme systems that generate and metabolize the gases. As previously noted, one example is the direct inhibition of CBS by NO and CO via binding to its heme group. Although multiple previous studies have explored the potential role of NO in cancer, its exact role remains unclear. Since all three gaseous transmitters can travel through cell membranes, in theory, all of them have the potential to affect cell–cell communications in the tumor microenvironment. The interactions of the three gaseous transmitters, both in the regulation of physiological processes in general and in cancer biology remains to be explored in the future.
Outlook: The Therapeutic Potential of H2S Biosynthesis Inhibition in Cancer
The realization of the functional importance of CBS-derived H2S production in the biology of the cancer cell is a very recent one. Our current model, supported by several lines of experimental data, is shown in Figure 20. CBS up-regulation results in enhanced H2S production in cancer cells, which, in turn, stimulate its mitochondrial function and activate proliferative and pro-inflammatory signaling. In addition, H2S acts as a mitochondrial antioxidant and cytoprotectant for the cancer cell. Moreover, in a paracrine fashion, H2S diffuses out from the cancer cells into the tumor microenvironment, where it stimulates angiogenesis and local vasodilatation, which, in turn, provides the tumor with nutrients and oxygen.
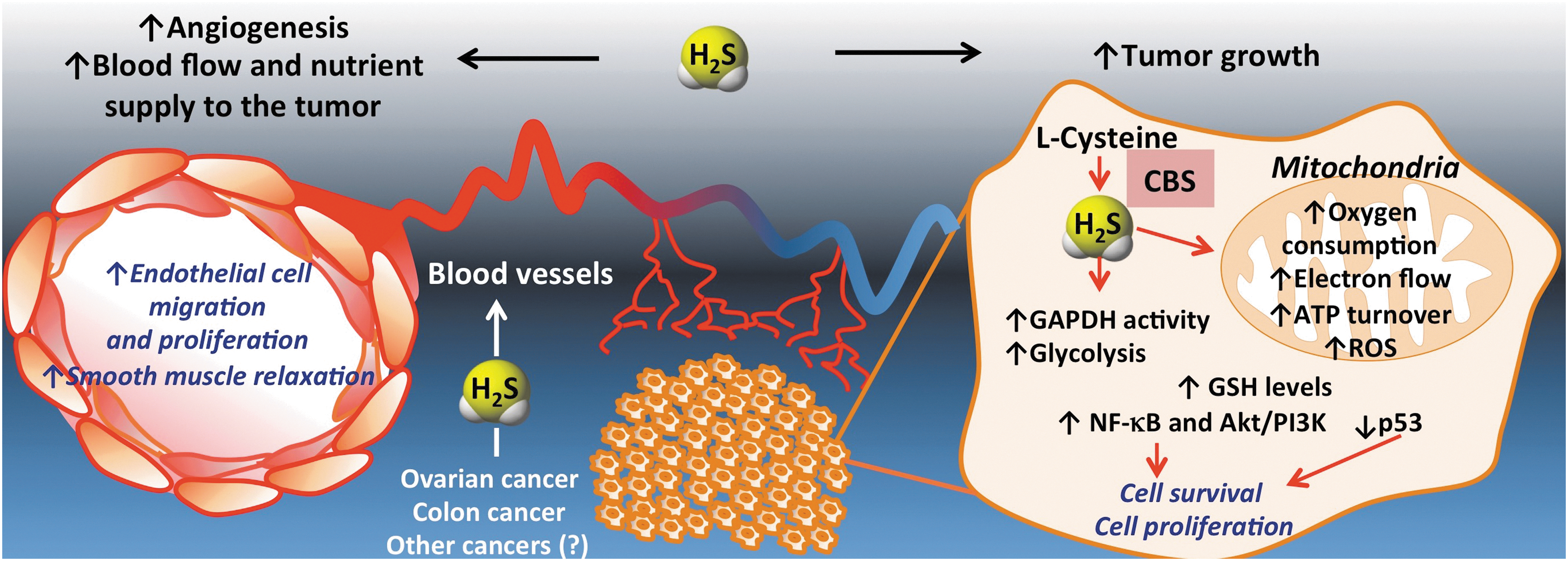
To progress this novel concept into clinical testing, significant further basics, as well as translational research are required. Colorectal and ovarian cancer are the two diseases with promising preclinical data. Thus far, the role of CBS/H2S in the translationally highly predictive PDTX model has only been confirmed in the colorectal cancer model. In these experiments, the CBS inhibitor AOAA showed consistent and potent anti-tumor growth effects independent of the cancer's KRAS status (Fig. 13) (117). Based on preclinical data (10, 16a), a combination of CBS inhibitors with existing antitumor agents is a distinct possibility, even though additional work is needed to identify the most effective combinations and the best sequence of their administration.
Finally, further development of pharmacological inhibitors of CBS are needed, including work to improve the potency and the selectivity of the currently known CBS inhibitors. All available published data on screening and medicinal chemistry studies in this area are covered in the current review.
Even with the most selective and most potent inhibitors, we predict that some mechanism-based side effects will remain unavoidable, as CBS is physiologically expressed in some non-tumor tissues, such as the liver, the kidney, and the nervous system. The main physiological role of CBS is to metabolize homocysteine. Thus, we predict that treatment with any pharmacological CBS inhibitor will be associated with some accumulation of plasma homocysteine, a known cytotoxic molecule and cardiovascular risk factor (12, 24, 27, 50, 104). Accordingly, CBS knockout animals have a limited life span, which is a consequence of severe hyperhomocysteinemia (3, 74). Patients who completely lack CBS exhibit severe pathophysiological alterations, including thrombosis, osteoporosis, and mental retardation (82). However, while extremely high levels of circulating homocysteine are dangerous, there may be a threshold effect with homocysteine. Moderate homocysteinemia is well tolerated in animal models (40). For example, CBS−/− mice that receive a transgene expressing a catalytically deficient form of human CBS which only partially normalizes circulating homocysteine levels are viable (129). In addition, CBS+/− heterozygote mice have significantly elevated homocysteine levels, but are fully viable (69, 126, 145).
Correlative data linking hyperhomocysteinemia and cardiovascular risk may be of concern with the use of CBS inhibitors. Recent clinical trials studied patients taking vitamin B12 and folate to facilitate the conversion of homocysteine to methionine. Although the intervention successfully reduced circulating levels of homocysteine, it was not effective in reducing the cardiovascular adverse events in the patients (2, 88), questioning whether hyperhomocysteinemia causes cardiovascular disease.
Taken together, in our view, CBS is a partially selective novel target for cancer therapy. Although we have shown that the CBS inhibitor AOAA was tolerated in mice at therapeutically effective doses, this does not imply that CBS inhibitors will be well tolerated by cancer patients. We predict that administration of CBS inhibitor anticancer drugs will present a risk of hyperhomocysteinemia, which will have to be monitored and clinically managed. Perhaps measurement of circulating homocysteine levels may be used in future anticancer clinical trials as a biomarker to estimate the extent of CBS inhibition and/or to assess patient compliance. Finally, a possible way of reducing the therapeutic dose of CBS inhibitors in human trials may be to use them in combination with appropriately selected standard anticancer agents.
Footnotes
Acknowledgments
The work was supported by National Institutes of Health (R01CA175803, K08CA125209) and the Shriners Hospitals for Children, the John Sealy Memorial Foundation, the Institute for Translational Sciences at the University of Texas Medical Branch, and a Clinical and Translational Science Award (8UL1TR000071).
Author Disclosure Statement
M.R.H., C. Chao, and C.S. have stock ownership in CBS Therapeutic Inc., a for-profit organization involved in the development of CBS inhibitor therapies for cancer.