
Abstract
Introduction
A
Reactive oxygen species (ROS) not only are well known to induce outer hair cell death but also have been shown to play a dual role in sensory hair cells under different noise exposure conditions. Temporary threshold shift noise induces low levels of oxidative stress, activating the autophagy process, while severe permanent threshold shift noise induces excessive oxidative stress, resulting in oxidative damage. This study demonstrates a relationship between oxidative stress and autophagy in sensory hair cells and reveals that autophagy is an intrinsic cellular process that attenuates noise-induced hearing loss (NIHL) by reducing oxidative stress. This novel finding thus supports, via an independent approach, the notion of a causal relationship between ROS and NIHL.
ROS also have the ability to induce cellular defense pathways such as autophagy (48), a protective process that delivers damaged cellular components to lysosomes for degradation (50). This process is mediated via the formation of autophagosomes (2, 12) that fuse with lysosomes to enzymatically degrade the engulfed components (33). The orderly disposal of these potentially damaged cellular constituents, including impaired organelles and misfolded proteins, plays a protective role limiting pathological alterations (4, 41). In neuronal cells, the activation of autophagy ameliorates brain injury and cortical neuron apoptosis (51). The pharmacological upregulation of autophagy increases the number of surviving retinal ganglion cells, while the deletion of autophagy genes reduces cell survival during optic nerve degeneration (42). Microtubule-associated light chain 3 protein (LC3) is an essential component associated with the autophagosome membrane and is involved in the mechanism of autophagy at a molecular level. In humans, three genes encode highly homologous LC3 proteins (LC3A, B, and C), two of which (LC3A and LC3B) are conserved in mice. The splicing variant LC3B has the broadest specificity and acts as a suitable marker for autophagy activity in cells (1).
Although the complex relationship of autophagy with ROS levels suggests a potential role for autophagy in NIHL, this has yet to be established. We hypothesize that lower levels of oxidative stress, as seen with temporary threshold shifts (TTS), stimulate autophagy, protecting against NIHL, while excessive oxidative stress induces oxidative damage in OHCs, leading to permanent threshold shifts (PTS). To investigate this hypothesis, we first characterized noise-exposure conditions for TTS, PTS, and severe PTS (sPTS) in male CBA/J mice at 3 months of age. We then assessed oxidative stress as indicated by products of lipid oxidation and protein nitration and the expression of the autophagy marker LC3B in OHCs under these three noise conditions. Healthy transgenic mice systemically expressing green fluorescent protein (GFP) fused to LC3 (a mammalian homologue of yeast Atg8) were generated (23, 32). We used GFP-LC3 mice to confirm the augmentations of cellular autophagy after TTS noise. The relationship between autophagy and oxidative stress, as well as auditory thresholds, was further explored using siRNA silencing and pharmacological modulators, specifically the autophagy activator rapamycin and the autophagy inhibitor 3-methyladenine (3MA).
Results
Noise exposures induce temporary and permanent hearing loss in adult CBA/J mice
Previous studies by our laboratory demonstrated that exposure of 12-week-old male CBA/J mice for 2 h to 2–20 kHz broadband noise (BBN) at 92, 94, or 96 dB SPL induces TTS, while intensities of 98, 100, 102, 104, and 106 dB SPL result in hair cell death and PTS without altering supporting cell and spiral ganglion cell structure as per light microscopy evaluation (6). In this study, we characterized the effects of noise exposure at 96, 98, and 106 dB SPL in detail.
Exposure to BBN at 96 dB SPL for 2 h resulted in significant auditory threshold shifts of 20, 50, and 50 dB at 8, 16, and 32 kHz, respectively, 3 h postexposure (F 2,90=101.377, p<0.001). The auditory thresholds measured by auditory brainstem response (ABR) at all three frequencies partially recovered 1 day after the exposure with full recovery by 3 days after exposure, confirming that the noise condition leads to TTS only (F 3,45=33.633, p<0.001) (Fig. 1A). Exposures at 98 dB SPL created significant threshold shifts of 30, 50, and 55 dB at 8, 16, and 32 kHz, respectively, at 3 days after the exposure (F 2,112=213.382, p<0.001), which remained stable for 7 to 14 days at 16 and 32 kHz, but recovered partially at 8 kHz (F 2,56=1.857, p=0.166) (Fig. 1B). Exposure to 106 dB SPL resulted in even larger threshold shifts at 8, 16, and 32 kHz at 3 days after exposure (F 2,98=150.876, p<0.001) that remained stable for 7 to 14 days at all three frequencies (F 2,49=0.415, p=0.663) (Fig. 1C). Detailed post-hoc tests for comparison of auditory threshold shifts from all three noise conditions are listed in Table 1. Two weeks after noise exposure, hair cell loss was quantified for all three conditions. There were no OHC or inner hair cell (IHC) losses after 96-dB TTS noise (Fig. 1D). The 98-dB SPL exposure resulted in OHC losses only, beginning around 3.5 mm from the apex and increasing toward the base, while IHCs remained intact (Fig. 1E, F). The 106-dB SPL noise exposure resulted in losses of both OHCs and IHCs. OHC losses began around 2.5 mm from the apex and IHC losses began around 4.5 mm from the apex, and both increased as a function of distance from the apex (Fig. 1E, F). The amount of OHC loss induced by 106 dB SPL was significantly higher than by 98 dB SPL (F 1,8=189,986, p<0.001, Fig. 1E). We therefore refer to the 98 dB SPL noise conditions as “PTS noise” and the 106 dB SPL noise conditions as severe, or “sPTS noise.” Age-matched sham-exposed control mice showed no elevation of auditory thresholds 2 weeks after exposure.
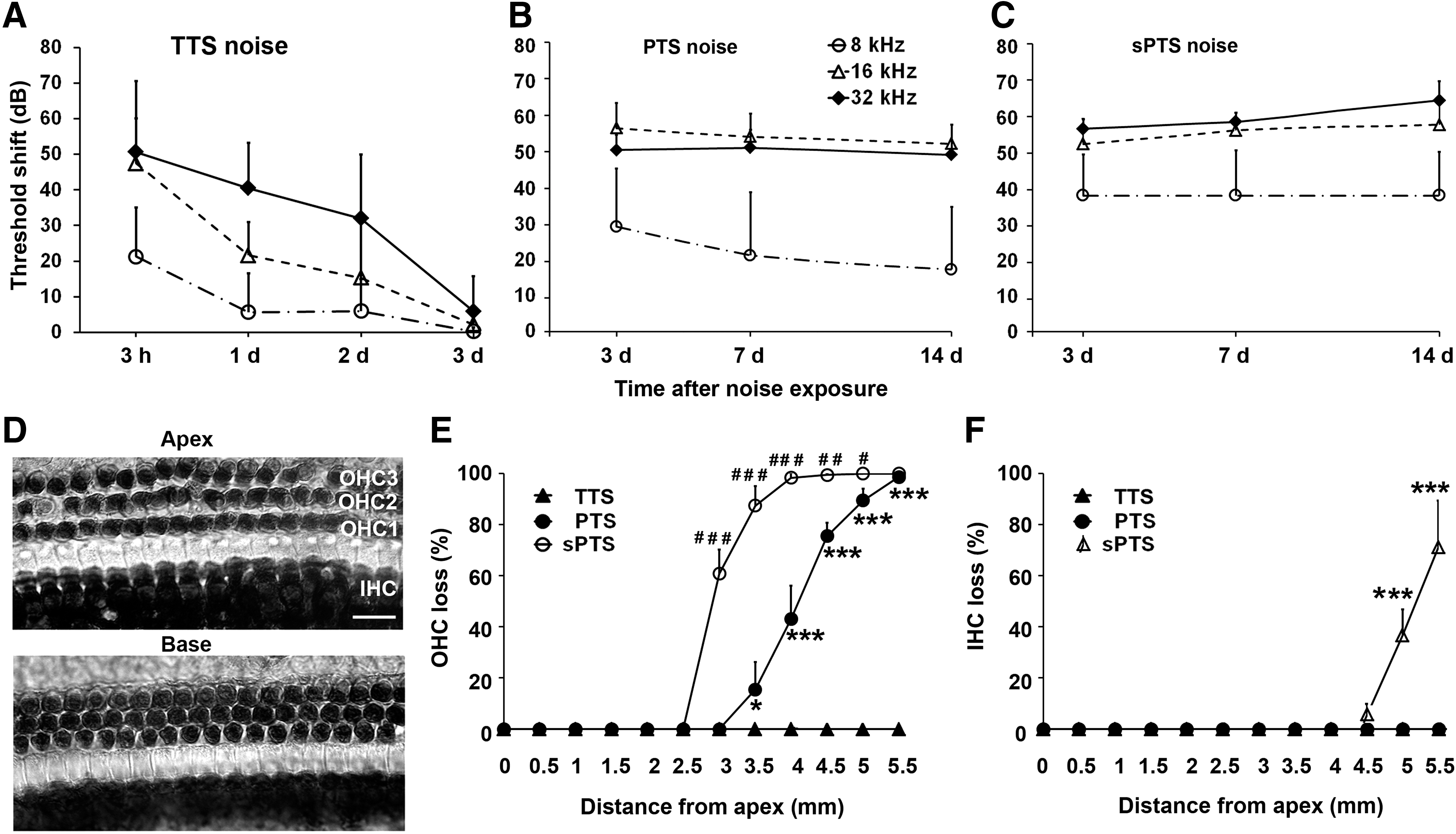
PTS, permanent threshold shift; sPTS, severe PTS; TTS, temporary threshold shift.
Noise-induced elevations of 3-NT and 4-HNE levels in OHCs are noise-dose dependent
Increased oxidative stress, as indicated by products of lipid oxidation, and protein nitration, determined by increased 4-HNE and 3-NT levels, have been well documented after noise exposure (53). We first assessed 3-NT and 4-HNE levels under control conditions without noise exposure and PTS-noise conditions in whole cochlear tissue homogenates by Western blot. In agreement with a previous report, incubation with 3-NT and 4-HNE antibodies each revealed a 70-kDa band (18). The 3-NT band density increased significantly 1 h after PTS-noise exposure compared with controls (t3 =4.805, p=0.017) (Fig. 2B). However, the levels of 4-HNE were unchanged (Fig. 2D). Next, we analyzed the expression of 3-NT and 4-HNE on surface preparations 1 h after exposure to the three noise conditions. Immunolabeling of both 3-NT and 4-HNE increased in OHCs and supporting cells compared with controls (Fig. 2A, C). Quantitative analysis of 3-NT immunolabeling in OHCs revealed that sPTS noise induces a 100% increase (t 2=8.660, p=0.013), PTS induces a 70% increase (t 2=8.111, p=0.015), and TTS induces a marginal 20% increase, relative to age-matched controls without noise exposure (t 2=4.305, p=0.050) (Fig. 2A′). Quantitative analysis of 4-HNE immunolabeling in OHCs also showed a 150% increase after sPTS (t 5=2.911, p=0.03) and a 100% increase after PTS (t 5=3.861, p=0.012), but not after TTS-noise exposure (t 5=1.998, p=0.102) compared with controls (Fig. 2C′). Furthermore, treatment with the antioxidant N-acetylcysteine (NAC) reduced PTS-noise-induced immunolabeling of 3-NT levels by 40% (t 2=−8.246, p=0.014) and of 4-HNE levels by 90% in OHCs (t 2=−62.794, p<0.001) (Fig. 3A, A′ for 3-NT; Fig. 3B, B′ for 4-HNE).


Moderate noise induces increased formation of autophagosomes in OHCs
First, the levels of autophagosome marker LC3B were assessed in whole cochlear tissue homogenates by Western blot. LC3B band densities at both 16 kDa (LC3B-I) and 14 kDa (LC3B-II) were unchanged after TTS-noise exposure compared with age-matched controls (Fig. 4E). Next, we evaluated the autophagosome marker LC3B in sensory hair cells in response to TTS-, PTS-, and sPTS-noise treatment compared with age-matched control mice. LC3B immunofluorescence showed a punctuate pattern in OHCs (Fig. 4A, enlarged images) that was stronger 1 h after TTS-noise exposure compared with PTS and control preparations (Fig. 4A, panels: control, TTS noise 1 h, PTS noise 1 h). Quantitative analysis of LC3B immunolabeling in OHCs confirmed a statistically significant increase for both TTS- (t 3=14.679, p<0.001) and PTS-noise conditions (t 4=5.570, p=0.005), but not for sPTS (t 4=2.075, p=0.107), with significantly higher fluorescence intensity under TTS (2.3 times control) than PTS conditions (1.5 times control) (t 8=9.349, p<0.001) (Fig. 4B).
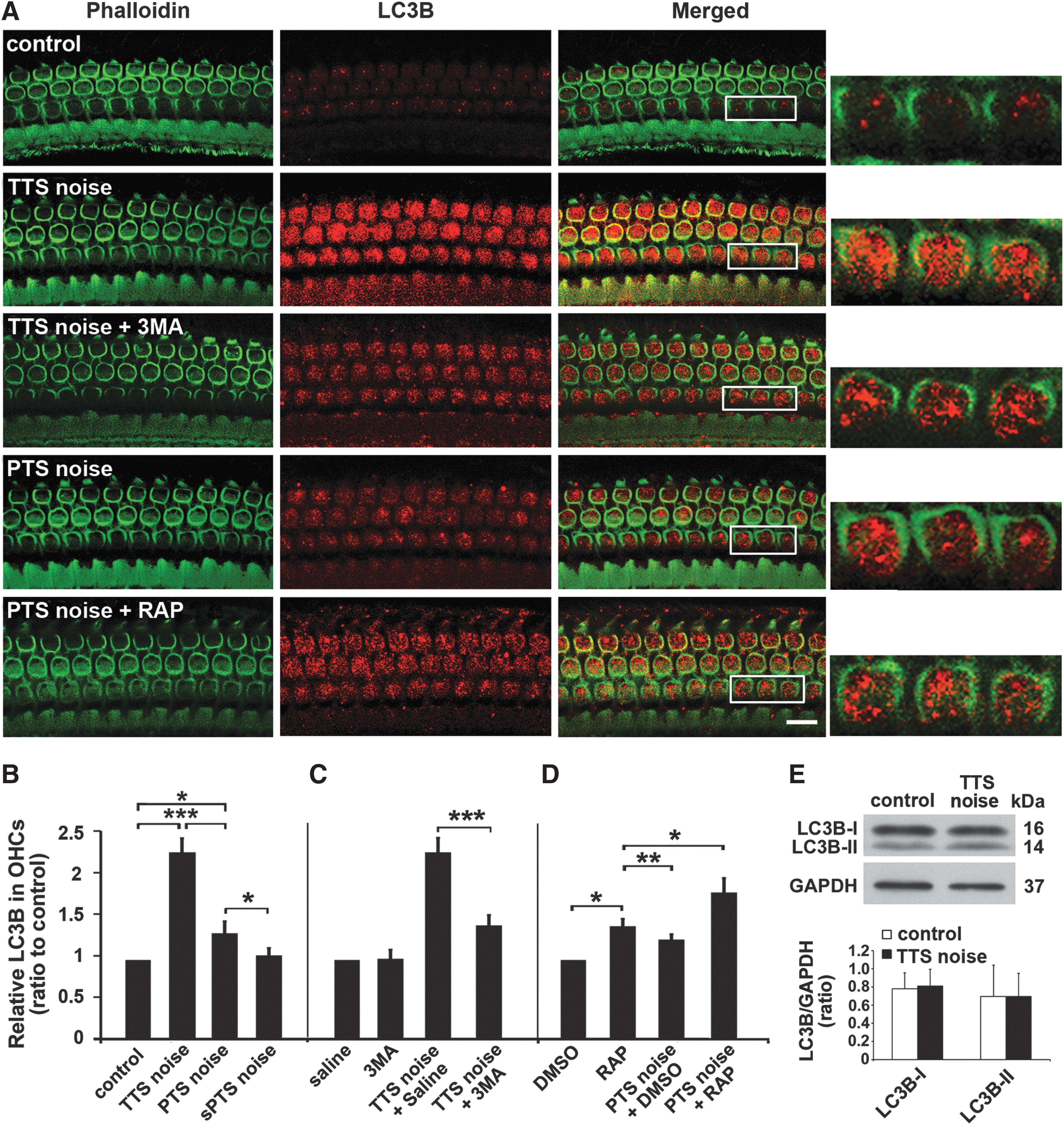
We then examined the effects of 3MA, an autophagy inhibitor, and rapamycin, an autophagy activator, on autophagosome formation in response to noise exposure. Since levels of LC3B were greater after TTS than after PTS noise, the TTS condition was used to examine the possible blocking effect of the autophagosome inhibitor 3MA, while the PTS condition was exploited to assess potential rapamycin-mediated elevation of LC3B. Surface preparations showed a decrease in LC3B immunolabeling in OHCs of TTS-noise-exposed mice treated with 3MA compared with mice exposed to TTS noise only (Fig. 4A, panels: TTS noise+3MA 1 h, TTS noise 1 h). In contrast, rapamycin treatment potentiated the induction of LC3B by PTS noise in OHCs (Fig. 4A, panels: PTS noise+rapamycin [RAP] 1 h, PTS noise 1 h). Quantitative analysis of LC3B-associated immunofluorescence confirmed that the reduction in intensity by 3MA treatment in TTS was about 40% (t 7=8.957, p<0.001) (Fig. 4C) and enhancement of intensity by rapamycin treatment in PTS was 25% (t 12=5.584, p<0.001) (Fig. 4D). The administration of 3MA alone did not influence the levels of LC3B (t 2=0.962, p=0.055) nor did vehicle (dimethyl sulfoxide [DMSO]) alone (t 2=0.693, p=0.561). In contrast, the administration of rapamycin alone increased levels of LC3B compared with controls (t 3=9.708, p=0.002), although levels did not reach those achieved by rapamycin with PTS-noise exposure (Fig. 4D).
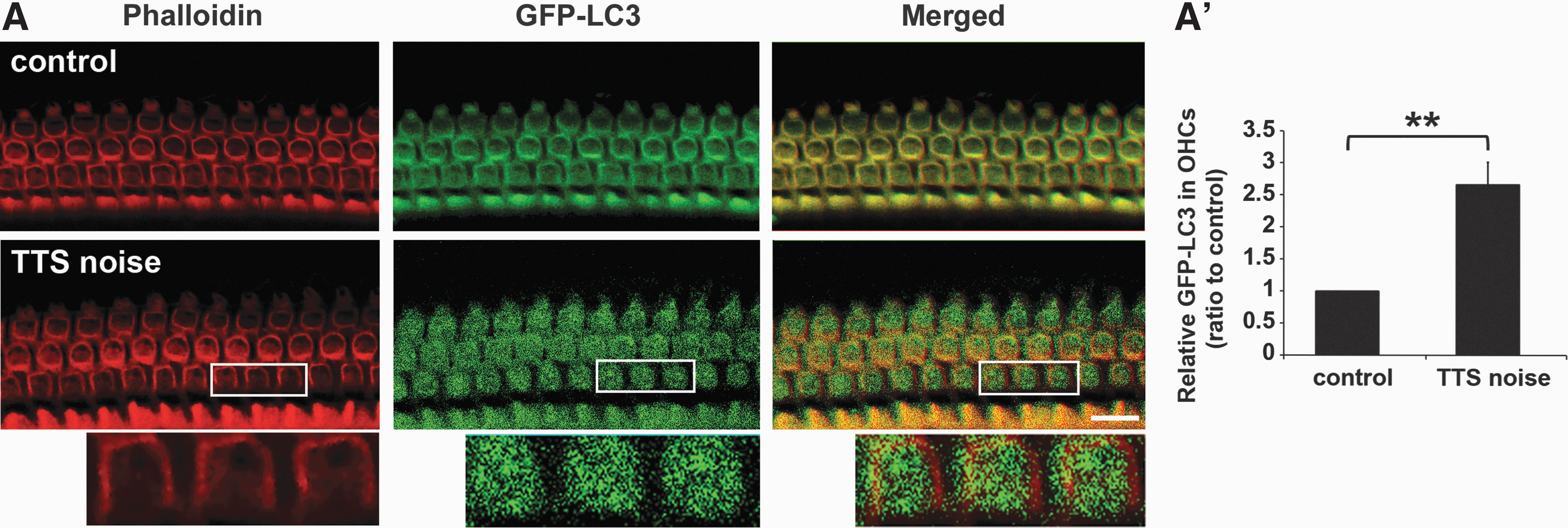
To confirm the increase in autophagy after noise exposure, we used GFP-LC3 mice. Five-week-old GFP-LC3 mice were exposed to 90-dB SPL noise, resulting in TTS (data not shown). Surface preparations showed that punctuate GFP fluorescence in OHCs (Fig. 5A, enlarged images) was much greater 1 h after the noise exposure compared with age-matched GFP-LC3 littermates not exposed to noise (Fig. 5A). Quantification of GFP fluorescence revealed a ratio of 1:2.5 for controls versus TTS-noise-exposed mice (t 3=9.255, p=0.003) (Fig. 5A′).
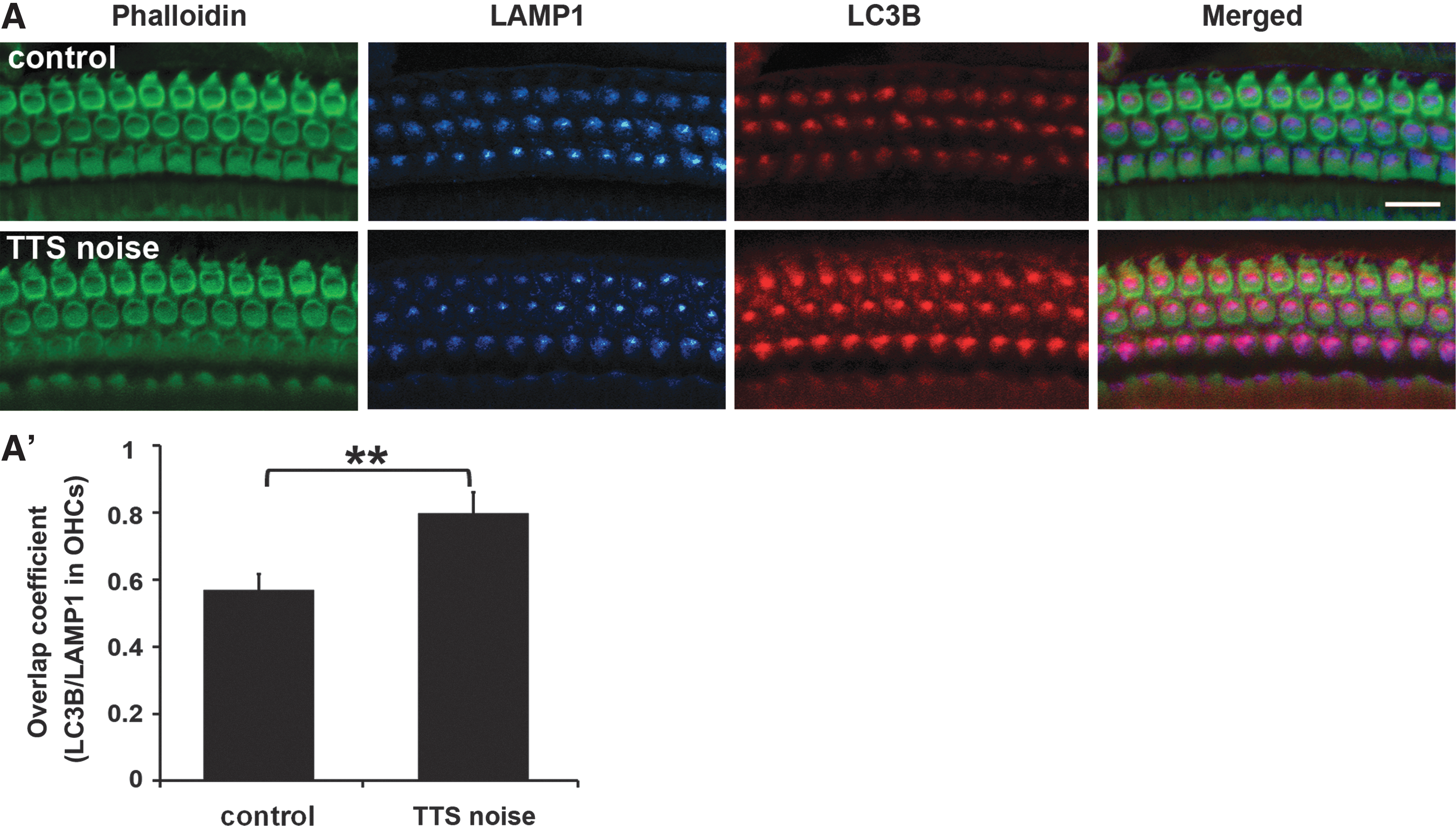
TTS-noise exposure induces formation of autolysosomes in OHCs
The formation of autophagosome/lysosome complexes (autolysosomes) was assessed by analyzing the co-localization of the autophagosome marker LC3B with the lysosome marker LAMP1 on surface preparations. Because TTS conditions induce a marked increase in LC3B immunolabeling, the co-localization of LC3B and lysosome marker LAMP1 in OHCs was analyzed 1 h after TTS noise. LAMP1 immunolabeling did not change after TTS noise, but co-localization of LC3B and LAMP1 fluorophores was increased in OHCs (Fig. 6A). Quantification of the overlap coefficient for LC3B/LAMP1 fluorophores using Zeiss Zen software from original confocal images showed a 23% increase in autolysosome abundance in OHCs after TTS noise (t 10=7.246, p<0.001) (Fig. 6A′).
Potentiation of autophagy attenuates oxidative stress in OHCs
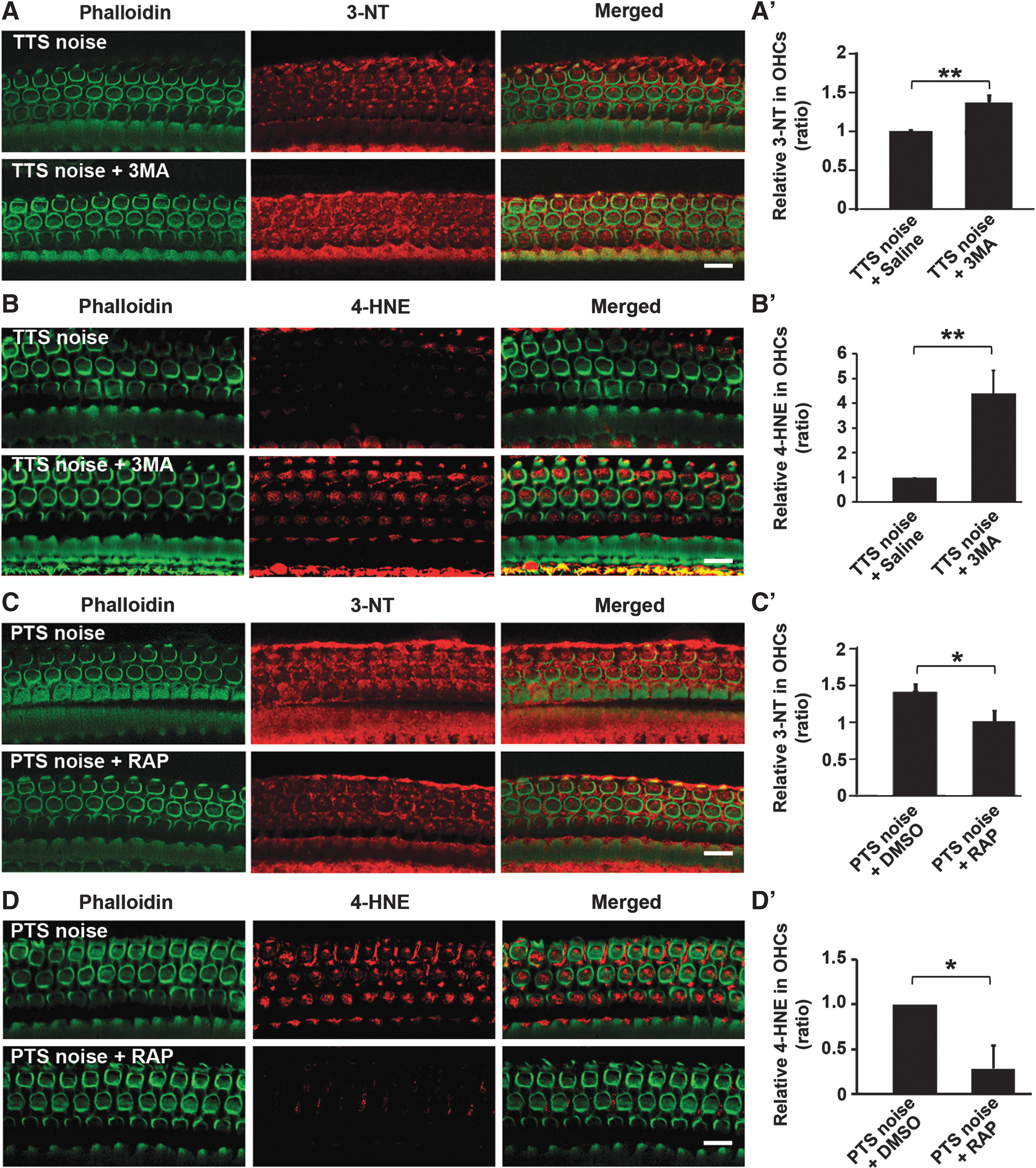
To determine the relationship of autophagy with oxidative stress, we first assessed the effects of the pharmacological autophagy inhibitor 3MA on 3-NT and 4-HNE levels in OHCs. Treatment with the autophagy inhibitor 3MA markedly increased TTS-noise-induced 3-NT and 4-HNE immunolabeling in OHCs and Deiters cells compared with TTS noise alone (Fig. 7A, B, panels: TTS noise 1 h, TTS noise+3MA 1 h). Quantification of 3-NT and 4-HNE immunolabeling in OHCs 1 h after TTS-noise exposure confirmed significant increases with 3MA treatment compared with saline controls (3-NT, t 4=4.478, p=0.011; 4-HNE, t 2=6.543, p=0.023) (Fig. 7A′, B′). We then assessed the effects of increased autophagy on 3-NT and 4-HNE levels using the pharmacological autophagy stimulator rapamycin. Treatment with rapamycin markedly reduced 3-NT and 4-HNE immunolabeling in OHCs in PTS-noise-exposed cochleae 1 h after the exposure (Fig. 7C, D, panels: PTS noise, PTS noise+RAP 1 h). Quantification of 3-NT and 4-HNE immunolabeling in OHCs confirmed significant attenuation of the PTS-noise-increased 3-NT levels (t 7=3.632, p=0.008) (Fig. 7C′) and 4-HNE levels (t 6=2.960, p=0.026) (Fig. 7D′) after treatment with rapamycin. Treatment with DMSO, the vehicle control of rapamycin, altered neither 3-NT nor 4-HNE levels in OHCs.
Finally, we assessed the ability of LC3B siRNA (siLC3B) to decrease autophagosome formation under TTS-noise conditions. siLC3B was delivered locally to the round window (7, 37). Two concentrations of siLC3B, 0.3 and 0.6 μg, were tested. Surface preparations showed a significant reduction of around 30% in LC3B expression in OHCs at 72 h after pretreatment with 0.6 μg siLC3B compared with treatment with scrambled siRNA (siControl) (t 2=5.5120, p=0.0314) (Fig. 8A, A′). Exposure of siLC3B-pretreated mice to TTS noise resulted in a 40% reduction in LC3B levels in OHCs relative to siControl-treated mice 1 h after the exposure (t 5=8.666, p<0.001) (Fig. 8B, B′).
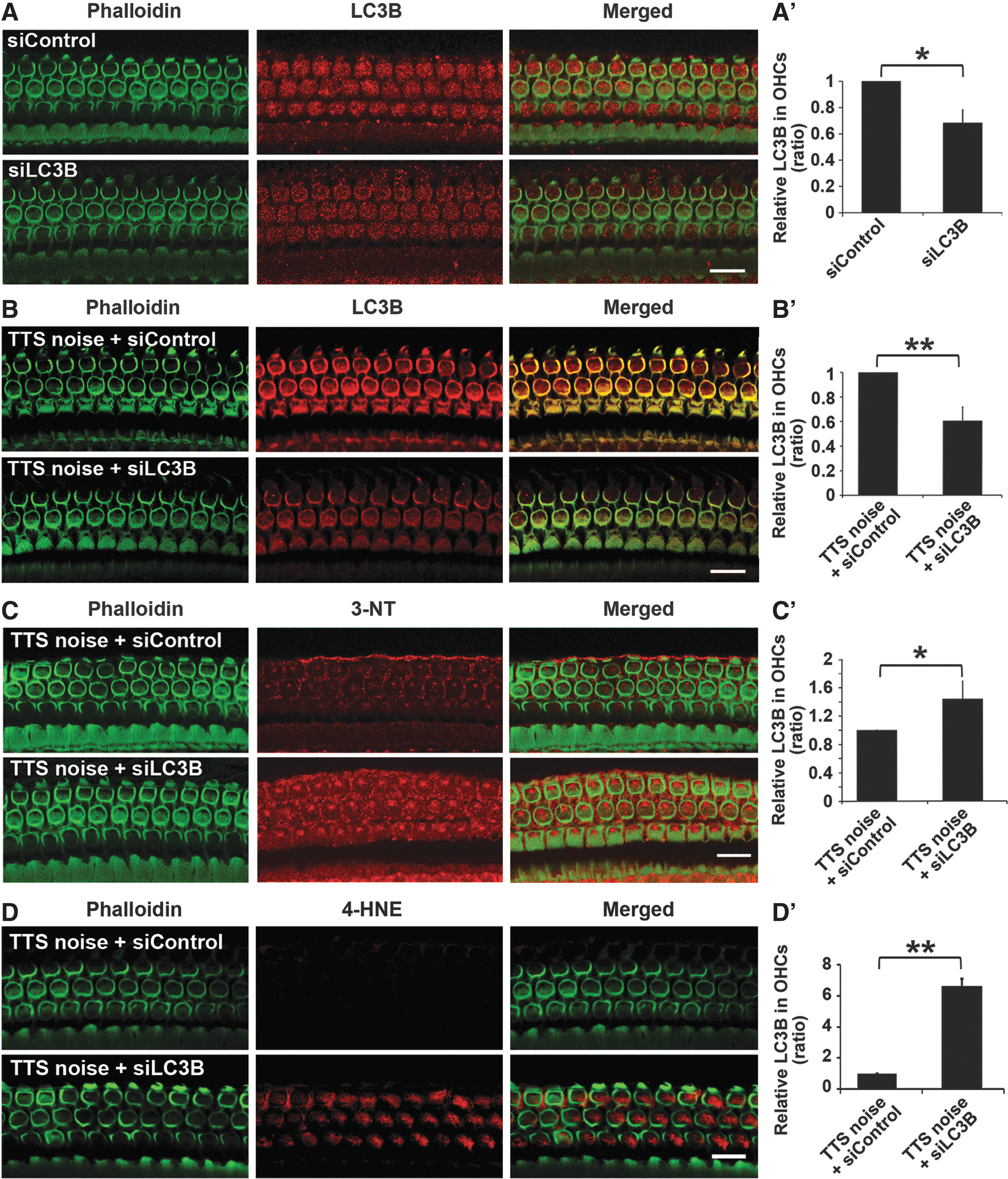
In addition, pretreatment with siLC3B resulted in increased immunolabeling of both 3-NT and 4-HNE in OHCs compared with siControls 1 h after TTS noise (Fig. 8C, D). Levels of 3-NT and 4-HNE immunolabeling in OHCs of siLC3B-treated cochleae were significantly higher than in siControls (3-NT, t 4=3.981, p=0.0164; 4-HNE, t 2=21.683, p=0.002) (Fig. 8C′, D′).
Manipulation of autophagy modulates the magnitude of NIHL and hair cell death
The preceding experiments showed that exposure to the pharmacological regulators 3MA and rapamycin, as well as LC3B siRNA, alters noise-induced autophagosome formation in OHCs. Here, we addressed the questions of whether treatment with these agents affects noise-induced auditory thresholds in vivo.
Treatment with 3MA before TTS exposure converts TTS to PTS at both 16 and 32 kHz 14 days after the noise exposure. Under TTS-noise conditions, noise-induced auditory threshold shifts recover to baseline by 3 days after the exposure. However, treatment with the autophagy inhibitor 3MA prevented recovery of auditory thresholds to baseline and instead resulted in PTS with thresholds elevated about 25 dB at 16 kHz (p=0.011) and 40 dB at 32 kHz (p<0.001) (Fig. 9A). Moreover, TTS-noise exposure did not induce any OHC or IHC losses, while the addition of 3MA treatment resulted in the loss of OHCs in the basal region. All IHCs remained intact with or without 3MA treatment (Fig. 9B). Quantification of OHC losses along the entire length of the cochlear sensory epithelium showed that treatment with 3MA induced OHC loss following a base-to-apex gradient after TTS-noise exposure. OHC loss increased progressively from 20% at 4.5 mm (t 7=3.895, p=0.006), to 30% at 5 mm (t 7=5.657, p<0.001), and to 40% at 5.5 mm from the apex (t 7=6.477, p<0.001) (Fig. 9C). Furthermore, 3MA treatment also significantly increased auditory threshold shifts at 16 kHz (t 10=4.025, p=0.009) and 32 kHz (t 10=3.536, p=0.017) 14 days after PTS-noise exposure (Fig. 9D).
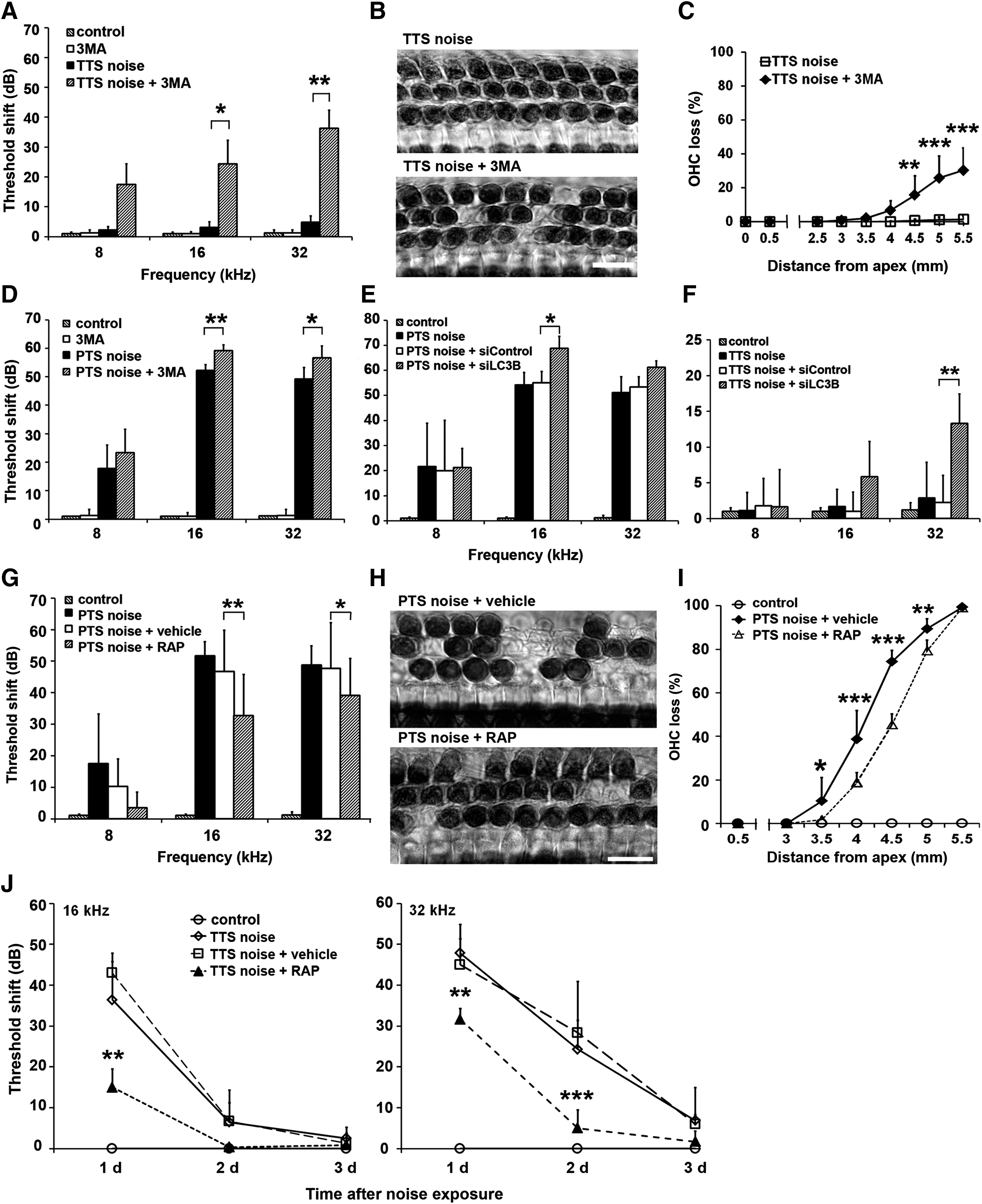
Similarly, the depression of LC3B levels by pretreatment with siLC3B exacerbated NIHL under PTS noise conditions (Fig. 9E). Auditory threshold shifts of siLC3B-treated groups (PTS noise, PTS noise+siControl, PTS noise+siLC3B) were significantly different at both 16 kHz (F 2,30=5.23, p=0.012) and 32 kHz (F 2,30=3.801, p=0.035), but not at 8 kHz (F 2,30=0.308, p=0.688). The threshold shifts of siLC3B-pretreated mice significantly increased by about 15 dB at 16 kHz (p=0.033) and marginally increased at 32 kHz (p=0.058) compared with siControl treatment. Remarkably, pretreatment with siLC3B escalated threshold shifts from TTS to PTS at 32 kHz. Auditory threshold shifts of the four groups (control, TTS noise, TTS+siControl, TTS+siLC3B) were significantly different at 32 kHz (F 3,18=10.548, p<0.001). Pretreatment with siLC3B increased auditory threshold shifts to 15 dB at 2 weeks after TTS-noise exposure (p=0.001) (Fig. 9F).
In contrast to the earlier results, treatment with the autophagy inducer rapamycin significantly reduced PTS-noise-induced auditory threshold shifts at both 16 (F 2,31=33.538, p<0.001) and 32 kHz (F 3,48=23.895, p<0.001), but not at 8 kHz 2 weeks after noise exposure compared with vehicle (DMSO)-treated groups. Treatment with rapamycin reduced auditory threshold shifts at 16 kHz by 15 dB (p<0.01) and 10 dB at 32 kHz (p<0.01) (Fig. 9G). Meanwhile, rapamycin treatment also reduced PTS-noise-induced OHC losses (Fig. 9H). Quantification of OHC loss along the length of cochlear epithelium revealed significant reduction with addition of rapamycin treatment (F 1,13=39.321, p<0.001) (Fig. 9I). OHC loss was reduced by 15% at 3.5 mm (p=0.045), 20% at 4 mm (p<0.001), 25% at 4.5 mm (p<0.001), and 10% at 5 mm (p=0.001) from the apex. In addition, rapamycin treatment accelerated hearing recovery from TTS-noise exposure at both 16 kHz (F 2,9=16.35, p=0.001) and 32 kHz (F 2,9=15.269, p=0.001) (Fig. 9J). TTS-noise-induced auditory threshold shifts returned to baseline within 2 days with rapamycin treatment instead of 3 days, as for controls. The auditory threshold shifts were reduced by 30 dB at 16 kHz on day 1 (p=0.003) and 15 dB at 32 kHz on day 1 (p=0.008) and by 25 dB on day 2 (p<0.001) after noise exposure.
Discussion
The salient finding of this study is that oxidative stress, as indicated by products of lipid oxidation (4-HNE) and protein nitration (3-NT) levels, is significantly elevated in a noise-dose-dependent fashion, whereas autophagy (based on LC3B levels) is significantly increased after TTS-, mildly increased after PTS-, and at control levels after sPTS-noise exposure. Thus, activation of autophagy appears to decrease noise-induced OHC death and NIHL by attenuating oxidative stress. Specifically, induction of autophagy by rapamycin reduces noise-induced oxidative stress (as measured by 4-HNE and 3-NT) and subsequent NIHL. Conversely, reduction of autophagy with the inhibitor 3MA or through pretreatment with siLC3B leads to increased levels of 3-NT and 4-HNE, exacerbating NIHL and hair cell death.
The findings in this mouse model that we have extensively characterized here are consistent with an earlier report using rats, in which hearing loss and the magnitude of cochlear lesions correlate with the intensity of noise exposure (8). OHCs play a significant role in auditory performance via their activity as motor units that amplify movements of the basilar membrane in response to sound stimuli. OHCs are more susceptible to noise trauma than IHCs. The correlation of NIHL and OHC loss was seen in the basal regions of the cochlea, but not in the apical regions. This partial correlation is in good agreement with previous reports (3, 8). Based on the frequency maps of Mϋller (34), the area coding to 8 kHz is located around 1 mm from the apex of cochlear sensory epithelium. However, roughly 40-dB threshold shifts at 8 kHz occur under sPTS-noise conditions (106 dB SPL) without OHC losses in that region. This may be due, in part, to surviving hair cells with damaged stereocilia, since the process of mechanical transduction occurs at the tips of these stereocilia (17). In addition, we found that young adult mice were more sensitive than mature adult mice to noise damage, as 96 dB SPL induces TTS in 3-month-old CBA/J mice, while only 90 dB SPL is required to produce TTS in 5-week-old C57/B6 GFP-LC3 mice, in agreement with the report by Ohlemiller and colleagues (36).
Noise-induced oxidative stress, as indicated by products of lipid oxidation (4-HNE) and protein nitration (3-NT) in cochlear tissues, including OHCs, is well documented (35, 52, 53). The results presented here are consistent with the literature demonstrating that noise exposure results in oxidative stress, as detected by the elevated protein nitration levels of 3-NT both in OHCs and in whole cochlear tissue homogenates, and by the elevated products of lipid oxidation 4-HNE levels in OHCs, but not in the cochlear tissue homogenates. The discrepancy in 4-HNE levels between the immunolabeled OHCs and immunoblots using whole cochlear tissue homogenates is intrinsic to such studies due to the fact that multiple cell types, including spiral ganglion, stria vascularis, and supporting cells are included in the homogenates, masking changes occurring only in OHCs. Increases in 3-NT and 4-HNE levels in OHCs are noise-dose dependent, providing further support for the concept of oxidative damage inciting hair cell death and NIHL (10, 16, 38). Interestingly, the increased levels of 3-NT and 4-HNE in Deiters cells after noise exposure in the absence of supporting cell death observed in this study has been previously reported in animal models such as guinea pigs (53). The potential effects, if any, of oxidative damage after noise exposure in Deiters cells are not known, but the protection of hair cells from aminoglycoside treatment by secretion of HSP70 from supporting cells has been recently reported (27).
The formation of ROS, such as superoxide, hydroxyl radicals, and nitric oxide, may occur by different mechanisms depending on the stress. The precise origin of ROS in the cochlea after noise exposure remains speculative. Several sources are likely to contribute. A surge of superoxide radicals followed by chain reaction formation of other free radicals (via Fenton-type reactions) may be due to increased mitochondrial activity, prolonged tissue hypoxia due to vasoconstriction (29), and rebound hyperperfusion (46). RNS may also arise from the activation or induction of the enzyme nitric oxide synthase. When produced in excess, not only is nitric oxide potentially damaging as a free radical, but it can also combine with superoxide to produce peroxynitrite, which is one of the more reactive and destructive free radicals. Nitric oxide levels can rise after noise exposure (44, 53), perhaps as a consequence of an enhanced release of excitatory amino acids (39, 40), and may contribute to the overall pathophysiology of noise-induced hair cell loss.
In contrast, the levels of LC3B in OHCs are markedly higher under TTS-noise conditions and unaltered with greater noise intensity (sPTS). The opposing behaviors of autophagy (LC3B) and oxidative stress, as indicated by products of lipid oxidation (4-HNE) and protein nitration (3-NT) under these noise conditions, suggest that a balance between oxidative stress and autophagy exists in sensory hair cells. The enhanced co-localization of LC3B with the lysosomal marker LAMP1 in OHCs under TTS-noise conditions demonstrates the fusion of autophagosomes and lysosomes to form autolysosomes, presumably to mediate the degradation of cellular constituents damaged by oxidative stress. We can conclude from these results that exposure to TTS noise induces low levels of oxidative stress and activates the autophagy process, whereas sPTS noise induces excessive oxidative stress resulting in oxidative damage.
Another line of evidence supporting the earlier conclusions comes from the pharmacological manipulation of autophagy in OHCs and pretreatment of LC3B siRNA in CBA/J mice. The autophagy-inducer rapamycin enhances the levels of LC3B and blocks PTS-noise-induced 3-NT and 4-HNE formation, while the autophagy inhibitor 3MA enhances noise-induced 3-NT and 4-HNE formation and diminishes LC3B. In addition, silencing of LC3B produces the same effects as 3MA pretreatment, further substantiating the idea that autophagy inhibits oxidative stress incurred during noise stress. These data are compatible with the general concept that small to moderate amounts of ROS promote autophagy to recycle damaged cellular constituents and maintain cellular homeostasis (2). Furthermore, activation of autophagy contributes to survival of spiral ganglion neurons in the cochlea of young SAMP9 mice, but promotes cell death in spiral ganglia of older animals (28). In addition, the early upregulation of autophagy protects against brain injury by removing damaged mitochondria (22). Since we predict that noise-induced oxidative stress may originate from mitochondria, mitophagy, the targeted lysosomal degradation of mitochondria, might also play a relevant role in maintaining the balance between the generation of ROS and antioxidant detoxification. This speculation deserves investigation in future studies.
Numerous genetic and biochemical studies have shown that mTORC1 negatively regulates autophagy (30). Treatment with rapamycin induces LC3B expression, in line with the concept that rapamycin can induce autophagy by blocking activation of the mTORC1 pathway (5, 49). However, the two pharmacological manipulators used in this study to modulate autophagy, rapamycin and 3MA, may have other pharmacological effects on the cells in addition to their effect on autophagy. For example, protection against NIHL by rapamycin may be only partly due to its effects on autophagy since rapamycin treatment also regulates other signaling molecules in OHCs. We speculate that rapamycin may stimulate Akt pro-survival signaling pathways in OHCs that attenuate PTS, as rapamycin treatment is known to promote Akt activation (5, 15, 47). In contrast, decreased levels of Akt pro-survival molecules in OHCs are found in aminoglycoside-induced hearing loss and age-related hearing loss (9, 19, 20, 43). In addition, inhibition of the TOR signaling pathway by feeding mice rapamycin extends their lifespan (14). On the other hand, the similar effect of the two different compounds (with different secondary actions) supports our contention of autophagy as a common target.
In summary, this study demonstrates that the oxidative stress, as indicated by products of lipid oxidation (4-HNE) and protein nitration (3-NT) in OHCs, is noise-dose dependent. The low levels of oxidative stress induced by TTS noise increased LC3B in OHCs, blocking noise-induced oxidative damage. In contrast, the overproduction of 3-NT and 4-HNE by PTS noise resulted in oxidative damage. Treatment with rapamycin increased LC3B levels in OHCs by blocking mTORC1, reducing the amount of 3-NT, and preventing NIHL and hair cell death. On the other hand, reduction of LC3B by the autophagy inhibitor 3MA or LC3B siRNA increased the levels of 3-NT in OHCs and promoted noise-induced hair cell loss and NIHL (Fig. 10). Our results suggest that autophagy is an intrinsic cellular process that protects against NIHL by attenuating oxidative stress. Furthermore, these novel observations suggest an untried yet potentially valuable approach to the amelioration of NIHL by activation of autophagy.

Materials and Methods
Animals
Male CBA/J mice at the age of 11 weeks (Harlan Sprague – Dawley, Indianapolis, IN) or male C57BL6 GFP-LC3 mice (23, 31) at 5 weeks of age (kindly provided by Dr. John Lemasters at MUSC) had free access to water and a regular mouse diet (Purina 5025; Purina, St. Louis, MO) and were kept at 22°C±1°C under a standard 12:12 h light-dark cycle to acclimate for 1 week before the experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina (MUSC). Animal care was under the supervision of the Division of Laboratory Animal Resources at MUSC.
Noise exposure
Mice were exposed in the same sound chamber as previously reported (6, 54). Briefly, unrestrained CBA/J mice at the age of 12 weeks were exposed to BBN with a frequency spectrum from 2 to 20 kHz for 2 h at 106-dB SPL to induce sPTS, 98-dB SPL to induce PTS, and 96-dB SPL to induce TTS. C57BL6 GFP-LC3 mice at 5 weeks of age were exposed to 90-dB SPL to induce TTS, since young adult mice are more sensitive to noise damage than older mice (36). As C57BL6 mice carry two copies of the age-related hearing loss gene (Ahl), and begin to lose high-frequency hearing at 2 months of age (13, 24), we used GFP-LC3 mice at the age of 5 weeks to avoid such a confounding factor.
Based on our previous studies, we focused on the molecular events of autophagy (using LC3B as a marker) and oxidative stress (3-NT and 4-HNE as markers) in OHCs using immunolabeling on surface preparations at the time point of 1 h after noise exposure (6).
Auditory brainstem responses
We followed the ABR measurement procedure that has been previously described in detail (6, 54). Thresholds were estimated between the lowest stimulus level where a response was observed and the highest level without response. All ABR measurements were conducted by the same experimenter. The ABR scores were assigned by an expert who was blinded to the treatment conditions.
Drug administration via intra-peritoneal route
Rapamycin (LC Laboratories, Woburn, MA) was dissolved in DMSO as a stock solution (75 mg/ml) and stored at −20°C. 3MA (Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% saline as a stock solution (30 mg/ml) and stored at −20°C. The solution was heated to 60°C to completely dissolve the 3MA and then allowed to cool at room temperature before being used for injections. NAC was dissolved in 0.2 M sodium hydroxide (NaOH) as a stock solution (650 mg/ml) and stored at −20°C. The stock solutions of rapamycin and 3MA were diluted with saline, and NAC was diluted with 0.2 M NaOH immediately before being injected into animals. Each animal received a total of three intra-peritoneal (IP) injections of rapamycin at a dose of 7.5 mg/kg per injection, 3MA at a dose of 30 mg/kg per injection, or NAC at a dose of 325 mg/kg based on conditions reported in the literature (21, 25, 42). IP injections were administered 24 h before, 2 h before, and immediately after noise exposure. The animals were euthanized 1 h after noise exposure and the temporal bones were removed to dissect the cochleae for immunofluorescence assays. The mice used for the experiments designed to observe the evolution of ABR thresholds after noise exposure received a fourth IP injection 24 h after noise exposure.
Intratympanic delivery of siRNA
siLC3B (Invitrogen, Carlsbad, CA) or siControl (Invitrogen) was locally delivered via intra-tympanic application. We followed the procedures as previously described (7, 37).
Two concentrations of siLC3B, 0.6 and 0.3 μg, were tested for preliminary study, and 0.6 μg of siLC3B or siControl was selected due to relative high efficiency for the described experimental studies.
Immunocytochemistry for cochlear surface preparations and quantification of the immunofluorescence signals from surface preparations
The detailed procedures of immunocytochemistry for cochlear surface preparations and quantification of the immunolabeling signals in OHCs were described in our previous reports (6, 54). The concentrations of primary antibodies used in these studies were polyclonal rabbit anti-LC3B at 1:50 (Cell Signaling Technology, Danvers, MA), polyclonal rabbit anti-nitrotyrosine at 1:50 (EMD Millipore, Billerica, MA), and polyclonal rabbit anti-4-HNE at 1:50 (Abcam, Inc., Cambridge, MA). Alexa-Fluor-594- or 350-conjugated secondary antibodies were at a concentration of 1:200, and Alexa Fluor 488 phalloidin was at a concentration of 1:100 (Life Technologies, Grand Island, NY). For the co-localization of LC3B and LAMP1, the specimens were first incubated with primary monoclonal mouse anti-MAP LC3B (G-2) (Santa Cruz Biotechnology, Dallas, TX) at 1:50 and the secondary antibody and then with primary rabbit anti-LAMP1 antibody at 1:50 (Sigma-Aldrich) and the secondary antibody.
The immunolabeling of LC3B, 3-NT, and 4-HNE was quantified from original confocal images, each taken with a 63×-magnification lens under identical conditions and equal parameter settings for laser gains and photomultiplier tube gains, using ImageJ software (National Institutes of Health, Bethesda, MD). The cochleae from the different groups were fixed, labeled simultaneously with identical solutions, and processed in parallel. All of the surface preparations were counter-labeled with Alexa-Fluor-488 phalloidin (green) to label hair cell structure to identify the comparable parts of the OHCs in confocal images. The relative fluorescence was quantified by normalizing the ratio of average fluorescence of noise-exposed OHCs to the average fluorescence of the unexposed OHCs.
Surface preparations and diaminobenzidine staining of cochlear epithelia for hair cell counts
We followed the detailed procedures for surface preparations and diaminobenzidine (DAB) staining of myosin VII-labeled cochlear sensory epithelia for hair cell counts described in our previous reports (6, 54).
Extraction of total cochlear proteins for Western blot analysis
The detailed procedures for extraction of total cochlear protein for Western blot and quantification of the band densities were described in our previous publication (6, 54). Tissues from the cochleae of an individual mouse were homogenized in ice-cold RIPA lysis buffer (Sigma-Aldrich) plus Phosphatase Inhibitor Cocktails II and III (Sigma-Aldrich) and Roche Protease Inhibitor (Roche Diagnostic, Indianapolis, IN). The concentrations of primary antibodies were anti-LC3B (1:1000), anti-3-NT (1:1000), anti-4-HNE (1:1000), or anti-GAPDH (1:10,000) (EMD Millipore, Billerica, MA).
X-ray films of Western blots were scanned and analyzed using Image J software. The band densities were first normalized to background. Next, the probing protein/GAPDH ratio was calculated from the band densities run on the same gel. Finally, the difference in the ratio of the control and experimental bands was analyzed for statistical significance.
Statistical analysis
Data were analyzed using IBM SPSS Statistics Premium V21 and GraphPad software for Windows. The group size (n) in vivo was determined by the variability of measurements and the magnitude of the differences between groups. Based on our previous and current preliminary studies, we determined that six to eight animals per group provide sufficient statistical power. Statistical methods used included one-way analysis of variance (ANOVA) with Tukey's multiple comparisons, repeated-measures ANOVA with post-hoc testing, unpaired t-tests, and one-sample t-tests. All tests were two tailed, and a p-value<0.05 was considered statistically significant.
Footnotes
Acknowledgments
The research project described was supported by grant R01 DC009222 from the National Institute on Deafness and Other Communication Disorders, National Institutes of Health. This work was conducted in the MUSC WR Building in renovated space supported by grant C06 RR014516. Animals were housed in MUSC CRI animal facilities supported by grant C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. The authors thank Drs. Jochen Schacht and Bradley Schulte for their valuable comments on this article. They also thank statistician Dr. Fu-Shing Lee for statistical analysis consultation on the results in this article.
Author Disclosure Statement
There are no conflicts of interest for any of the authors.
The data contained in this article have not been previously published.
All research protocols were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (MUSC). Animal care was under the supervision of the Division of Laboratory Animal Resources at MUSC.
All authors have reviewed the contents of this article, approve of its contents, and validate the accuracy of the data.