
Abstract
Introduction
NO
BH4 plays a key role for normal NOS function inasmuch as it inhibits the generation of superoxide instead of NO by auto-oxidation of the intermediate oxyferrous complex (14). Endothelial BH4 deficiency has been shown to be associated with eNOS-dependent vascular oxidative stress in several models of cardiovascular disease, such as hypertension, atherosclerosis, and diabetic endothelial dysfunction (10, 13, 17, 61). Moreover, supplementation of BH4 was reported to improve endothelial function in human studies (38), although clinical trials with BH4 analogs revealed contradictory results (6, 13).
The results of this study provide the first in vivo evidence that oxidative stress generated by an impairment of endothelial NO synthase (eNOS) activity is unlikely to be an initial cause of impaired endothelium-dependent vasodilation and/or a pathologic factor promoting intimal hyperplasia. Therefore, other important enzymatic sources of vascular oxidative stress such as the many known oxidases appear to be more important. Our findings might be explained by concomitant generation of NO by eNOS, which is a fundamental difference from oxidases. They are in line with beneficial effects of therapeutic strategies improving eNOS expression and/or activity and substantiate the importance of vasoprotection conferred by endothelial NO.
Many oxidative enzymes have been implicated in vascular pathologies and in a variety of cardiovascular diseases, including those mentioned above and stroke (18, 22, 29, 35, 43). This makes it difficult to define the relative role for eNOS-dependent formation of superoxide and peroxynitrite in cardiovascular pathologies using animal models. The aim of this study was to investigate the impact of eNOS-dependent vascular oxidative stress on endothelial function and on neointimal hyperplasia. To accomplish this, a mutant of bovine eNOS where cysteine 101 was replaced by alanine was cloned and introduced into eNOS-KO in an endothelial-specific manner.
Results
C101A mutation of eNOS increases oxidative stress
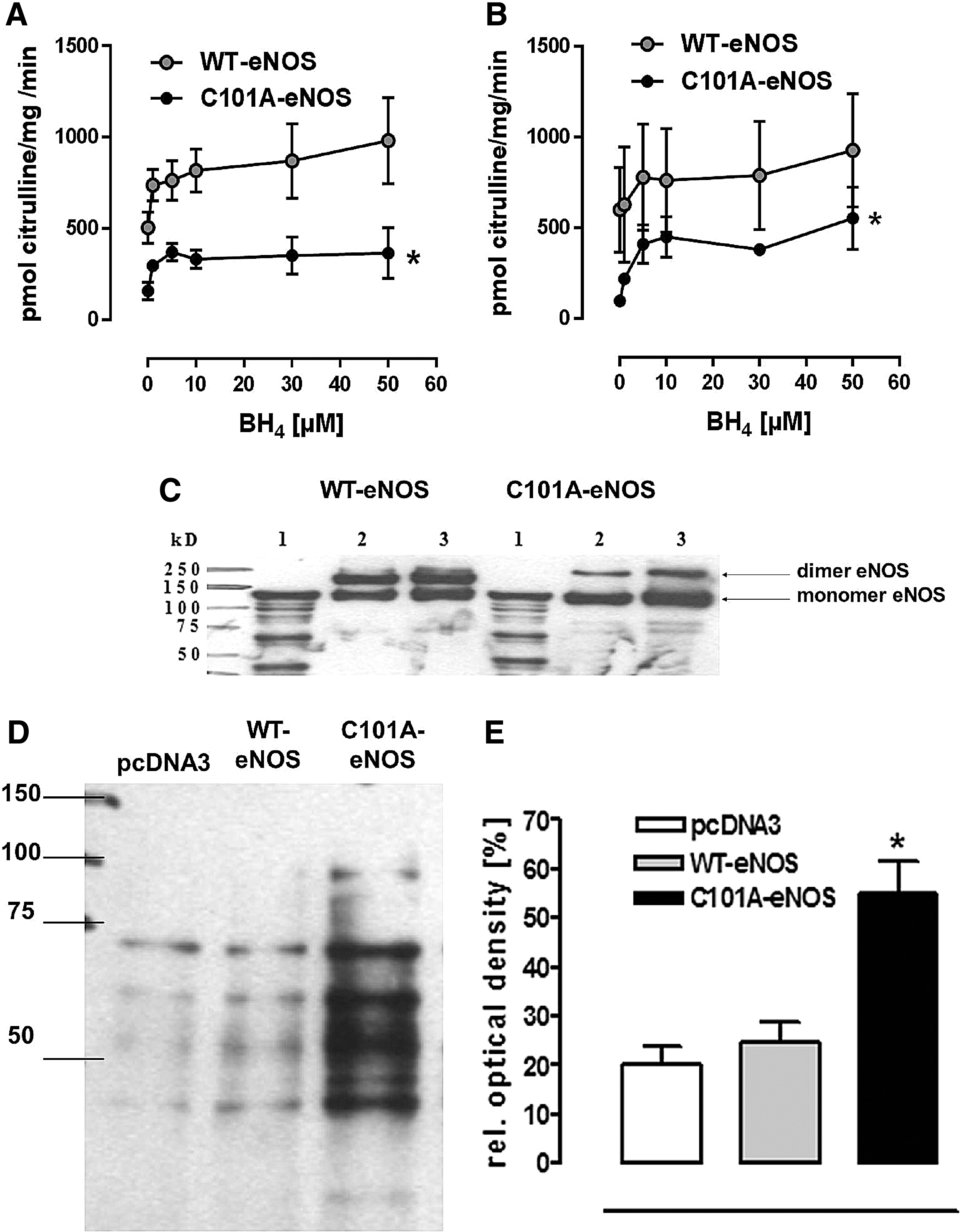
Despite having similar protein levels of the respective wild-type bovine eNOS (WT-eNOS) and C101A-eNOS following transfection to human embryonic kidney (HEK) cells (Supplementary Fig. S1A; Supplementary Data are available online at
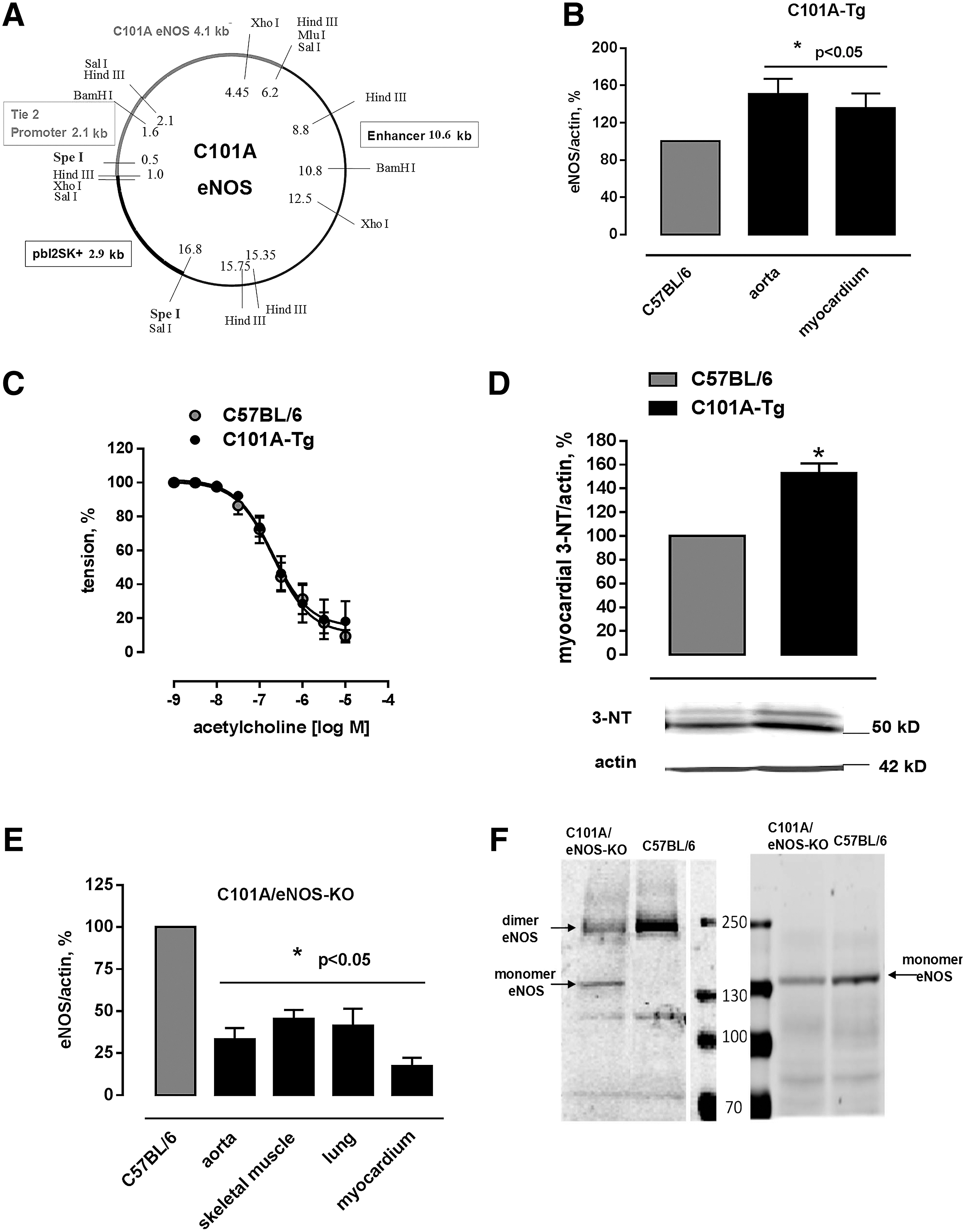
C101A-eNOS activity induces vascular oxidative stress in vivo
Mice expressing C101A-eNOS (C101A-Tg) were generated by cloning C101A-eNOS in a construct that allows endothelium-specific expression (Fig. 2A). Western blot analysis demonstrated increased eNOS protein signals in the aorta (151%±16%) and the left myocardium (135%±16%) of C101A-Tg (Fig. 2B and Supplementary Fig. S2A, B). Aortic endothelium-dependent relaxation of C101A-Tg and their transgene-negative littermates was indistinguishable (Fig. 2C), despite a significant increase of tyrosine nitration in C101A-Tg (Fig. 2D), which confirms the data obtained in HEK cells. Double transgenic mice expressing endothelial C101A-eNOS only (C101A/eNOS-KO) were obtained by crossing C101A-Tg with eNOS-deficient mice (eNOS-KO, backcrossed to C57BL/6) (51). The genotype of C101A/eNOS-KO was confirmed by sequencing the C101A mutation, by polymerase chain reaction (PCR) using mutation-specific primers, by primers binding in the neomycin cassette, and by primers detecting the Tie-2-C101A-eNOS construct (Supplementary Fig. S2C–E). On visual inspection, C101A/eNOS-KO do not appear different from C57BL/6. However, compared with C57BL/6, their litter size is lower and they show increased blood pressure, decreased heart rate, and myocardial hypertrophy just like eNOS-KO (Supplementary Fig. S2F–H). Furthermore, 3 weeks of treatment with the NOS inhibitor, Nω-nitro-L-arginine (L-NA), evoked an increase of blood pressure in C57BL/6 to the level observed in eNOS-KO, while L-NA had no effect on blood pressure in eNOS-KO and C101A/eNOS-KO (Supplementary Fig. S2F). The localization of C101A-eNOS in vivo was restricted to endothelial cells as evidenced by fluorescence microscopy (Supplementary Fig. S3). Compared with C57BL/6, C101A/eNOS-KO expressed lower levels of eNOS in the aorta, the lung, the myocardium, and the skeletal muscle (Fig. 2E and Supplementary Fig. S4A–D) and increased eNOS phosphorylation on Ser 1176 (Ser 1179 in bovine eNOS) (Supplementary Fig. S4E). Similar to HEK cells, we observed increased eNOS monomer formation during separation of lung homogenates on a native SDS gel (Fig. 2F).
Generation of superoxide was assessed by following the cumulative photon emission from thoracic aortic segments (Fig. 3A) and the left ventricular myocardium (Fig. 3B) incubated with 5 μM lucigenin at 37°C and found a 2.5- to 3-fold stronger formation of superoxide in tissues of C101A/eNOS-KO compared with C57BL/6 and eNOS-KO, while there was no difference between the latter two strains (Fig. 3A, B). Furthermore, this increase was dependent on C101A-eNOS as demonstrated by complete inhibition following in vitro incubation with L-NAME in the aortic (Fig. 3C) and myocardial tissues (Fig. 3D). In striking contrast, L-NAME tended to increase aortic and myocardial superoxide formation in C57BL/6, while having no effect in eNOS-KO (Fig. 3C, D). To investigate peroxynitrite formation, we used Western blot detection of tyrosine nitration in aortic and myocardial homogenates. In the aorta, myocardium, and skeletal muscle of C101A/eNOS-KO, we observed a significantly higher nitrotyrosine level compared with C57BL/6 and eNOS-KO (Supplementary Fig. S4F–H). The increase of nitrotyrosine levels was completely inhibited by oral treatment with L-NA, demonstrating the dependence on C101A-eNOS (Fig. 3E, F).
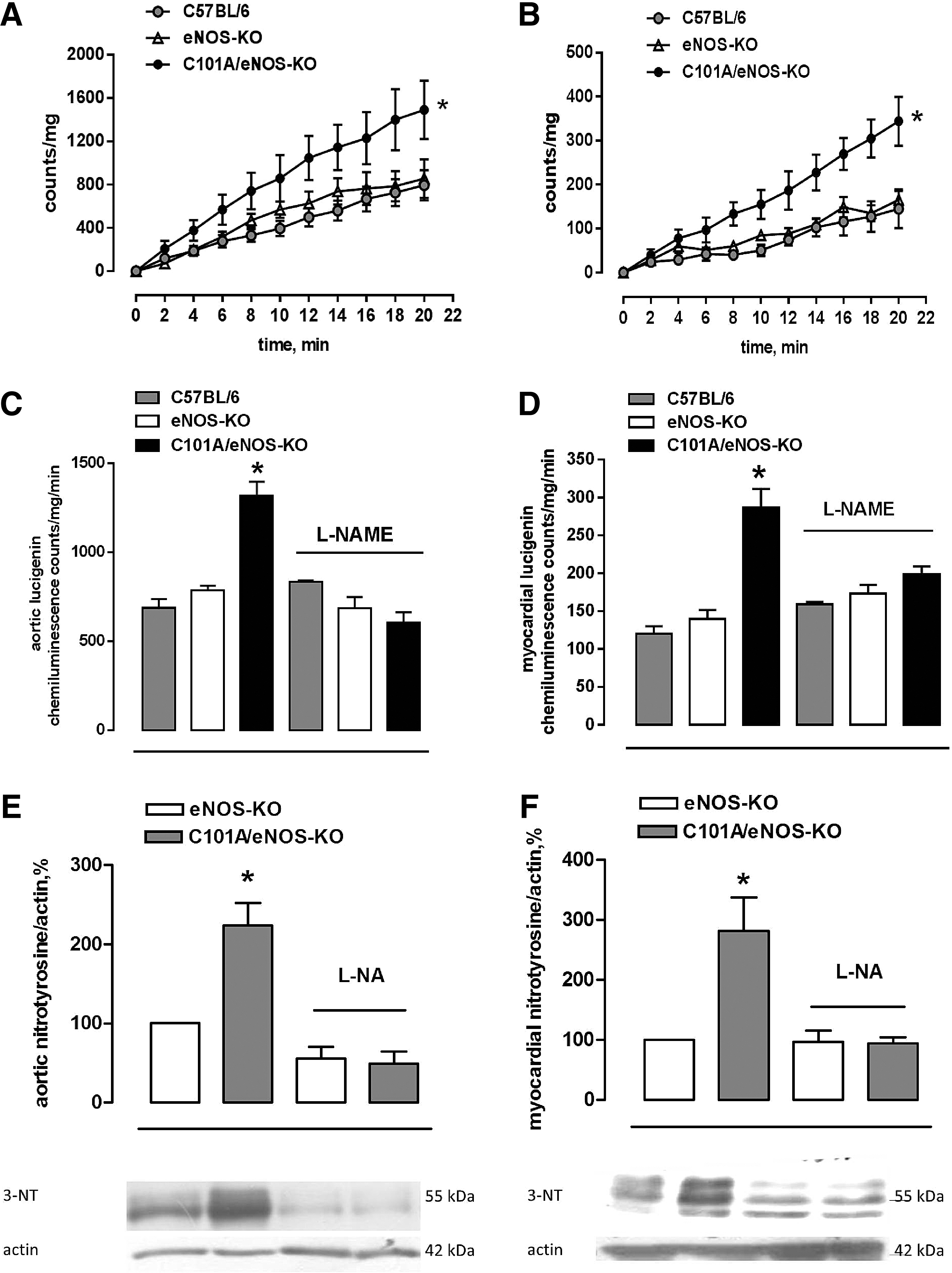
Similar to increase of protein carbonylation in C101A-eNOS HEK cells, a significantly higher amount of amino acid carbonyl groups was detected in aortic and myocardial tissues of C101A/eNOS-KO compared with wild type and eNOS-KO (Supplementary Fig. S5).
C101A-eNOS activity restores endothelial function in eNOS-KO
Endothelial function was studied using acetylcholine-induced endothelium-dependent dilation of aortic rings, as described previously (11). We found a concentration-dependent reduction of vascular tone in C101A/eNOS-KO, while there was just a slight vasoconstriction in eNOS-KO (Fig. 4A). These data suggest that the expression level of eNOS in the aortic endothelium may fall by as much as 60% without impairing maximal endothelium-dependent vasodilation. Furthermore, this vasorelaxation occurred despite a strong increase of vascular oxidative stress as shown in Figure 3A, C, and E. There was, however, a significant rightward shift of the concentration–response curves to acetylcholine in C101A/eNOS-KO versus C57BL/6 (Fig. 4A). The corresponding pD2 values were 6.85±0.08 (n=14) and 6.58±0.1 (n=11, p=0.0358), respectively. To study whether the rightward shift of the acetylcholine concentration–response curve is induced by increased superoxide generation, aortic rings of C57BL/6 and C101A/eNOS-KO were preincubated with 100 U/ml of pegylated superoxide dismutase (SOD), but the pD2 values for C57BL/6 (7.01±0.126) and C101A/eNOS-KO (6.573±0.07) remained significantly different (*p<0.05, n=4, Fig. 4B). Likewise, a direct comparison revealed that 100 U/ml of pegylated SOD did not change acetylcholine-dependent vasodilation in C57BL/6 or C101A/eNOS-KO (Supplementary Fig. S6A, B). Rubbing off the endothelial layer completely inhibited acetylcholine-induced vasodilation in C101A/eNOS-KO (data not shown).
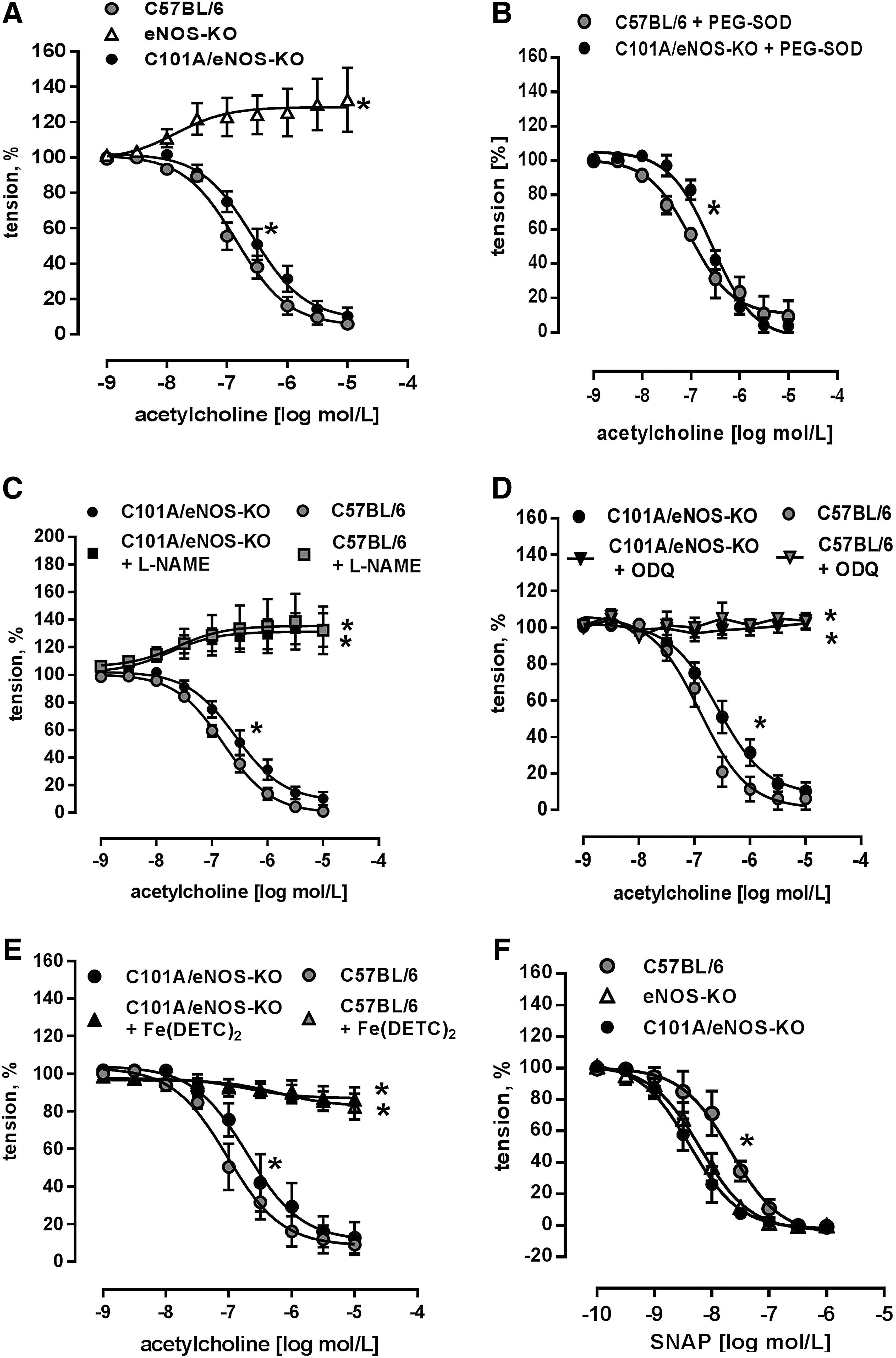
Several approaches were accomplished to investigate the molecular mechanism of aortic vasodilation to acetylcholine in C101A/eNOS-KO. First, it was observed that L-NAME reversed endothelium-dependent vasodilation to vasoconstriction in C101A/eNOS-KO and C57BL/6 and that vasoconstriction did not differ between the strains (Fig. 4C). Thus, endothelium-dependent vasodilation to acetylcholine in C101A/eNOS-KO is dependent on the activity of C101A-eNOS. Second, incubation of aortic rings with 1H-[1,2,4] oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) to inhibit soluble guanylyl cyclase (sGC) completely abolished the vasodilator response to acetylcholine with no difference between both strains (Fig. 4D). These data demonstrate the crucial involvement of the NO/cGMP pathway. Third, these data were confirmed by subjecting aortic rings to the spin trap Fe(DETC)2. As shown in Figure 4E, scavenging of NO completely inhibited acetylcholine-induced vasodilation in both strains. Finally, it was investigated whether hydrogen peroxide might be involved in endothelium-dependent vasodilation. However, neither conventional nor pegylated catalase (Supplementary Fig. S6C, D) had any influence on the vasodilation. Investigation of the sensitivity of the NO/cGMP pathway confirmed the previously reported sensitization in eNOS-KO (25) and demonstrated that a similar hypersensitivity to NO donors, such as S-nitroso-N-acetyl-D,L-penicillamine (SNAP) (Fig. 4F) and diethylamine/nitric oxide (DEA/NO) (Supplementary Fig. S6E), occurred in C101A/eNOS-KO. There was no change of the expression of sGC subunit β1 (Supplementary Fig. S6F). In addition, aortic and skeletal muscle expression of nNOS was not changed in C101A/eNOS-KO (Supplementary Fig. S7A–C). Taken together, these data demonstrate that C101A-eNOS-dependent vascular oxidative stress had no influence on complete vasodilation to acetylcholine in aortic rings of C101A/eNOS-KO. The relaxation was entirely endothelium dependent and mediated by the same pathway as in C57BL/6, that is, generation of NO by eNOS and activation of the NO/cGMP pathway.
C101A-eNOS activity inhibits increased neointima formation in eNOS-KO
In an attempt to investigate whether increased vascular oxidative stress present in C101A/eNOS-KO may play a role in the development of vascular pathologies such as neointimal hyperplasia, we subjected C57BL/6, eNOS-KO, and C101A/eNOS-KO to ligation for 4 weeks of the left common carotid artery proximal to the bifurcation. In C57BL/6, this procedure resulted in a strong formation of neointima, which declined with increasing distance from the ligation as reported previously (Fig. 5A–C) (16). Likewise, the lumen area was largely decreased, while the media were enlarged (Supplementary Fig. S8A, B). This pronounced development of neointimal hyperplasia resulted in an increased intima/media ratio, which declined with increasing distance from the ligation (Fig. 5D, E). In eNOS-KO, neointimal hyperplasia was much more pronounced as evidenced by a significantly increased neointima area (Fig. 5A–C) and intima/media ratio (Fig. 5D, E). In striking contrast, neointimal hyperplasia in ligated carotid arteries of C101A/eNOS-KO resembled that observed in C57BL/6. The small increase of neointima and intima/media ratio versus C57BL/6 did not reach statistical significance (Fig. 5C, E). In addition, neointimal hyperplasia in C101A/eNOS-KO significantly differed from that in eNOS-KO. Thus, despite strongly increased vascular oxidative stress, mild hypertension, and a low level of C101A-eNOS expression, neointimal hyperplasia was not accelerated in C101A/eNOS-KO. These data suggest that the lack of NO rather than increased levels of eNOS-derived vascular oxidative stress accelerates neointimal hyperplasia in response to ligation.
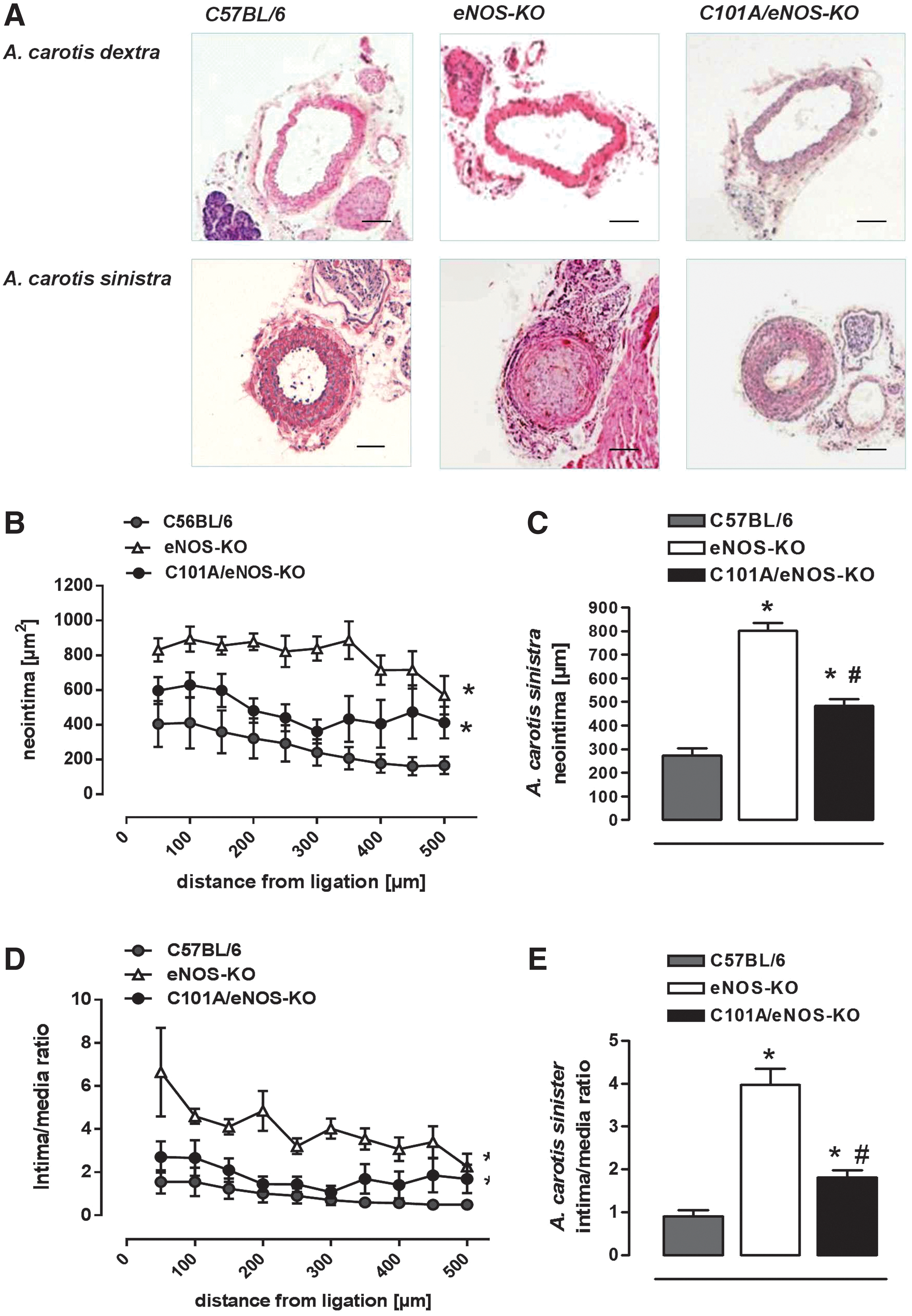
C101A-eNOS activity partially restores vascular remodeling in eNOS-KO
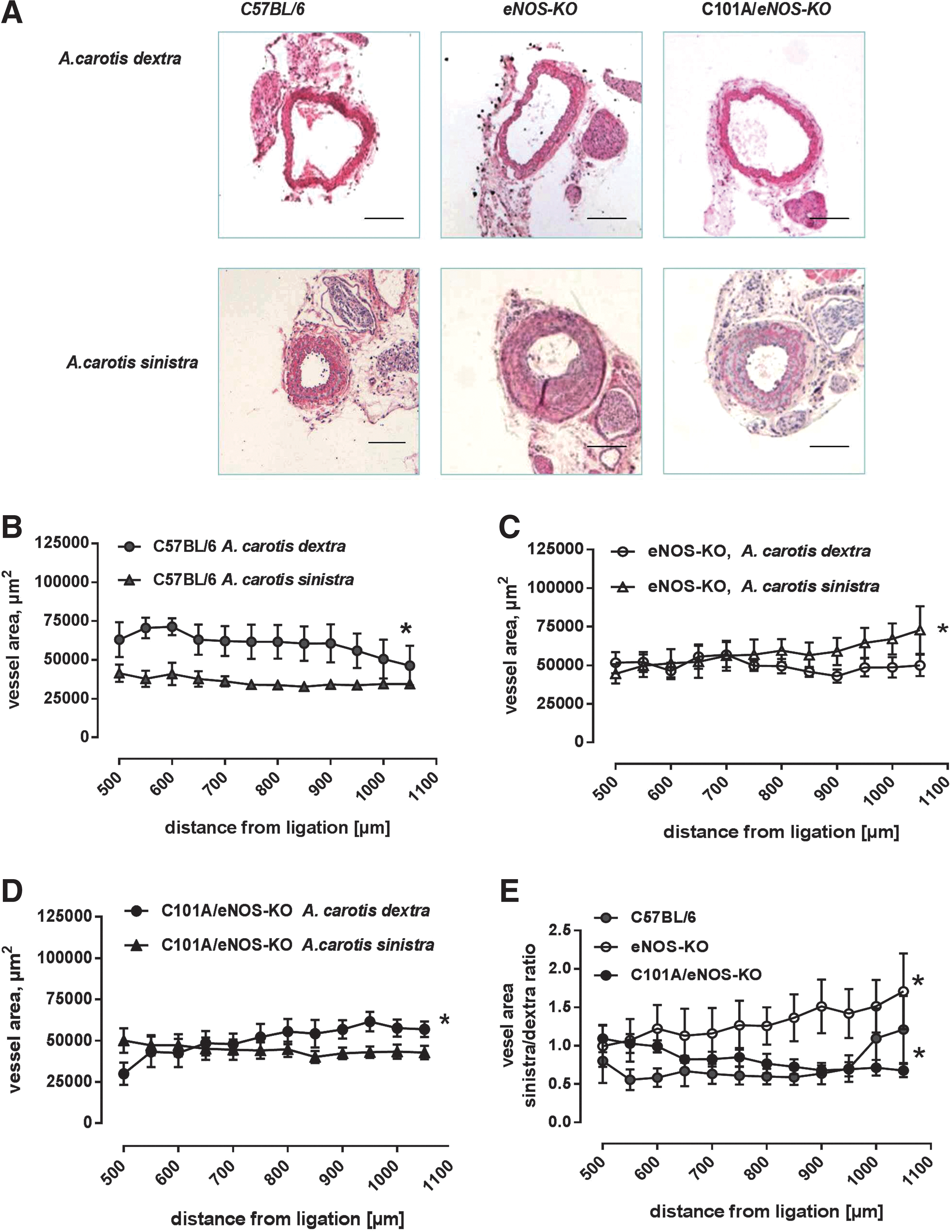
To estimate remodeling following the ligation procedure, the left common carotid vessel area was measured 500–1000 μm distal to the ligation and the averaged values were compared with those obtained from the unligated right common carotid artery. As shown in Figure 6A, there was still considerable neointimal hyperplasia in this region of the ligated artery. In C57BL/6, there was a statistically significant difference between the vessel area of the left and right common carotid artery, suggesting the occurrence of vascular remodeling induced by the reduction of flow in the ligated and/or an increase of flow in the unligated vessels (Fig. 6B). In striking contrast, in eNOS-KO, the vessel area distal to the ligation was significantly increased over the vessel area of the unligated right common carotid artery (Fig. 6C), suggesting that vascular remodeling was inversed. In C101A/eNOS-KO, this abnormal vascular response was partially restored, that is, the vessel area of the ligated carotid artery was smaller than that of the unligated right common carotid artery, although the difference was not as pronounced as in C57BL/6 (Fig. 6D). In accordance, the ratio of the vessel area of the ligated versus the unligated carotid artery, which was calculated in each separate animal and then averaged for each distance point, clearly demonstrated a mouse strain-dependent significant increase over the investigated distance from the ligation in eNOS-KO, while C57BL/6 and C101A/eNOS-KO showed a decrease (Fig. 6E).
Discussion
This study aimed to investigate the impact of eNOS-dependent vascular oxidative stress on endothelial function and on neointimal hyperplasia. A mutant destabilized eNOS, which was introduced in eNOS-KO in an endothelial-specific manner, (i) largely increased vascular generation of superoxide and protein nitrotyrosine formation, but (ii) had little impact on aortic endothelium-dependent vasodilation by acetylcholine, and (iii) corrected the accelerated neointimal hyperplasia and the abnormal vascular remodeling observed in eNOS-KO. These data suggest that vascular oxidative stress generated by an impairment of eNOS activity has a limited effect on endothelial function of conduit arteries, presumably because of concomitant generation of NO by eNOS.
As predicted by the crystal structure of the oxygenase domain of eNOS, genetic disruption of its Zn ion complex disturbs dimer formation and thereby binding and correct orientation of BH4 (44). BH4 deficiency promotes the transfer of an electron to molecular oxygen and thereby the generation of the superoxide (14). Thus, a replacement of cysteine 101 by alanine destabilizes eNOS, resulting in reduced NO formation and increased generation of superoxide in transfected HEK cells and in different tissues of C101A-Tg and C101A/eNOS-KO. Our data on decreased catalytic activity of C101A-eNOS are consistent with previous reports describing eNOS mutations at one or both Zn-binding cysteines (5, 9). Of note, mutation of eNOS by replacing both Zn-binding cysteines with serine did not abolish formation of L-citrulline from L-arginine (9), suggesting partial preservation of NO generation, despite decreased dimer stability as reported in this study with C101A-eNOS. The destabilization of C101A-eNOS was also evident by the decrease of the dimer/monomer ratio following separation on native SDS gels, and similar findings have been reported previously (45). Moreover, increased superoxide generation induced by C101A-eNOS in transfected HEK cells and in aortic and myocardial tissues of C101A/eNOS-KO was abolished by L-NAME, identifying C101A-eNOS as the underlying source both in cells and in transgenic mice.
Previous studies with purified eNOS have demonstrated that BH4 deficiency strongly increases the eNOS-derived superoxide electron spin resonance (ESR) signal and that BH4 supplementation inhibits this oxidase activity in a concentration-dependent manner (57, 58). In striking contrast, citrulline formation by purified C101A-eNOS never reached the maximal rate that was achieved with WT-eNOS, not even at BH4 concentrations of 50 μM in the reaction tube, demonstrating that the ability of C101A-eNOS to generate citrulline and hence NO cannot be improved by excess BH4. Taken together, these results are in line with previous investigations on the importance of BH4 (5, 14, 17, 44) and strongly suggest that an impairment of binding and correct orientation of BH4 underlies the increase of superoxide production observed in cells and tissues harboring C101A-eNOS.
A surprising finding of this study is that activation of C101A-eNOS by acetylcholine in aortic rings resulted in complete endothelium-dependent vasodilation in C101A-Tg and C101A/eNOS-KO, despite reduced NO formation and increased vascular oxidative stress induced by C101A-eNOS. The relaxation by acetylcholine in aortic rings of C101A/eNOS-KO was found to be mediated by NO and developed at tissue levels of superoxide and/or peroxynitrite, resembling those occurring in various animal models with clearly detectable impairment of eNOS function and/or endothelial-dependent vasodilation (2, 7, 15, 21, 32, 33, 61). However, in each of these studies, concomitant conditions, such as atherosclerosis, severe hypertension, diabetes, or disruption of certain genes such as interleukin 10 or Bmal1, were present, which might have contributed to endothelial dysfunction beside oxidative and/or nitrosative stress. Nevertheless, there was a significant rightward shift of the acetylcholine concentration–response curve, indicating a reduced efficacy of eNOS activation in aortic rings of C101A/eNOS-KO. A contribution of increased generation of superoxide is most likely not involved since pegylated SOD had no impact on acetylcholine-induced aortic dilation. It is also possible that the reduction of eNOS expression and its ability to generate NO in C101A/eNOS-KO compared with C57BL/6 is the underlying cause. According to the data obtained from the carotid-binding experiments, this small shift appears to be rather unimportant for NO bioavailability in vivo. Taken together, our data suggest that oxidative stress generated by eNOS appears to have little impact on endothelium-dependent vasodilation in conduit arteries.
It might be argued that the lack of effect of C101A-eNOS on mild hypertension of eNOS-KO demonstrates an effect of oxidative stress generated by an impairment of eNOS activity on resistance vessels and blood pressure. One would expect a blood pressure-lowering effect of endothelial expression of eNOS as reported previously (40, 41, 54); however, data of a further study have shown that even endothelial-specific expression of normal eNOS does not reduce the hypertension of eNOS-KO (55). Thus, the persistence of hypertension in C101A-eNOS-KO is probably not provoked by C101A-eNOS-dependent oxidative stress and is unlikely to represent a specific phenotype caused by pronounced vascular oxidative stress in this animal model. On the other hand, our data certainly do not exclude that oxidative stress due to an impairment of eNOS activity may have an impact on the regulation of blood pressure, for example, in more severe hypertensive cardiovascular disease states (34).
Smooth muscle proliferation is an obvious event during the development of atherosclerosis and restenosis (36, 39, 46). It is generally believed that oxidative stress plays a crucial role in this process (35, 48) and that this pathologic role of oxidative stress is closely linked to a downregulation of vascular NO bioavailability (56). Furthermore, inhibition of oxidative stress is considered to be a key component of the overall vasoprotective effect of endothelial eNOS activity (12). In this study, neointima formation following carotid ligation (26) was used to estimate the vascular bioavailability and vasoprotective activity of endothelial NO in vivo. In this model, neointima formation occurs in the presence of intact endothelial lining and is directly related to the alteration of the blood flow pattern, that is, turbulent rather than laminar flow, as well as to the infiltration of monocytes (20). Consequently, neointima formation is largest close to ligation and becomes smaller with increasing distance (26) and a similar pattern was observed in C57BL/6, C101A/eNOS-KO, and eNOS-KO. However, neointima formation was significantly more pronounced in eNOS-KO than in C57BL/6, confirming that vascular eNOS elicits protective effects in the setting of carotid ligation (59). Neointima formation in C101A/eNOS-KO tended to increase in comparison with C57BL/6, but the difference did not reach statistical significance, while the difference compared with eNOS-KO was obvious. Furthermore, this difference was evident, despite increased oxidative stress in C101A/eNOS-KO, which does not occur in eNOS-KO, suggesting that C101A/eNOS-KO generate bioavailable NO sufficiently to inhibit smooth muscle proliferation as observed in C57BL/6. Finally, these data obtained from the carotid ligation experiments indicate that increased neointima formation in eNOS-KO appears to be caused by the lack of endothelial NO formation and not by the presence of mild hypertension.
Another important role of NO is to mediate vascular remodeling in response to flow (28, 47). Although carotid ligation is not a good model to investigate flow-dependent vascular remodeling due to the neointima formation, the ratio of ligated to unligated vessel area, which indicates vascular remodeling, was almost similar in C57BL/6 and C101A/eNOS-KO. In these strains, the ratio decreased with increasing distance from the ligation and reached values well below one, while in eNOS-KO this was not the case. These data again suggest a limited impairment of NO bioavailability in C101A/eNOS-KO.
The alteration of activity of C101A-eNOS might resemble the features of uncoupled eNOS, a term which has been used to describe early findings with purified NOS protein showing increased production of superoxide and hydrogen peroxide in the absence of important cofactors such as BH4 (19). In contrast, mutant C101A-eNOS produces less NO and more superoxide in response to dimer destabilization and subsequent impairment of binding and correct orientation of BH4. Hence, this approach shows a direct relationship between a reduced stability of homodimers and eNOS function. In contrast, there is no evidence that the presence of monomers and a reduced dimer/monomer ratio are directly related to uncoupling of the enzyme since only the dimeric form has been shown to be active and generate NO and superoxide (46). Moreover, we believe that an advantage of this approach is the possibility to study the importance of eNOS dimer destabilization and impaired BH4 binding for vascular physiology and pathophysiology in otherwise healthy animals. This largely reduces unpredictable confounding factors, such as complex disease processes occurring in hypertension, atherosclerosis, and/or diabetes, which are associated with an impairment of eNOS cofactor availability or proven increases of other components such as the eNOS inhibitor, ADMA (3). Nevertheless, concomitant generation of NO and superoxide by C101A-eNOS probably closely resembles the situation in the endothelium of diseased vessels when uncoupled eNOS molecules and coupled eNOS molecules can exist in the same cell at the same time (35).
Taken together, the results of this study suggest that vascular oxidative stress generated by an impairment of eNOS activity might not be as important as estimated previously and rather highlight the contribution of oxidative stress to vascular pathologies caused by other enzymatic sources (18, 22, 29, 36, 43, 52). A reasonable explanation for the limited importance of eNOS-dependent oxidative stress for functional vascular damage might be the concomitant generation of NO by eNOS, which is a fundamental difference from oxidases. Hence, our findings substantiate the importance of vasoprotection conferred by endothelial NO (12), which appears to be able to cope with even larger increases of vascular oxidative stress. Similar findings have been reported for the vasoprotective activity of eNOS in murine atherosclerosis (42). Although it is difficult to extrapolate our data to clinical conditions as the transgenic approach used does not directly match clinically occurring disease pathologies, our data support previous observations on interventions associated with increased eNOS protein expression and improvement of endothelial NO production, such as regular exercise training (24), pharmacotherapy with statins (31), and treatment with inhibitors of the renin–angiotensin system (12) have provided superior cardiovascular prevention than targeting vascular oxidative stress (4, 18a, 37, 50, 60).
Materials and Methods
Stable transfection of HEK 293 cells
Using primer overlap extension PCR, we genetically destabilized bovine eNOS by replacement of Cys 101 to Ala (C101A). The mutation was confirmed by sequencing. The cDNAs for wild-type bovine eNOS (WT) and the mutant (MT) C101A eNOS were cloned into the pcDNA3 vector (Invitrogen Corporation, Carlsbad, CA) and stably transfected in HEK 293 cells.
ESR measurements
ESR measurements were performed as described previously (8, 27). Cellular basal and Ca2+ ionophore A23187-stimulated NO production was measured using 0.2 M of the spin trap iron/diethyldithiocarbamic acid, Fe(DETC)2. CMH (1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine) at 100 μM was used for superoxide detection in the absence and presence of the NOS inhibitor, L-NAME (1 M).
eNOS activity
The specific activity of WT- and C101A-eNOS in cell homogenates from stably transfected HEK cells and in isolated enzyme preparations was determined by conversion of 14C-L-arginine to 14C-L-citrulline as described previously (49) in the presence of increasing concentrations of BH4 (0–50 μM).
Detection of oxidative protein modification
Protein modifications caused by additional carbonyl groups were detected in C101A-eNOS and WT-eNOS-transfected HEK cells and aortic and myocardial tissue of C57BL/6, eNOS-KO, and C101A/eNOS-KO using the Oxyblot™ system (Intergen, Burlington, MA).
Transgenic C101A-eNOS mice
We generated a DNA construct, in which C101A-eNOS cDNA (4.1 kb) was inserted between the murine Tie-2 promoter (2.1 kb) cDNA and a 10 kb Tie-2 intron fragment, designated as Tie-2 enhancer. This construct was used to target C101A-eNOS gene expression to the vasculature, as described previously (30). Double transgenic mice expressing endothelial C101A-eNOS only (C101A/eNOS-KO) were obtained by crossing C101A-Tg with eNOS-deficient mice (eNOS-KO, backcrossed >20 times to C57BL/6J) (51). Additional groups of C57BL/6J, eNOS-KO, and C101A/eNOS-KO received L-NA (100 mg L-NA/kg BW/day; Sigma, Munich, Germany) for 3 weeks before the experiments. Permission for the animal studies was provided by the regional government of Germany (AZ 50.05-230-3-94/00, AZ 50.05-230-18/06, AZ 8.87-51.04.20.09.383), and the experiments were performed according to the guidelines for the use of experimental animals, as given by the German “Tierschutzgesetz” and the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health.
Vasorelaxation studies
Function of the endothelium was examined in aortic rings of C57BL/6, eNOS-KO, and C101A/eNOS-KO by cumulative addition of acetylcholine (0.01 to 10 μM) after submaximal precontraction with 0.2 μM phenylephrine, as described previously (53). Thereafter, vasorelaxation to the NO donor, SNAP (1 nM to 10 μM) or DEA/NO (1 nM to 10 μM), was studied by cumulative application. In some experiments, the NOS inhibitor, L-NAME (1 mM), or the sGC inhibitor, ODQ (10 μM), was administrated 30 min before application of acetylcholine. To determine the contribution of hydrogen peroxide to aortic relaxation in C101A/eNOS-KO, acetylcholine concentration–response curves were recorded in the absence and presence of either conventional (1200 U/ml) or pegylated catalase (1400 U/ml). To investigate a possible interaction between endothelium-dependent relaxation in C101A/eNOS-KO and increased aortic superoxide formation, we studied the acetylcholine concentration–response curve in aortic segments of wild type and C101A/eNOS-KO preincubated for 30 min with pegylated SOD (100 U/ml). In another experimental set, aortic rings of C101A/eNOS-KO and C57BL/6 were incubated with the colloidal Fe(DETC)2 spin trap solution before application of the increasing acetylcholine concentrations. The spin trap was freshly prepared by mixing equal amounts of deoxygenated 1.6 mM FeSO4 and 3.2 mM diethyldithiocarbamate (DETC) solutions (Noxigen, Elzach, Germany). In all experiments, only one acetylcholine concentration–response curve per aortic ring was recorded and analyzed.
SDS-polyacrylamide gel electrophoresis and immunoblotting
Western blots for eNOS, nNOS, immunoprecipitated eNOS phosphorylated at Ser1176 (Ser 1179 in bovine eNOS), sGC-β1, nitrotyrosine residues, von Willebrand factor, and actin were performed in different mouse tissues using standard techniques, as described previously (25, 41) (please also refer to Supplementary Materials and Methods section).
Immunohistochemistry
Colocalization of eNOS and endothelium-specific marker, CD31, in aortas of C57BL/6, eNOS-KO, and C101A/eNOS-KO was performed as described previously (41) (please also refer to Supplementary Materials and Methods section).
Lucigenin superoxide detection
Lucigenin-enhanced chemiluminescence detection of superoxide in the intact aorta and left ventricular myocardium of C101A/eNOS-KO, eNOS-KO, and C57BL/6 was measured as described previously (23).
Measurement of systolic blood pressure
Systolic blood pressure and heart rate were measured in awake, 3–4-month-old male C57BL/6, eNOS-KO, and C101A/eNOS-KO using an automated tail-cuff system (Visitech Systems, Apex, NC), as described previously (25). In some experiments, establishment of basal blood pressure was followed by L-NA treatment with the drinking water, and blood pressure recordings were continued for another 21 days.
Neointimal hyperplasia
Ten-week-old C57BL/6, eNOS-KO, and C101A/eNOS-KO (n=9–10) were anesthetized and the left common carotid artery was dissected and ligated near the carotid bifurcation, as described previously (26). All animals were sacrificed 28 days after ligation. After excision of the left and right common carotid arteries, the vessels were fixed in 4% p-formaldehyde for 6 h, dehydrated, and embedded in paraffin. Serial 5-μm sections were cut, and sections of every 50 μm were used for morphometric analysis using hematoxylin–eosin staining (vessels with thrombus formation were excluded). Morphometric analysis was carried out using image analysis software (Leica Microsystems, Wetzlar, Germany). The circumference of the lumen, elastic lamina interna, and elastic lamina externa were determined by tracing along the different vessel layers.
Statistics
All data were analyzed by standard computer programs (GraphPad Prism PC Software, version 4.0) and are expressed as mean±SEM of n individual samples. Statistical comparisons between groups were performed by t-tests, the Newman-Keuls multiple comparisons post-hoc test following one-way analysis of variance (ANOVA, for more than two groups) or two-way ANOVA (concentration–response curves). p<0.05 was considered statistically significant.
Materials and reagents
All chemicals were purchased from Sigma or Merck (Darmstadt, Germany), except otherwise stated.
A more detailed description of the Materials and Methods section is provided in the online Supplementary Materials and Methods section.
Footnotes
Acknowledgments
This study was supported by the Forschungskommission of the Heinrich Heine University Düsseldorf (Project 9772 272 to G.K. and 9772 446 to T.S.) and by the DFG research grant, SU 783/1-1, to T.S.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.