
Abstract
The high intrinsic radiosensitivity of the lung tissue precludes the use of curative radiation doses and fosters a therapeutic failure by local tumor recurrence and metastasis. In this study, we demonstrate that therapy with bone marrow-derived or vascular wall-derived mesenchymal stromal cells (MSCs) protects the lung tissue from radiation-induced vascular dysfunction through inhibition of matrix metalloproteinase 2 (Mmp2) and antagonizes increased metastasis of circulating tumor cells to previously irradiated lungs. MSC therapy also reversed radiation-induced senescence of epithelial cells and suppressed associated secretion of senescence-associated secretory phenotype factors and immune changes. Thus, MSC therapy is a promising strategy to prevent radiation-induced local and systemic effects with suggested tumor-promoting potential.
Introduction
R
Others and we showed in preclinical studies that radiation-induced normal tissue toxicity is closely linked to vascular endothelial cell (EC) damage and dysfunction of the blood–air barrier (9, 20, 22, 71). Moreover, investigations in murine models suggest that radiation-induced EC damage favors the transmigration of tumor cells through the endothelium—which is a critical step in the metastatic cascade—resulting in increased formation of micrometastasis (5, 7, 29).
Dysregulated proteolysis has been implicated in tumor invasion and metastasis by promoting tumor cell transendothelial migration. In particular, matrix metalloproteinases (Mmps) have previously been shown to degrade the extracellular matrix and the basement membrane in the process of metastasis (49). Interestingly, invasive breast cancer cells foster endothelial barrier dysfunction by the activation of endothelial Mmp2 production and thereby facilitate tumor cell extravasation through lung microvascular ECs (36, 54). We therefore speculated that the primary damage and functional impairment of the EC facilitate the formation of micrometastasis in the irradiated lung tissue.
Further investigations, including own previous work, revealed that IR induces complex immune changes such as secretion of cytokines/chemokines and recruitment of cells from the innate and adaptive immune system (11, 70). Interestingly, tumors grown in previously irradiated tissues also recruit large numbers of bone marrow (BM)-derived CD11b+ myeloid cells to support their growth (1, 70). However, the role of immune cells infiltrating irradiated tissues for treatment outcome is still controversial as they can either enhance the antitumor effects of RT or provide paracrine signals that facilitate tumor cell survival and growth, including the formation of tumor blood vessels (1, 3, 44, 69).
Up to now, the mechanisms driving immune cell recruitment or tumor cell recruitment to irradiated tissues are still unclear. Herein, a biological process that can be initiated in various cells in response to diverse stressors, including IR, and has the potential to generate paracrine signals affecting the recruitment of tumor cells and immune cells, is cellular senescence. Senescence is aimed to remove irreparably damaged cells, and therefore potentially harmful cells from the proliferative pool. Although senescent cells do not proliferate, they acquire a particular phenotype characterized by complex gene expression changes and increased expression of diverse secreted proteins termed the senescence-associated secretory phenotype (SASP).
The SASP includes inflammatory cytokines, growth factors, and proteases with potential immunomodulatory function, and some of the SASP proteins even have a suspected tumor-promoting effect (8, 14, 15, 43). It has been demonstrated that single SASP factors regulate key processes that facilitate tumor progression and metastasis, including proliferation, senescence, angiogenesis, epithelial mesenchymal transition, and immune evasion (2, 53, 55). However, the involvement of senescence in radiation-induced adverse effects has not yet been investigated.
Stem cell therapy is a promising option for the prevention or treatment of radiation-induced normal tissue injury as it can promote survival and repair of damaged cells (16). Moreover, transplantation of BM-derived mesenchymal stem cells, also referred as multipotent mesenchymal stromal cells (MSCs), has established itself as a potential strategy for the treatment of lung diseases (46, 63). However, there is a lack of preclinical and clinical studies of stem cell therapy for radiation-induced adverse effects in the lung (46, 58), and there are only few ongoing clinical trials with MSCs in chronic lung disease, including their therapeutic applications in patients with idiopathic pulmonary fibrosis (61, 62).
We hypothesized that therapeutic application of MSCs may be suited to promote repair of radiation-induced damage to resident lung cells, particularly vascular ECs, thereby reducing settlement of tumor cells to previously irradiated lung tissues. A common source of MSCs is the BM (51). However, the procedure to collect BM-derived MSCs is very invasive and only 0.01 to 0.001 percent of mononuclear cells in the BM are MSCs, so other more accessible sources would represent good alternatives. In addition, cord blood, placenta, blood, fetal liver, and even adipose tissue can be used to obtain human MSCs (26, 34, 37, 75). Above all, cord blood and peripheral blood could be good and easily accessible sources (30, 31). However, the percentage of hMSCs in peripheral blood is very low. Therefore, the donor must be stimulated with granulocyte-macrophage colony-stimulating factor to increase this percentage by a washout of MSCs from the BM, but this is very distressing and stressful for the patient.
We showed in previous studies that nestin-positive multipotent MSCs reside in the wall of adult blood vessels and may serve as source for tissue-specific MSCs (vascular wall-resident MSCs [VW-MSCs]). In pathophysiological conditions, for example, the neovascularization of tumors, these VW-MSCs are involved in the stabilization of newly formed vessels and exhibit classical behavior of MSCs in vitro (40, 41). To obtain sufficient material for experimental studies, these cells can be isolated directly ex vivo from the largest blood vessel of the murine system, the aorta, and cultured upon immunomagnetic separation using the stem cell antigen 1 (Sca1). For human use, VW-MSCs can be isolated from small vessel pieces (excess material) obtained during surgery (e.g., vena saphena or arteria radialis).
Importantly, tissue-specific stem cells differentiate mainly to the tissue type from which they derive, indicating that there is a certain code (priming) within the cells as determined by the tissue of origin. Besides the relatively simple extraction of these cells for autologous transplantation, this might be a particular advantage for the therapeutic application of VW-MSCs for improving vascular function or preventing vascular damage. We therefore hypothesized that therapeutic application of VW-MSCs might be particularly well suited for the radioprotection of ECs in a murine model of radiation-induced lung injury.
In this study, we used MSCs isolated either from the BM or from the aorta (Ao) to investigate their potential to counteract lung injury and the resulting microenvironmental changes in response to whole-thorax irradiation (WTI) and explore the underlying mechanisms in a murine model. We show that thorax irradiation induces vascular damage, senescence of bronchial epithelial cells, and secretion of SASP factors with suspected tumor-promoting potential. These changes were associated with an enhanced metastasis of intravenously injected tumor cells or subcutaneously growing tumors to previously irradiated lungs. Adoptive transfer of MSCs normalized vascular dysfunction through inhibition of endothelial Mmp2, reversed epithelial cell senescence and certain aspects of the associated SASP, and reduced the risk of lung metastasis. We speculate that MSC-mediated protection of the lung tissue from the damaging effect of IR precludes the initiation of a tumor-promoting signaling network between damaged resident cells recruited immune cells and tumor cells that support invasion and growth of invading tumor cells.
Results
Impaired vascular function after WTI is accompanied by increased tumor cell extravasation and metastasis in experimental murine models
To investigate a suggested general metastasis-promoting effect of IR, we studied seeding and growth of intravenously injected tumor cells in previously irradiated lung tissue using two different murine cell lines. For this, C57BL/6 mice and BALB/c mice were irradiated with 15 Gy WTI, and syngeneic tumor cells (B16F10 melanoma cells for C57BL/6 mice and TS/A mammary adenocarcinoma cells for BALB/c mice) were injected into the tail vein at 21 days postirradiation (Fig. 1A, B). Micrometastasis formation and subsequent formation of macrometastasis significantly increased in lungs 14 days after tumor cell injection in both mouse strains (C57BL/6/15 Gy: 26.5 per lung cross section±4.6 [standard error of the mean, SEM], n=10; 0 Gy: 12.2±4.1, n=13; p<0.05; BALB/c 15 Gy: 24.7±3.5, n=11; 0 Gy: 10.6±2.9, n=9; p<0.05 as analyzed by two-way analysis of variance [ANOVA], followed by post hoc Bonferroni test: F-factors 0.00297 [interaction], 11.64 [column factor, cf/Gy], 0.2247 [row factor, rf/mouse strain]).
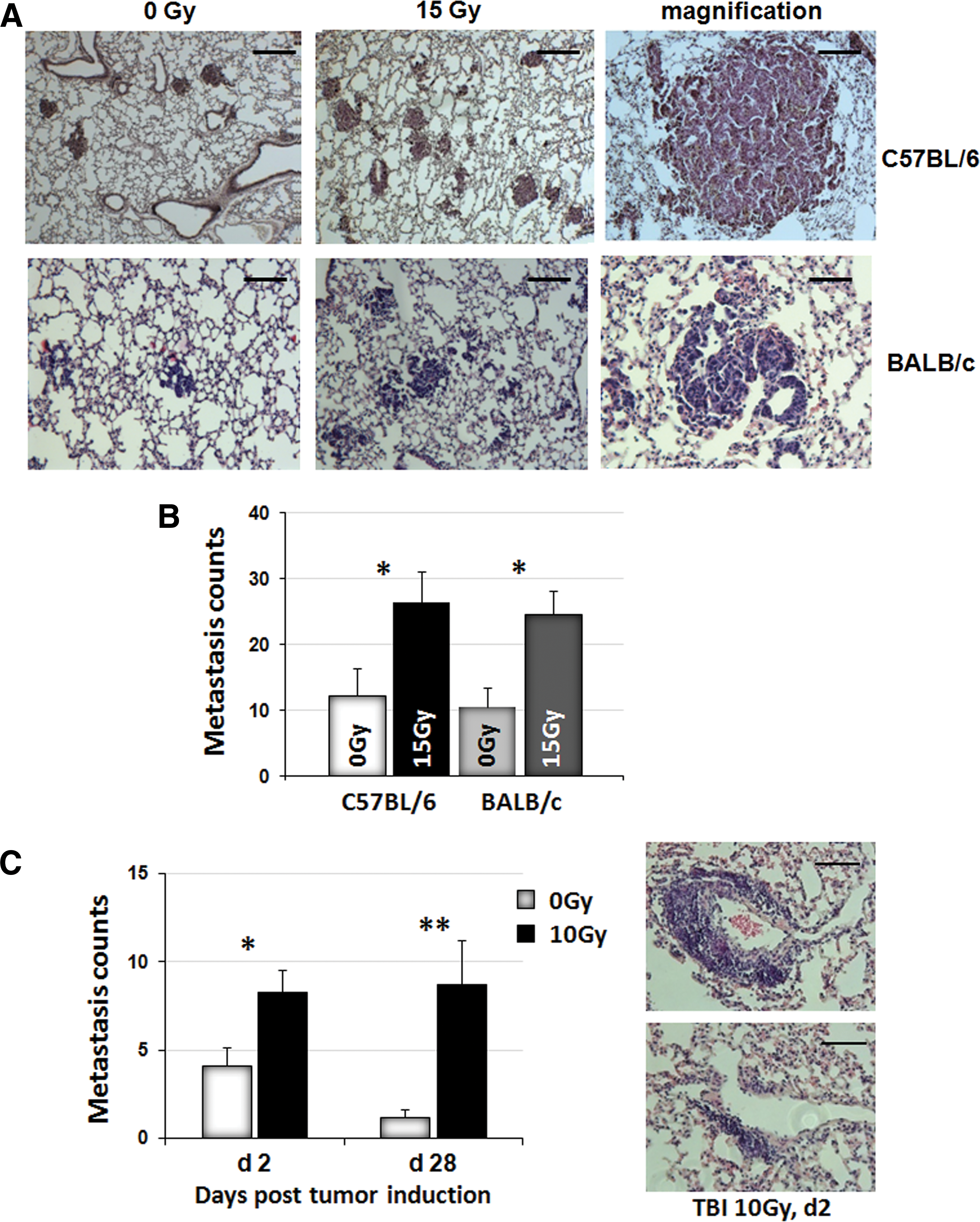
Although tail vein injection of cancer cells is a well-established model of experimental metastasis (66), we next aimed to exclude that the observed effects may be artifacts of injecting large tumor cell numbers into the circulation. Therefore, we performed additional experiments measuring metastasis formation of subcutaneously growing C57BL/6 xenograft flank tumors. For this, C57BL/6 mice received a lethal total body irradiation (TBI) with a splitting dose of 7+3 Gy, followed by BM reconstitution and subcutaneous implantation of B16F10 tumor cells into the flank at 4–6 weeks postirradiation. Two or 28 days later, tumor cell extravasation (here designated as micrometastasis) and subsequent growth of macrometastasis were quantified in whole lung sections (Fig. 1C). These data revealed that metastatic spread to previously irradiated lungs is also increased in mice bearing subcutaneously growing tumors (28 days/10 Gy: 8.2±1.3, n=22; 28 days/0 Gy: 14.1±1.0, n=11; p<0.01; 2 days/15 Gy: 8.7±2.5, n=18; 2 days/0 Gy: 1.2±0.4, n=11; p<0.05 as analyzed by two-way ANOVA, followed by post hoc Bonferroni test: F-factors 1.026 [interaction], 12.17 [cf/Gy], 0.5355 [rf/days]). Because the observed effects in the different models were rather similar, all subsequent experiments were only performed in the B16F10 melanoma model using C57BL/6 mice.
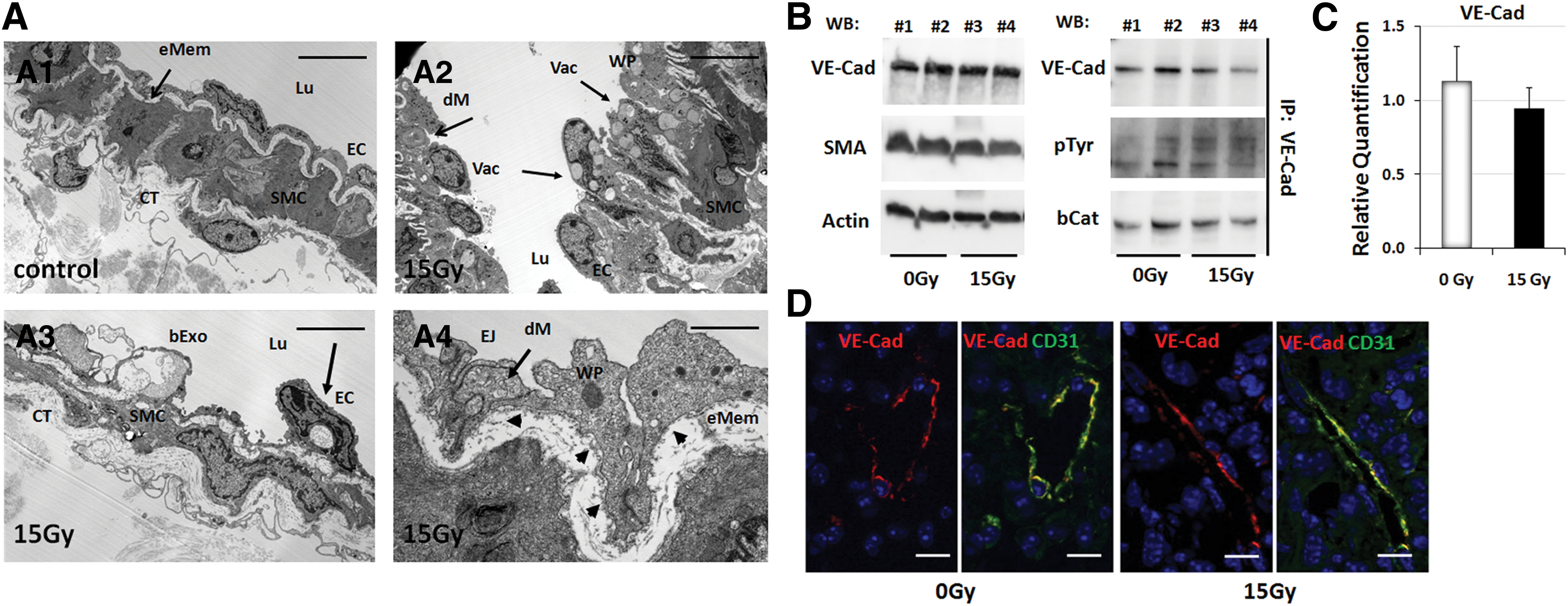
Next, we aimed to gain more detailed insight into the effects of WTI on the vascular compartment. An intensive morphological analysis of the lung tissue at 21 days after WTI corroborated the suggested link between the increased radiation-induced seeding of circulating tumor cells and a functional impairment of lung ECs (Fig. 2A). Indeed, arterial ECs showed numerous vacuoles, partially degraded mitochondria (Fig. 2A2, A3), and a defective and irregular basement membrane (bMem) lining arterial ECs (Fig. 2A 4, arrow heads) at day 21 postirradiation, although EC junctions were not affected. The regular arrangement of EC junctions was further analyzed by Western blot analysis of protein cell lysates obtained from whole mouse lung (Fig. 2B). The total amount of VE-cadherin (VE-Cad), as well as its low phosphorylation status and the proper association with beta-catenin, was not altered after WTI. Real-time reverse transcription polymerase chain reaction (RT-PCR) quantification of VE-Cad mRNA levels in whole lung RNA isolates further confirmed no alterations of VE-Cad expression after WTI (Fig. 2C). Immunocolocalization of VE-Cad and endothelial CD31 (PECAM1) expression confirmed cell surface localization of VE-Cad in lung ECs and a regular arrangement after WTI (Fig. 2D). These results suggest that IR-induced EC damage facilitates the transmigration of tumor cells through the endothelium—a critical step in the metastatic cascade.
MSC therapy improves vascular dysfunction and reduces lung metastasis to previously irradiated lungs
Next, we explored a novel treatment strategy based on therapeutic application of MSCs to limit the radiation-induced vascular dysfunction and the associated risk of increased growth of metastatic tumor cells in previously irradiated lungs. Enhanced green fluorescent protein (EGFP)-tagged MSCs were routinely isolated and cultured from aorta (Ao) of EGFP-Nagy mice and from the BM. C57BL/6 mice received 15 Gy WTI, followed by intravenous transplantation of single cell suspensions of cultured EGFP-positive (EGFP(+)) MSCs into the tail vein at 24 h or 14 days after irradiation (Fig. 3). RT-induced vascular dysfunction resulted in increased albumin leakage (Fig. 3A: difference in mean mg Evans blue dye (EBD) per mg lung tissue=0.13; control irradiated mice=0.06, n=5, and WTI mice=0.19, n=5, p<0.05 (95% confidence interval [CI]=0.01 to 0.24). IR-induced impairment of EC function as determined by EBD extravasation was restored 21 days after irradiation in MSC-treated animals at both 24 h and 14 days after RT, independently from the origin of the applied MSCs (mean EBD extravasation=−0.13 of WTI and BM24h, n=5, 95% CI=−0.24 to −0.002, p<0.05; Ao24h=−0.10, 95% CI=−0.2 to 0.006, p=0.06; BM14d=−0.14, 95% CI=−0.26 to −0.02, p<0.05; Ao14d=−0.10, 95% CI=−0.24 to 0.04, p=0.15). Electron microscopy of lung blood vessels demonstrated a regular vessel structure as well as restored regular EC morphology in the lungs of MSC-treated animals (Fig. 3B). Immunofluorescent analysis of EGFP expression in isolated lung sections revealed that only a few preferably single donor cells can be detected in lung sections, indicating that nearly no therapeutically applied EGFP(+) MSCs homed to the injured lung tissue 21 days after WTI. In-stead circulating EGFP(+) MSCs could still be detected in peripheral blood several weeks after transplantation (Fig. 3C).
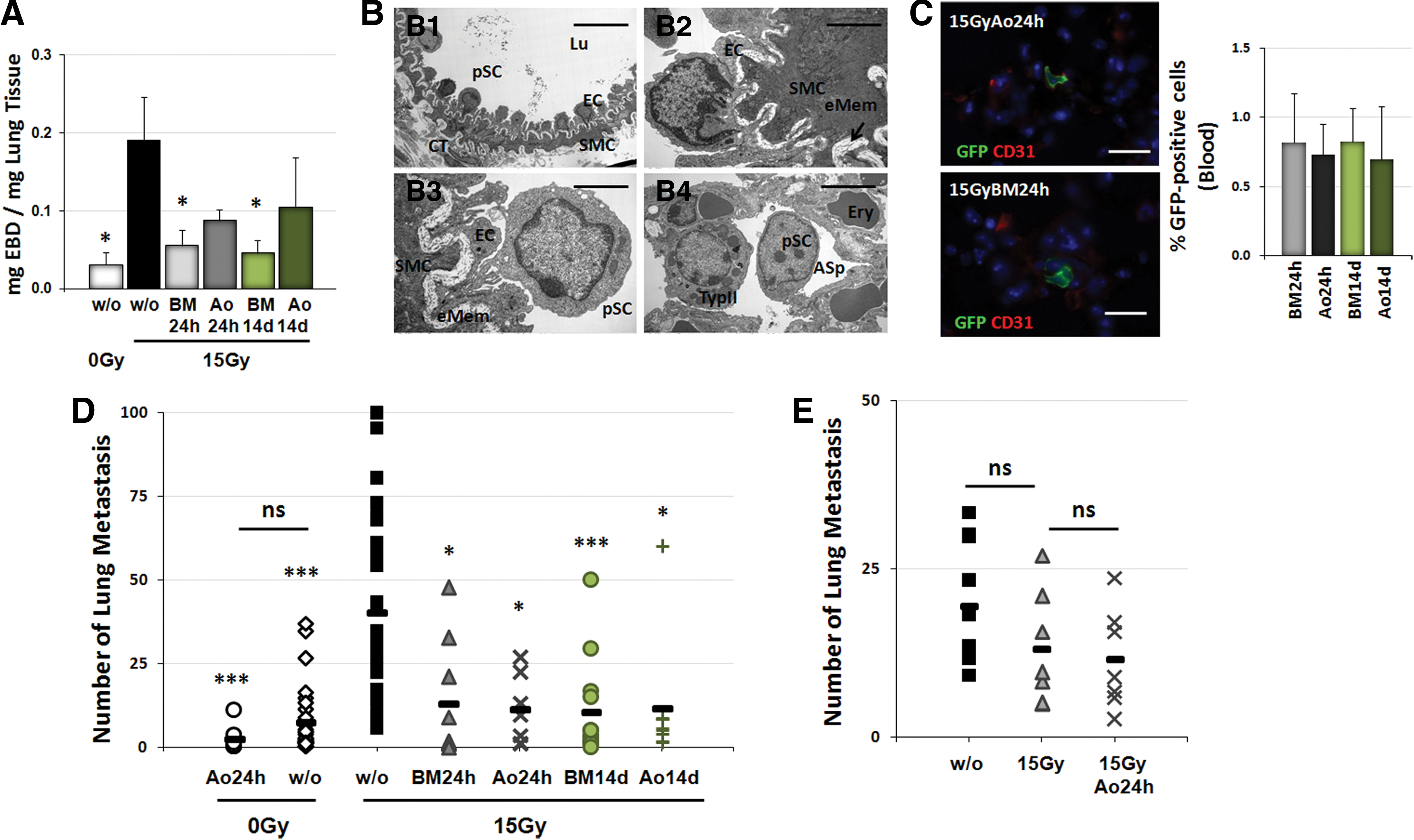
Quantification of lung metastasis formation (Fig. 3D) demonstrated that application of MSCs derived from BM or Ao within 24 h after irradiation (15 Gy: 36.9 metastasis per lung cross section±4.80 [SEM], n=33; 0 Gy: 7.4±1.9, n=10, p<0.001; 0GyAo24h: 2.4±1.3, n=8, p<0.001; 15GyBM24h: 13.0±5.8, n=9, p<0.05; Ao24h: 11.3±3.9, n=7, p<0.05) or 14 days later (BM14d: 10.3±4.1, n=13, p<0.001; Ao14d: 11.4±6.1, n=9, p<0.05 as analyzed by two-way ANOVA, followed by post hoc Bonferroni test: F-factors 13.297 [interaction], 11.44 [cf/Gy], 7.248 [rf/treatment]) significantly reduced seeding of circulating tumor cells and subsequent metastasis formation. A similar tendency to a pronounced but not significantly reduced metastasis formation was also detected in MSC-treated animals after sham irradiation. These results demonstrate that therapeutic application of MSCs has a high potential to counteract seeding and growth of circulating tumor cells in previously irradiated lungs. To exclude that MSC therapy has itself a metastasis-promoting effect on established tumors, untreated C57BL/6 mice were first intravenously injected with B16F10 cells into the tail vein and metastasis formation and growth were allowed for 14 days (Fig. 3E). Thereafter, animals received a 15 WTI without or with additional adoptive transfer of cultured MSCs (0.5×10(6) cells) derived from the aorta 24 h after irradiation (15GyAo24h). Quantification of lung metastasis at 3 weeks after MSC therapy revealed that MSC therapy did not increase the number or size of established lung metastasis. These results indicate that MSC therapy reduces the risk of lung metastasis in irradiated lungs without promoting growth of established metastatic lesions.
MSC treatment reduces radiation-induced endothelial Mmp2 expression, thereby normalizing vascular function
Invasion and metastasis of tumor cells require proteolytic activity (e.g., Mmps) to degrade components of the extracellular matrix, thereby facilitating tumor cell migration as well as metastatic dissemination of malignant cells. Therefore, we analyzed expression levels of Mmp2 as well as of its cofactors, Mmp14 and Timp2, in more detail in irradiated lung tissue (Fig. 4). Real-time RT-PCR quantification of these metastasis-associated genes revealed a significant upregulation of the Mmp2-Mmp14-Timp2 axis in the lungs of WTI animals after WTI (Fig. 4A; p-values as indicated: *p<0.05, **p<0.01, ***p<0.001 as analyzed by two-way ANOVA, followed by post hoc Bonferroni test: F-factors 2.311 [interaction], 9.589 [cf/Gy and treatment], 5.721 [rf/genes]). Of note, the mRNA levels of these enzymes were almost reduced to normal levels when animals were treated with MSCs from BM from Ao within 24 h after irradiation (Fig. 4A). Increased radiation-induced Mmp2 expression was further confirmed on the protein level as shown by using Western blot analysis in whole protein lysates and in paraffin sections using immunohistochemistry (IHC) (Fig. 4B).

Increased amounts of Mmp2 protein levels were confined to arterial blood vessels of lungs compared after WTI; notably, Mmp2 levels were reduced in lungs of animals treated with IR plus subsequent MSC therapy (Fig. 4C). To confirm the endothelial origin of Mmp2, mouse lung endothelial cells (MLECs) were purified and irradiated in vitro. Interestingly, IR of cultured MLECs induced an upregulation of Mmp2 and an increased secretion of Mmp2 to the medium (not shown). These results suggest that endothelial Mmp2 contributes to radiation-induced EC damage and can be targeted by MSC therapy.
To gain insight into the role of Mmp2 in the adverse effects of RT to the lung, we examined potential protective effects of a specific Mmp2 inhibitor in our experimental model. For this, C57BL/6 mice received repeated intraperitoneal injections of the selective Mmp2 inhibitor, ARP100 (15 μg/g bodyweight), twice a week for 3 weeks after WTI (Fig. 5). As shown in Figure 5A and B, ARP100 treatment normalized RT-induced bovine serum albumin (BSA) extravasation at day 21 postirradiation (15 Gy: 6.8±2.9 μg BSA/g total lung protein, mean±SEM, n=4; 15 Gy/ARP100: 0.5±0.5 μg BSA/g total lung protein, mean±SEM, n=6, p<0.05). Moreover, Mmp2 inhibitor treatment restored regular EC morphology and vessel structures in the lungs of WTI mice (Fig. 5C). However, Mmp2 inhibition was not sufficient to significantly reduce colonization of previously irradiated lungs by circulating tumor cells (Fig. 5D,E; difference in mean counts [ME] of sham-irradiated mice [n=10] and WTI mice [n=15]=35.7 [95% CI=22.5 to 48.8], p<0.001 [comparison with 15 Gy] as determined by one-way ANOVA, followed by post hoc Bonferroni test; ME of WTI mice and Mmp2 inhibitor-treated WTI mice [n=6]: not significant]). These data indicated that MSC inhibition of vascular dysfunction involves downregulation of endothelial Mmp2 in irradiated lung tissue.

MSC therapy counteracts radiation-induced senescence of resident epithelial cells and recruited immune cells
As Mmp2 is not only linked with vascular dysfunction and tumor cell invasion but also a well-known factor associated with senescence, we next investigated whether WTI might induce cellular senescence (Fig. 6). Therefore, senescence-associated beta-galactosidase (SAbetagal) activity was assessed in frozen lung tissue sections at 21 days postirradiation in lung tissue from control and WTI animals with or without additional MSC therapy. Interestingly, increased SAbetagal activity was detected in bronchial–alveolar epithelial cells in lungs after WTI when compared with lung sections from control animals (Fig. 6A). In lungs of MSC-treated animals, SAbetagal activity showed a staining pattern comparable with lung sections from control animals. Apart from epithelial cells, single infiltrating cells were also SAbetagal positive, suggesting IR-induced immune senescence. Indeed, coimmunostainings with CD45 antibody identified these cells to be leukocytes (Fig. 6A, upper right panel).
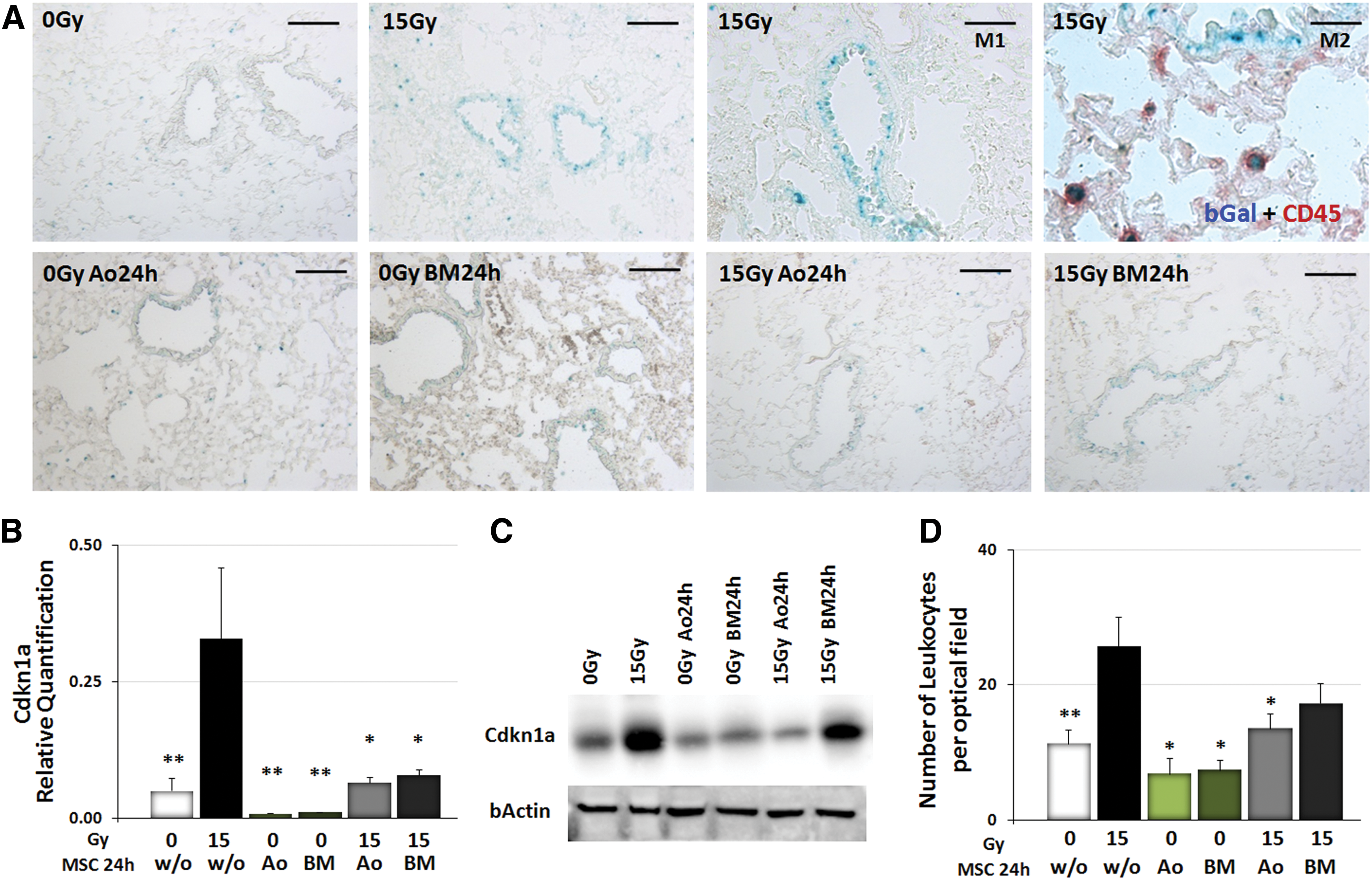
WTI-induced senescence induction was further confirmed on the mRNA level by real-time RT-PCR quantification of the cellular senescence mediator gene, cyclin-dependent kinase inhibitor 1 (Cdkn1a, p21), as well as on the protein level (Fig. 6B, C). Notably, Cdkn1a expression levels were significantly decreased in lungs of irradiated animals treated with MSCs.
To gain insight into the resulting effects of MSC therapy on immune cell recruitment, infiltrating CD45+ leukocytes were quantified by counting numbers of CD45-immunoreactive structures in frozen sections of lung tissue from control and irradiated animals with or without additional MSC therapy. The numbers of infiltrating CD45+ cells were increased in tissue sections of irradiated animals at 21 days after WTI and their numbers were significantly reduced upon MSC therapy (Fig. 6D; p-values as indicated: *p<0.05, as analyzed by two-way ANOVA, followed by post hoc Bonferroni test: F-factors 0.5583 [interaction), 14.32 [cf/Gy], 3.645 [rf/treatment]). These findings indicate that WTI induces senescence of bronchial–alveolar epithelial cells as well as leukocyte infiltration and that both effects are antagonized by MSC treatment.
MSC therapy counteracts certain aspects of the secretome of irradiated lungs and the recruitment of myeloid cells with reported tumor-promoting potential
Since SASP factors such as the chemokine (C-C motif) ligand 2 (Ccl2) and urokinase-type plasminogen activator (Plau/uPA) are known to be involved in both immune regulation and metastasis, we next examined whether WTI would increase the levels of these factors in the lung tissue. Quantification of Plau/uPA and Ccl2 mRNA expression levels showed increased Plau/uPA and Ccl2 mRNA expression levels in total lung RNA isolates of irradiated lungs when compared with lung tissue from sham-irradiated control mice (Fig. 7A). Of note, adoptive transfer of MSCs normalized the expression levels of Ccl2 and to a lesser extent of Plau/uPA in lungs of irradiated animals. A similar tendency of decreased expression levels of Ccl2 and Plau/uPA mRNA was also detected in MSC-treated animal sham controls compared with sham controls without MSC treatment (Fig. 7A; p-values as indicated: *p<0.05, ***p<0.001 as analyzed by two-way ANOVA, followed by post hoc Bonferroni test: F-factors 2.560 [interaction], 4.422 [cf/Gy and treatment], 1.990 [rf/gene]). IHC analysis of Ccl2 and Plau/uPA proteins further revealed that radiation-induced Ccl2 and Plau/uPA expression was confined to the bronchial–alveolar epithelial cells in irradiated lungs (Fig. 7B). Notably, these levels were reduced in lungs of animals treated with IR plus subsequent MSC therapy.
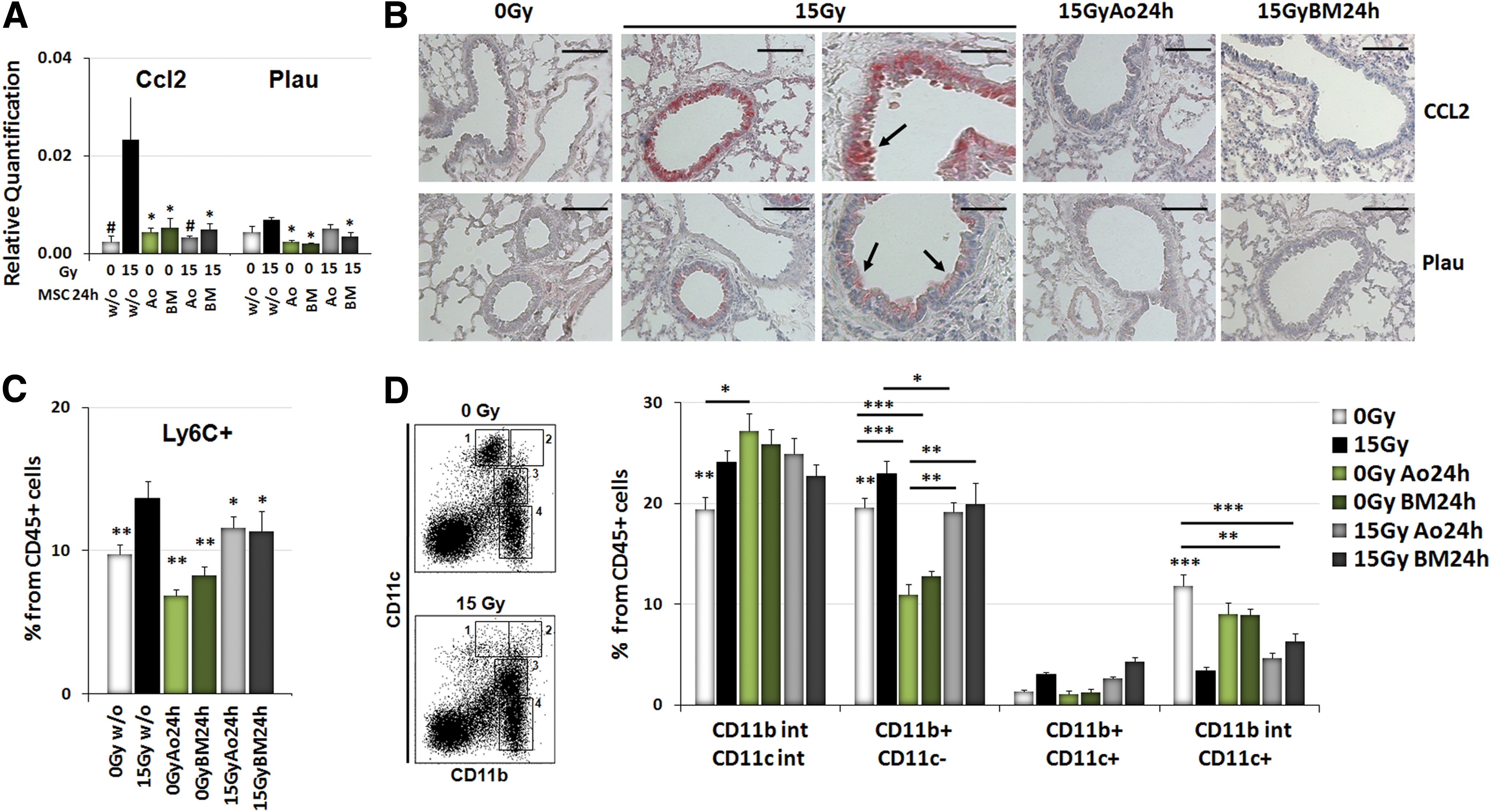
Since Ccl2 has been linked to immune modulation, we further investigated whether RT-induced alterations in the lung secretome may result in altered recruitment of myeloid cells. For this, we analyzed the composition of the myeloid cell compartment by FACS analysis of cell extracts generated from freshly isolated lung tissue. Interestingly, the percentage of CD11b+CD11c− myeloid cells (monocytes/granulocytes) from CD45+ leukocytes, particularly of Ly6C+ (myelomonocytic cells), was significantly increased after WTI, whereas the percentage of CD11bintCD11c+ cells (alveolar macrophages) was decreased when compared with sham controls (Fig. 7C, D; p-value as indicated: p≤0.05, **p≤0.01, ***p≤0.001 as analyzed by two-way ANOVA, followed by post hoc Bonferroni test (for Fig. 7C: F-factors 0.48 [interaction], 11.77 [cf/Gy], 1.268 [rf/treatment]; for Fig. 7D: F-factors 9.026 [interaction], 1.025 [cf/Gy], 327.5 [rf/marker]). Even more important, MSC therapy antagonized infiltration of Ly6C+ and CD11b+CD11c− myeloid cells with high efficiency (Fig. 7C, D). These data suggest that abrogation of certain aspects of the secretome of irradiated resident lung cells and the resulting immune deviation may participate in MSC-mediated protection from RT-induced lung injury and metastasis.
Discussion
In this study, we demonstrate for the first time that MSC therapy has the potential to protect the highly radiosensitive lung tissue from radiation-induced damage to vascular structures and the associated increase in lung metastasis in experimental murine models. Moreover, we identified enhanced senescence of lung epithelial cells, secretion of the SASP factors, Mmp2, Plau/uPA, and Ccl2, as well as increased recruitment of CD11b+CD11c− myelomonocytic cells as novel factors participating in the response of the normal lung tissue to IR. Importantly, we identified inhibition of Mmp2 as the mechanism underlying MSC-mediated normalization of vascular dysfunction. The observation that Mmp2 inhibition did not significantly reduce increased lung metastasis indicates that further cellular or paracrine effects of MSCs participate in their protective effects against lung metastasis. These may include protection of bronchial epithelial cells from radiation-induced senescence, suppression of tumor-promoting SASP factors, and/or modulation of radiation-induced lung inflammation.
Our data corroborate earlier reports about a delayed disturbance of endothelial barrier function in the lung tissue at 21 days postirradiation (9) and the resulting increase in transmigration of tumor cells through the endothelium (5, 7, 29). In our hands, the prometastatic effect of RT was observed for two distinct cell lines (B16F10 melanoma and TS/adenocarcinoma) injected into the tail vein of the two distinct respective syngeneic mouse strains (C57BL/6 and BALB/c). Furthermore, the enhancement of tumor cell seeding to previously irradiated lung tissue was observed with tumor cells injected into the tail vein as well as with subcutaneously growing tumors. These observations point to a more general cell line and mouse strain-independent phenomenon, presumably initiated by radiation-induced microenvironmental changes.
Although the reason for the formation of micrometastasis in specific organs is still unknown, this process requires adhesion of the tumor cells to the vessel wall, followed by the emigration of the tumor cells into the surrounding tissue (32, 52). We show here that radiation-induced tissue damage activates proinvasive and prometastatic signals, such as upregulation of endothelial Mmp2, as well as Mmp14, and Timp2, suspected to promote the invasion of circulating tumor cells to irradiated lung tissue. However, although administration of a selective Mmp2 inhibitor restored EC morphology and function in the lungs of irradiated mice, Mmp2 inhibition was not sufficient to significantly reduce seeding and growth of circulating tumor cells in irradiated lungs. Although we cannot exclude that the inhibition of tumor cell extravasation and subsequent metastatic growth might require higher Mmp2 inhibitor concentrations, these findings strongly suggest the contribution of additional mechanisms. Nevertheless, our data demonstrate a role of Mmp2 for radiation-induced vascular dysfunction in the lung and offer novel perspectives for the treatment of lung pathologies associated with radiation-induced EC damage such as pulmonary arterial hypertension (PAH). In this study, a functional impairment or even partial loss of the EC as a result of selective lung irradiation was detected long before the clinical manifestation of the disease (24).
Interestingly, our further investigations revealed that WTI triggers a pronounced senescence of bronchial epithelial cells. These findings corroborate observations made by Le and coworkers about the induction of senescence markers in murine brain, lung, and liver tissue exposed to a sublethal dose of IR in vivo (43). Cellular senescence is a tumor-suppressive mechanism that permanently arrests cells at risk for malignant transformation. However, accumulating evidence shows that senescent cells can have deleterious effects on the tissue microenvironment and may even fuel the development of secondary cancers (14, 48).
A potential link between cellular senescence and metastasis has recently been established by Shimizu et al. who found higher numbers of lung metastasis from intravenously injected K1735M2 melanoma cells in aged compared with young senescence-accelerated mouse prone 10 (SAMP10) mice (55). The authors concluded that aging-associated senescence facilitates metastasis due to the decrease in immune surveillance in the aging microenvironment. Vice versa, oncogene-induced senescence in proliferating breast cancer cells through expression of p95HER2, a 80 to 115 kDa carboxy-terminal and constitutively active fragment of the tyrosine kinase receptor HER2, increased their metastatic potential presumably through the senescence secretome of the cancer cells (2). Thus, the protumor effects of senescent cells are linked to the senescence-associated secretory pattern, or SASP, which seems to be shaped by the respective stimulus, for example, IR or oncogenic transformation (2, 14).
We show here that senescence of bronchial epithelial cells from irradiated mice is associated with increased secretion of further SASP factors such as Ccl2 and Plau/uPA. There is accumulating evidence that senescent cells secrete various proinflammatory cytokines and chemokines known to play a vital role in tumor progression and metastasis (42, 53, 59). As an example, Ccl2 has been associated with poor clinical outcomes in several cancers, including myeloma, breast cancer, and prostate cancer (17). Furthermore, Ccl2-induced recruitment of Ly6C(+) myeloid cells promoted cancer cell metastasis to the lung (64). Myelomonocytic cells were also shown to be essential for the establishment of a premetastatic niche and survival of metastatic cells in mice (25).
Vice versa, Plau/uPA was reported to contribute to cancer cell invasion and metastasis (47). Plau/uPA catalyzes the transformation of plasminogen to plasmin, which degrades extracellular matrix molecules and basement membranes directly or indirectly through activating promatrix metalloproteinases (proMmps), thereby facilitating cancer cell invasion and metastasis (65).
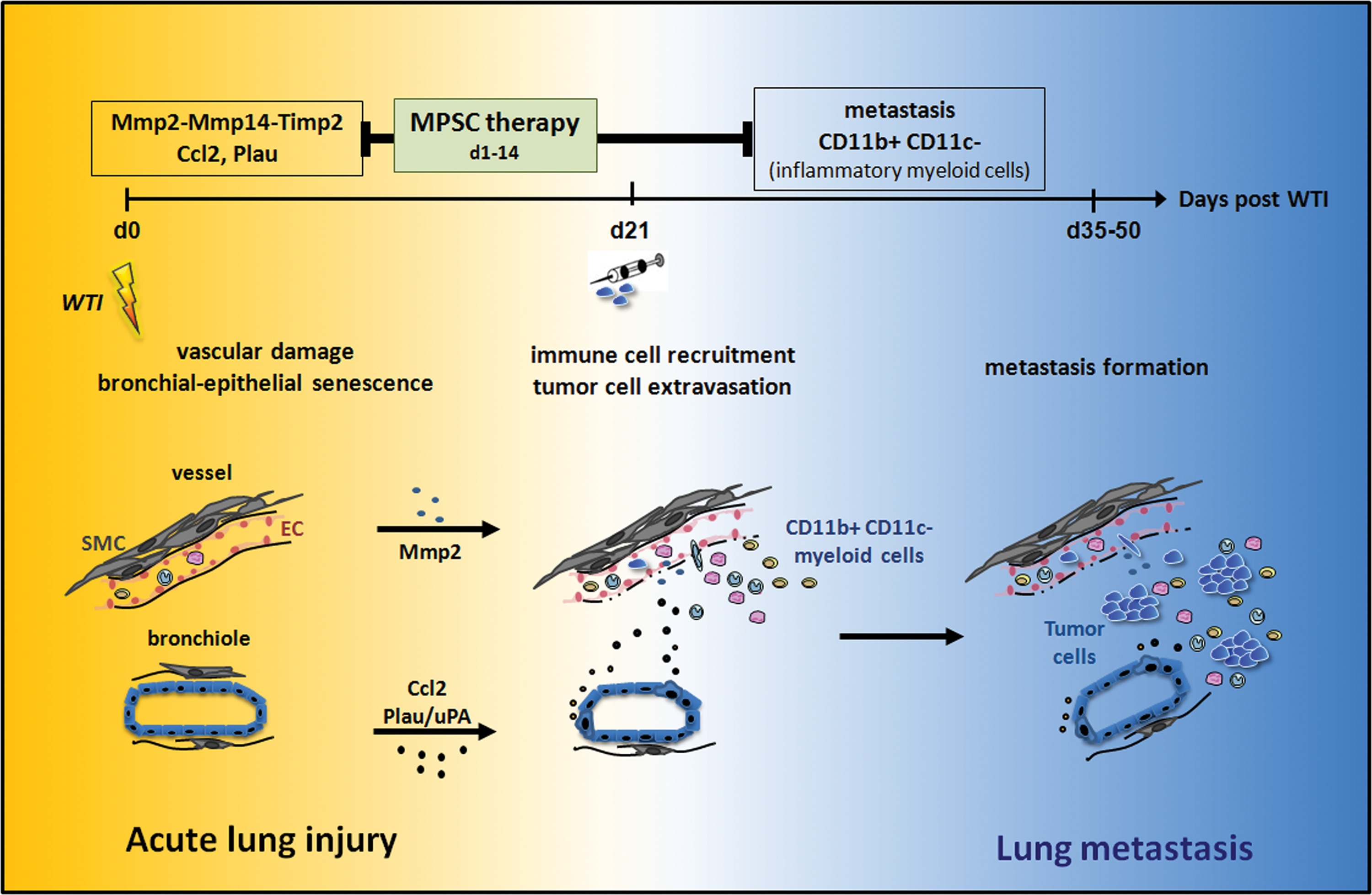
From the vast majority of cancer cells that enter the circulatory system, only very few successfully engraft, survive, and proliferate at secondary sites (35, 67). Therefore, it is tempting to speculate that radiation-induced lung injury, including senescence of lung epithelial cells, and the resulting increase in secreted factors, such as Mmp2, Ccl2, and Plau/uPA, participate in the generation of a receiving microenvironment (niche) that enables seeding, survival, and growth of metastatic tumor cells (Fig. 8). However, the functional relevance of specific secreted factors and the resulting immune changes for increased lung metastasis remain to be demonstrated.
Current research efforts are aimed at protecting adjacent tissues to minimize the risk of recurrence. It is assumed that adult MSCs may be a valuable therapeutic option for the prevention of lung diseases or the regeneration of diseased lung tissue because these cells are relatively easily available, have immunomodulatory effects, and have the capacity for cell differentiation (6, 62), although engraftment in the lung as structural epithelium or endothelium is not currently considered the mechanism by which BM-MSCs can repair lung tissue (63).
In this study, we investigated the therapeutic potential of MSCs derived classically from BM or from aorta to protect the lung tissue from radiation-induced injury and the resulting increase in lung metastasis from circulating tumor cells. We demonstrate that MSCs derived from BM and aorta both efficiently counteract RT-induced vascular damage. The high activity of the aorta-derived MSCs for EC protection might be due to the fact that tissue-specific stem cells mainly support the tissue type from which they originate (21).
Several studies have shown that MSCs provide an important contribution to tissue neovascularization by migrating to the site of damage and differentiating to restore damaged cell types (13, 68). However, the greatest potential of MSCs in terms of neovascularization is attributed to the trophic effects, for example, to prevent fibrosis and apoptosis, or to promote angiogenesis and arteriogenesis due to the production of cytokines for a paracrine action (33, 50, 73). A similar observation could be made in the animal model of PAH. In this study, the therapeutic use of BM-MSCs led to a reduced vascular remodeling and increased expression of VEGF (45).
We found here that treatment with an inhibition of Mmp2 mimics the beneficial effect of MSC therapy on vascular function in irradiated lungs. This implicates that upregulation of Mmp2 participates in the pathogenesis of radiation-induced vascular dysfunction. Moreover, previous studies have shown that BM-MSCs migrate to injured tissues, communicate with injured parenchyma cells, and function in wound healing through the production of paracrine-soluble cytokines/chemokines and growth factors, which modulate the regeneration of the epithelium and endothelium and modulate the activation, proliferation, and downstream effects of inflammatory and immune cells in both the innate and adaptive immune systems (12, 63, 74). Therefore, it is highly likely that the therapeutically applied MSCs exert their beneficial effects in the irradiated lungs by the secretion of multiple paracrine factors or the modulation of the secretory activity of damaged resident cells and/or recruited immune cells that have yet to be identified.
Even more important, we demonstrate here for the first time that therapeutically applied MSCs also prevent the seeding of circulating tumor cells. The inhibitory effect of MSCs on lung metastasis was associated with a reduction in RT-induced senescence of bronchial epithelial cells and expression of SASP factors such as Ccl2. As outlined above, Ccl2 and Plau/uPA may be linked to the recruitment of CD11b+CD11c− myeloid cells to the lung and these cells have reported tumor-promoting activity (Fig. 8). This assumption is supported by the finding that metastatic cancer cells can co-opt chemokine pathways to migrate to distant sites, in particular those chemokines that normally regulate the migration of immune cells (53). Moreover, it has been shown in rats that therapeutically applied MSCs can downregulate inflammatory mediators through paracrine effects and by reducing alveolar cell apoptosis and lung inflammation responses (72). However, the functional relevance of our observations for the mechanisms of MSC-mediated tissue protection needs to be addressed in future studies.
Our data reveal that MSCs have a high potential for the development of protective treatment strategies in irradiated patients. First, clinical applications of stem cell transplantation indicate a potential of these cells in bone regeneration as well as immunosuppressive effects after allogeneic stem cell transplantation. Isolated from the BM, MSCs are already used in the clinic in patients with dilated myopathy, cartilage disorders, stroke, and autoimmune diseases (4, 10, 57). In line with these findings, we show here that intravenously applied MSCs lingered in the circulation and only single cells homed to irradiated lung tissue when cells were applied in the early phase of irradiation. Fortunately, no adverse effects on progression of lung pathology were found (not shown).
In summary, the high radiosensitivity of lung resident cells plays a crucial role for radiation-induced lung disease and the metastatic colonization of previously irradiated lungs by intravenously injected tumor cells or subcutaneously growing tumors. Adoptive transfer of MSCs counteracts radiation-induced vascular damage, bronchial epithelial senescence, and metastasis of circulating tumor cells to the irradiated lungs. We speculate that downregulation of radiation-induced expression of endothelial Mmp2 and of the SASP factors Ccl2 and Plau/uPA, as well as a reduced infiltration of metastasis-promoting myeloid cells, is involved in the protective action of MSCs. However, the role of specific SASP factors for the niche-promoting effects of RT in the suspected network between damaged cells, immune cells, and invading tumors cells remains to be demonstrated. These investigations will be performed in further studies using mouse models deficient for single SASP factors or the respective specific inhibitors. Our results are of direct clinical relevance since they contribute to an improved understanding of the mechanisms of the dose-limiting side effects of RT. This is a necessary step in the development of protective treatment strategies.
Materials and Methods
Tumor cells and mouse models
B16F10 (ATCC® CRL-6475; ATCC, Manassas, VA) mouse melanoma and TS/A murine mammary adenocarcinoma cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/10% fetal calf serum (FCS; 5% CO2 at 37°C). The TS/A cell line is a moderately differentiated and immunogenic mammary adenocarcinoma of spontaneous BALB/c origin (28). Wild-type C57BL/6 and BALB/c mice received 15 Gray of WTI in a single dose of a Cobalt 60 source (60Co γ-rays at 0.5 Gy/min) as previously described (69). Single cell suspensions of cultured MSCs (0.5×10(6) cells) were intravenously transplanted into the tail vein of WTI mice 24 h or 14 days after irradiation or in sham-irradiated (0 Gy) control animals. Seeding of circulating tumor cells into the lungs was initiated 21 days after irradiation. Preliminary tests for cell numbers were performed to determine the optimal tumor cell number for intravenous injection and resulting quantifiable metastasis. Consequently, 1×10(6) B16F10 cells or TS/A cells were intravenously transplanted via the tail vein in the subsequent experiments. Fourteen days after tumor cell injection, animals were sacrificed and lungs were isolated and subjected for IHC, RNA, or protein isolation.
Additionally, we examined seeding of lung metastases in mice exposed to TBI and subsequent implantation of subcutaneous tumors (40). In brief, C57BL/6 mice were lethally irradiated with a split dose (7+3 Gy) of an X-ray source (3 Gy/min) and were intravenously transplanted with 2×106 unfractioned murine EGFP-expressing BM cells from C57BL/6-Tg(CAG-EGFP)1Osb/J transgenic donor mice (Jackson Laboratory, Bar Habor, ME). BM cells were harvested aseptically by flushing the tibias and femurs of adult animals and subjected to erythrocyte lysis. After BM reconstitution (4–6 weeks postirradiation), B16F10 cells were subcutaneously implanted into the flanks of the mice. Two or 28 days later, animals were sacrificed and lungs were isolated and lung histology was performed. All procedures involving mice were approved by the local institutional Animal Care Committee (Regierungspräsidium Düsseldorf Az84-02.04.2012.A137; 84-02.04.2012.A034). The Mmp2 inhibitor ARP100 (sc203522, CAS 704888-90-4) was from Santa Cruz (Santa Cruz, CA). Stock solutions of ARP100 were prepared by dissolving 5 mg ARP100 in 200 μl sterile dimethyl sulfoxide (DMSO). The stock solution was further diluted with sterile phosphate-buffered saline (PBS) for in vivo application. Intraperitoneal injections of ARP100 were given twice weekly at a final concentration of 15 μg/g bodyweight (e.g., 15 μl ARP100 stock solution+85 μl PBS per injection per mouse with a bodyweight of 25 g).
Isolation and purification of aortic MSCs and BM-MSCs
Vascular wall-resident MSCs were isolated from aortas of C57BL/6-Tg(CAG-EGFP)1Osb/J mice (Jackson Laboratory) as previously described (40). In brief, tissue pieces were mechanically minced and dissociated for 15 min at 37°C in OptiMEM I medium containing 0.2% type 2 collagenase (CLS2, 43J14367B, ≥125 U/mg dry weight; Worthington, Lakewood, WA). Cells were washed twice in PBS/5% FCS. Pure MSCs were generated using the Sca-1 antibody (130-092-529) and MACS technology (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Primary MSCs were cultivated on plastic plates in DMEM/20% FCS. The medium was removed 24 h after initial plating and nonadherent cells were washed away. Primary cultures were clonally expanded under limiting dilution conditions. BM cells were harvested and cultured using complete DMEM/20% FCS as previously described (40).
Vascular leakage
Twenty-one days after irradiation, vascular leakage was determined by EBD (E2129; Sigma-Aldrich, St. Louis, MO) or Alexa555-labeled BSA (A34786; Life Technologies, Carlsbad, CA) extravasation from the blood stream into the lung interstitium. Therefore, 100 μg EBD or 50 μg Alexa555-BSA/100 μl PBS was intravenously injected into the tail vein. Two to 4 h after injection, animals received deep anesthesia and were sacrificed by transcardial perfusion with PBS to remove the blood from the vascular system. Lungs were isolated and subjected for protein isolation and EBD extraction. Lung pieces were weighed and EBD exuded in the lung interstitium was extracted by incubating the tissue in 200 μl formamide for 24 h at 65°C, and dye concentrations were measured by absorption at 620 nm and related to the weight of lung tissue (μg of EBD per mg wet weight of the lung). Alexa555-BSA concentrations were determined by measuring fluorescence at 555 nm.
Western blot
Whole cell lysates were generated by scraping cells into ice-cold RIPA-P buffer (150 mM NaCl, 1% NP40, 0.5% sodium desoxycholate, 0.1% sodium dodecylsulfate, 50 mM Tris/HCL pH8, 10 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4) supplemented with complete protease inhibitor cocktail (04693159001, Hoffmann-La Roche, Basel, Switzerland) and performing two to three freeze–thaw cycles. Protein samples (50–100 μg total protein) were subjected to SDS-PAGE electrophoresis and Western blots were done as previously described using indicated antibodies (39). Mmp2 (H76, sc10736), p21 (F8, sc271610), and VE-Cad (C19, sc6458) antibodies were from Santa Cruz, and pTyr (P-Tyr-100, 9411S) antibody was from New England Biolabs (Ipswich, MA).
Real-time RT-PCR
RNA was isolated using the RNeasy Mini Kit (74106; Qiagen, Hilden, Germany) according to the manufacturer's instructions and as previously described (38, 39). Expression levels were normalized to the reference gene (beta-actin; set as 1) and are shown as relative quantification. Specific primers were synthesized based on available sequences for each listened gene. Primer design was done with the program Primer 3 (
IHC and electron microscopy
Paraffin-embedded tissue sections were hydrated using a descending alcohol series, incubated for 10–20 min in target retrieval solution (DAKO, Glostrup, Denmark), and incubated with blocking solution (2% FCS/PBS). After permeabilization, sections were incubated overnight at 4°C with primary antibodies (Mmp2 [H76, sc10736], Plau/uPA [uPa; H-140, sc14019] both from Santa Cruz and Ccl2 [AA102-130, ABIN1108186] from antibodies-online [Atlanta, GA]). Antigen was detected with an alkaline phosphatase-conjugated secondary antibody (1/250) and alkaline phosphatase (ALP) staining (DAKO). Nuclei were counterstained using hematoxylin. For fluorescence analysis (VE-Cad [C19, sc6458 from Santa Cruz], CD31 [DIA-310; Dianova, Hamburg, Germany]), the antigens were detected with anti-rat Alexa488 and anti-mouse Alexa555-conjugated secondary antibodies (1/500) as previously described (41). Hoechst 33342 (H1399; Life Technologies) was used for staining of nuclei. Electron microscopy was done as previously described (41).
SAbetagal activity
SAbetagal activity was detected as previously described using frozen sections of lung tissue at pH 6.0 (19). In brief, for the X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside, B4252; Sigma-Aldrich) staining procedure, all working solutions were freshly prepared. Slides were fixed in 4% PFA for 30 min at 4°C, rinsed with wash buffer for 4×5 min, and developed by incubation of slides with X-gal staining solution (containing 0.1% X-gal, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM sodium chloride, and 2 mM magnesium chloride in 40 mM citric acid/sodium phosphate solution, pH 6.0) for more than 12 h in a 37°C incubator. The staining container was impermeable to normal light. After overnight staining, slides were washed with PBS 3×5 min. All chemicals were from Sigma-Aldrich if not otherwise indicated.
Lung histopathology
For lung histology, mice were narcotized using isoflurane (2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane) and killed by transcardial perfusion with PBS. Whole lungs were taken out and lung tissue was fixed in 4% formalin and subsequently embedded in paraffin. Three to four 5-μm paraffin longitudinal cross sections were taken per mouse lung at the midpoint through the lung block depth. Sections were stained with hematoxylin and eosin for histological evaluation. Samples were then analyzed microscopically with a 20×objective. Metastatic lesions/foci were quantified by counting numbers of nodules in at least three whole cross sections per lung and averages for individual animals were calculated. Depicted data represent the mean values of all mice per group (mean of single average number for each mouse/mouse number) as indicated.
Phenotyping of lung leukocytes by flow cytometry
Crude cell extracts of freshly isolated lungs were generated and FACS analysis was performed as previously described (69). Lung cell suspensions were stained with anti-mouse CD45 (30-F11; Cat. 103126) for determination of leukocytes in the lung tissue. Lung cells were further fluorochrome labeled with anti-mouse Ly6C (HK1.4; Cat. 128006), CD11b (M1/70; Cat. 101228), and CD11c (N418; Cat. 117309). All antibodies used in this study were obtained from BioLegend (San Diego, CA), respectively. Flow cytometric measurements were performed on a BD LSRII flow cytometer using FACS DIVA software. Analysis of data sets was done using BD FACS DIVA software (all from BD Bioscience, Franklin Lakes, NJ).
Statistical analysis
If not otherwise indicated, data were obtained from three independent experiments with at least three mice each. Mean values were calculated and used for analysis of SEM as indicated by error bars. Statistical significance was evaluated by one-way or two-way ANOVA, followed by Bonferroni's multiple comparison post-test. Statistical significance was set at the level of p≤0.05. Data analysis was performed with Prism 5.0 software (GraphPad, La Jolla, CA).
Footnotes
Acknowledgments
The authors thank L. Lüdemann and M. Groneberg for their support with the irradiation of mice and Mohamed Benchellal, Inge Spratte, and Eva Gau for their excellent technical assistance. This work was supported by grants of the DFG (GRK1739/1; JE275/1), the BMBF (ZISS 02NUK024-D), and the UK Essen/IFORES (D/107-81040).
Author Contributions
D.K., A.S., R.I., F.W., and K.U. performed experiments; H.J. performed electron microscopy; D.K. supervised and analyzed results and made the figures; K.U. and M.S. provided materials; and D.K. and V.J. designed the research and wrote the article. All authors read and approved the manuscript.
Author Disclosure Statement
No competing financial interests exist.