
Abstract
Introduction
D
The role of AhR in the pathophysiological processes of diabetic nephropathy (DN) remains unclarified. The present study elucidates for the first time that AhR plays an important role in MC activation, macrophage infiltration, and ECM accumulation in DN conferred by oxidative stress using AhR gene knockout (AhRKO) and pharmacological inhibitor a-naphthoflavone mouse models.
Aryl hydrocarbon receptor (AhR) is a ligand-activated basic helix-loop-helix transcription factor, also referred to as the specific binding site for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and is known to regulate xenobiotic-metabolizing enzymes such as cytochrome P450 (3, 28). Recent epidemiologic studies suggest a possible association between endocrine-disrupting chemicals such as polychlorinated biphenyls, dioxins, and diabetes in human populations (2, 31). Wu et al. have recently reported that treatment of macrophages with TCDD leads to AhR-dependent activation of inflammatory mediators, the formation of cholesterol-laden foam cells, and atherosclerotic lesions, suggesting that activation of AhR cascade contributes to the development of atherosclerosis through the induction of a vascular inflammatory response (43). Kerley-Hamilton et al. have also found that toxicant-activated AhR may disrupt fat metabolism and contribute to Western diet-induced obesity in mice (17).
Paradoxically, the study of Baglole et al. has demonstrated that lung fibroblasts from AhR−/− mice produce and strengthen the inflammatory effect in response to cigarette smoke, typified by raised levels of cyclooxygenase-2 (COX-2) and prostaglandins (PGs), when compared with AhR wild-type fibroblasts, suggesting that AhR may protect obstructive pulmonary disease (4). An AhR−/− animal study by Alexander et al. has shown that AhR is a constitutive inhibitor of triglyceride synthesis and an early regulator of adipocyte differentiation (1). A recent report has shown elevated numbers of fibroblasts and increased collagen contents during wound healing in AhR−/− animals (8). These findings imply that AhR may participate in either cellular perturbation or protection, depending on the pathophysiological conditions.
Sequestered leukocytes exaggerate cell damage by further generating more reactive oxygen species (ROS) and eicosanoids, aggrandizing inflammation and vascular tone (7). Oxidative stress is also known to be involved in the renal injury. Wang et al. have shown that the AhR deficiency enhances insulin sensitivity and reduces PPAR-α activity in mice (42). Moreover, Thackaberry et al. have also found that AhR is required for normal insulin regulation in pregnant and older mice and for cardiac development in embryonic mice (41). However, the role of AhR in MC activation, macrophage infiltration, and extracellular matrix (ECM) deposition in DN remains to be clarified.
In the present study, we hypothesized that AhR deficiency may suppress inflammatory responses and further prevent the oxidative stress cascade-induced microvascular complication during DN. We investigated the role of AhR in DN using both in vivo and in vitro models. We demonstrate that AhR deficiency attenuates oxidative stress, MC activation, macrophage infiltration, and matrix accumulation during DN in vivo and in vitro. These findings suggest that the AhR may be a useful target in treating diabetes-related vascular events.
Results
AhR deficiency improves glucose intolerance, renal fibrosis, and macrophage infiltration in streptozotocin-induced diabetic mice
First, we performed genotyping for AhR from wild-type and AhR knockout (AhRKO) (Fig. 1A) mice. We recorded their body weight (Fig. 1B) and monitored the blood sugar (Fig. 1C) per week and found a significant mitigating effect in AhRKO mice by induced streptozotocin (STZ). Similarly, fasting glucose levels were monitored in wild-type and AhRKO mice. STZ-induced AhRKO mice have improved glucose tolerance as measured by an intraperitoneal (i.p.) glucose tolerance test (Fig. 1D, E). Simultaneously, an insulin tolerance test was performed, which indicated that AhR deficiency enhanced insulin sensitivity in a mouse model of type 1 diabetes (data not shown). We also detected serum albumin and that a low albumin level might reflect a kidney disease condition. We found that serum albumin levels were 4.25 ± 0.65, 2.27 ± 0.19, and 3.72 ± 0.64 g/dl (n = 6) in the control group, STZ group, and AhRKO+STZ group, respectively. There was a reduction in serum albumin of STZ-induced diabetic mice (p < 0.05 vs. control group). Furthermore, renal function parameters were assessed. As shown in Table 1, there was remarkably increased chow and water intake, urine output, urine albumin, daily urine albumin, daily urine total protein, and albumin-to-creatinine ratio (ACR) in STZ-induced diabetic mice. Importantly, the increased urine albumin, daily urine total protein, and ACR were significantly decreased in AhRKO+STZ mice.

Functional renal parameter was measured at 8 weeks after induction with different groups. All values are expressed as mean ± standard error of the mean of experimental number (N = 5–13), as indicated in separate experiments. Statistical analysis was performed using the SAS system (SAS Institute, Inc.). Multiple comparisons were performed where one-way ANOVA was significant using Fisher's least significant difference test or Tukey's honestly significant difference test at an alpha level equal to 0.05.
Tests are better categorized as a, b, c, d. The same letters indicate non-statistical difference.
ACR, albumin-to-creatinine ratio; AhRKO, aryl hydrocarbon receptor knockout; STZ, streptozotocin.
Moreover, we evaluated the pathological morphology in the kidneys. The STZ-induced diabetic mouse model for DN that encompassed the salient features of fibrosis was stained with Masson's trichrome staining, periodic acid-Schiff (PAS) staining, and fibronectin accumulation, which were efficiently suppressed in the AhRKO mice (Fig. 2 and Supplementary Fig. S1; Supplementary Data are available online at
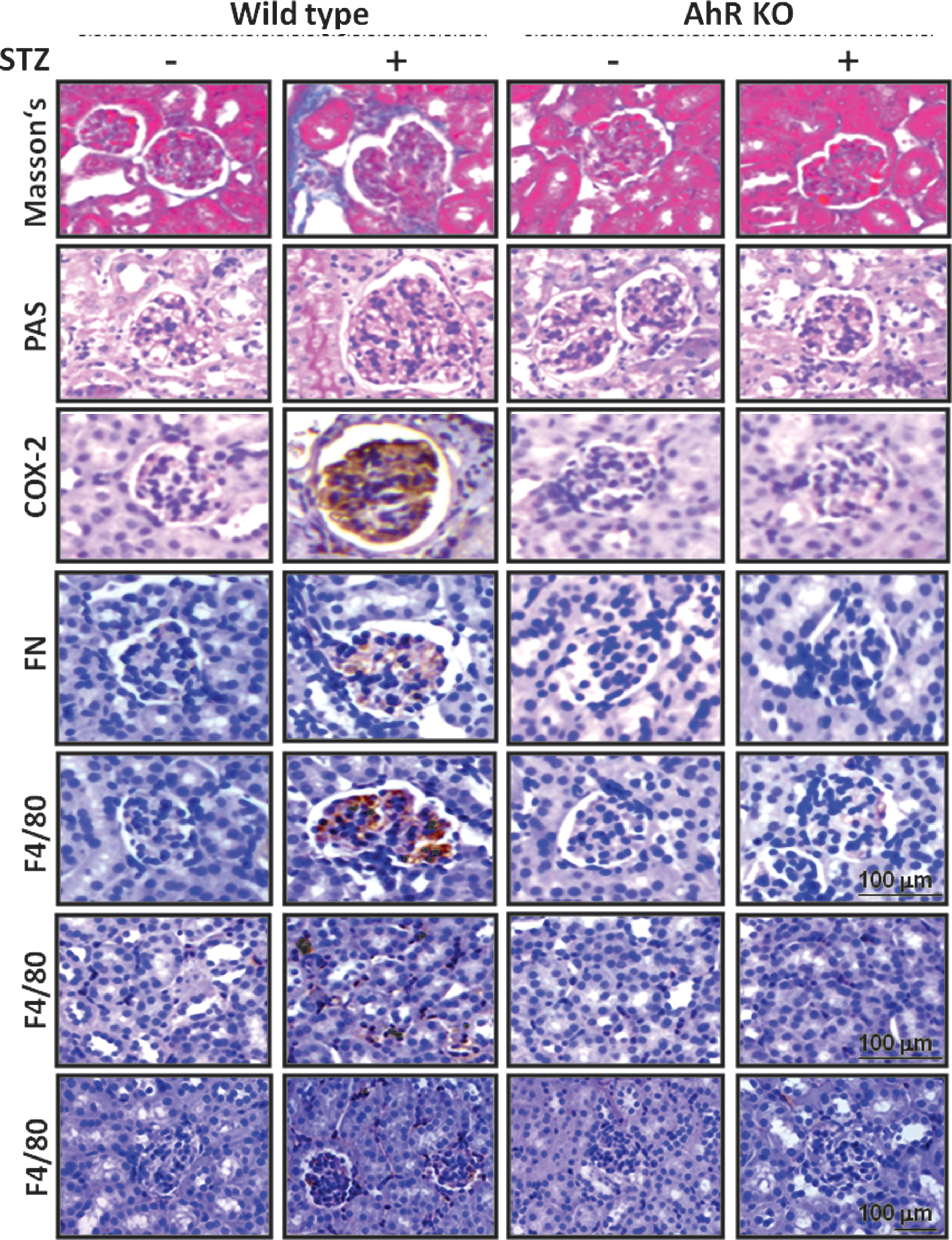
Pharmacological AhR inhibitor, α-naphthoflavone, suppresses renal injury and macrophage infiltration in the STZ diabetic mice
As shown in Figure 4, treatment of α-naphthoflavone (α-NF) significantly restored the collagen deposition (Masson's trichrome staining), glycogen deposition (PAS staining), and fibronectin accumulation in the kidneys of STZ diabetic mice. Treatment of α-NF could also alleviate the pathologic alterations in the kidneys, as revealed by histologic examination, from STZ-induced diabetic mice (data not shown).

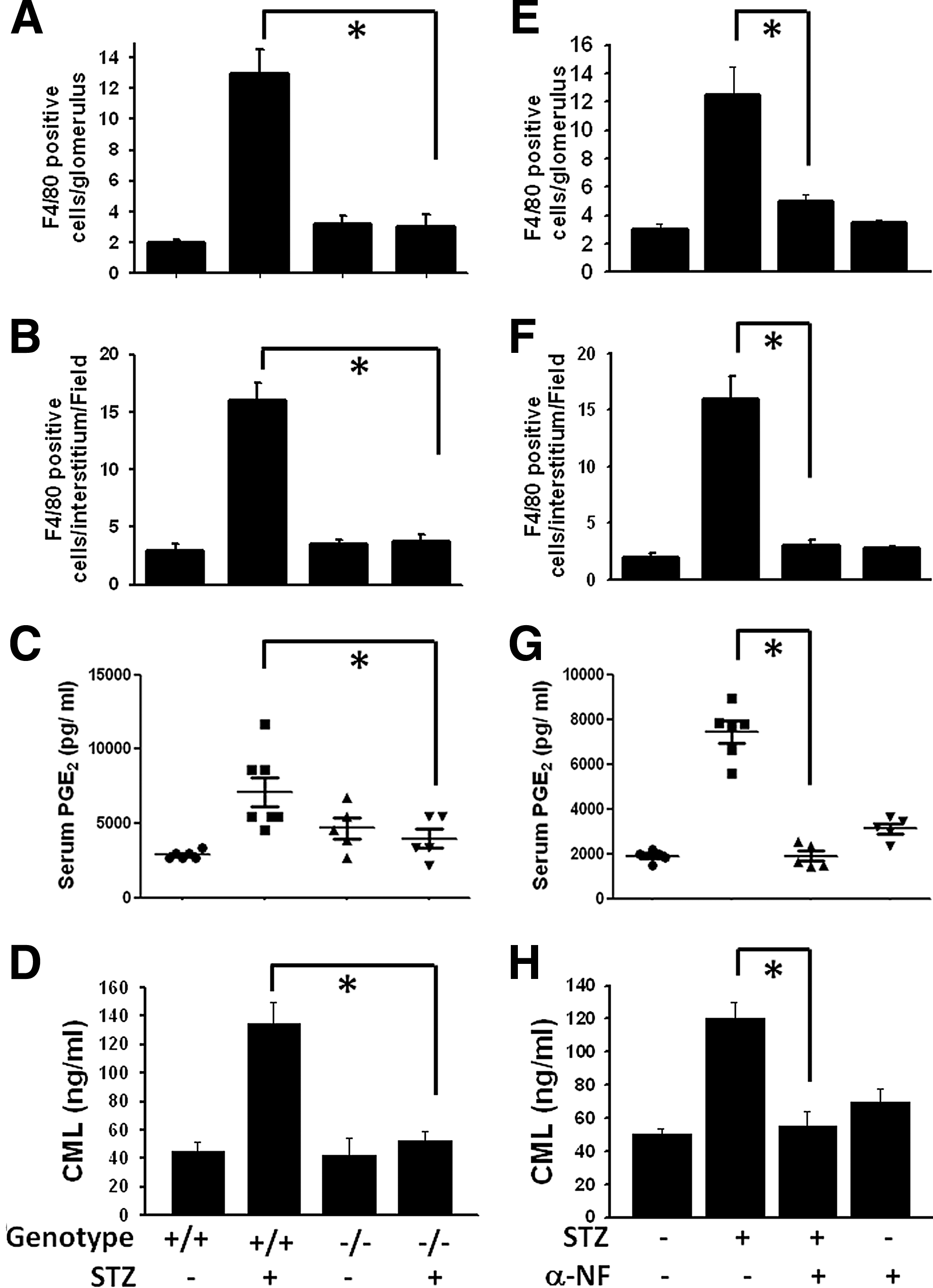
A criterion in glomerulosclerosis for macrophage infiltration was also assessed. The number of F4/80-positive cells (macrophages) in the areas of the glomerulus and interstitium was obviously higher in the STZ diabetic group than in the control group. Overtly, macrophage infiltration was attenuated in α-NF-treated STZ mice (Figs. 3E, F and 4). Based on the quantification analysis, α-NF treatment suppressed diabetic pathologic alterations of renal function and structure (Supplementary Fig. S1). α-NF could also efficiently abolish the COX-2 expression in the kidneys (Fig. 4) and serum PGE2 levels (Fig. 3G) of STZ diabetic mice. Apparently, the CML evaluations in the circulatory system via α-NF therapy for diabetic mice also significantly reduced (Fig. 3H).
AhR deficiency diminishes oxidative stress, renal cell injury, and NADPH oxidase activity in STZ diabetic mice
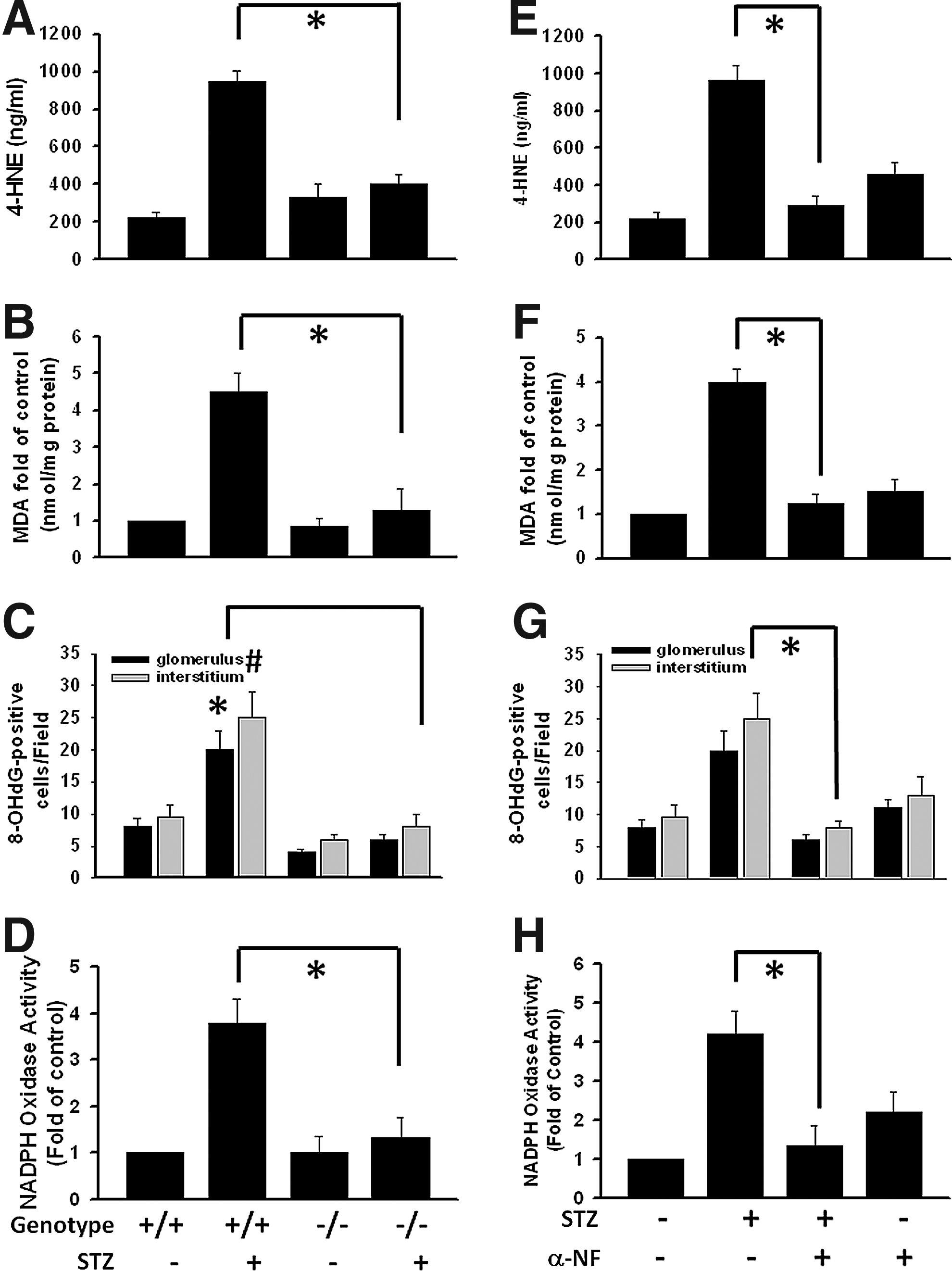
The pathological process of DN may occur, in part, as a result of intrarenal oxidative stress. Lipid peroxides are unstable indicators of oxidative stress in cells that decompose to form more complex and reactive compounds such as 4-hydroxy-2-nonenal (4-HNE), which has been shown to be capable of binding to proteins and forming stable adducts with possible signaling functions. Lipid peroxidation products, 4-HNE and malondialdehyde (MDA) contents, were significantly decreased in the kidneys of AhRKO mice with STZ-induced diabetes (Fig. 5A, B) as well as α-NF-treated STZ diabetic mice (Fig. 5E, F). Moreover, oxidative DNA damage marker was measured using immunochemical analysis with an anti-8-OHdG antibody. Nuclear staining was detected in glomeruli and interstitium from STZ diabetic mice, which could be mitigated by AhRKO and α-NF treatment (Fig. 5C, G). STZ-induced diabetes increased renal NADPH oxidase activity, but not in the kidneys of AhRKO or α-NF-treated mice (Fig. 5D, H). Moreover, we also detected the expression of NOX2 and NOX4 in the renal tissue sections of wild-type and AhRKO mice with or without STZ treatment. As shown in Supplementary Figure S2A and B, the expression of NOX4, but not NOX2, was markedly increased in wild-type mice with STZ treatment and significantly decreased in AhRKO mice with STZ treatment. These results indicate that blockade of AhR activation reduces oxidative stress signaling and renal cell injury under diabetic condition.
AhR knockdown or α-NF treatment alleviates CML-induced COX-2/PGES-1 and ECM deposition
To clarify the relationship between oxidative stress and AhR in DN, we used CML to further elaborate the complex mechanisms in vitro and in hyperglycemic mouse models. The increase of CML, AhR, and COX-2 protein expression and fibronectin deposition in the glomeruli were shown in STZ-induced type 1 diabetic mice, high-fat diet (HFD)-induced obese mice, and db/db type 2 diabetic mice (Fig. 6A, B). Moreover, abundant macrophage infiltration assessed by positive F4/80 staining was observed in the renal interstitium and mesangium (Figs. 2 and 6A, C). Activated MC marker alpha-smooth muscle actin exhibited high fluorescence intensity in the glomeruli of diabetic mice (Fig. 6A).

Furthermore, primary rat mesangial cells (RMCs) cultured in a medium containing high glucose (33 mM) and CML (25 μg/ml) for 24 h had higher levels of AhR and COX-2 protein expression compared with the control group (Fig. 6B). The protein expression of AhR and COX-2 was also markedly higher in renal cortical tissue from STZ-induced diabetic mice and diabetic db/db mice compared with control mice. Exposure of RMCs and HK2, a human proximal tubule epithelial cell line, to CML for 24 h significantly increased production of PGE2 (Fig. 6D).
CML increases the expression of AhR and COX-2/PGE2, but not AhR nuclear translocator
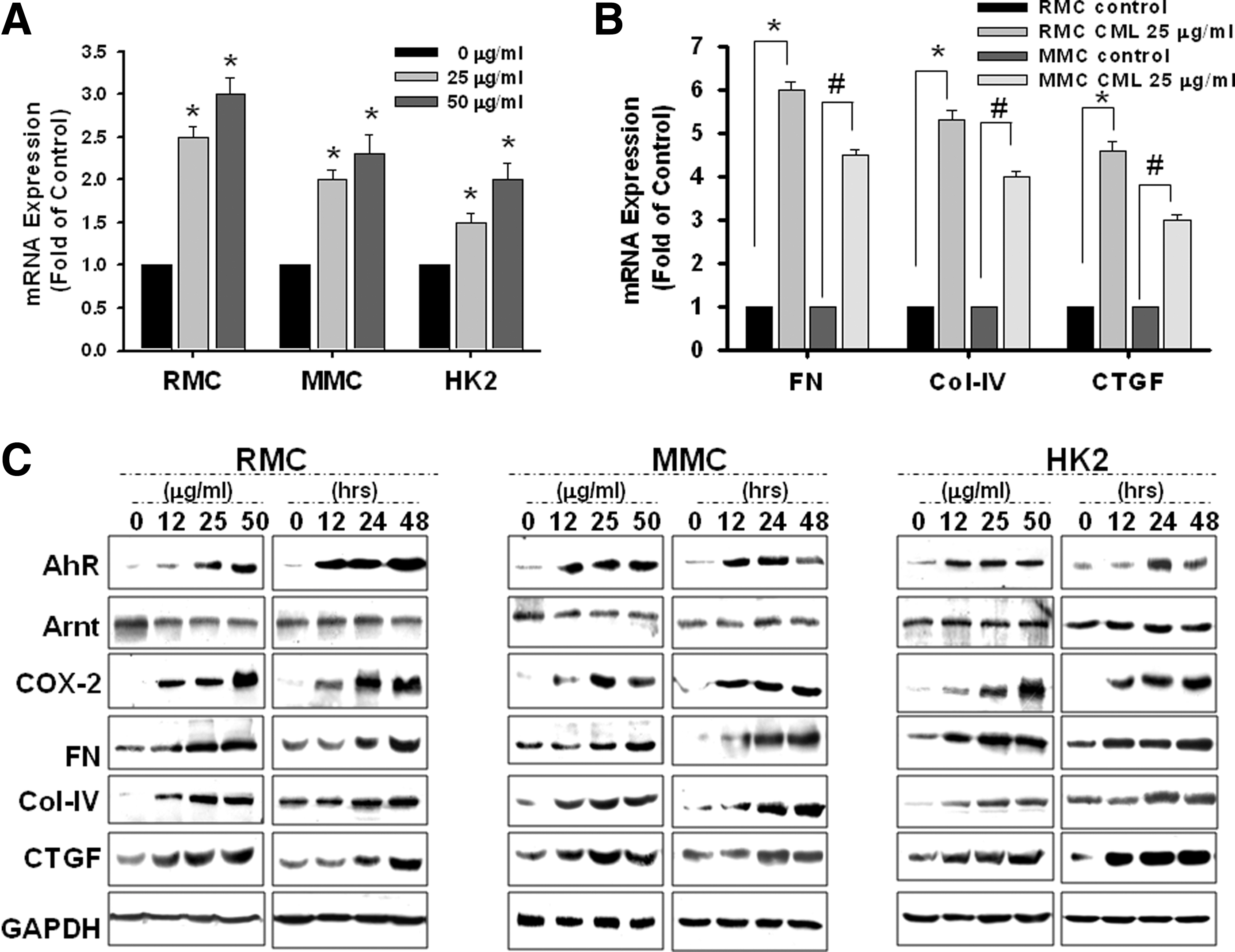
CML increased AhR mRNA expression in MCs and HK2 cells in a dose-dependent manner (Fig. 7A), which paralleled the expression of ECM accumulation markers, such as fibronectin, collagen-IV, and connective tissue growth factor (CTGF) (Fig. 7B). To determine whether this increase in mRNA level was associated with an increase in AhR protein expression and fibrosis signals as indicated, Western blotting with specific antibodies was performed. As shown in Figure 7C, CML increased the protein expression of AhR, COX-2, and ECM markers in MCs and HK2 cells in a time- and dose-dependent manner. Noticeably, the protein expression of AhR nuclear translocator (Arnt) did not have any change. Based on these findings, CML could specifically increase the expression of AhR and COX-2.
Blocked AhR activity abolished COX-2/mPGES-1 expression in MCs or PTC
According to laboratory in silico prediction, the promoter regulation in the COX-2 promoter-flanking region (−158 to −171) containing the cis-acting elements for AhR DNA-binding activity was predictable. Exposure of MCs (RMC and murine glomerular mesangial cell [MMC]) and HK2 cells to CML significantly increased the expression of AhR, COX-2, mPGES-1, fibronectin, and CTGF, which could be abolished by α-NF (Fig. 8A, B) and transfection of shRNA-AhR (shAhR) (Fig. 8A). Scrambled RNA per se did not have any effect. Moreover, protein expression by nuclear extraction (Fig. 8B) or fluorescence labeling (Fig. 8D) revealed that α-NF or 3′,4′dimethoxy-aNF (DiMNF) inhibited the AhR translocation into the nucleus by CML stimulation. Simultaneously, pretreatment with antioxidants, apocynin (Apo), N-acetyl-cysteine (NAC), or vitamin C, markedly annihilated CML-induced AhR nuclear translocation (Fig. 8C, D). CML also triggered the increase of DCF-sensitive ROS production in MMCs in a time- and dose-dependent manner, which could be reversed by apocynin and vitamin C (Fig. 8E).

To clarify whether AhR directly regulated COX-2 expression, the DNA-binding activity in COX-2 promoter was examined. The results of electrophoretic mobility shift assay (EMSA) showed that CML activated the AhR-binding activity on the COX-2 promoter binding site (Fig. 9A). Furthermore, transfection with shAhR or treatment with antioxidant NAC thwarted CML-induced AhR binding to the promoter of COX-2 (Fig. 9A). We further investigated the functional relationship between AhR and COX-2; the chromatin immunoprecipitation (ChIP) assay was carried out in the region −201 to −222. As shown in Figure 9B, shAhR transfection or α-NF could thwart AhR binding to the promoter of COX-2 and repress transcription expression. CML treatment significantly increased the PGE2 production in the cell supernatants after 24 h, which could be efficiently blocked by α-NF treatment or shAhR transfection (Fig. 9C). These results indicate that CML activates MCs or proximal tubular cells via an AhR-COX-2-PGE2 constraint axis mechanism. Moreover, the AhR promoter-flanking region contained the transcriptional and DNA-binding activity on mesangial matrix molecules (fibronectin, collagen-IV, and CTGF). Gene silencing AhR or antioxidant vitamin C treatment dampened the DNA-regulated activation phenomena (Fig. 9D). This finding suggests that AhR is unique in its ability to induce renal fibrosis via a gene regulation axis mechanism.

Discussion
Although AhR has a relevant role in the pathogenesis of diabetes, there is paucity of information on its role in improving oxidative stress-derived renal dysfunction, fibrosis, and macrophage infiltration. A previous report has shown a pivotal role for NADPH oxidase NOX4-derived ROS in the development and progression of DN (13). Consideration of pharmacological intervention and the nonspecific inhibitors of ROS formation, such as NAC, have also been shown to activate nuclear factor erythroid-related factor 2 (Nrf2) and protect from STZ-induced DN (14). Interestingly, glucuronidation by UDP-glucuronosyltransferase, which mediates antioxidant and cytoprotective effects, has been found to be regulated by the AhR and Nrf2 by cis-acting antioxidant and xenobiotic response elements (16). Therefore, we postulated that genetic deficiency or pharmacology therapy study for AhR is potentially more relevant with respect to clinical target validation, although their role in diabetic kidney injury has not been previously determined. In the present study, a pathological role of AhR-meditated DN is clearly demonstrated through several cellular and animal models. This work provides evidence that oxidative stress and AhR are causatively linked to the development and progression of fibrosis marker. The evidence is based on the direct comparison of AhR deletion in STZ-induced diabetic mice. The STZ-induced diabetic mouse model is a type 1 diabetic animal model, which is a well-characterized model of advanced renal injury with prominent ECM accumulation. The increased blood glucose, glucose intolerance, MC activation, macrophage infiltration, and ECM accumulation in diabetic mice were effectively improved by genetic AhRKO or pharmacological AhR inhibitor treatment. The current study also demonstrated the AhR induction in the rodent DN experimental models, including STZ, HFD feeding, and genetic db/db mice, which correlate with increase in CML expression. Moreover, the present study also found that CML-induced AhR induction regulates the COX-2/PGE2 axis, resulting in ECM accumulation in the activation of MCs and proximal tubular cells. The signaling cascade for hyperglycemia/CML-induced renal cell dysregulation and the role of AhR are shown in Figure 10. These findings suggest a pivotal role for AhR in the pathogenesis of DN and provide evidence for AhR inhibition as a potential therapeutic modality in DN.
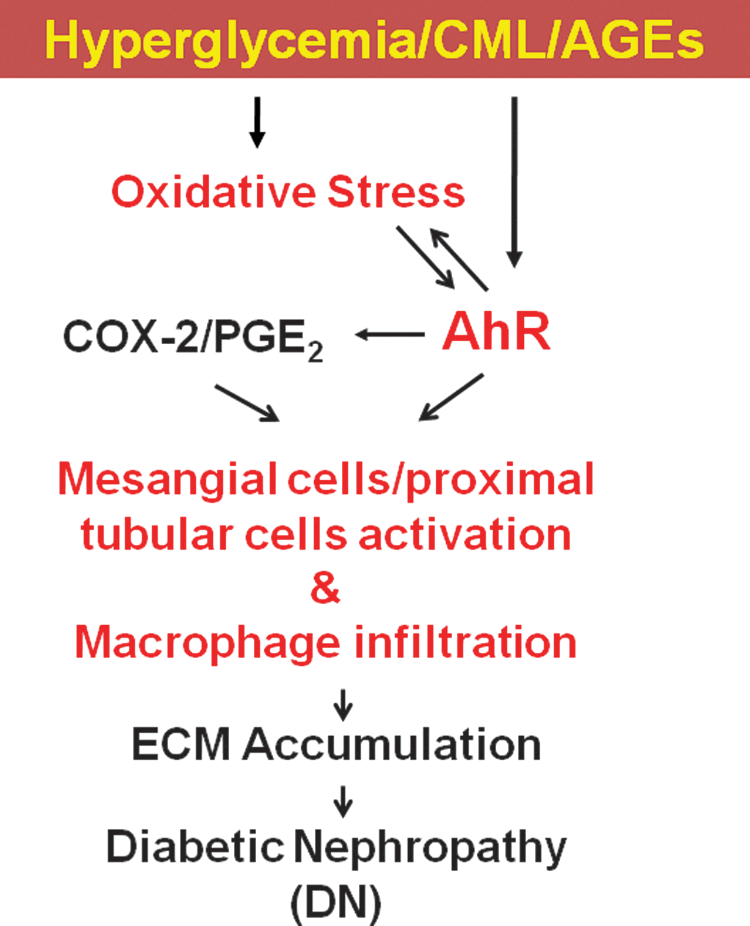
NADPH oxidase-derived ROS may play a pivotal role in the development and progression of renal injury in animal models of type 1 and type 2 diabetes (20, 33, 38). Our previous study has shown that AGEs can induce the expression of NOX4, an isoform of NADPH oxidase, in vascular endothelial cells (21). Recently, Jha et al. have shown that NOX4 is as a key source of ROS responsible for diabetes nephropathy and an innovative avenue to remedy and/or prevent chronic kidney injury (13). The evidence is based on the first direct comparison of NOX4 or NOX1 deletion in ApoE−/− mice. Yong et al. have also shown that transforming growth factor beta 1 (TGFβ1) increases NOX4 mRNA expression, and NOX4 siRNA significantly inhibits TGFβ1-induced fibronectin and collagen IV expression in human HK2 cells (44). In the present study, we also found that NOX4 expression markedly increased in glomeruli of STZ-induced diabetic mice, which could be reversed by AhRKO. These observations pointed out that NOX4 is a key source of ROS, and genetic targeting or pharmacologic inhibition of NOX4 provides renal protection in DN. On the other hand, we also found that genetic targeting or pharmacologic inhibition of AhR significantly diminished the oxidative stress, renal cell injury, and NADPH oxidase activity during STZ-induced diabetic condition. This finding indicated that AhR could regulate the STZ-induced oxidative stress. Simultaneously, we also found that pretreatment with antioxidants, apocynin, NAC, or vitamin C, markedly inhibited CML-induced AhR protein expression and nuclear translocation. This finding indicated that oxidative stress was involved in the CML/AGE-triggered AhR expression and activation. Taken together, these observations indicated the existence of a reciprocal regulation of AhR and oxidative stress to enhance renal cell injury under diabetic hyperglycemia/AGE exposure.
In the present study, both genetic AhRKO and α-NF significantly ameliorated diabetic COX-2/PGE2 production and were associated with reduced renal macrophage infiltration and glomerular pathology. The transcriptional activity of AhR could regulate the expression of COX-2 in the diabetic kidneys. As a transcription factor, AhR can regulate many related genes, such as CYP1A1, CYP1A2, CYP4A13, CYP1B1, AIP, ATP6V0C, Arnt, hsp90, CDK2/4, NQO1, and RB1 (3, 6, 12, 15, 28, 37). Interestingly, in silico prediction has shown that AhR can regulate arginase-2 (−179 to −184 and −69 to −74). This finding is consistent with those findings by Morris et al. wherein targeting arginase-2 activity or expression may be a novel therapeutic intervention in the treatment of DN (27, 45).
One of the major aims in the current study is the investigation of the regulatory relationship between AhR and COX-2/PGE2. Serum PGE2 levels were significantly increased in diabetic wild-type mice and retarded in compared AhRKO mice, suggesting a possible contribution of COX-2 to reductions in mPGES-1 bioavailability and to the development and progression of DN. More experiments are required to examine this practicability. The kidney protective effects of AhR inhibition are consistent with significant reduction in kidney macrophage recruitment. Whether the suppression of macrophage accumulation is regulated directly by AhR inhibition or indirectly by reducing diabetic renal injury warrants further elucidation. Further studies are needed to verify the role of AhR on macrophage infiltration of the glomerulus or interstitium. Moreover, the expression of COX-2 in glomerular cells, such as MCs, proximal tubular epithelial cells, podocytes, and capillary endothelial cells is an important aspect that warrants investigation. Cheng et al. have identified COX-2 expression in podocytes (9), while Kiritoshi et al. have reported that hyperglycemia increases mitochondrial ROS production, resulting in NF-κB activation, COX-2 expression, and PGE2 synthesis in human MCs (19). On the other hand, in silico prediction has shown that AhR regulates the COX-2 promoter binding site. AhR inhibition results in reduced expansion of the mesangial matrix and in glomerular hypercellularity, which ultimately occluded the glomerular capillaries in diabetes, indicating a possible contribution of AhR to the initiation and/or progression of diabetic renal fibrosis.
Clinical manifestation of endothelial cell dysfunction leading to DN is an expression of diabetic microangiopathy (5, 10, 30, 36). Emerging evidence contributes exciting insights into the pathophysiological role of renal endothelial dysfunction in the initiation and progression of renal disease (32, 35). Noticeably, systemic and renal endothelial dysfunctions have been reported in diabetic patients with normal urine albumin excretion and glomerular filtration rate. This implies that endothelial injury or dysfunction precedes overt nephropathy (11). According to hyperglycemic pathological progression, endothelial cells are particularly vulnerable. A previous study has shown that tyrosine phosphatase SHP-1-regulated VEGFR-2 dephosphorylation through NADPH oxidase-derived ROS is involved in the CML-triggered endothelial cell dysfunction/injury (22). Moreover, CML-induced, ROS-triggered, MKP-3-regulated extracellular signal-regulated kinase dephosphorylation may be involved in triggering ER stress (21). The current study provided evidence to support the importance of endothelial alterations related to CML in the progression of kidney damage. The prevention of renal injury emerges as a promising treatment strategy in kidney diseases.
In conclusion, consistent evidence supports that AhR, but not Arnt, is a recipient pathological renal oxidative stress in a diabetic mouse model, leading to MC activation, macrophage infiltration, and ECM accumulation, which relatively advance DN. The present study elucidates AhR as an attractive mechanism-based therapeutic target for the treatment of DN. Furthermore, we contribute the proof of principle that this molecular mechanism can be translated into a novel pharmacologic therapy with influential future clinical implications. Therefore, the present study strengthens the need to develop AhR-specific inhibitors that can be used for prevention and treatment of this major diabetic complication.
Materials and Methods
Cell culture
Cell culture primary MCs (RMC) from Sprague-Dawley rats were obtained by enzymatic digestion as described previously and cultured in RPMI 1640. MMCs (MES-13) were cultured in DMEM/F12. HK2 cells (human renal proximal tubular epithelial cells immortalized by transduction with human papilloma virus 16 E6/E7 genes) were cultured in keratinocyte serum-free medium (KSFM Kit Catalog No. 17005042; Invitrogen, Gibco). Then, bovine pituitary extract, 0.05 mg/ml, and epidermal growth factor, 5 ng/ml, supplemented by 10% fetal bovine serum were added. The cells were routinely passed by trypsinization after they reached 80% confluence using 10-cm culture dishes. They were then incubated at 37°C in a humidified chamber with 5% CO2/95% air mixture.
Animals
Diabetic mouse models containing STZ-induced type 1 diabetic and HFD-induced obese diabetic models and db/db mice (C57BLKS/J-leprdb/leprdb) as control were performed as described previously. Experimental AhRKO mice from the Jackson Laboratory (Strain Name: B6.129-Ahrtm1Bra /J and Stock No. 002831) were bred and AhR+/+ and AhR−/− C57BL/6 mice were obtained by breeding AhR+/−. All of the animals received water and food ad libitum.
Eight-week-old mice received either sodium citrate (control) or STZ (50 mg/kg, pH 4.5, dissolved in sodium citrate) through i.p. injection for 5 consecutive days. Two weeks following STZ injection, fasting glucose levels were measured and the mice with a fasting glucose level >350 mg/dl were considered diabetic and used for this study. In α-NF treatment experiment, STZ-induced diabetic mice were given α-NF (5 mg/kg) i.p. for 8 weeks. The AhRKO mice were randomly allocated into two groups (n = 8–15/group) to receive STZ treatment. At 12 weeks, the mice were sacrificed and their kidneys and blood were isolated for analysis. In addition, the urine samples on days 3–5 were individually collected and verified.
Glucose tolerance test (IPGTT)
After fasting overnight, the mice were injected i.p. with glucose (1 mg/kg). Blood glucose was monitored at different time points (15–120 min) after glucose administration.
Urinary analyses
Urine was continuously collected and recorded from individual mice in metabolic cages. Both urinary albumin and creatinine excretion were examined using Albuwell M kits (Exocell, Inc.). The ACR was used to evaluate renal function. Total urinary protein was analyzed by a Dade Behring Dimension RxL clinical chemistry analyzer or Siemens ADVIA 1800 analyzer.
Masson's trichrome staining, PAS staining, and immunohistochemistry analysis
Sections (4–5 μm thick) cut from 10% formalin-fixed, paraffin-embedded kidney samples were used for PAS staining and Masson's trichrome staining. The immunohistochemistry (IHC) analysis was performed as described previously (21 –24). The antibodies used in the present study are listed in Table 2.
AGEs, advanced glycation end products; Arnt, AhR nuclear translocator; CML, N-ɛ-carboxymethyllysine; COX-2, cyclooxygenase-2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IF, immunofluorescence; IHC(P), immunohistochemistry (paraffin); HRP, horseradish peroxidase; PGE, prostaglandin E; WB, Western blot.
Quantitative real-time polymerase chain reaction and Western blotting
Quantitative polymerase chain reaction (PCR) and Western blotting were performed as previously described. Primer sequences are shown in Table 3 and available from the authors upon request. The antibodies for Western blotting are listed in Table 3. Detection was performed by electrochemiluminescence detection kits (Amersham Biosciences) and by chemiluminescence using Kodak X-Omat film.
CTGF, connective tissue growth factor.
Transient transfection
The delivery of AhR shRNA into cells was performed using Lipofectin. Cell transfection was conducted for 24 h at a final shRNA concentration of 1–5 μg/ml. Normal growth medium control cells were mock transfected without control-shRNA. All experimental results were confirmed using scrambled shRNA (RNAi Core Laboratory, National RNAi Core Facility platform, Academia Sinica, Taiwan).
Electrophoretic mobility shift assay
The EMSA was performed as described previously (23, 24, 29). The oligonucleotide with the AhR consensus binding sequence used was AhR/COX-2 (5′-CTCATTTGCGTG-3′) −163 to −174 mouse; −165 to −176 rat; −161 to −172 human; DNA-protein complexes were resolved on 6% nondenaturing polyacrylamide gels and visualized by exposure to autoradiographic films.
ChIP assay
The ChIP assay protocol was modified from previous descriptions (23, 24, 29) and a fragment (270 bp) of the COX-2 promoter primer used was 5′-GAGCTTTTAGGCCCCCACTG-3′; 5′-GCGGAAAGACAGAGTCACCA-3′. The fibronectin promoter primer used was mouse 5′-GCGGTCAGCTATCAGTCCC-3′; rat 5′-GGGGAAGGCTGGAGG-3′; human 5′-GGGGGGAAAGGCAG-3′. The collagen-IV promoter primer used was mouse 5′-GGGCGGCTTACCTGGGG-3′; rat 5′-GGGGGATGTTGGACCTCAAGG-3′; human 5′-CGCCCGGCGCGCCTCCCGC-3′. The CTGF promoter primer used was mouse 5′-GGCGAGCTAAAGTGTGCCAGC-3′; rat 5′-GGCCAGCTAAAGTGTGCCAGC-3′; human 5′-GTGAGCTGGAGTGTGCCAGC-3′.
Immunofluorescence and laser scanning confocal microscopy
Cells were prepared and immunofluorescence was determined by laser scanning confocal microscopy (LSCM, TCS SL; Leica), as previously described (23, 24, 29). Images were background subtracted and merged using the Confocal Assistant and MetaMorph software and processed with Adobe Photoshop software.
ELISA detection kits for PGE2, MDA, 4-HNE, and CML production
PGE2 levels in cell culture supernatants were determined using the PGE2 enzyme immunoassay kit (Cayman Chemical). MDA was detected through a quantified colorimetric TBARS assay (Cayman Chemical). The quantity of 4-HNE in protein samples is determined by comparing its absorbance with that of a known standard curve (Cell Biolabs, Inc.). For analysis of serum CML level, a CML ELISA kit (OxiSelect ELISA Kit; Cell Biolabs) was used.
Statistical analysis
The values are presented as mean ± standard error of the mean. Analysis of variance, followed by Fisher's least significant difference test, was performed for all data. Statistical significance was set at p < 0.05.
Footnotes
Acknowledgments
This work was supported by funding from the National Science Council of Taiwan (NSC102-2628-005-001-MY3), National Chung Hsing University (NCHU102S0902, 102-5037), and Taichung Veterans General Hospital in Taiwan (TCVGH-NCHU1037602; TCVGH-1030105D; TCVGH-1033108D; TCVGH-1033608D; TCVGH-1023506D; TCVGH-1023107D; TCVGH-1013610D; TCVGH-1013002D; TCVGH-1013108D; and TCVGH-1013504D).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.