
Abstract
Introduction
A
Our previous studies have developed high-fluence, low-power laser irradiation (HF-LPLI) as a novel cancer treatment modality using a laser (633 nm). In this study, we focused on the immunological effects of this phototherapy and explored its antitumor immune regulatory mechanism. Our results demonstrate that HF-LPLI mediates tumor-killing effects via targeted photoinactivation of respiratory chain oxidase to trigger superoxide anion burst, leading to generation of a high level of oxidatively modified moieties, which contributes to phenotypic changes of macrophages and mediates antitumor immune response. We conclude that the mitochondria-targeting HF-LPLI is an effective cancer treatment modality that both eradicates the solid tumors and induces an antitumor immune response.
The ideal treatment modality for cancer should achieve tumor destruction via a minimally invasive local intervention that can induce an antitumor immunological response. Low-power laser irradiation (LPLI) is a modality that uses either coherent or noncoherent low-level light in the red to near-infrared wavelengths (λ = 630–1000 nm) to induce nondestructive and nonthermal biological reactions with therapeutic effects. In a previous study, we reported that 632.8 nm LPLI at 60 J/cm2, which was referred to as high-fluence, low-power laser irradiation (HF-LPLI), induces cancer cell apoptosis (40). A series of mechanistic studies have demonstrated that as a mitochondria-targeted cancer phototherapy, HF-LPLI selectively photoinactivates its endogenous photoacceptor, cytochrome c oxidase (COX), to generate a mitochondrial superoxide anion burst that results in oxidative damage to tumor cells (43). HF-LPLI initiates the mitochondrial pathway via the induction of a reactive oxygen species (ROS)-mediated mitochondrial permeability transition (41). Another proapoptotic signaling pathway comprising the inactivation of protein kinase B/glycogen synthase kinase 3 beta by HF-LPLI was also explored (14). These studies provide evidences for the potential phototherapy using high-intensity red light for cancers, but whether this phototherapy modulates antitumor immunity is unclear.
Macrophages are a prominent component of the stroma and leukocyte infiltrates in tumors (11). The role of macrophages in cancer is controversial and many aspects remain unresolved. Studies propose that macrophages can have conflicting roles in cancer depending on their phenotype (25). The classically activated (M1) macrophages promote antitumor immunity, whereas the alternatively activated (M2) macrophages inhibit antitumor immunity. Macrophages that infiltrate tumor tissues are driven by tumor-derived and T-cell-derived cytokines to acquire a polarized M2 phenotype (24). Therefore, a large number of studies support the concept of macrophage reprogramming as a sufficient and feasible approach to initiate T-cell-mediated antitumor immunity (27). The recognition and phagocytosis of apoptotic tumor cells by tumor-associated macrophages can affect the phenotype of these immune cells (12). Historically, apoptotic corpses have been thought to suppress the transcription of proinflammatory cytokine genes, promote the secretion of anti-inflammatory cytokines by phagocytes, and cause antigen-presenting cells to present dead cell antigens in a manner that promotes immunological tolerance. Nevertheless, emerging data support the notion that ACs that express proinflammatory danger-associated molecular patterns (DAMPs) trigger efficient antitumor immune responses (8, 28). A recent study has demonstrated that ACs generate oxidatively modified moieties, which can affect immune responses and the local inflammatory response by recruiting monocytes (3). We hypothesize that HF-LPLI-induced ACs (HF-LPLI-ACs) generate oxidatively modified moieties, and we investigate whether these moieties modulate antitumor immunity by influencing the phenotype of macrophages.
In this study, we report the antitumor efficacy of HF-LPLI phototherapy and explore the immunologic mechanisms responsible for the outcome of this therapy. We clarify that HF-LPLI-induced tumor cell apoptosis boosts antitumor immunity in vitro and in vivo. The local and systemic antitumor immune responses are further evaluated using a mouse mammary tumor model treated with HF-LPLI in vivo. The roles of oxidatively modified moieties on the surface of the AC in activating immune cells and the potential mechanisms of antitumor immunity are also studied.
Results
In vitro macrophage activation by HF-LPLI-induced apoptotic tumor cells
To validate the effects of HF-LPLI on cell apoptosis in vitro, we first performed chromatin staining with Hoechst 33258 to observe the changes in cell morphology in response to HF-LPLI treatment. As indicated in Figure 1A (upper panel), chromatin condensation was generally observed at 6 h after HF-LPLI treatment, indicating that HF-LPLI induces EMT6 and 4T1 cell apoptosis. Similar results were also obtained from flow cytometric analysis with Annexin V/propidium iodide (PI) double staining (Fig. 1A, lower panel). We also observed that caspase-3 activity was significantly increased following HF-LPLI treatment (Fig. 1B). These results provide evidences for the potential phototherapy using high-intensity red light for cancers.
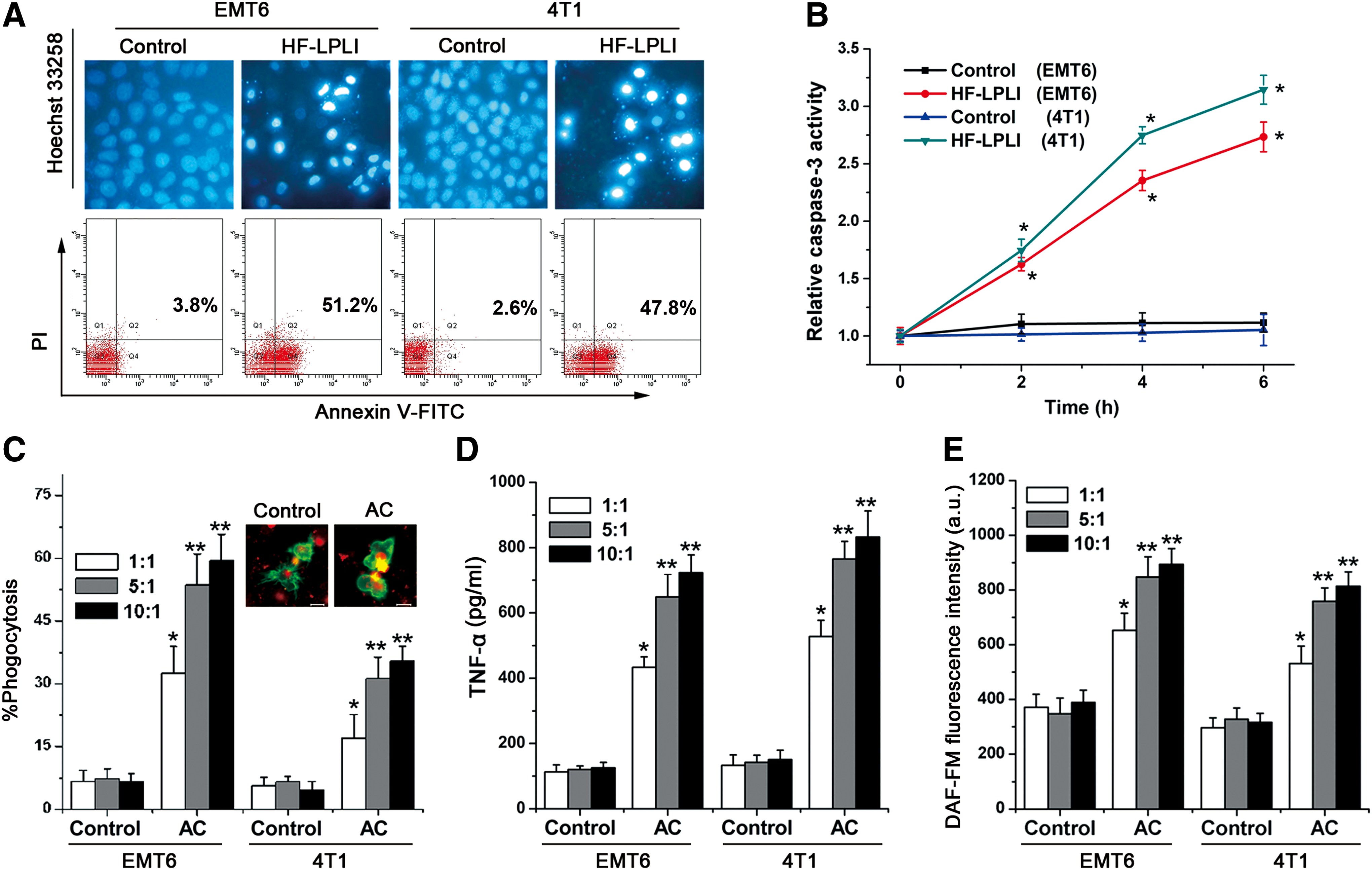
To determine whether HF-LPLI-ACs are capable of activating macrophages, the phagocytosis was measured after macrophages were incubated with HF-LPLI-ACs at different ratios (1:1, 5:1, and 10:1). The results indicate that macrophages recognize ACs (Fig. 1C and Supplementary Fig. S1; Supplementary Data are available online at
Secretion of type 1 cytokines and generation of cytotoxic T lymphocytes by the vaccination of mice with HF-LPLI-ACs
To further determine whether HF-LPLI-AC is capable of initiating systemic immune responses, mice were subcutaneously injected with HF-LPLI-ACs. Five days after immunization with the HF-LPLI-ACs, mice splenocytes were harvested and coincubated with mitomycin C-treated EMT6 cells. We found that splenocytes primed in vivo by the vaccination with HF-LPLI-ACs exhibited a significantly increased secretion of IFN-γ (Fig. 2A), interleukin (IL)-2 (Fig. 2B), and CD69 (Fig. 2C) upon restimulation in vitro, indicating that vaccination with HF-LPLI-AC induces splenocytes to display a higher expression of antitumor T helper 1 (Th1) markers. These markers may involve in Th1 immune responses and induce cell-mediated immunity. To test whether potent cytotoxic T lymphocyte (CTL) activity against EMT6 cells is generated after vaccination with HF-LPLI-ACs, EMT6 cells were labeled with CFSE and used as target cells. As shown in Figure 2D, the proportion of CFSE/PI double-positive EMT6 cells was higher when the splenocytes were primed in vivo by the vaccination with HF-LPLI-ACs, suggesting that HF-LPLI-ACs markedly increase the cytotoxic activity of activated T cells. These results demonstrate that vaccination with HF-LPLI-ACs in vivo activates Th1/CTL-mediated immune responses.

To further substantiate that injection of ACs leads to mice immunization, we evaluated tumor growth and survival rates after vaccination with HF-LPLI-ACs. HF-LPLI-ACs were subcutaneously injected into the left flanks of the mice, and after 7 days, the right flanks of the mice were rechallenged with viable EMT6 cells. As shown in Figure 2E and F, compared with the control group, the tumor growth of the HF-LPLI-AC injection group was slower and 60% mice were still alive on day 60 after the rechallenge with the tumor. Taken together, these data suggest that HF-LPLI-AC is capable of initiating systemic antitumor immune responses.
Characterization of contributing factors in HF-LPLI-AC-induced macrophage activation
To characterize the molecule(s) derived from ACs that confer(s) the immunological effects induced by HF-LPLI-ACs, the proteins and lipid factors of HF-LPLI-ACs were extracted, respectively. When macrophages were incubated with the proteins from ACs, NO generation changed to a small extent, whereas the lipids of ACs induced a large amount of NO production (Fig. 3A). These results suggest that the lipid fractions of HF-LPLI-ACs may play an important role in activating macrophages. To further determine which lipid fractions of the cells are critically involved in macrophage activation, we focused on two major phospholipids of the cell membrane, phosphatidylcholine (PC) and phosphatidylserine (PS). EMT6 cells containing different mixtures of phospholipids (PC+PS, PC+oxPS, or PS+oxPC) were incubated with macrophages. We found that NO generation was significantly increased when macrophages were incubated with oxPS-EMT6 cells, while the presence of PC+PS or PS+oxPC induced only a small amount of NO generation (Fig. 3B). Similar results were also obtained with the secretion of TNF-α by macrophages (Fig. 3C). These results indicate that PS and oxPS may differentially modulate the functional responses in macrophages. Compared with PS, PC, or oxPC, oxPS plays more important roles in activating macrophages.

HF-LPLI-induced PS oxidation is attributed to the superoxide anion burst triggered by the photoinactivation of COX
Our previous studies have demonstrated that HF-LPLI-induced apoptosis was initiated from mitochondrial ROS (42). To explore the mechanism of oxPS production on the surface of HF-LPLI-ACs, first, we used fluoresceinyl cypridina luciferin analog (FCLA)-based chemiluminescence (CL) to detect the levels of superoxide anion (O2 −•) and singlet oxygen (1O2) generation after HF-LPLI treatment. Compared with histidine (singlet oxygen scavengers), SOD (superoxide dismutase, superoxide anion scavengers) significantly reduced the HF-LPLI-triggered CL signal, suggesting that HF-LPLI induces a selectively superoxide anion (Fig. 4A), and the apoptosis ratio was reduced by 28.5% after pretreatment with SOD, but by 11.2% following exposure to histidine. These results indicate that HF-LPLI exerts its effects through inducing superoxide anion burst (Fig. 4B).

Then, to determine the effect of superoxide anion on oxPS production, we used cytochrome c oxidase III knockdown cells (RNAi COX III cells, Supplementary Fig. S2) and respiration-deficient EMT6 cells (ρ0EMT6 cells, Supplementary Fig. S3). The lipid fractions of these cells were extracted, and the levels of lipid oxidation were analyzed. The fluorescence-activated cell sorting (FACS) results showed that HF-LPLI induced a striking increase of mitochondrial superoxide anion, whereas this phenomenon was significantly inhibited in the RNAi COX III and ρ0EMT6 cells (Fig. 4C). Malondialdehyde, a marker of lipid oxidation, was significantly increased at 6 h after HF-LPLI treatment. Pretreatment with SOD prevented the effect of HF-LPLI-induced oxidative damage on lipids (Supplementary Fig. S4). Furthermore, the levels of PS oxidation were analyzed by LC/MRM. There was a gradually increased content of oxPS in the lipid fractions of wild-type EMT6 cells in response to HF-LPLI treatment, but in neither the RNAi COX III nor ρ0EMT6 cells (Fig. 4D). These results indicate that the HF-LPLI-induced PS oxidation is attributed to the superoxide anion burst triggered by the photoinactivation of COX.
Macrophage activation by HF-LPLI-induced oxPS
To investigate whether these respiratory chain-triggered various events affect macrophage function (Fig. 5) and the following other immunological endpoints (Fig. 7), we choose ρ0EMT6 cells to compare the immunological difference with wild-type EMT6. As shown in Figure 5A–D, when macrophages were incubated with HF-LPLI-ACs, the levels of phagocytosis (Fig. 5A, B and Supplementary Fig. S5), TNF-α secretion (Fig. 5C), and NO production (Fig. 5D) were significantly increased. We then used etoposide to inhibit lipid antioxidant (Supplementary Fig. S6) or used Annexin V, which specifically binds to PS/oxPS to block the recognition of PS/oxPS by macrophages (Supplementary Fig. S7) (20, 31, 39). We found that AC-induced macrophage activation was inhibited in the presence of etoposide or Annexin V (Fig. 5A–D). Moreover, HF-LPLI-ACs derived from ρ0EMT6 cells had almost no effect on macrophage activation (Fig. 5C, D). Therefore, these data suggest that oxPS is indeed responsible for the macrophage activation caused by HF-LPLI-ACs.

In vivo tumor-killing efficacy of HF-LPLI
To better determine whether the mouse mammary tumor model treated with HF-LPLI induces efficient antitumor effects, a preliminary in vivo study was performed. The power intensity and irradiation time of HF-LPLI at a fluence of 1200 J/cm2 (500 mW/cm2 for 40 min) were screened by evaluating the mean tumor volume (data not shown). The results demonstrated that a high scathe level was observed in cells treated with HF-LPLI based on hematoxylin and eosin (H&E) staining (Fig. 6A) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Fig. 6B), and the tumor burden was significantly smaller than that of the control group (Fig. 6C). For the survival studies, mice were monitored for 100 days after tumor inoculation. Kaplan–Meier survival curves showed that 80% of the HF-LPLI-treated mice survived beyond day 100, whereas no mice in the control group survived beyond day 60 (Fig. 6D). Furthermore, we investigated the tumor-killing efficacy of HF-LPLI using a ρ0EMT6 tumor model. Compared with the control group, there was no scathing of the tumor tissue, no reduction of tumor size, and no survival in the HF-LPLI-treated group (Fig. 6A–D). These results suggest that HF-LPLI triggers tumor killing by targeting the respiratory chain and may be an efficacious treatment modality for cancer.

In vivo antitumor immunological effects of HF-LPLI
The in vivo antitumor immunological effects of HF-LPLI treatment in the mouse mammary tumor model require further investigation. To clarify this issue and explore whether PS oxidation mediates the HF-LPLI-induced antitumor immunity in vivo, both a wild-type EMT6 model and a ρ0EMT6 tumor model were used. We found that the oxidative stress induced by HF-LPLI resulted in PS oxidation in situ, while no obvious changes in the oxPS level were detected in the ρ0EMT6 tumor model (Fig. 7A). HF-LPLI treatment caused a statistically significant increase in tumor-infiltrating macrophages in the wild-type EMT6 tumor model, but not in the ρ0EMT6 tumor model. Moreover, when the ρ0EMT6 tumor-bearing mice were injected intratumorally with oxPS (5 mM, 20 μl), the level of macrophage infiltration into the tumors was increased, indicating that oxPS regulates the macrophage response in vivo (Fig. 7B). Similar results were obtained with the generation of cytokines (Fig. 7C) and the level of tumor-infiltrating CTLs (Fig. 7D) in tumor-bearing mice 1 week after HF-LPLI treatment. The results in the ρ0EMT6 tumor model indicate that the level of oxPS of tumor cells may influence the phenotype of macrophages and the various following immunological endpoints, and HF-LPLI may regulate the immune response in vivo by inducing PS oxidation in situ.
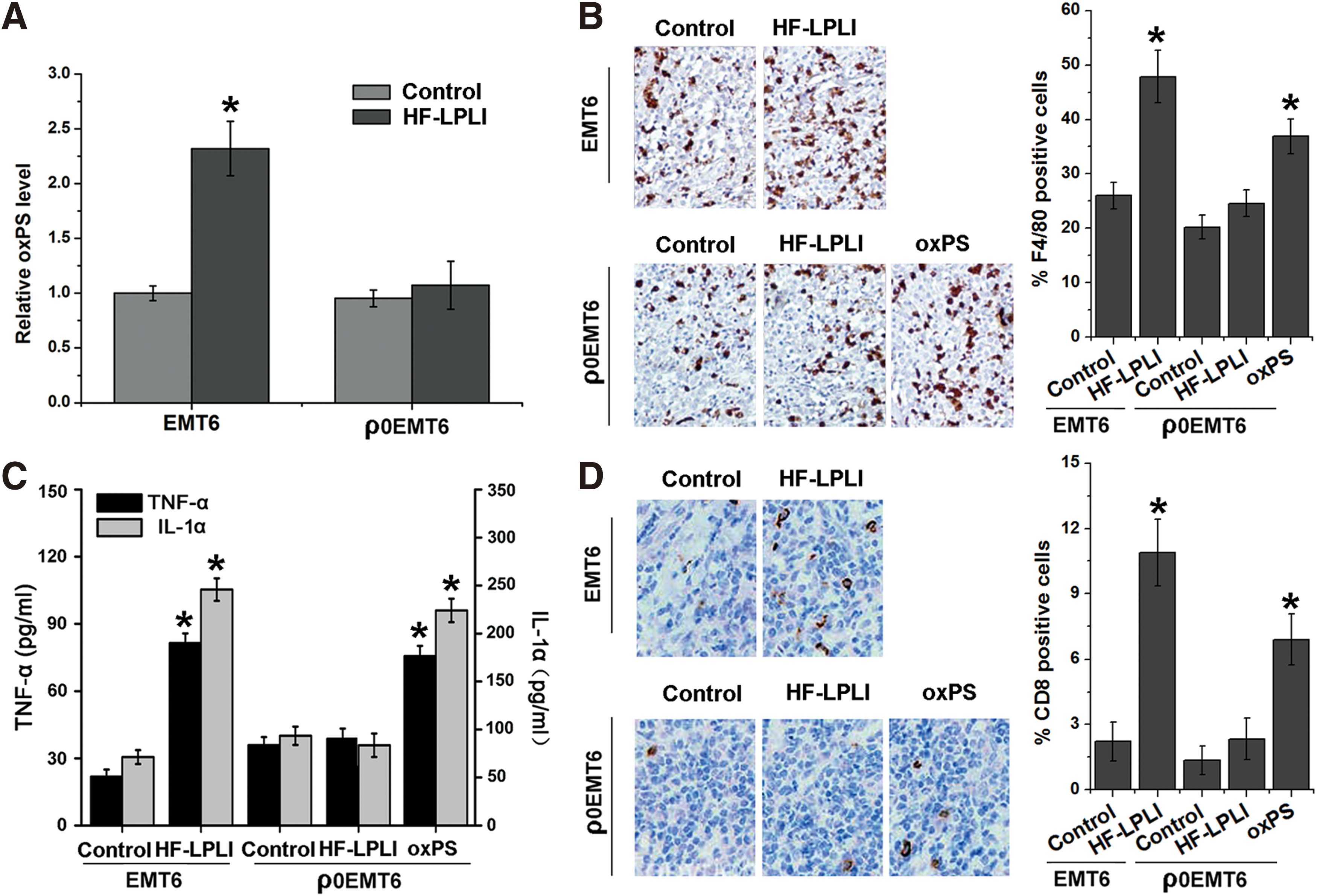
Taken together, in vivo results suggest that HF-LPLI treatment causes the inflammatory reaction in tumor tissue by polarizing the newly invading macrophages toward the M1 phenotype acting as a proinflammatory factor. Following such a tumor-localized insult, extensive phagocytosis of large numbers of rapidly appearing dead cancer cells by macrophages facilitates processing and presentation of tumor antigens to T lymphocytes, leading to the development of adaptive antitumor immune response.
Long-term antitumor effects of HF-LPLI treatment
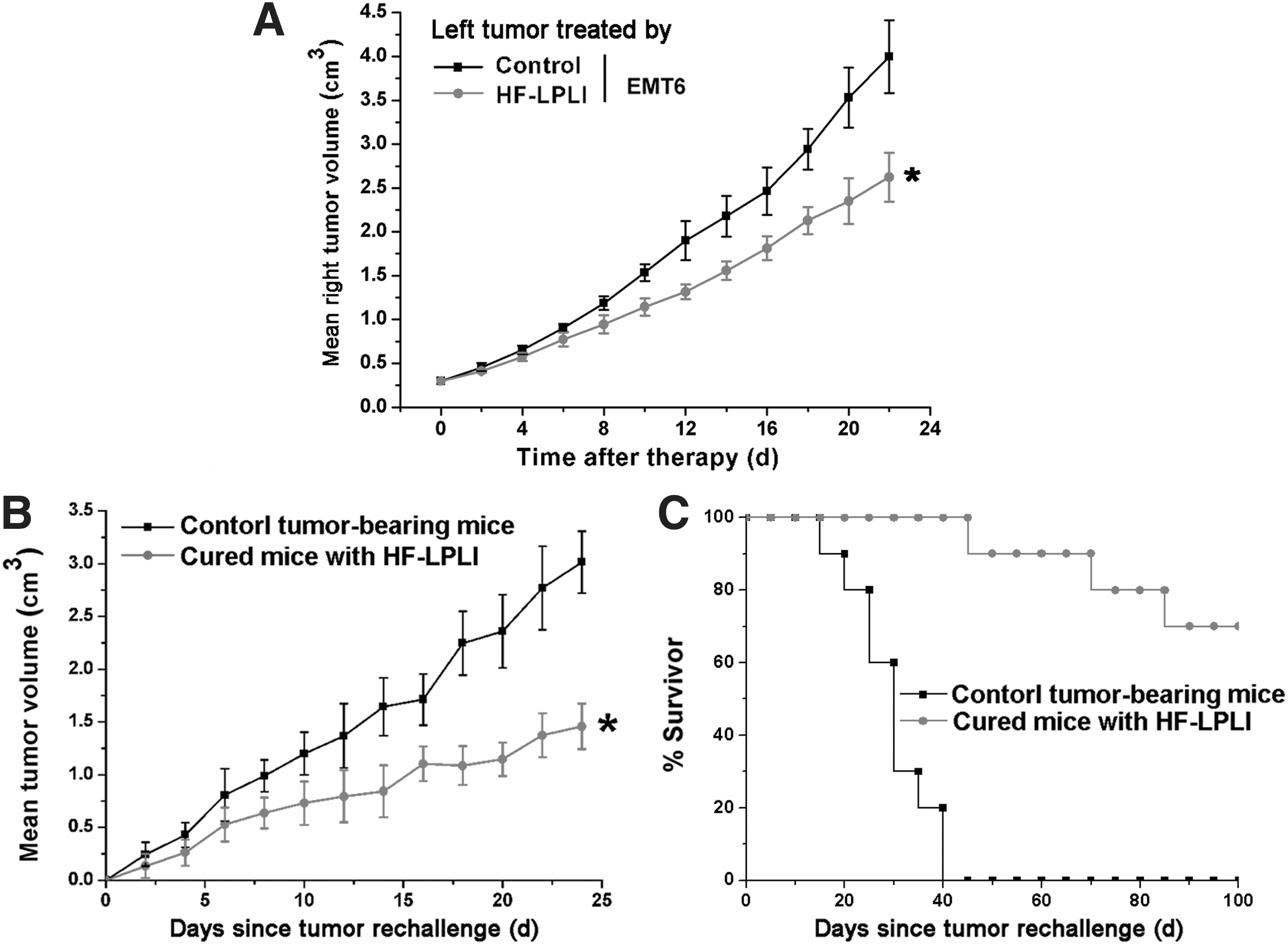
To evaluate whether HF-LPLI treatment induces systemic antitumor immunological effects to suppress tumor growth, EMT6 tumor cells were implanted on both the right and left flanks of the mice. Only the tumor on the left side was treated with HF-LPLI, while the tumor on the right side served as the observation points without treatment. Our results revealed that the growth of the untreated tumor on the right was significantly inhibited when the tumor on the left side was treated with HF-LPLI (Fig. 8A). These in vivo results suggest that in a mouse mammary tumor model, treatment with HF-LPLI in vivo may induce efficient systemic antitumor immunological effects to suppress tumor growth. To evaluate whether HF-LPLI phototherapy induces long-term antitumor effects, mice that were successfully treated with HF-LPLI were rechallenged with 2 × 106 viable EMT6 cells 60 days after the initial tumor inoculation. Mice of the same age were inoculated with 2 × 106 viable tumor cells per mouse as the controls. In the HF-LPLI-cured group, the tumor burden was significantly smaller and 70% of the HF-LPLI-cured mice were still alive on day 100 after the rechallenge with the tumor (Fig. 8B, C). Taken together, the in vivo data suggest that the HF-LPLI treatment induces long-term antitumor immune responses in tumor-bearing mice.
Discussion
The relationship between apoptosis and immune responses is currently becoming a target of intense investigation that may be beneficial for cancer therapy. Our previous studies have shown that HF-LPLI generates a mitochondrial superoxide anion burst, resulting in oxidative damage to tumor cells (43). In this study, we demonstrate that tumor cell lines (EMT6 and 4T1) treated with HF-LPLI in vitro are able to activate macrophages, and based on the mouse mammary tumor model, treatment with HF-LPLI in vivo initiates local and systemic antitumor immune responses. Furthermore, results reveal that HF-LPLI mediates its effects via a targeted respiratory chain COX to induce a superoxide anion burst, followed by PS oxidation, which mediates the antitumor immune response. This provides new insights into the mechanisms by which ACs induce antigen-presenting cell activation and suggests that as a potential antitumor therapy, HF-LPLI treatment not only kills tumor cells but also causes efficient immune responses.
The immunomodulatory effects of apoptotic tumor cells on the functions of antigen-presenting cells are currently being debated. Therefore, an increasing number of studies are focused on investigating the immunological interactions between apoptotic tumor cells and immune cells (9, 22). Although dendritic cells (DCs) are the most effective antigen-presenting cells, macrophages are major immunoregulatory cells in tumors and play crucial roles in initiating antitumor immunity. Activated macrophages can kill tumor cells by releasing high levels of TNF-α, NO, and related reactive nitrogen species, such as nitroxyl and peroxynitrite (30). Importantly, as an agent of inflammation and cell-mediated immunity, NO also plays an important role in host antitumor immunity (32). In this study, we found that HF-LPLI induced tumor cell apoptosis (Fig. 1A, B); increased levels of NO and TNF-α were detected when macrophages were incubated with HF-LPLI-treated tumor cells (Fig. 1D, E). The cytotoxic effect of the proinflammatory cytokines on tumor cells was suggested as a possible mechanism of the antitumor effect of HF-LPLI. In vivo results showed that tumor tissue was damaged 1 day after HF-LPLI treatment (Fig. 6A, B), subsequent tumor-infiltrating macrophages were significantly increased 3 days after treatment (Fig. 7B), and meanwhile the levels of TNF-α and IL-1α were increased (Fig. 7C). These in vivo results suggest that HF-LPLI treatment causes the inflammatory reaction in tumor tissue by polarizing the newly invading macrophages toward the M1 phenotype acting as a proinflammatory factor.
Besides HF-LPLI treatment that inflicts an immediate trauma at the treated site, these newly invading macrophages increase damage by producing inflammatory mediators and exhibiting antitumor activity. Following such a tumor-localized insult, extensive phagocytosis of large numbers of rapidly appearing dead cancer cells by macrophages facilitates processing and presentation of tumor antigens to T lymphocytes, leading to the development of adaptive antitumor immune response. To examine whether HF-LPLI treatment is capable of activating systemic immune responses, we investigated T-lymphocyte activity in vitro and in vivo. Vaccination with HF-LPLI-treated tumor cells in vivo activates Th1/CTL-mediated immune responses (Fig. 2). Moreover, based on the mouse mammary tumor model, treatment with HF-LPLI in vivo increases the level of tumor-infiltrating CTLs (Fig. 7D) and initiates systemic antitumor immune responses (Fig. 8).
These remarkable results led us to study the molecular mechanisms responsible for antigen-presenting cell activation after incubation with HF-LPLI-ACs. Our results suggest that both the proteins and lipid factors of HF-LPLI-ACs contribute to the activation of macrophages (Fig. 3A). It has been demonstrated that ACs that express proinflammatory DAMPs trigger efficient immune responses (8, 28). In addition to the proteins, the lipids of dying cells may affect the maturation of the antigen-presenting cells (46). The exposure of PS on the cell surface during apoptosis acts as an eat-me signal. Studies have shown that PS suppresses inflammation and provides a signal to inhibit antigen-presenting cell maturation, thereby avoiding activation of an immune response perhaps through the generation of regulatory T cells (4, 7, 15). However, ACs generate oxidatively modified phospholipids, which can affect immune responses and produce a local inflammatory response by recruiting monocytes (3). Studies have suggested that synthetic oxPS, which is generated under Fenton reaction conditions, induces proinflammatory cytokine secretion in monocytes and DCs (5). Investigations have also demonstrated that increased oxPS content is generated by ROS during oxidative stress-induced apoptosis (19, 38). However, whether oxPS on ACs modulates antitumor immunity is still unclear. Our results indicate that macrophages generate the M1 marker when incubated with EMT6 cells integrated with oxPS liposomes, but not with PS liposomes (Fig. 3B, C). These results indicate that PS and oxPS may differentially modulate the functional responses in macrophages.
Whether HF-LPLI-AC generates oxidatively modified moieties to modulate antitumor immunity requires investigation. Consistent with a previous study that PS is oxidized by ROS generated from disrupting mitochondrial electron transport (17), our results demonstrate that HF-LPLI-generated ROS contribute to PS oxidation (Fig. 4). In our current study, low levels of ROS and PS oxidation were produced when the activity of COX was inhibited by RNAi (Fig. 4C, D). This indicates that HF-LPLI has a potentially significant oxidative effect on the components of cells with higher COX activity. We hypothesize that a high level of oxidatively modified moieties in the HF-LPLI-treated tumor cells influences the phenotype of macrophages. Oxidative stress by HF-LPLI resulted in PS oxidation in situ, while no obvious changes in the oxPS level were detected in the ρ0EMT6 tumor model (Fig. 7A). In the wild-type EMT6 tumor model, but not in the ρ0EMT6 tumor model, HF-LPLI treatment caused a statistically significant increase in tumor-infiltrating macrophages and CD8+ T cells (Fig. 7B, D). Moreover, when the ρ0EMT6 tumor-bearing mice were injected intratumorally with oxPS, increased levels of leukocytes infiltrated into the tumors, indicating that oxPS regulates the immune response in vivo.
We speculate that the ratio of PS and oxPS on the ACs may influence the ability of antigen-presenting cells to produce cytokines or other mediators. Another possibility is that PS and oxPS may be recognized by different specific receptors and may activate different downstream cascade responses. A number of different studies have shown that macrophages recognize ACs via the interaction between membrane-associated oxPS and CD36 (10, 29). The signaling cascades link CD36 to cellular responses related to scavenger receptor activity, NF-κB activation, and cytokine and ROS secretion (34). It has been reported that CD36 cooperates with members of the Toll-like receptor (TLR) family to activate the immune response (37). Therefore, whether CD36 signaling cascades are involved in the immune responses induced by oxPS on the surface of ACs and whether there is cooperation between CD36 and TLRs require further investigation.
Previous studies have shown that enhanced immunity was observed after photodynamic therapy using light-activated photosensitizers (2, 36, 44). Our previous study has reported that HF-LPLI may be a novel modality for cancer phototherapy without any additional photosensitizers in an experimental model and explored the mechanism of cell death induced by HF-LPLI (43). In this study, in addition to the tumor-killing efficacy of HF-LPLI, the immunological effects of HF-LPLI treatment in an experimental model were investigated. Our results indicate that HF-LPLI phototherapy induces tumor cell damage in situ, and a high level of oxidatively modified moieties (such as oxPS) in the HF-LPLI-treated tumor cells is generated due to a superoxide anion burst triggered by the photoinactivation of a respiratory chain oxidase, which then, in turn, contributes to the phenotypic changes in macrophages. In addition, the release of cytokines by Th1 T cells especially by CTLs polarizes the newly recruited macrophages into M1-like cytotoxic macrophages, thus initiating a feed-forward loop that enhances antitumor immunity. This study reveals that HF-LPLI can be potentially applied as a mitochondria-targeted cancer treatment.
Materials and Methods
Cell lines, tumor models, and reagents
The EMT6 and 4T1 were obtained from Jinan University (Guangzhou, China). These cell lines were authenticated based on viability, recovery, growth, morphology, and isoenzymology by their suppliers. The cells were grown in RPMI 1640 medium (GIBCO) supplemented with 15% fetal bovine serum (FBS) (Gibco-BRL), 50 U/ml penicillin, and 50 μg/ml streptomycin in 5% CO2 at 37°C in a humidified incubator. For the isolation of mouse peritoneal macrophages, 8- to 10-week-old female BALB/c mice were euthanized, and 5–10 ml of Ringer solution containing 5% FBS was injected into the peritoneum. The cells were recovered, pooled, seeded in complete RPMI medium, and left to adhere to culture dishes. Respiration-deficient EMT6 cells (ρ0EMT6 cells) were generated by incubating wild-type cells with ethidium bromide for 4 weeks in medium supplemented with pyruvate and uridine.
EMT6 or ρ0EMT6 cells (1 × 106) in a 100 μl solution were injected into the flank region of female Balb/c mice, aged 6–8 weeks. The animals were used in experiments 7–10 days after the tumor cell inoculation when the tumors had reached a size of ∼300 mm3. Our study was performed in accordance with the guidelines of the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council). It was approved by the Institutional Animal Care and Use Committee of our University (South China Normal University, Guangzhou, China).
MitoSOX™ (5 μM) was purchased from Molecular Probes, Inc. NAC (250 μM), PKH26 (5 μM), PKH67 (5 μM), and MnSOD (50 units/ml) were purchased from Sigma-Aldrich. DAF-FM DA (0.5 mM) was purchased from Beyotime.
HF-LPLI treatment
For in vitro HF-LPLI treatment, tumor cells were irradiated with the He-Ne laser (632.8 nm; HN-1000) at a fluence of 0.2 W/cm2 for 10 min. To minimize the ambient light interference, the entire procedure was carried out in a dark environment at room temperature. For in vivo HF-LPLI, the tumor-bearing mice were directly irradiated with the 632.8 nm laser. The total fluence delivered to the tumors was 1200 J/cm2 (500 mW/cm2, 40 min). The mice were anesthetized with an intraperitoneal injection (i.p.) of pentobarbital sodium (2%) and were restrained in a specially designed holder. The control samples were maintained on the laser table for the same amount of time used in the irradiated groups, but the laser source was not activated (sham irradiation).
Cell death assays
To assess apoptotic changes in the nuclear morphology, the cells were stained with Hoechst 33258 for 20 min and washed twice with phosphate-buffered saline (PBS) at 6 h after HF-LPLI treatment. The cell samples were visualized with a Nikon fluorescent microscope (mercury lamp, Ex. 330–380 nm, Em. BA 435 nm). To assess cell apoptosis via flow cytometry (Becton Dickinson FACScan), 6 h after treatment, the cells were stained with 5 μl Annexin V–fluorescein isothiocyanate (FITC) and 5 μl PI for 15 min using an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences Pharmingen). The activity of caspase-3 was determined using a caspase-3 activity kit (Sigma), and the OD405 was read with a 96-well plate reader (INFINITEM200; Tecan). For in situ tumor cell death analysis, H&E staining was performed 1 day after the indicated treatments. The middle sections of each tumor were formalin fixed and paraffin embedded following a standard routine. For TUNEL staining analysis, mice from each treatment group were sacrificed 1 h after treatment. Individual tumors were fixed in 10% neutral buffered formalin, processed routinely into paraffin, sectioned at 5 μm, stained with TUNEL fluorescence dye (FITC; Genmed), and examined by fluorescence microscopy.
NO measurement
Macrophages were incubated for 30 min in RPMI 1640 containing a low concentration (i.e., 0.5 mM) of the fluorescent probe DAF-FM DA (excitation/emission maxima of 495/515 nm). After loading, the cells were rinsed thrice with RPMI 1640 and incubated for 10 h with EMT6 cells that had undergone different treatments. NO production by the macrophages was detected with confocal microscopy (LSM510/ConfoCor2), flow cytometric analysis (Becton Dickinson FACScan), or a 96-well plate reader (INFINITEM200; Tecan).
Preparation of PS- and oxPS-containing liposomes
The liposomes were prepared essentially as described by Kagan et al. (19). Small unilamellar liposomes containing 50% PC and 50% PS were produced as described by Fadok et al. (6). EMT6 cells (5 × 107 cells) were incubated with N-ethylmaleimide (10 μM, 10 min at 37°C) to inhibit aminophospholipid translocase, and then incubated in the presence of PC+PS, oxPC+PS, or PC+oxPS liposomes (50 μM) for 30 min at 37°C. The liposome-EMT6 cells represent those EMT6 cells containing different mixtures of phospholipids (PC+PS, PC+oxPS, or PS+oxPC).
Phagocytosis assay
Tumor cells were labeled with PKH26 (5 μM; Sigma-Aldrich). These cells were collected at 10 h after HF-LPLI treatment. Macrophages were incubated with PKH67 (5 μM; Sigma-Aldrich) and rinsed thrice. Then, these macrophages (green) were cultured with PKH26-labeled, HF-LPLI-treated tumor cells (red) for 3 h and visualized with confocal microscopy. Macrophages incubated with tumor cells without HF-LPLI treatment were used as the controls. The yellow color indicates colocalized pixels and suggests that macrophages uptake the tumor cells. The phagocytosis percentage was quantitatively analyzed based on 10 randomly selected fields (∼500 cells). Due to unique properties of PKH26 and PKH27, phagocytosis assay using flow cytometry was also performed. Double-stained population indicates phagocytosis of tumor cells by the macrophages.
Extraction of proteins and lipids
To clarify the effect of AC-derived unknown factors on macrophage activation, the proteins and the lipid factors of HF-LPLI-induced ACs were, respectively, extracted. The proteins and the lipid factors of HF-LPLI-induced ACs (5 × 106 cells) were incubated with macrophages (1 × 106 cells). The extraction method is as follows: total lipids were extracted from HF-LPLI-induced ACs by chloroform/methanol as described by Hörkkö et al. (13). In brief, lipid factors of cells were extracted with chloroform: methanol (2:1, v/v). Then, the organic phase was evaporated under nitrogen and reconstituted in PBS containing 1 mg/ml BSA. The proteins of cells were extracted using a CelLytic™ MEM Protein Extraction Kit (Sigma).
ROS measurement
Cells were loaded at 37°C with 5 μM MitoSOX for 10 min in the dark, and then rinsed with PBS before the fluorescence measurement with a flow cytometer (excitation at 488 nm, emission at 560–700 nm); the data were analyzed using Cell Quest software (Becton Dickinson).
Assay of phospholipid peroxidation
To measure the level of PS oxidation, total lipids were extracted from the EMT6 cells with chloroform/methanol as described by Hörkkö et al. (13). The oxPS in these lipid extracts was analyzed by liquid chromatography–multiple reaction monitoring (LC/MRM) as described by Chang et al. (3). The internal standard 1-palmitoyl-2-arachidonoyl-sn-glycero-3-PS (PAPS) was added to the samples.
Enzyme-linked immunosorbent assay
Cell culture supernatants were collected, and the levels of TNF-α, IL-2, and IFN-γ were measured with the enzyme-linked immunosorbent assay (ELISA) kit from eBioscience following the manufacturer's instructions.
In vivo cytotoxicity assay
The mice were immunized with the treated tumor cells. Approximately 1 × 106 treated tumor cells in a volume of 0.1 ml were injected subcutaneously into the flanks of the animals. Five days after the immunization, splenocytes were harvested and cocultured with mitomycin C-treated tumor cells. IFN-γ and IL-2 secretions by the splenocytes were detected 2 days later with ELISA. CD69 expression was detected with FACS. Stimulated effector cells were tested for cytolytic activity against EMT6 cells 5 days later using a CFSE-PI-based cytotoxicity assay. Briefly, the targeted cells were prestained with CFSE (5 μM; Invitrogen) and incubated with splenocytes. The cocultured cells were maintained for 4 h in RPMI 1640 (1% FBS). PI was added before the flow cytometric analysis, and the percentage of lysed EMT6 cells was calculated using the formula (CFSE+ PI+)/(CFSE+ PI+) + (CFSE+ PI−) × 100%.
Immunohistochemical analysis
Tumor-infiltrating mononuclear cells were visualized in optimal cutting temperature-embedded tumor samples with immunohistochemical methods. Tissue sections (5 μm) were air-dried overnight, immersed in ice-cold acetone for 10 min, and treated with hydrogen peroxide and Superblock (ScyTek Laboratories). Anti-mouse antibodies, CD8 and F4/80 (1/200; Santa Cruz Biotechnology), were applied overnight at 4°C. After washing, the slides were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG for 30 min. The HRP reaction was developed using amino-9-ethylcarbazole (Scytec), and the slides were counterstained with hematoxylin.
Statistical analysis
Standard Student's t-test and one-way or two-way ANOVA were performed to evaluate the significance of differences between experimental groups. The differences were considered statistically significant at p < 0.05. Data are presented as mean ± SEM.
Footnotes
Acknowledgments
The authors acknowledge the financial support from the National Basic Research Program of China (2011CB910402; 2010CB732602), the Program for Changjiang Scholars and Innovative Research Team in University (IRT0829), and the National Natural Science Foundation of China (81101741; 31101028; 31470072).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.