
Abstract
Introduction
E
The study identified that electronic cigarette vapor (eCV) exposure leads to aggresome formation via proteostasis/autophagy impairment and serves as a mechanism to induce inflammatory–oxidative stress, apoptosis, and senescence that can be ameliorated by an autophagy inducer, carbamazepine, or cysteamine. Thus, it suggests the mechanisms by which eCV exposure can potentially induce chronic obstructive pulmonary disease–emphysema upon chronic exposures.
In addition, recent preliminary studies demonstrate that e-cigarette vapor (eCV) exposure not only induces inflammatory–oxidative signaling but also compromises the immunity increasing the susceptibility to bacterial and viral infections (36, 49, 63). These studies suggest that eCV induces toxicity, inflammatory response and oxidative stress similar to regular cigarettes; this suggests its potential role in lung cancer and chronic obstructive pulmonary disease (COPD)–emphysema upon chronic vaping, warranting further investigation. Therefore, we designed this study to evaluate whether the central mechanism(s) known to induce inflammatory–oxidative stress and COPD-emphysema pathogenesis are modulated by eCV exposure. We recently identified proteostasis/autophagy impairment as a novel central mechanism leading to aggresome formation and subsequent downstream effects such as apoptosis in COPD-emphysema (41, 51). We and others have identified the global impact of first- and second-hand smoke exposure on overall protein synthesis, ubiquitination, and aggregation of misfolded proteins in perinuclear spaces as aggresome bodies (AB) leading to a unique pathology associated with severity of emphysema in COPD subjects (21, 23, 35, 51, 56, 58). In addition, we identified that clearance of these AB using an autophagy inducer carbamazepine not only reduces apoptosis but also controls cigarette smoke (CS)-induced emphysema (51). Hence, recent studies from our group and others (27, 51) have identified the potential of autophagy inducer carbamazepine in rescuing both smoke-induced and alpha-1-antitrypsin Z mutation-associated emphysema in COPD.
Moreover, cysteamine drugs such as Procysbi (Food and Drug Administration [FDA] approved for other clinical conditions) and Lynovex are promising candidates for COPD as they can restore autophagy while inhibiting ROS activity, bacterial infection, and mucus obstruction due to their antioxidant, antibacterial, and mucoactive properties.
The present study was designed to evaluate if short-term effects of eCV exposure modulate mechanisms known to be involved in CS-induced COPD-emphysema. Based on the historical studies on nicotine toxicity and recent preliminary literature on eCV exposure (1, 9, 10, 20, 36, 40, 46, 49, 64), we anticipated that it would modulate inflammatory–oxidative responses. However, it was not apparent if eCV exposure can significantly impair central mechanism(s) associated with inflammatory–oxidative responses and COPD-emphysema progression. Hence, we first evaluated whether eCV exposure could modulate proteostasis/autophagy as a potential mechanism for inflammatory–oxidative stress observed to be induced by nicotine and other eCV components (3, 26, 36). In addition, we evaluated if modulating autophagy via FDA approved drug carbamazepine and/or antioxidant cysteamine can regulate eCV-induced inflammatory–oxidative stress. Since e-cigarette vaping is marketed as a safer alternative to tobacco cigarette smoking, our experimental design was focused on sequentially dissecting the potential role of eCV exposure and its impact on proteostasis, specifically overall protein ubiquitination and autophagy, in initiating a lung disease by quantifying its impact on mechanisms involved in inflammatory–oxidative stress, apoptosis, and/or senescence.
Results
eCV induces ubiquitinated protein accumulation and impaired autophagy
To quantitate the impact of eCV upon proteostasis, we first aimed to evaluate overall protein ubiquitination and autophagy. eCV-exposed media (5 or 15 min, as described in the Materials and Methods section) were used to treat Beas2b cells for 1, 3, or 6 h. Next, we isolated total cellular soluble and insoluble protein fractions that were first subjected to immunoblotting (IB) for ubiquitin. Compared to the air exposure control, we found that eCV (1 h) exposure of Beas2b cells showed a significant (p < 0.001) increase in accumulation of polyubiquitinated proteins in soluble protein fractions. Accumulation of these ubiquitinated proteins in soluble fractions was significantly (p < 0.01) decreased in longer 3-h eCV exposures of Beas2b cells, and no significant detection of ubiquitinated proteins was observed in Beas2b cells exposed to eCV for 6 h. In insoluble protein fractions, eCV exposure led to significantly (p < 0.001) increased accumulation of polyubiquitinated proteins at 6 h compared to 3 h (p < 0.001) and 1 h (p < 0.01) or control samples suggesting translocation of these ubiquitinated proteins from soluble to insoluble fractions upon longer eCV exposures may induce AB. To check activity of autophagy in Beas2b cells exposed to eCV, IB of aberrant autophagy marker, p62 showed significant increase at 1 h (p < 0.05) and 3 h (p < 0.01) followed by its decline at 6 h (p < 0.01), as compared to air-exposed controls. Moreover, significantly (p < 0.05) increased p62 translocation and accumulation were observed in the insoluble protein fraction with 6 h of eCV exposure suggesting impaired autophagy. Next, we evaluated the AAA+ ATPase molecular chaperone valosin-containing protein (VCP)/p97 as it is implicated in critical cellular proteostasis mechanisms, including aggresome formation and autophagy (7, 28, 30, 45). The eCV exposure of Beas2b cells led to significant increase in VCP at 1 h (p < 0.001) followed by its significant decrease in 3 h (p < 0.001) and 6 h (p < 0.001), as compared to air-exposed controls. Similar to p62, VCP is significantly (p < 0.05) localized in the insoluble fraction upon longer eCV exposure, potentially as an early cellular protective mechanism to control chronic accumulation of ubiquitinated proteins by participating in autophagy (Fig. 1A). These findings suggest that longer 6-h eCV exposure may be inducing aggresome formation by inducing VCP-mediated retrograde translocation and autophagy impairment (p62-VCP accumulation in insoluble protein fractions).
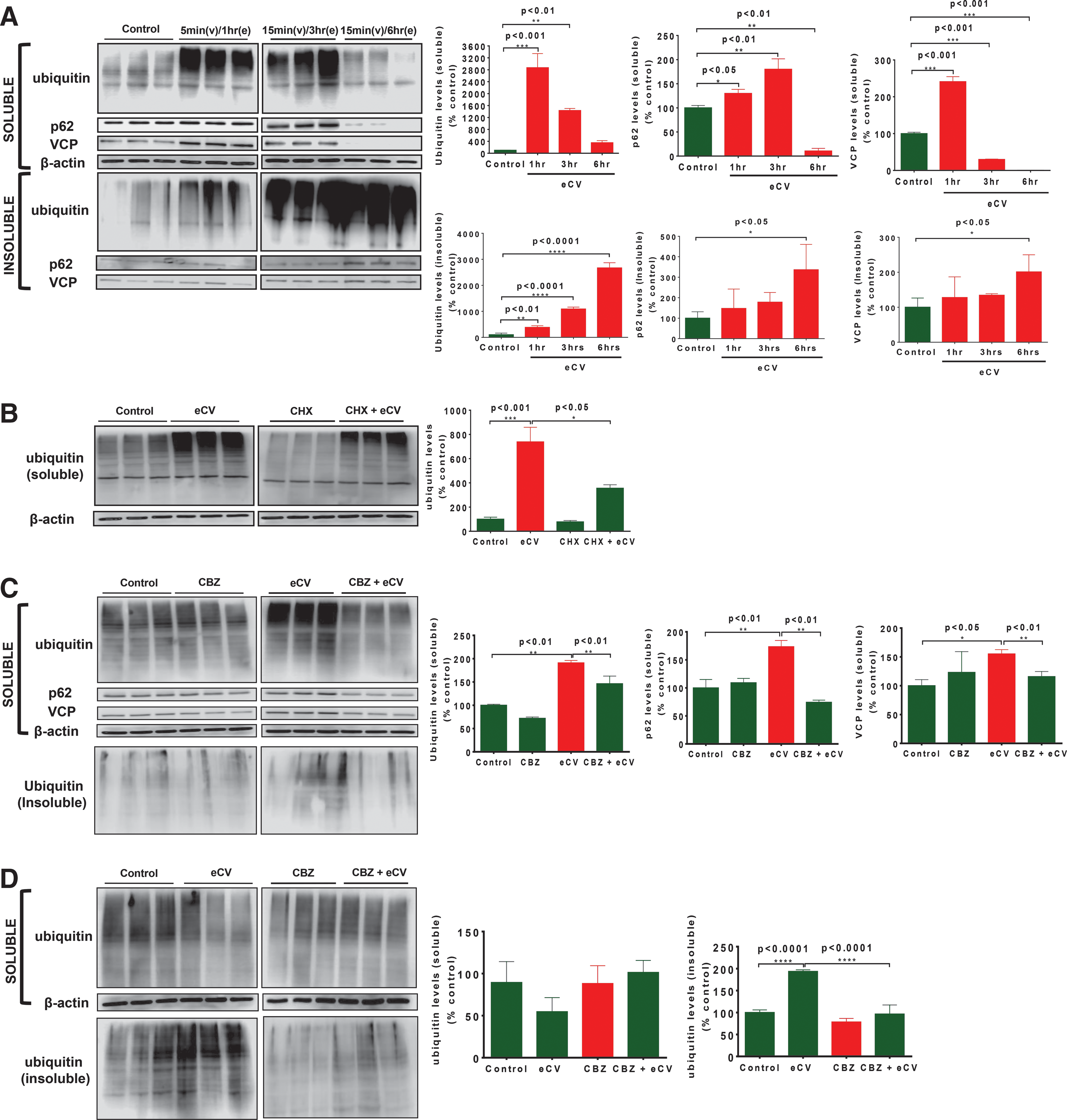
We next sought to determine whether eCV modulates protein synthesis as a mechanism for increased ubiquitinated protein accumulation. We recently found that CSE induces aggresome formation by modulating cellular protein turnover (51). Hence, Beas2b cells were similarly pretreated with 50 μg of cycloheximide (CHX) for 6 h followed by eCV exposure for 1 h. Significant (p < 0.001) accumulation of ubiquinated proteins was observed in the soluble protein fraction at 1 h of eCV exposure similar to Figure 1A. Preincubation with CHX significantly (p < 0.05) attenuated the accumulation of ubiquitinated proteins (Fig. 1B), suggesting that eCV is not able to impact protein synthesis unlike CS and may only be inducing subsequent steps of protein misfolding and aggresome formation.
We also observed that eCV induced a significant increase in the aberrant autophagy marker p62 (insoluble fraction), suggesting autophagy impairment on longer eCV exposures. Hence, we used an autophagy inducer carbamazepine to verify our hypothesis if eCV-induced autophagy impairment can be rescued by carbamazepine treatment. Beas2b cells were preincubated with 20 μM carbamazepine for 6 h followed by eCV exposure for 1 h. Similar to Figure 1A, significant (p < 0.01) accumulation of ubiquitinated proteins, p62 (p < 0.01) and VCP (p < 0.05), was observed in soluble protein fractions of Beas2b cells exposed to eCV for 1 h. Accumulation of ubiquitinated proteins was significantly (p < 0.01) attenuated when carbamazepine preincubated cells were exposed to eCV, suggesting the potential of carbamazepine in inhibiting aggresome formation by inducing autophagy clearance/degradation mechanisms. Preincubation of Beas2b cells with carbamazepine alone showed no significant impact on ubiquitinated protein levels compared to air exposure. Carbamazepine induced a significant (p < 0.01) decrease in p62 levels on eCV exposure for 1 h. Similarly, carbamazepine treatment significantly (p < 0.01) decreased eCV (1 h)-induced VCP protein levels in Beas2b cells. Slight accumulation of ubiquitinated proteins was observed in insoluble protein fractions of Beas2b cells exposed to eCV (1 h) as anticipated based on previous experiments, but this was not seen for carbamazepine pretreated eCV-exposed cells (Fig. 1C).
Next, we sought to verify whether carbamazepine could rescue accumulation of ubiquitinated proteins from the insoluble protein fraction on longer 6 h eCV exposures. Beas2b cells were preincubated with 20 μM carbamazepine for 6 h followed by eCV exposure. Fractionation of soluble and insoluble protein lysates was made and assessed for ubiquitinated protein accumulation through IB as shown previously. No significant accumulation of ubiquitinated proteins was observed in Beas2b cells pretreated with carbamazepine and/or exposed to eCV (6 h) in comparison to carbamazepine treatment and air exposure controls. Analysis of the insoluble fraction showed eCV-induced significant (p < 0.001) accumulation of ubiquitinated proteins (as seen in Fig. 1A as well) and that carbamazepine could significantly (p < 0.001) abolish the eCV (6 h)-induced accumulation of ubiquitinated proteins (Fig. 1D). To further verify the role of autophagy in eCV-induced accumulation of ubiquitinated proteins, Beas2b cells were preincubated with an autophagy inhibitor, N2,N4-dibenzylquinazoline-2,4 diamine (DBeQ; 25 μM), for 6 h and/or exposed to eCV (6 h). We observed a significant increase in accumulation of ubiquitinated proteins in the insoluble fraction as anticipated upon DBeQ treatment or eCV exposure, and cotreatment of DBeQ with eCV exposure amplified aggregation of ubiquitinated proteins verifying that eCV impairs autophagy to induce aggresome formation (Supplementary Fig. S1; Supplementary Data are available online at
eCV induces autophagy impairment
To further substantiate the eCV-induced accumulation of ubiquitin, and p62 as AB and the potential of carbamazepine in clearance of AB in response to eCV treatment, we transfected Beas2b cells with ubiquitin-RFP and LC3-GFP fusion proteins. Transfected cells were air exposed and/or treated with carbamazepine as controls. In addition, transfected cells, pretreated with 20 μM carbamazepine (6 h) and/or exposed to eCV (1 h), showed significant increase in red ubiquitin-positive (p < 0.01) bodies and green LC3-positive autophagy bodies (p < 0.001); both were found to be significantly (p < 0.001) colocalized upon eCV exposure. Significant (p < 0.01) decrease in colocalization of red ubiquitin-positive bodies and green LC3-positive autophagy bodies was observed after eCV-exposed cells were pretreated with carbamazepine for 6 h (Fig. 2A). To evaluate the effect of carbamazepine in clearance of aggresome bodies, cells were immunostained and fluorescence microscopy was performed to visualize the localization of ubiquitin and the aberrant autophagy marker p62 in aggresome bodies upon eCV exposure. Our microscopic analysis showed significant increase in ubiquitin (p < 0.001) and p62 (p < 0.001) levels on eCV exposure. Further observation revealed significant colocalization of ubiquitin and p62 (p < 0.001) as aggresomes around the perinuclear region. Preincubation of carbamazepine decreased (p < 0.001) ubiquitin and p62 as AB around the perinuclear region. This observation was compared to air-exposed or carbamazepine-treated cell control groups that showed no significant increase in aggresome bodies (Fig. 2B). This study verifies the efficacy of carbamazepine's autophagy induction in alleviating eCV-induced aggresome formation.

An autophagy inhibitor chloroquine is a lysosomotropic agent that prevents fusion of autophagosomes to lysosomes. To affirm that eCV induced aberrant autophagy, Beas2b cells were air exposed, pretreated with 90 μM chloroquine for 12 h, and/or exposed to eCV for 1 h. Beas2b cells were immunostained with the aberrant autophagy marker p62. Fluorescence microscopic analysis showed significant increase (p < 0.001) in p62 immunostaining in Beas2b cells exposed to eCV (1 h) in comparison to air-exposed control cells as anticipated. Treatment with an autophagy inhibitor chloroquine showed significant increase (p < 0.001) in p62 immunostaining in comparison to room air-exposed control. Moreover, a 1.4-fold increase (p < 0.05) in p62 immunostaining was observed when Beas2b cells were pretreated with chloroquine (90 μM; 12 h) and exposed to 1 h eCV (Fig. 2C) in comparison to eCV exposure. This result was supported by fluorescence microscopic visualization of autophagosome and autolysosome formation by autophagy flux assay using the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B Kit. By transducing Beas2b cells with 30 particles per cell of the BacMam reagent, containing LC3B tagged to acid-sensitive green fluorescent protein (GFP) and an acid-insensitive red fluorescent protein (RFP), we were able to dissect the formation of autophagosomes (emission of RFP and GFP fluorescence, or yellow) and autolysosomes (emission of RFP fluorescence) upon eCV exposure and/or treatment with 90 μM chloroquine. We observed significantly bright RFP and GFP fluorescence as puncta bodies and their colocalization in Beas2b cells upon 1 h eCV exposure (p < 0.01) in comparison to air-exposed cells. Treatment with an autophagy inhibitor, chloroquine, showed significant (p < 0.001) increase in colocalization of RFP and GFP fluorescence puncta bodies (yellow). Moreover, a significant (p < 0.01) 2.3-fold increase in bright RFP and GFP fluorescence as puncta bodies was observed when Beas2b cells were pretreated with chloroquine (90 μM; 12 h) and exposed to 1 h of eCV (Fig. 2D). Next, we selected cysteamine, the antioxidant (19, 61) and autophagy inducer (37), to investigate its efficacy in controlling eCV-induced impaired autophagy. As shown in Figure 2E, brighter GFP and RFP fluorescence puncta bodies and their colocalization (p < 0.001, yellow) was observed upon eCV exposure (1 h). Pretreatment of Beas2b cells with 250 μM cysteamine for 6 h significantly (p < 0.001) reduced colocalization of GFP and RFP fluorescence puncta bodies. These results validate our previous observation that eCV impairs autophagy that can be rescued by cysteamine, an antioxidant and autophagy inducer.
eCV induces colocalization of protein aggresomes, p62 and VCP, as aggresome bodies
Since we observed that eCV (1 h) exposure increased p62 and VCP protein levels in Beas2b cells and these were translocated to the insoluble fraction upon 6 h eCV exposure, we evaluated the mechanism and if eCV actually induces p62-VCP protein–protein interaction. Immunoprecipitation (IP) of VCP from eCV-exposed (1 h) soluble protein lysates showed significant (p < 0.05) increase in p62-VCP protein–protein interaction through Western blots of IP samples. Similarly, in reverse IP by p62, eCV-exposed (1 h) soluble protein lysates showed significant (p < 0.05) increase in VCP-p62 protein–protein interaction through Western blot of IP samples (Fig. 3A), verifiying that eCV induces protein–protein interaction of p62-VCP in AB upon autophagy impairment and not just colocalization. This experiment was further supported by microscopic visualization of aggresome bodies using the ProteoStat® Aggresome Detection dye. Beas2b cells were exposed to air or pretreated with cysteamine and/or exposed to eCV (1 h). The increase in aggresome formation (red fluorescence), p62 levels (green fluorescence), and their colocalization was observed in eCV (1 h) exposed Beas2b cells as compared to air exposure. As a positive control, treatment of Beas2b cells with 5 μM MG132 (12 h) showed significant increases in aggresome bodies that were p62 positive. Moreover, pretreatment of eCV-exposed (1 h) cells with cysteamine (250 μM; 6 h) resulted in a significant decrease in colocalization of aggresome/p62 that resembled air-exposed and cysteamine controls (Fig. 3B), verifying the antioxidant potential of cysteamine in restoring autophagy and inhibiting aggresomes in eCV exposed cells.
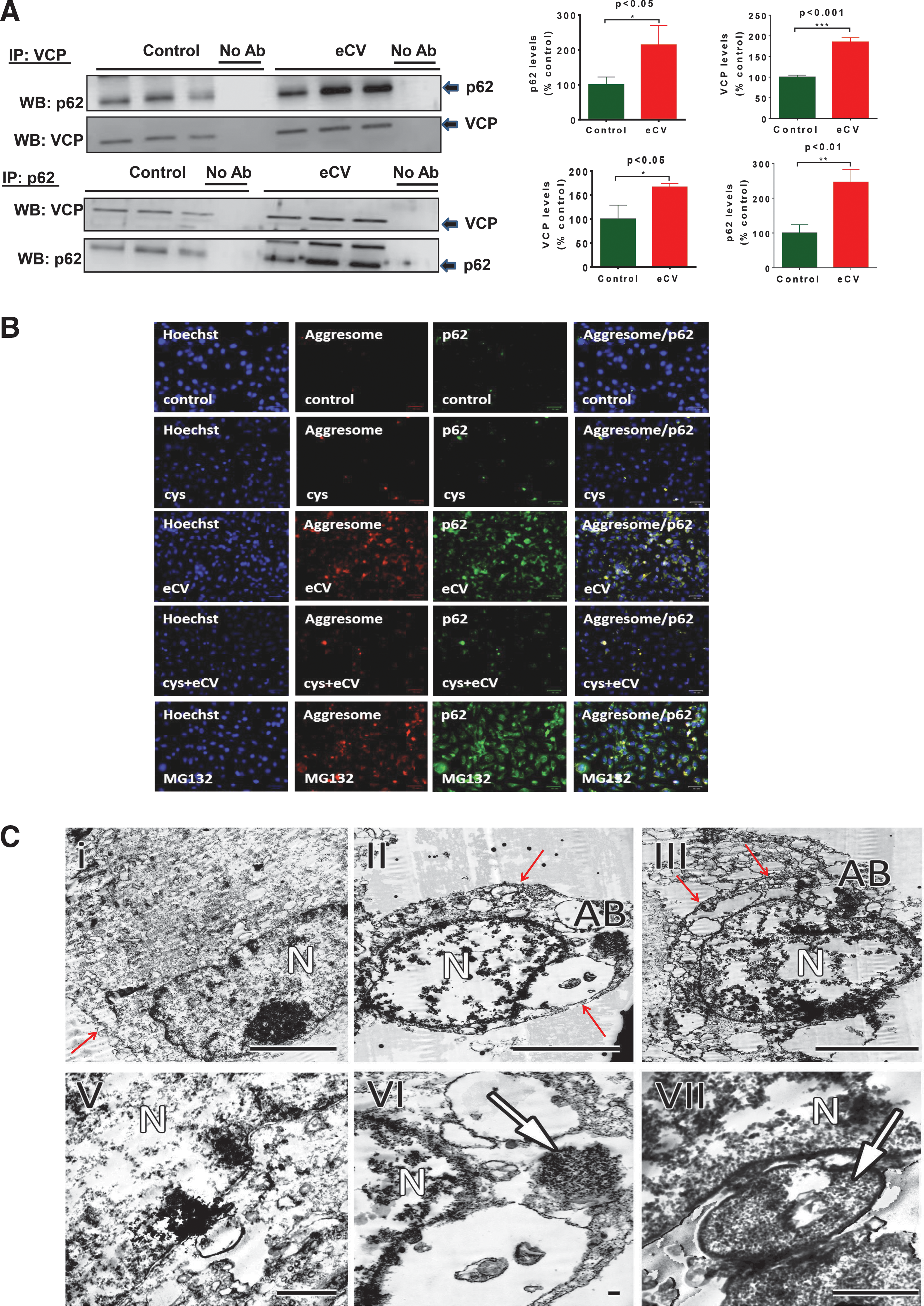
Next, we used transmission electron microscopy (TEM) analysis, and observed AB around the nuclei and verified that 1 and 3 h of eCV exposure induced aggresome formation in contrast to room air-exposed cells (Fig. 3C, I–VI). We also observed that greater eCV exposure initiated cytoplasmic thinning and membrane disintegration subsequent to aggresome formation, indicative of apoptotic morphology (Fig. 3C, III and VII). Data verified the presence of eCV-induced AB and showed the state of cells undergoing apoptosis.
eCV exposure induces aggresome formation in murine lungs
To confirm the in vitro results for our preliminary studies, we used acute eCV-exposed (three times 1-h eCV exposure over 24 h; Fig. 4A) C57BL/6 mice to verify if eCV-induced aberrant autophagy and aggresome formation in murine lungs are also the mechanism for decreased cell viability and inflammation. In a preliminary analysis using flow cytometry, we first standardized the eCV dose that induces levels of CD4+ T cells and macrophages (Mac-1) while inhibiting cell viability (propidium iodide [PI] staining) in broncheoalveolar lavage fluid (BALF). Moreover, the selected carbamazepine dose was able to control eCV-induced inflammation (CD4+ T cells and macrophages) while restoring cell viability (Supplementary Fig. S2). eCV-exposed lungs were harvested, and soluble and insoluble protein lysates were separated from lung protein homogenates in this experiment. Significant (p < 0.01) increase in ubiquitin accumulation was observed in the soluble protein fraction from eCV-exposed mice in comparison to phosphate-buffered saline (PBS)-vaped mice. In addition, a significant increase in p62 (p < 0.01) and VCP (p < 0.01) protein levels was observed in the soluble protein fraction of acute eCV-exposed mouse lungs (Fig. 4B). However, only mild ubiquitin accumulation was observed in insoluble protein fractions (Supplementary Fig. S3). Although, in vitro data support our hypothesis that eCV-induced aggresome formation and aberrant autophagy would aggravate upon chronic eCV exposures of murine lungs.

Since CD-1 is an outbred mouse that is commonly used in bioassays to mimic variability in human subjects, we sought to use this mouse strain to confirm variability in impact of acute eCV exposure in inducing aggresome formation and the effectiveness of carbamazepine in the clearance of AB. Similar to C57BL/6 mice, significant (p < 0.05) accumulation of ubiquitinated proteins was observed in the soluble protein fraction of eCV-exposed mouse lung lysates. Before acute eCV exposure, intratracheal administration of carbamazepine significantly (p < 0.01) decreased the accumulation of ubiquitinated proteins compared to eCV-exposed mice (Fig. 4C). No significant change in accumulation of ubiquitinated proteins was observed in the insoluble protein fraction (Supplementary Fig. S3), as anticipated for acute exposures.
To determine if acute eCV exposures can modulate protein–protein interaction of p62-VCP, soluble protein fractions were used for IP of VCP followed by p62 IB. Data showed significant (p < 0.05) increase in p62-VCP protein–protein interaction even with acute eCV exposure. Similarly, IP of p62 showed significant (p < 0.05) increase in VCP-p62 protein–protein interaction on acute eCV exposure (Fig. 4D), thus supporting our in vitro finding that eCV induced protein–protein interaction of p62-VCP as AB via autophagy impairment. Taken together, these findings substantiate our preliminary in vitro results and indicate the central role of eCV-mediated autophagy impairment that induces aggresome formation as a potential mechanism for inflammatory–oxidative stress, apoptosis, and/or senescence that warrants future validation in a chronic eCV exposure murine model.
eCV exposure induces apoptosis and cellular senescence via autophagy impairment
Flow cytometry was performed to verify whether carbamazepine attenuates the eCV-induced ubiquitin and p62 accumulation in Beas2b cells. As anticipated, we found a significant attenuation in accumulation of ubiquitin and p62 upon carbamazepine pretreatment of Beas2b cells exposed to eCV for 1 h (Fig. 5A). We evaluated whether eCV-induced accumulation of ubiquitinated protein could induce proteasomal dysfunction and cellular apoptosis. We found that Beas2b cells exhibited a time- and dose-dependent decrease in proteasomal activity upon 1 h (p < 0.05), 3 h (p < 0.001), and 6 h (p < 0.001) of eCV exposures (Fig. 5B). Data suggests that accumulation of ubiquitinated proteins is a result of eCV-induced proteosomal dysfunction. The proteasomal inhibitor MG132 (1 μM), used as a positive control, showed significant decrease in proteasomal activity (p < 0.001), verifying the efficacy of the assay. We anticipated that eCV-induced autophagy/proteasome dysfunction and aggresome formation can impact cell viability. Moreover, it is known that CS exposure leads to decline in cell growth by inducing apoptosis and cellular senescence (5, 41, 43, 50, 56, 60, 66). We suspected that eCV exposure results in cellular cytotoxicity via similar mechanisms, based on a recent study (36). To determine the cytotoxic effect of eCV, Beas2b cells were exposed to eCV for 1, 3, and 6 h. Cell viability was measured by monitoring the reduction of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) to colored formazan compounds. Compared to air exposure, significant decrease in cell viability was observed at 1 h (p < 0.01), 3 h (p < 0.001), and 6 h (p < 0.001) (Fig. 5C) in a time- and dose-dependent manner; inhibition of cell viability indicates the significant cytotoxicity of eCV. We were not confident whether this eCV-induced decrease in cell viability was the result of apoptotic cell death. Hence, we investigated caspase-3/7 activity after eCV exposure of Beas2b cells and found only a modest increase (p < 0.01) in caspase-3/7 activity resulting from 1 h of eCV treatment (Fig. 5D). Nonetheless, eCV-induced caspase-3/7 activity was significantly elevated (p < 0.001) with longer 3-h exposures, as anticipated. These results clearly indicate that eCV induces apoptotic cell death (caspase-3/7) and inhibit cell proliferation (MTT) as a mechanism to decrease cell growth.
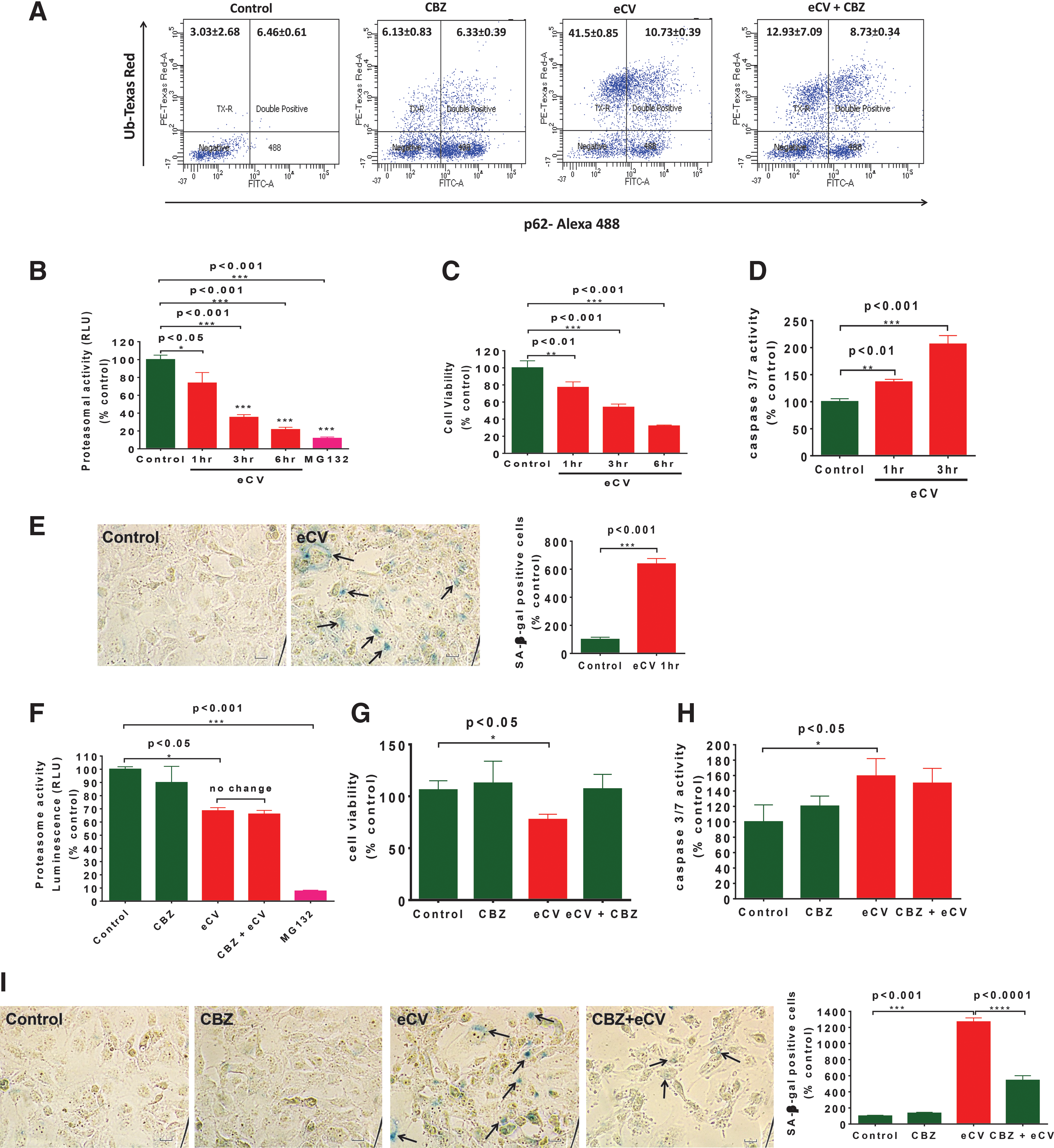
To determine its impact on cell viability or survival, we evaluated whether exposure to eCV induced senescence-associated β-galactosidase (SA-β-gal) activity. We observed a significant (p < 0.001) increase in the percentage of SA-β-gal-positive cells after eCV exposure (1 h) in comparison to air-exposed control cells, suggesting that even short (1 h) exposures to eCV are enough to initiate senescence (Fig. 5E). We had previously observed (Fig. 1C, D) that the autophagy inducer carbamazepine rescued eCV-induced aggresome formation and aberrant autophagy, although we questioned whether carbamazepine could restore eCV-impaired proteasomal activity. To quantify changes in proteosomal activity, Beas2b cells were preincubated with carbamazepine followed by eCV (1 h) exposure. We observed that carbamazepine pretreatment resulted in no significant restoration of the eCV mediated decrease in proteasomal activity (p < 0.05). The proteasomal inhibitor MG132, used as a positive control, showed a significant (p < 0.001) decrease in proteasomal activity (Fig. 5F) as anticipated. Although eCV induced significant (p < 0.05) decrease in Beas2b cell viability (MTT), pretreatment with carbamazepine before eCV exposure restored cell viability to levels seen in air-exposed controls (Fig. 5G). Next, we sought to determine the potential of carbamazepine to inhibit apoptotic cell death and senescence. We found that short-term eCV exposure slightly induces caspase-3/7 activity; carbamazepine treatment exhibited a trend toward decrease that was not significant. Moreover, no significant difference in caspase-3/7 activity was observed among air-exposed or carbamazepine-treated control groups (Fig. 5H). To confirm that eCV induced apoptosis, flow cytometry was used to quantitatively analyze the number of PI-stained apoptotic Beas2b cells that had been preincubated with carbamazepine and/or exposed to eCV (6 h). As anticipated, significant cell death was observed upon eCV (6 h) exposure and carbamazepine treatment attenuated eCV-induced cell death. The proteasomal inhibitor MG132, used as a positive control, showed a significant increase in cell death as expected (Supplementary Fig. S4). eCV exposure (1 h) significantly increased (p < 0.001) the number of SA-β-gal-positive cells, and preincubation of eCV-exposed (1 h) Beas2b cells with carbamazepine significantly (p < 0.001) attenuated SA-β-gal-positive staining (Fig. 5I). Overall, these initial observations suggest that autophagy plays an important role in eCV-induced cytotoxicity.
eCV induces inflammatory–oxidative stress via autophagy impairment
The transcription factor NFκB is activated by inflammatory stress or response (33) and is thereby used as an indicator to quantify the impact of eCV exposure upon inflammation. Peroxynitrate-mediated nitration of tyrosine produces nitrotyrosine adduct(s), an oxidative stress marker (15); this was used together with NFκB to evaluate whether eCV induces inflammatory–oxidative stress via autophagy impairment. Beas2b cells, preincubated with carbamazepine for 6 h, were exposed to eCV for 1 h. IB analysis showed significant increase in total protein NFκB levels (p < 0.001) and protein bound nitrotyrosine (p < 0.001) adduct, a product formed by peroxynitrite-mediated nitration of protein tyrosine residues, (Fig. 6A) upon eCV exposure, as anticipated based on a recent study (36). The specificity of the 3-nitrotyrosine antibody was tested using whole-cell protein lysates from Beas2b cells exposed to air or 10% cigarette smoke extracts (CSE) for 24 h (Supplementary Fig. S5); the result was a single protein-bound nitrotyrosine adduct similar to the vendor's datasheet (Santa Cruz Biotech). We also observed the attenuation of NFκB (p < 0.05) and protein-bound nitrotyrosine adduct levels (p < 0.01) in carbamazepine preincubated cells exposed to eCV, confirming the potential of carbamazepine in regulating eCV-induced inflammatory–oxidative stress.
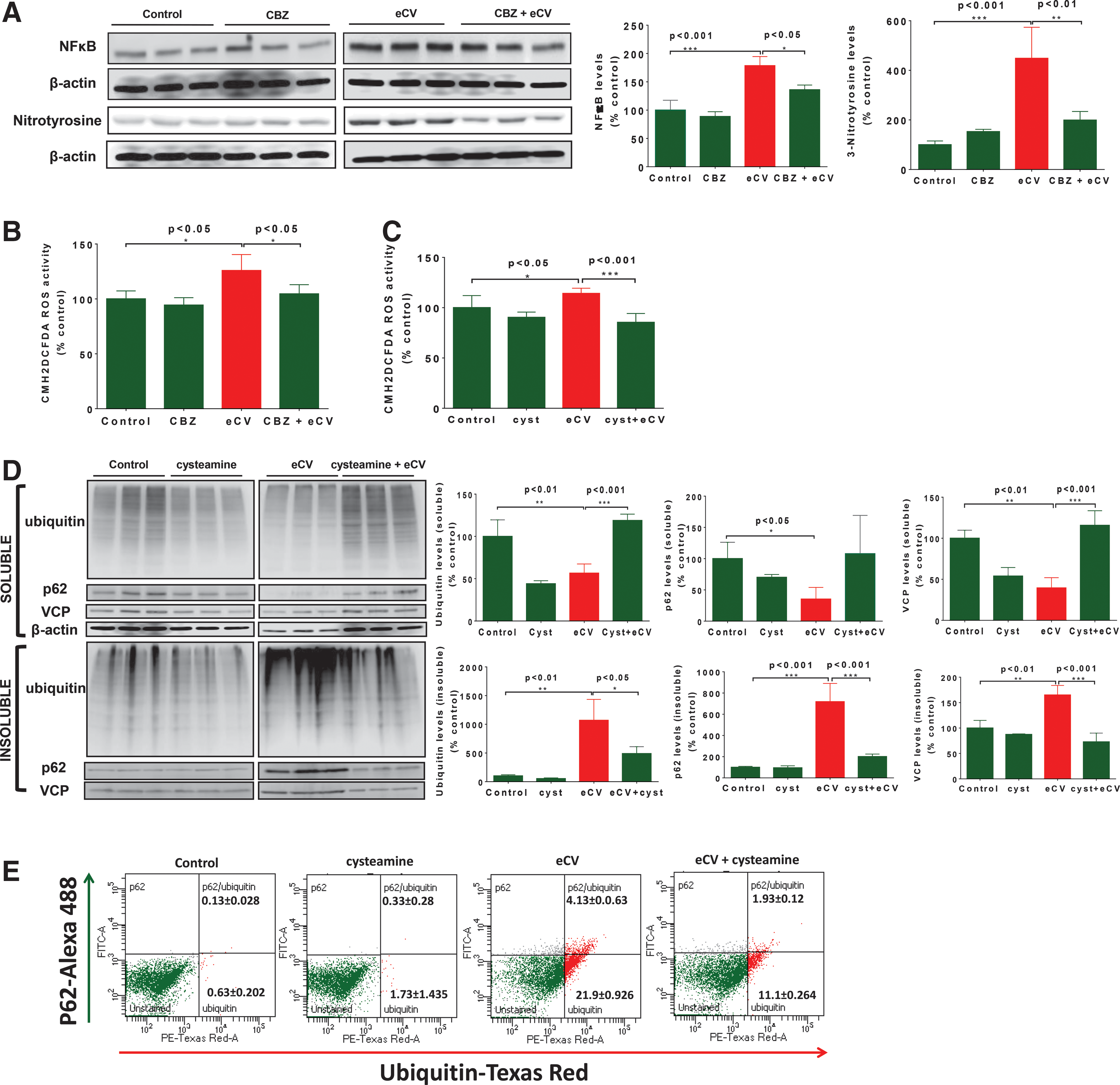
The cellular oxidative stress was analyzed to quantify intracellular ROS using an ROS indicator, CMH2DCFDA. After exposing Beas2b cells to eCV for 1 h, we observed a significant (p < 0.05) increase in intracellular ROS levels in comparison to air-exposed control. Preincubation with carbamazepine significantly (p < 0.05) decreased eCV-induced ROS production to air-exposed control levels (Fig. 6B). Next, we determined the efficacy of cysteamine in controlling eCV-induced oxidative stress. As illustrated in Figure 6B, eCV significantly (p < 0.05) increased intracellular ROS levels and preincubation with cysteamine significantly (p < 0.001) reduced eCV-induced (1 h) intracellular ROS levels in Beas2b cells. We observed a 1.5-fold reduction in intracellular ROS levels upon cysteamine pretreatment (Fig. 6C) before eCV exposure. We next investigated whether cysteamine treatment results in rescuing eCV-induced aggresome formation and aberrant autophagy. In parallel, Beas2b cells were incubated with cysteamine and exposed to eCV for 6 h. The significant accumulation of ubiquitinated proteins (p <0.01) and an increase in p62 (p < 0.001) and VCP (p < 0.01) levels were observed in the insoluble protein fraction upon eCV exposure (6 h); cysteamine pretreatment of eCV-exposed cells significantly inhibited ubiquitin (p < 0.05), p62 (p < 0.001), and VCP (p < 0.001) accumulation (Fig. 6D), supporting cysteamine's antioxidant and autophagy inducer potential to alleviate aggresome formation. We confirmed these results by flow cytometry-based quantitative analysis and observed that cysteamine can control eCV-induced accumulation of ubiquitinated proteins in p62-positive autophagy bodies (Fig. 6E).
Discussion
Electronic cigarettes (e-cigarettes) have been recently introduced to consumers of regular tobacco cigarettes as a safer alternative assuming that it has the potential to enable tobacco cigarette smokers quit smoking. One of the major components of e-cigarettes is nicotine; this accounts for around 3–5% of regular cigarette components and is responsible for inducing addiction in tobacco smokers. Since 2007, electronic nicotine delivery systems (e-cigarettes) have been sold in U.S markets as a medical device without regulation by the FDA, despite known nicotine potential to cause drug addiction.
Although electronic cigarettes are marketed as a safer alternative to conventional cigarettes, recent studies suggest that even if eCV has different composition and toxicity, it would not be correct to label one as safer in comparison to the other. In the present study, we designed in vitro and in vivo experimental models to specifically identify and evaluate the impact of eCV exposure upon central mechanism(s) known to regulate inflammatory–oxidative stress, apoptosis, and emphysema in COPD subjects.
Our group and others have recently identified that tobacco CSE induce proteostasis/autophagy impairment (41, 51, 56) and accumulation of ubiquitinated protein aggregates (51) in perinuclear spaces; both of these serve as regulatory mechanisms associated with inflammatory–oxidative stress responses, apoptosis, and emphysema pathogenesis in COPD. We validated the role of CS-induced autophagy impairment in COPD-emphysema progression in a recent study by using experimental models and an autophagy inducer or inhibitor (51). To evaluate eCV's potential risk of inducing COPD and lung cancer in comparison to tobacco cigarettes, we first evaluated whether the impact of eCV upon protein processing (proteostasis) is similar to CS exposure. We found that even acute eCV vaping (5 or 15 min) of media used to treat Beas2b cells resulted in accumulation of ubiquitinated proteins and increase in p62 and VCP levels that were translocated from soluble to insoluble protein fractions upon changes in eCV exposures (1 or 6 h; Fig. 1A); this suggests that eCV-induced autophagy impairment and induction of VCP-mediated retrograde translocation serve as a potential mechanism in initiating aggresome formation. Although modulation of protein synthesis in response to acute doses of eCV was not (Fig. 1B) similar to CS exposure (51), this does not rule out the impact of chronic eCV exposure upon protein synthesis. Furthermore, our recent findings showed an increase in the aberrant autophagy marker p62 upon CS exposure (5, 6, 51), suggesting that impaired autophagy serves as a mechanism to induce aggresome formation in COPD-emphysema. The studies from Fuji et al. (21) and Monick et al. (42) support our findings that p62 and Ub are accumulated after CS exposure in various models and confirm that CS impairs autophagy, contrary to earlier reports (11) suggesting that CS induces autophagy. We also evaluated and verified that eCV induces aggresome formation and that impaired autophagy can be controlled by the drug carbamazepine (Fig. 1C, D and Supplementary Fig. S1), an autophagy inducer that was approved by the FDA to treat epilepsy and bipolar disorder (18, 22). Based upon these preliminary findings, we performed experiments by using various short-term exposure models; we maintained the understanding that measurement of reference compounds will need to be conducted in media preparations to validate consistency of doses, requiring the standardization and evaluation of such compounds for future longitudinal studies.
Induction of autophagy leads to sequestration of cytoplasmic contents and organelles inside the autophagosome that fuse with the lysosome (autolysosome) for the degradation of cargo along with the inner membrane of autophagosomes. Microtubule-associated light chain protein-3 (LC3B) is localized in inner and outer membranes of autophagosomes and is considered to be an autophagosomal marker (31, 34). The aberrant autophagy marker p62 has been demonstrated to increase aggregation of ubiquitinated proteins for autophagy degradation (32, 62). Based upon our recent study with tobacco cigarettes (51), it was predicted that eCV induces colocalization of a critical proteostasis/autophagy mediator, VCP, and an aberrant autophagy marker, p62. We used a fluorescence microscopy-based autophagy reporter assay to verify the impact of eCV upon autophagy and aggresome formation. Our data indicate that eCV exposure resulted in accumulation of autophagosomes (LC3B) and increase in p62 levels that were colocalized with ubiquitin as AB (Figs. 2A, B and 3B). This was supported by p62-VCP protein–protein interaction in both Beas2b cells and murine lungs (Fig. 3A and C57BL6/7; Fig. 4D) and by TEM analysis (Fig. 3C), in which we observed AB and membrane disintegration, upon Beas2b cell exposure to high doses of eCV that is indicative of apoptotic morphology. In additional studies, we used chloroquine (Fig. 2C, E) with the aim of demonstrating impaired autophagy via the absence of autolysosomes that are important in the process of functional autophagy. To measure carbamazepine's relative potential to clear eCV-induced AB, we compared its efficacy to another autophagy inducer, cysteamine; this drug is under consideration for use as CF (cystic fibrosis) therapeutics and its potential appears to overshadow carbamazepine. Cysteamine has known mucolytic, antibacterial, antibiofilm, and antioxidant potential (37, 49), and we are currently evaluating it further in a follow-up study. As discussed above, cysteamine-based therapeutics, such as FDA-approved Procysbi and Lynovex, have been clinically evaluated in ΔF508-CF subjects. eCV-induced accumulation of autophagosomes was reduced upon both carbamazepine and cysteamine treatment, confirming cysteamine's role as an autophagy inducer (Fig. 2B, E).
In additional murine experiments, we found that even acute eCV exposures of mice can induce, aggregation of ubiquitinated proteins, and p62 and VCP protein levels in soluble protein fractions (Fig. 4B); these are potentially involved in sequestration of ubiquitinated proteins to AB upon repeated/chronic eCV exposures and we aim to validate this in a subsequent study. Considering the known differences in susceptibility to CS exposure in different murine strains, we also verified these findings in another murine strain (CD-1). We found that acute eCV exposure of the CD-1 strain results in relatively less accumulation of ubiquitinated proteins in soluble protein fractions (Fig. 4C). We did not observe the sequestration of ubiquitinated proteins to AB (insoluble fraction; Supplementary Fig S2) upon acute exposure. Based upon our studies with acute/chronic CS exposure murine models (41, 51), we anticipate the sequestration of ubiquitinated proteins upon repeated e-cigarette vaping (chronic exposure).
A growing body of evidence indicates the role of oxidative stress, apoptosis, and aging in COPD-emphysema pathogenesis (17, 38, 39, 52, 65). In this study, we evaluated the impact of secondhand eCV exposure on airway and found a time/dose-dependent decrease in proteasomal activity and cell proliferation (Fig. 5B, C); concurrently, we also observed induction of apoptosis and senescence (Fig. 5D, E). Furthermore, treatment with an autophagy inducer, carbamazepine, rescued cell growth (Fig. 5G) upon eCV exposure and moderately decreased senescence (Fig. 5I); however, complete inhibition of apoptosis was not observed at the selected carbamazepine dose (Fig. 5H). We also observed that carbamazepine had no impact upon restoration of eCV-impaired proteasomal activity (Fig. 5F); this suggests that to overcome insufficient proteasomal activity, carbamazepine induces autophagy to alleviate eCV-induced aggresome formation. However, significant reduction in accumulation of ubiquitinated proteins (Fig. 1C, D) via a modest dose of carbamazepine is sufficient to rescue downstream functional mechanisms such as cellular growth and senescence.
In a recent study, human subjects acutely exposed to acute eCV for 5 min were used to evaluate the impact of eCV upon lung function, oxidative stress, and exhaled nitric oxide (2, 44, 57). The study used 30 healthy e-cigarette-vaping subjects who were compared to 10 control subjects and found an immediate effect of eCV upon airway mechanics and a significant decrease in the fraction of exhaled nitric oxide, suggesting that even acute eCV exposures can induce pathophysiological changes similar to regular tobacco CS exposures (2, 57). The toxicity of nicotine in human subjects is known historically, and recent preliminary studies have specifically evaluated the potential of eCV doses used by subjects who vaped to induce inflammatory–oxidative stress and increase susceptibility to bacterial and viral infections commonly seen in COPD subjects (14, 36, 40, 48, 49, 63). In this study, we found that acute eCV-vaped (5 min) media, used to treat (1 h) Beas2b cells, significantly induced ROS production and increased total NFκB activity and nitrotyrosine protein adduct levels, suggesting their role in eCV-mediated inflammatory–oxidative stress. To verify the roles of autophagy and resulting aggresome formation in eCV-induced inflammatory–oxidative stress, we again used carbamazepine treatment of eCV-exposed cells. We found that carbamazepine can significantly decrease eCV-induced ROS production, total NFκB activity, and nitrotyrosine protein adduct (Fig. 6A, B), suggesting that the role of autophagy in regulating eCV-mediated inflammatory–oxidative stress responses is similar to that for regular tobacco CS. We next verified that eCV induces ROS activity even upon acute exposures (data not shown), similar to a recent study where the impact of eCV was evaluated for both ROS activity and inflammatory cytokines (36). Based on our recent study documenting autophagy impairment as a critical mechanism for CS-induced COPD-emphysema (51), we focused on evaluating whether eCV-induced accumulation of ubiquitinated protein modulates inflammatory–oxidative stress responses via autophagy impairment. We found that autophagy induction in response to carbamazepine can control NFκB- and ROS-mediated inflammatory responses and oxidative stress. Cysteamine is known to alleviate ROS activity via a mechanism that promotes autophagy-mediated clearance of ubiquitinated proteins, specifically ΔF508-CFTR (16, 37). As anticipated, we found that cysteamine can control eCV-induced (5 min, 1 h) ROS activity (Fig. 6C). We and others have recently demonstrated the role of CS exposure in acquired cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction (4, 5, 13, 15). Hence, we predict that the use of cysteamine as an autophagy inducer will allow clearance of ubiquitinated proteins and restoration of eCV-impaired cell membrane CFTR expression (data not shown), warranting further investigation via a chronic eCV exposure model (4, 5, 13, 16, 37, 59). In our preliminary analysis, we have found that cysteamine attenuates eCV-induced accumulation of ubiquitinated proteins, p62 and VCP, and their translocation from soluble to insoluble protein fractions in comparison to air-exposed control (Fig. 6D, E).
In summary, cysteamine can control eCV-impaired primary defect in proteostasis/autophagy and oxidative stress and can also alleviate obstructive lung disease by acting upon mucus buildup and bacterial (Pseudomonas aeruginosa) biofilms. Thus, we are currently further evaluating the efficacy of cysteamine in regulating both CS and chronic eCV-induced oxidative stress, autophagy impairment, and lung disease progression.
Materials and Methods
eCV exposure of cells and mice
The Kanger EVOD (Kangertech) electronic cigarette (e-cigarette) was used for the eCV exposure studies. The e-cigarette cartridge was filled with nicotine (25 mg/ml) prepared in 50:50 (v/v) propylene glycol and vegetable glycerin, as commonly used by e-cigarette smokers. For in vitro experiments, eCV was exposed on Dulbecco's modified Eagle's medium (DMEM) (without fetal bovine serum [FBS]) in a conical flask via a suction created by a motor. Using a standard EVOD e-cigarette device, eCV was vaped upon the media for 5 or 15 min similar to intermittent vaping session. Media were collected and supplemented with 10% FBS (BioExpress) and 1× PSA (penicillin, streptomycin, and amphotericin-B, diluted from 100×; Invitrogen); it was then used to treat Beas2b cells. For in vivo studies, groups of three to four mice were intratracheally (i.t.) instilled with a control vehicle, 0.5% carboxymethylcellulose (CMC; Acros Organics), or an autophagy inducer, carbamazepine (20 mg/kg body weight dissolved in 0.5% CMC; Sigma) (51), for 24 h. The mice were exposed to eCV generated from 4 ml of 25 mg/ml nicotine prepared in propylene glycol and vegetable glycerin (as indicated above in this section) via a nebulizer (Aeroneb® Lab nebulizer system) fixed upon a custom-designed mouse cage. For eCV exposure, operation of the nebulizer, via electric power, turns liquid nicotine mix into vapor that flows directly into the cage. After carbamazepine treatment (t = 0), eCV exposure was repeated thrice (t = 2, 8, and 21 h) for 1 h each and the experiment was terminated at t = 24 h as discussed later (time scale; Fig. 3A).
Murine experiments
The 5–6-week-old C57BL/6 or CD-1 mice (two males and two females for n = 4 groups or one male and two females for n = 3) were used for each experiment, in accordance with our CMU IACUC approved protocols. The mice were exposed to eCV (nicotine in propylene glycol and vegetable glycerin) or PBS, as described previously, and were housed in a clean environment during the course of these studies in our satellite animal housing facility. Carbamazepine [20 mg/kg body weight; (51) or 0.5% CMC control vehicle] was administered to the mice by intratracheal instillation (i.t.) followed by 3× PBS or eCV exposure, as described previously. Mice were sacrificed after 24 h, in accordance with our IACUC approved protocols, and both BALF and lung lysates were collected for Moxi Flow cytometry and IB.
Cell culture, transfection, and reporter assays
The human bronchial epithelial cell line (Beas2b) was cultured in DMEM/F12 media containing L-glutamine (Invitrogen), 10% fetal bovine serum (BioExpress), and 1% PSA (penicillin, streptomycin, and amphotericin B; Invitrogen) at 37°C in 5% CO2 until they were 80% confluent. Cells were preincubated with DBeQ (25 μM; 24 h), carbamazepine (20 μM; 6 h), or cysteamine (250 μM; 6 h); the cells were then exposed to eCV or room air for 1, 3, or 6 h, respectively, for IB and other experiments. For reporter assays, an equal number of cells were transiently transfected with plasmid constructs containing an autophagy-related protein, light chain 3 (LC3II), that was tagged with green fluorescent protein (LC3-GFP) and/or ubiquitin (Ub) that was tagged with red fluorescent protein (Ub-RFP) for 24 h using the K2® Transfection System (Biontex Laboratories GmBH). Images were captured via the ZOE™ Fluorescent Cell Imager (Bio-Rad) that is equipped with a monochrome digital camera, a 20× objective with a 4× field of vision and a 175× to 700× display magnification. Images were captured at a 315× display magnification, where display magnification = size of the image of the object on the LCD display screen/actual size of the object. Briefly, total or display magnification is how large the object appears on the LCD screen. Measurement of screen size (25.6 cm) divided by the actual physical size of the object gives a maximum standard display magnification of 700×. To calculate the size of an object, the measure of the captured image screen is divided by display magnification, 315× (in this case). Scales were autoembedded while capturing each image to enable size calculation from the print size.
Autophagy flux assay
The autophagy flux assay was carried out using the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3 Kit (Molecular Probes), following the manufacturer's instructions. Briefly, 40,000 Beas2b cells were grown overnight in six-well plates and were incubated with 12 μl of BacMam reagent (insect baculovirus with a mammalian promoter containing tandem RFP-GFP-LC3B DNA) for 16 h. Later, cells were treated with chloroquine (90 μM; 12 h) or cysteamine (250 μM; 6 h) and/or exposed to eCV for 1 h. The cells were washed twice with 1× PBS and visualized under the ZOE Fluorescent Cell Imager (Bio-Rad) as described previously. The change in number of LC3B-positive autophagosomes and autolysosomes in merged images was quantified using ImageJ 1.49o (NIH) software.
Western blot analysis
After eCV exposure, cell or lung tissues were harvested and lysates were prepared with the use of RIPA buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing 1× protease inhibitor cocktail (Pierce). Lung tissues were homogenized and sonicated on ice for 3–5 min and with 10–15-s pulses, respectively. Cultured cells were washed with ice-cold PBS and directly lysed with an ice-cold lysis buffer on plates constantly shaken for 10 min at room temperature, followed by gentle scrapping. Soluble and insoluble protein fractions were separated by centrifugation at 13,000 rpm for 15 min at 4°C. Total protein lysates were used to quantify protein using the Bradford Protein Assay Kit. Soluble (50 μg) and/or insoluble protein fractions (pellet, isolated from equal amount of protein ∼500 μg, for each sample) were separated in 10% SDS-PAGE and transferred to 0.45 μm nitrocellulose membranes (Bio-Rad) for IB. Nonspecific binding sites on membranes were blocked with 5% nonfat milk at room temperature for 1 h on a shaker. Membranes were incubated overnight at 4°C with mouse monoclonal ubiquitin (1:1000; Santa Cruz), rabbit polyclonal p62 (1:500; Santa Cruz), rabbit polyclonal VCP (1:500; Santa Cruz), rabbit polyclonal NFκB (1:500; Santa Cruz), and rabbit polyclonal Nitrotyrosine (1:500; Santa Cruz) as primary antibodies. Later, the membranes were washed several times (3×) with a PBS-Tween buffer (0.5% tween-20 in 1× PBS) and were incubated with 1:5000 goat horseradish peroxidase-conjugated anti-mouse antibody (Bio-Rad) or 1:5000 goat horseradish peroxidase-conjugated anti-rabbit antibody (Bio-Rad) for 1 h at room temperature, followed by 3× washing as described previously. The Clarity™ Western ECL substrate (Bio-Rad) was used for chemiluminescence detection of immunoblots using a LI-COR C-DiGit™ Blot Scanner. Images were captured with Image Studio Lite 5.0, and ImageJ 1.49o (NIH) software was used to quantify changes in immunoblots by densitometry analysis. The densitometry analysis of each protein is shown as a percentage change from the control group and is normalized to the average density of β-actin (soluble fraction only) for each selected sample group.
Immunofluorescence, microscopy, and flow cytometry
Beas2b cells were grown on sterile coverslips in a six-well plate, or plates, directly. After treatments, cells were washed once with PBS and fixed with 4% paraformaldehyde, followed by permeabilization with 0.2% Triton X-100. Cells were blocked with 5% normal goat or donkey serum for 1 h followed by incubation with primary antibodies, 0.8 μg mouse monoclonal Ubiquitin (clone AC-15; Santa Cruz), and 0.8 μg rabbit polyclonal p62 (Santa Cruz), prepared in 5% goat or donkey serum (Jackson Immunological), 0.4% BSA (Sigma), and 0.2% Triton X-100 (Sigma) solution overnight at 4°C. Detection of primary antibodies was performed using 0.4 μg goat anti-rabbit Alexa 488 (Santa Cruz) and 0.4 μg donkey anti-mouse Texas Red (Santa Cruz) secondary antibodies. Nuclei were counterstained with Hoechst 33342 (1 μg/ml) for 5 min and were washed with 1× PBS. Coverslips were mounted on slides with 30% glycerol, and images were acquired using the ZOE Fluorescent Cell Imager, as described previously. To count positively stained cells or bodies, fluorescence images were analyzed using ImageJ 1.49o (NIH) software. For flow cytometry analysis, Beas2b cells were preincubated with carbamazepine (20 μM) or cysteamine (250 μM) for 6 h and exposed to eCV or room air for 1 h, as described previously, followed by fixation and permeabilization via application of Fix–Perm solution (BD Biosciences) for 20 min at 4°C and an ice-cold 1× PBS wash. Next, cells were blocked for 20 min with 3% normal goat or donkey serum; this was followed by immunostaining for 20 min on ice with ubiquitin mouse monoclonal (1 μg; Santa Cruz) and p62 rabbit polyclonal (1 μg; Santa Cruz) antibodies prepared in 100 μl of cell staining buffer (BioLegend). Cells were washed with ice-cold 1× PBS, and ubiquitin and p62-positive cells were detected by incubating samples with Alexa 594 anti-mouse (1 μg; Santa Cruz) and Alexa 488 anti-rabbit (1 μg; Santa Cruz) secondary antibodies. The stained cells were washed with ice-cold 1× PBS and resuspended in 1% paraformaldehyde. For PI staining, 5 μl of PI staining solution (Biotool) was added to 100 μl of cell suspension and incubated at room temperature for 15 min, followed by addition of binding buffer. Results were acquired and analyzed using the BD FACSAria flow cytometer and BD FACSDiva 8.0 analysis software. Moxi Flow™ protocols for BALF cell analysis for macrophages, CD4+ T cells, and cell proliferation PI staining are described in Supplementary Methods.
Protein aggresome and p62 colocalization microscopy
The ProteoStat Aggresome Detection Kit (Enzo Lifesciences) was used to perform this assay. The procedure was followed according to the manufacturer's instruction. Briefly, Beas2b cells were grown on coverslips in six-well plates. After treatments, cells were washed twice with 1× PBS and fixed with 4% paraformaldehyde for 30 min. Cells were then washed twice with 1× PBS and permeabilized (0.5% Triton X-100 and 5 mM EDTA) for 30 min, followed by staining with ProteoStat Aggresome Detection reagent and Hoechst 33342 nuclear stain for 30 min. To study aggresome/p62 colocalization, cells were incubated with 5% normal goat serum for 1 h, followed by incubation with primary antibody, 0.8 μg rabbit polyclonal p62 (Santa Cruz), prepared in 5% goat serum (Jackson Immunological), 0.4% BSA (Sigma), and 0.2% Triton X-100 (Sigma) solution, for 1 h at room temperature. Detection of primary antibodies was performed by using 0.4 μg goat anti-rabbit Alexa 488 (Santa Cruz) antibody. Nuclei were counterstained with Hoechst 33342 (1 μg/ml) for 5 min and were washed with 1× PBS. Coverslips were mounted upon slides with 30% glycerol, and images were acquired by using the ZOE Fluorescent Cell Imager (Bio-Rad), as described previously.
Transmission electron microscopy
Beas2b cells grown on six-well plates were exposed to air or eCV for 1 or 3 h. Cells were washed with 1× PBS and were harvested by low-speed centrifugation (1250 rpm, 10 min). Next, cells were fixed with 1.15% glutaraldehyde and 1% tannic acid in a cacodylate buffer (pH 7.4) overnight in microcentrifuge tubes at 4°C while rotating. The following morning, samples were washed with cacodylate buffer and fixed in 1% osmium tetroxide in deionized water for 1 h at room temperature. The samples were also washed with deionized water for 5 min and stained with 2% uranyl acetate for 1 h at room temperature. Samples were then dehydrated in a graded series of ethanol concentrations (50%, 70%, 80%, 90%, 100%) and were embedded in Spurr's resin (Electron Microscopy Sciences). The polymerized resin blocks were mounted upon the RMC PowerTome to generate 70 nm thick sections that were collected on copper grids and stained with 2% uranyl acetate for 30 min and Reynold's lead citrate for 5 min. The Philips CM 10 Transmission Electron Microscope was used to capture images of stained sections.
Cell viability assays
Beas2b cells (10,000 cells per well) were grown on 96-well plates for 24 h and were preincubated with carbamazepine (20 μM; 6 h) and/or exposed to eCV as described previously. The cells were incubated with 1.2 mM MTT (Life technologies) for 4 h at 37°C and 5% CO2. Media containing MTT were replaced with SDS-HCl (0.01% SDS in 0.01M HCl) to solubilize formazan crystals; the absorbance was measured at 570 nm. The data were analyzed as a percent change from the control by using the SoftMax Pro 6.0 software.
Proteasomal activity and caspase-3/7 assay
Beas2b cells (10,000 cells per well) were grown overnight in 96-well plates. Cells were pretreated with 20 μM carbamazepine for 6 h and/or exposed to eCV or room air for 1, 3, or 6 h. Caspase-3/7 activity was assessed using the commercially available Caspase-Glo® 3/7 Kit (Promega) to measure the dissociation of immunogenic tripeptide z-DEVD. Proteasomal activity was measured by using the Proteasome-Glo™ Assay Kit (Promega) to evaluate cleavage of the chymotrypsin-like substrate Suc-LLVY-aminoluciferin by quantifying the resulting luminescent signal produced by the luciferase reaction. These assays were performed as per the manufacturer's (Promega Corp.) instructions, recently described (55).
Senescence assay
The Senescence Cells Histochemical Staining Kit (Sigma-Aldrich) was used according to the manufacturer's instruction. Briefly, 0.5 million Beas2b cells were plated on six-well plates overnight at 37°C and 5% CO2. After treatments and exposure, cells were fixed with 10% fixation buffer for 7 min at room temperature. Next, cells were washed with PBS and incubated with X-gal staining solution at 37°C overnight. SA-β-galactosidase-positive (blue) cells were counted under the Nikon ECLIPSE TS100 microscope equipped with the Infinity 2 camera (Lumenera Corporation) by using Infinity Analyze 6.4.1 software, and data are expressed as a percentage change in number of senescent cells in comparison to the control group.
ROS production assay
The commercially available dye, CMH2DCFDA (Invitrogen), was used to measure the production of reactive oxygen species (ROS) in eCV-exposed Beas2b cells. Briefly, 50,000 cells per well were grown overnight in 96-well plates. Cells were exposed to room air or eCV for 1 h and were preincubated with carbamazepine (20 μM, 6 h). Next, the cells were washed with Hank's Balanced Salt Solution (1× HBSS buffer containing calcium and magnesium) and incubated with 10 μM CMH2DCFDA for 20 min at 37°C in the dark followed by 2× HBSS wash. Changes in fluorescence were quantified using excitation (495 nm) and emission (515 nm) wavelengths recorded by the SpectraMax M5e (Molecular Probes) plate reader and SoftMax Pro 6.0 analysis software.
Data analysis
Data were analyzed using GraphPad Prism 4.0/6.0 software (GraphPad Prism). Results represent mean ± SEM and are shown as either percent change from the control group or actual concentration, as indicated. Statistical analysis was performed by using one-way analysis of variance followed by Tukey's post hoc test or one/two-tailed t tests to identify significant changes between the treatment groups, as indicated. When p-values were <0.05, this was considered a significant difference.
Footnotes
Acknowledgments
The authors would like to thank Philip Oshel, Director of the Electron Microscopy Core Facility, Central Michigan University, for help with the Transmission Electron Microscopy experiment. They also thank Carolyn Kaya, FACS technician, for help during the flow cytometry experiments.
Funding: NV, CVW & RH were supported by the Flight Attendant Medical Research Institute's (FAMRI) Young Clinical Scientist Award (YCSA_082131) to N.V. N.V. was also supported by the National Institute of Health (NIH, U54CA141868 and R0IHL59410) grant during this period. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.