
Abstract
Introduction
O
Dynamin-related protein 1 (Drp1) and NOX2 oxidase reciprocally regulate each other, contributing to NLRP3 inflammasome activation in endothelial dysfunction. Corosolic acid (CRA) inhibited Drp1 activation by phosphorylation of Drp1 (Ser637) in an AMP-activated protein kinase (AMPK)-dependent manner and attenuated oxidative stress by suppression of NOX2 expression. As a downstream result, CRA inhibited NLRP3 inflammasome activation, leading to reduction in cell apoptosis. These results presented another mechanism for mitochondrial fission-mediated cell apoptosis and suggested that pharmacological inhibition of Drp1 activation might be a potential therapeutic strategy for combating oxidative stress in the endothelium.
As the major sites for ROS production (15), mitochondria are particularly susceptible to oxidative damage (19). Enhanced oxidative phosphorylation in the mitochondrial electron transport chain increases the leakage of electrons, promoting ROS production through the NADPH oxidase pathway, and the produced ROS in turn impairs mitochondrial membrane through calcium accumulation, leading to excessive ROS generation, further exacerbating oxidative stress (39). In this forward cycle of ROS production, the defect in mitochondrial integrity is responsible for mitochondrial malfunction and exacerbated oxidative stress. Mitochondria are organized in a highly dynamic tubular network whose morphology is reshaped by dynamic processes of fusion and fission (12, 32). The balance between mitochondrial fusion and fission is essential for the maintenance of mitochondrial function (1, 30) and regulated by the cytoplasmic dynamin-related protein 1 (Drp1). In response to oxidative stress, ROS induces mitochondrial fission by phosphorylation of Drp1 at serine 616 residue, leading to mitochondrial dysfunction. By contrast, pharmacological inhibition of Drp1 activation can prevent mitochondrial fission and subsequent functional loss (28).
Although it is well documented that mitochondrial fission and malfunction promote cell vulnerability to apoptosis, NLRP3 inflammasome activation emerges as a key event linking oxidative stress to inflammation and cell apoptosis in endothelial cells (21, 22). NLRP3 inflammasome can be activated by a number of diverse triggers and ROS is considered to be the proximal signaling for NLRP3 inflammasome activation (8). In response to oxidative stress, the NLRP3 inflammasome-derived IL-1β secretion is responsible for inflammation and apoptosis in special tissues, including the endothelium (21, 27). Despite both mitochondrial fission and NLRP3 inflammasome activation being susceptible to ROS, their relevance to NADPH oxidase activation in endothelial dysfunction remains unknown.
Corosolic acid (CRA) is a triterpenoid abundant in Banaba (Lagerstroemia speciosa L.), which has been used as a folk medicine to treat diabetes in Southeast Asia. CRA regulates glucose homeostasis (26) and protects vessel function with anti-inflammatory and antioxidative activities in rats (6, 38). However, up to now, little is known about its molecular targets and working pathways in the protection of vessel function. For these, in the current study, we aimed to elucidate the connection between oxidative stress and NLRP3 inflammasome activation from the aspect of mitochondrial fission and NOX2 activation in endothelial cells. Furthermore, the effect of CRA on the protection of endothelial homeostasis was investigated in vitro and in vivo with focus on the regulation of oxidative signaling and mitochondrial function.
Results
CRA prevented Drp1 translocation and mitochondrial fission
As mitochondria are one of the major sites for ROS production and their dysfunction promotes excessive ROS production, we first examined the effect of CRA on the regulation of mitochondrial morphology in the primary rat aortic endothelial cells (RAECs). Drp1 is a mitochondrial fission protein, but its phosphorylation at Ser637 residue can prevent its activation (3). CRA enhanced Drp1 Ser637 phosphorylation in RAECs, a significant effect being observed after 2 h of incubation (Fig. 1A). Meanwhile, we also observed that CRA increased Drp1 Ser637 phosphorylation at concentrations ranging from 0.01 to 1 μM in cells exposed to palmitate (PA) challenge (Fig. 1B). These results indicated that CRA positively regulated Drp1 phosphorylation (Ser637) under basal and pathological conditions. Diphenyleneiodonium chloride (DPI) is a flavoprotein inhibitor with the ability to inhibit NADPH oxidase activation, and Mito-TEMPO is a mitochondria-targeted antioxidant. DPI and Mito-TEMPO treatment preserved Drp1 Ser637 phosphorylation against PA insult, indicating the involvement of ROS in Drp1 activation.

Mitochondria are organized in a highly dynamic tubular network, whereas cells bearing predominantly fragmented or spherical mitochondria undergo mitochondrial fission in response to stress. Therefore, we observed the changes in mitochondrial morphology by MitoTracker Red staining. The translocation of Drp1 from cytosol to mitochondria is an indicator for Drp1 activation. CRA reduced the location of Drp1 at mitochondria (Fig. 1C), indicating the inhibition of Drp1 recruitment. As a result, CRA prevented PA-induced mitochondrial fission in endothelial cells (Fig. 1D and Supplementary Fig. S1; Supplementary Data are available online at
CRA inhibited mitochondrial fission with regulation of AMPK
As AMP-activated protein kinase (AMPK) agonist is shown to regulate Drp1 phosphorylation (21), we wondered whether this regulation was involved in the action of CRA. We observed the effect of CRA on AMPK activity in RAECs. LKB1 is an AMPK upstream regulator for AMPK phosphorylation. CRA treatment increased LKB1 phosphorylation (Fig. 2A) and subsequent AMPK phosphorylation in endothelial cells (Fig. 2B). Consistently, we detected an increase in phosphorylation of AMPK substrates such as ACC in RAECs treated with CRA (Fig. 2C). These results demonstrated that CRA promoted AMPK activation in the LKB1-dependent pathway.
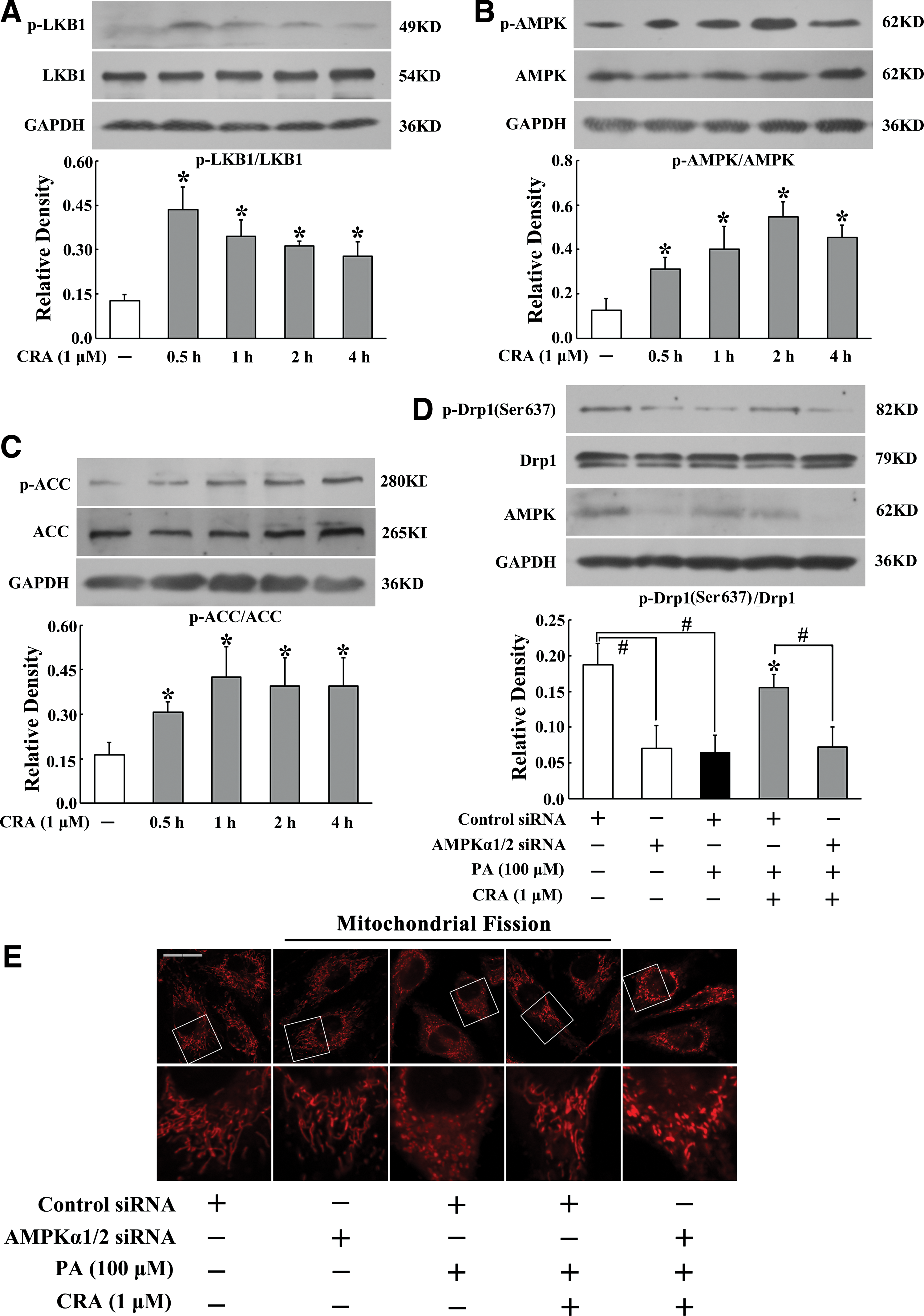
As RAECs are not available for transfection, we transfected human umbilical vein endothelial cells (HUVECs) with AMPKα1/2-specific siRNA and found that silencing of AMPK attenuated the positive effect of CRA on Drp1 phosphorylation at Ser637 (Fig. 2D). Meanwhile, AMPKα1/2 knockdown abolished the inhibitory effect of CRA on PA-induced mitochondrial fission (Fig. 2E and Supplementary Fig. S2). These results supported our hypothesis that AMPK activation was essential for the inhibitory action of CRA in Drp1 activation and mitochondrial fission.
CRA reduced ROS generation in endothelial cells
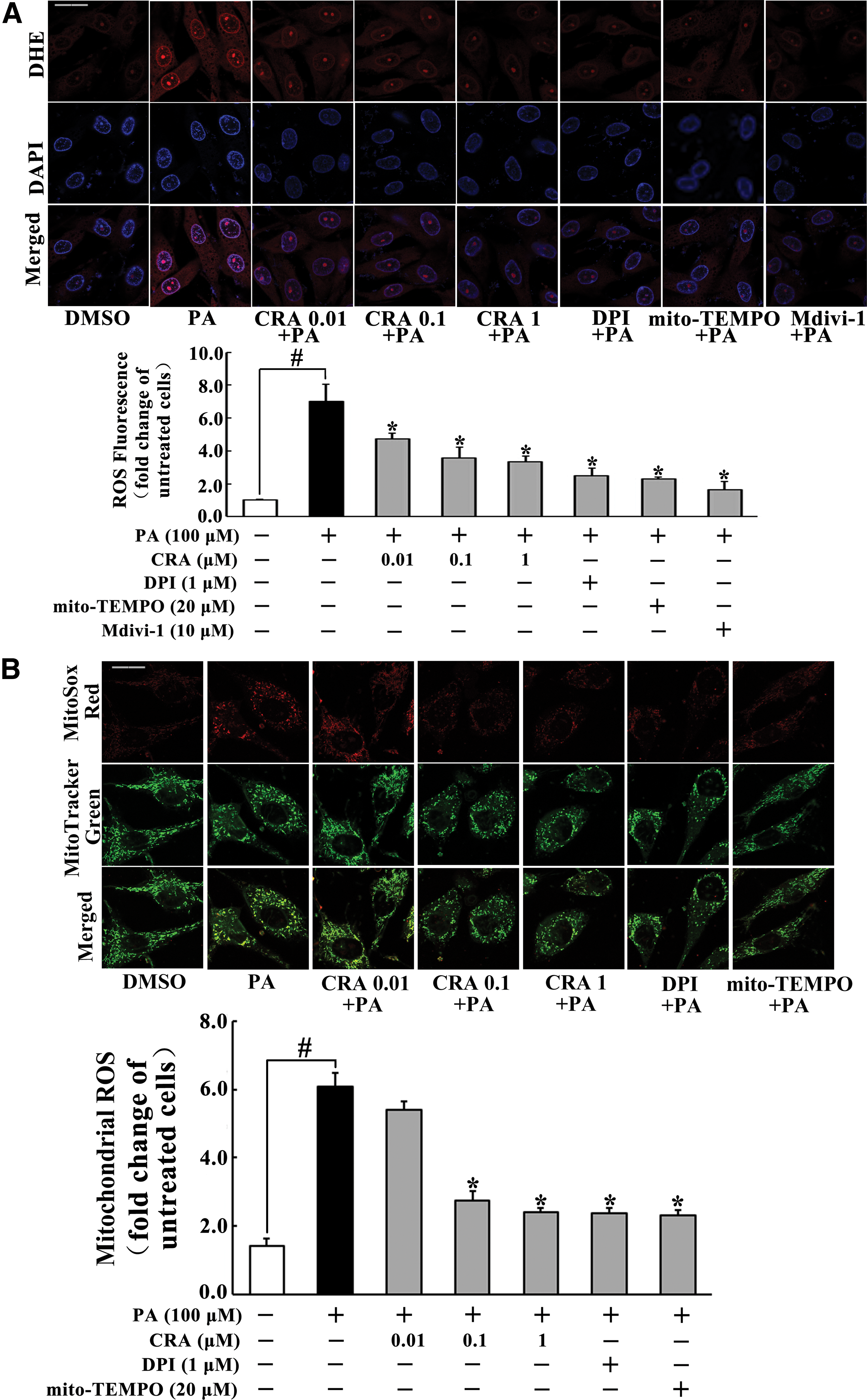
As NOX2 mediates superoxide generation, we assayed ROS production with fluorescent probe dye dihydroethidium specific for superoxide. PA stimulation induced intracellular ROS production, indicated by the increase in red fluorescence, while CRA treatment reduced the red fluorescence intensity (Fig. 3A). CRA also effectively reduced mitochondrial ROS production (Fig. 3B). Meanwhile, CRA treatment reduced PA-induced TNF-α production (Supplementary Fig. S3), suggesting the possible role of anti-inflammatory activity in the suppression of oxidative stress. Fission inhibitor, Midivi-1, also reduced intracellular ROS generation and TNF-α production, demonstrating the relevance of mitochondrial fission to oxidative stress and inflammation.
CRA inhibited p47phox and p67phox translocation and their binding to gp91phox
Intracellular ROS generation relies on the NADPH oxidases, a multisubunit enzymatic complex that consists of membrane proteins and cytosolic proteins. Assembly of the active NADPH oxidase complex requires the association of the cytosolic factors with the plasma membrane protein. The expression and coassociation of subunits are useful indices of NADPH oxidase activation. As p47phox, together with p67phox, is the cytosolic functional subunit of NOX2, we investigated the translocation of p47phox and p67phox in response to PA challenge. p47phox is distributed in the cytosol, and in response to PA stimulation, it colocalized in the cytosol, especially in the perinuclear region (Fig. 4A), and the recruitment to the membrane is increased (Fig. 4B). Together with p67phox, the abundance of p47phox protein in the membrane is increased (Fig. 4C), indicating the translocation of p47phox and p67phox. As a result of the recruitment of p47phox and p67phox, gp91phox protein expression in the membrane is increased (Fig. 4D, E), indicating the association of membrane subunit with cytosolic subunit of NOX2 oxidase. Pretreatment of RAECs with CRA inhibited p47phox and p67phox translocation, prevented the association with membrane gp91phox, and thus suppressed NOX2 induction. Similarly, PA-induced alternations were reversed by fission inhibitor, Mdivi-1.

CRA suppressed NLRP3 inflammasome activation
We further investigated NLRP3 inflammasome activation in response to oxidative stress. PA stimulation induced NLRP3 inflammasome activation evidenced by the induction of NLRP3 and cleaved caspase-1, whereas these alternations were reversed by treatment with CRA at concentrations of 0.01, 0.1, and 1.0 μM (Fig. 5A, B). NLRP3 inflammasome activation promotes IL-1β and IL-18 secretion. When endothelial cells were exposed to PA challenge, the increased IL-1β and IL-18 production was reduced by CRA treatment in a concentration-dependent manner (Fig. 5C, D). However, in resting cells, CRA alone did not affect basal NLRP3 expression (Supplementary Fig. S4), and no alternation of IL-1β and IL-18 secretion occurred (Fig. 5C, D). These results indicated that CRA effectively suppressed NLRP3 inflammasome activation. When endothelial cells were exposed to PA challenge, Mdivi-1 inhibited NLRP3 inflammasome activation, suggesting the involvement of mitochondrial dysfunction in NLRP3 inflammasome activation.

CRA suppressed NLRP3 inflammasome activation with regulation of Drp1 and NOX2
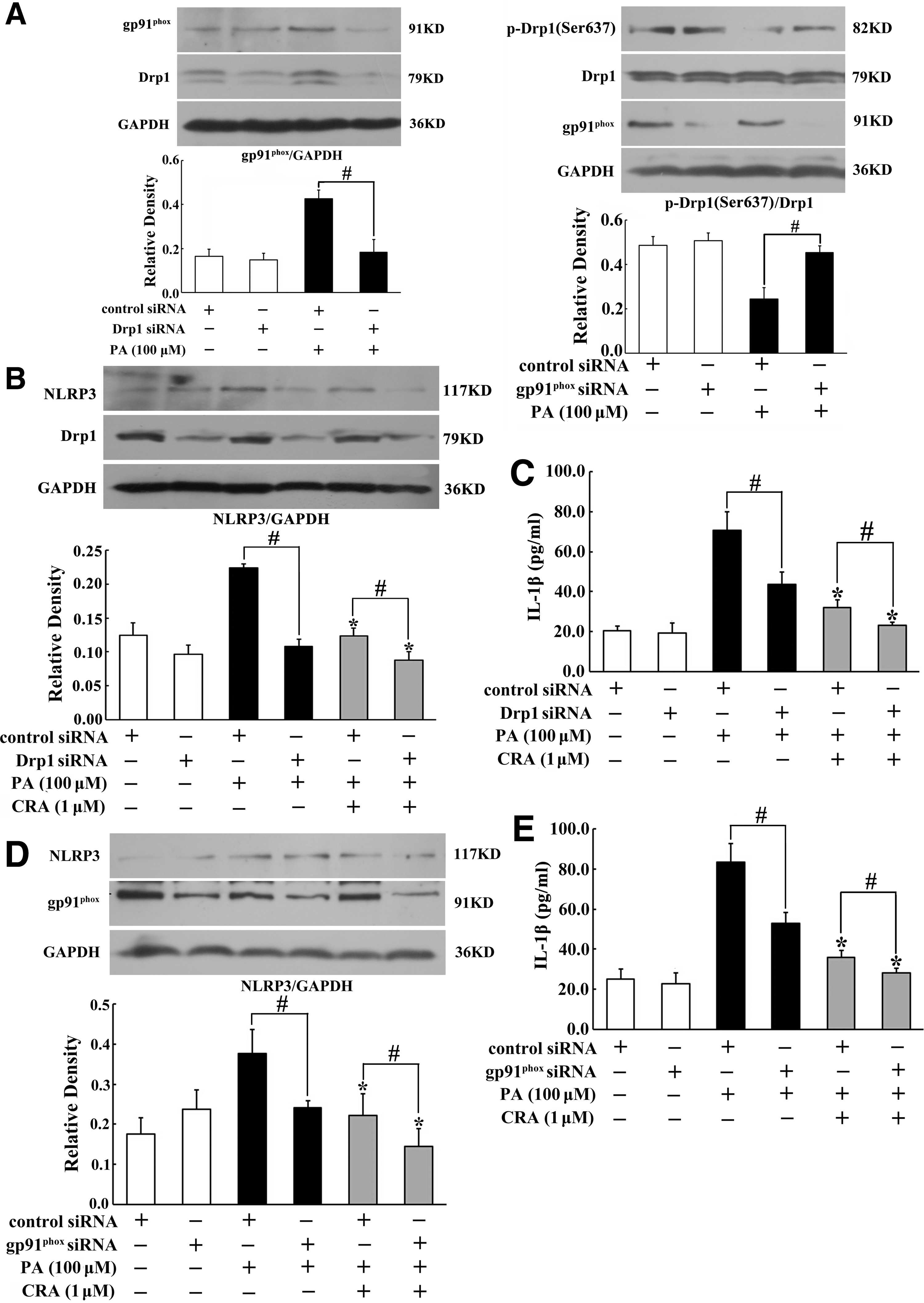
To elucidate the functional interaction between Drp1 and NOX2 activation, we investigated the regulation of gp91phox and Drp1 expression in cells transfected with siRNA for silencing of gp91phox or Drp1, respectively. Drp1 knockdown reduced PA-induced gp91phox expression (Fig. 6A), while Drp1 induction was also diminished by gp91phox knockdown (Fig. 6A). Our work also showed that silencing of gp91phox prevented mitochondrial fission (Supplementary Fig. S5). These results demonstrated that both Drp1 and NOX2 regulate each other. Knockdown Drp1 or gp91phox attenuated PA-induced NLRP3 induction and enhanced inhibitory effects of CRA (Fig. 6B, D), indicating that Drp1 and NOX2 contributed to NLRP3 inflammasome activation and the inhibitory effects of CRA were relevant to the suppression of Drp1 activation and NOX2 expression. Similar regulation action was also observed in IL-1β production (Fig. 6C, E).
CRA reduced mitochondria-dependent cell apoptosis
The mitochondria-dependent pathway is one of the mechanisms involved in cell apoptosis and therefore we observed the influence of CRA on mitochondria-dependent cell apoptosis. We first assayed mitochondria membrane potential (Δψm) by a method based on TMRE staining. As shown in Figure 7A, Δψm was greatly degraded by PA stimulation, shown by the reduction in red fluorescence, while CRA and Mdivi-1 effectively prevented the collapse of Δψm evidenced by increased red fluorescence intensity. Meanwhile, CRA also reduced PA-induced caspase-3 activity (Fig. 7B). Neutralizing IL-1β with anti-IL-1β antibody or Mdivi-1 treatment also reduced caspase-3 activity, indicating the necessity of NLRP3 inflammasome activation in the initiation of caspase-3 activity. Consistent with the inhibition of caspase-3 activation, flow cytometry analysis revealed that CRA as well as Mdivi-1 effectively reduced apoptotic cells in RAECs, demonstrating its protection of cell survival from PA insult (Fig. 7C). As a result of reduced cell damage, CRA effectively restored the loss of NO production in cells subjected to PA challenge (Fig. 7D). Mdivi-1 demonstrated a similar regulation to CRA treatment.

CRA inhibited mitochondrial fission and oxidative stress in the aorta from HFD-fed mice
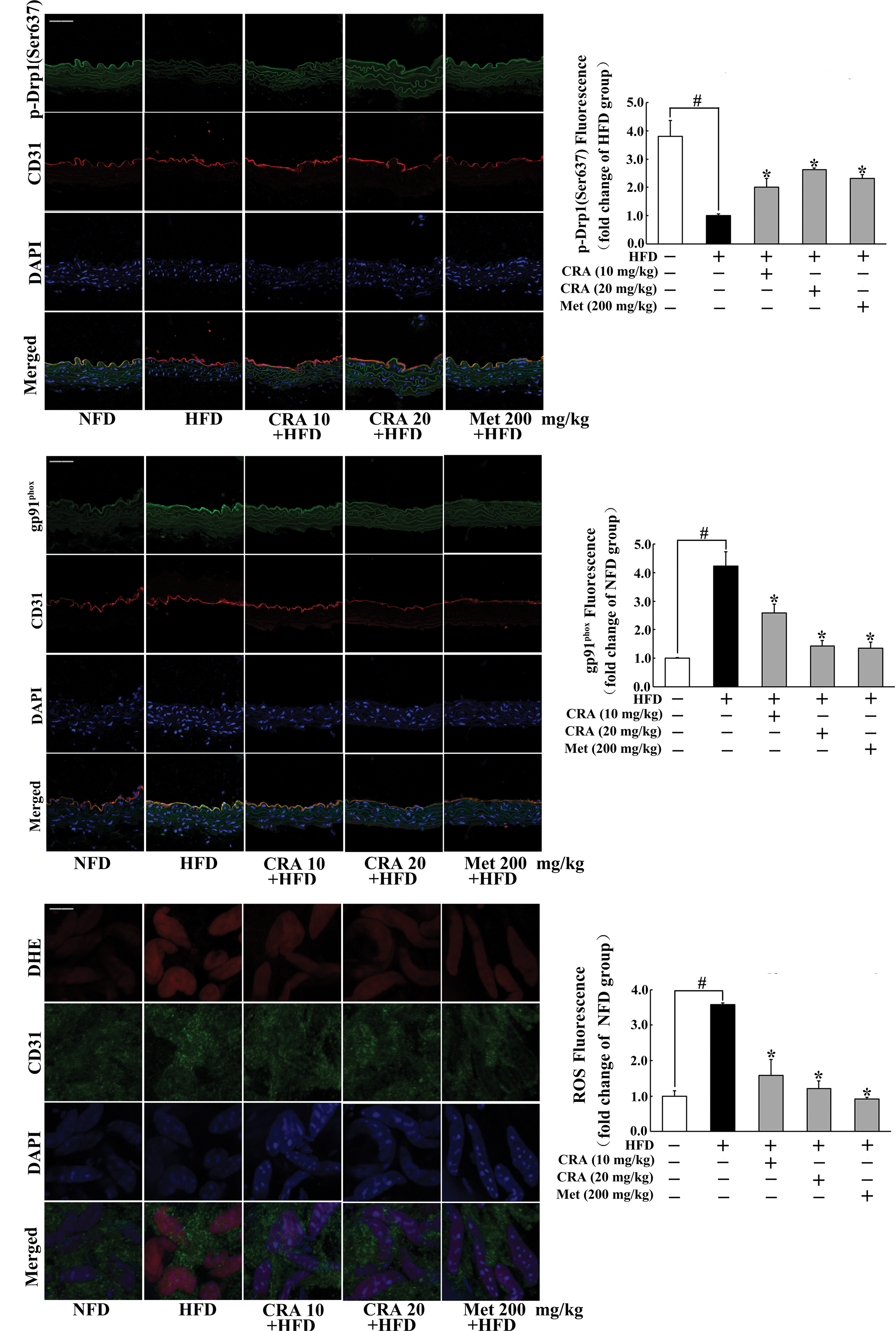
To confirm the protective effect of CRA on the vessel function in vivo, we observed the influence of CRA on the vessel endothelium in the aorta of high-fat diet (HFD)-fed mice. Immunofluorescence staining images showed that HFD feeding attenuated Drp1 phosphorylation (Ser637) and increased gp91phox expression in the vessel endothelium, whereas oral administration of CRA attenuated Drp1 phosphorylation at Ser637 and effectively prevented gp91phox expression in the endothelium (Fig. 8A, B). As a positive control, antidiabetic agent, metformin, also showed similar effect with CRA. As a result of suppression of gp91phox expression, immunofluorescence staining images further showed that both CRA and metformin effectively reduced ROS production in the vessel (Fig. 8C). These results demonstrated that CRA as well as metformin protected vessel function by suppression of Drp1 activation and oxidative stress.
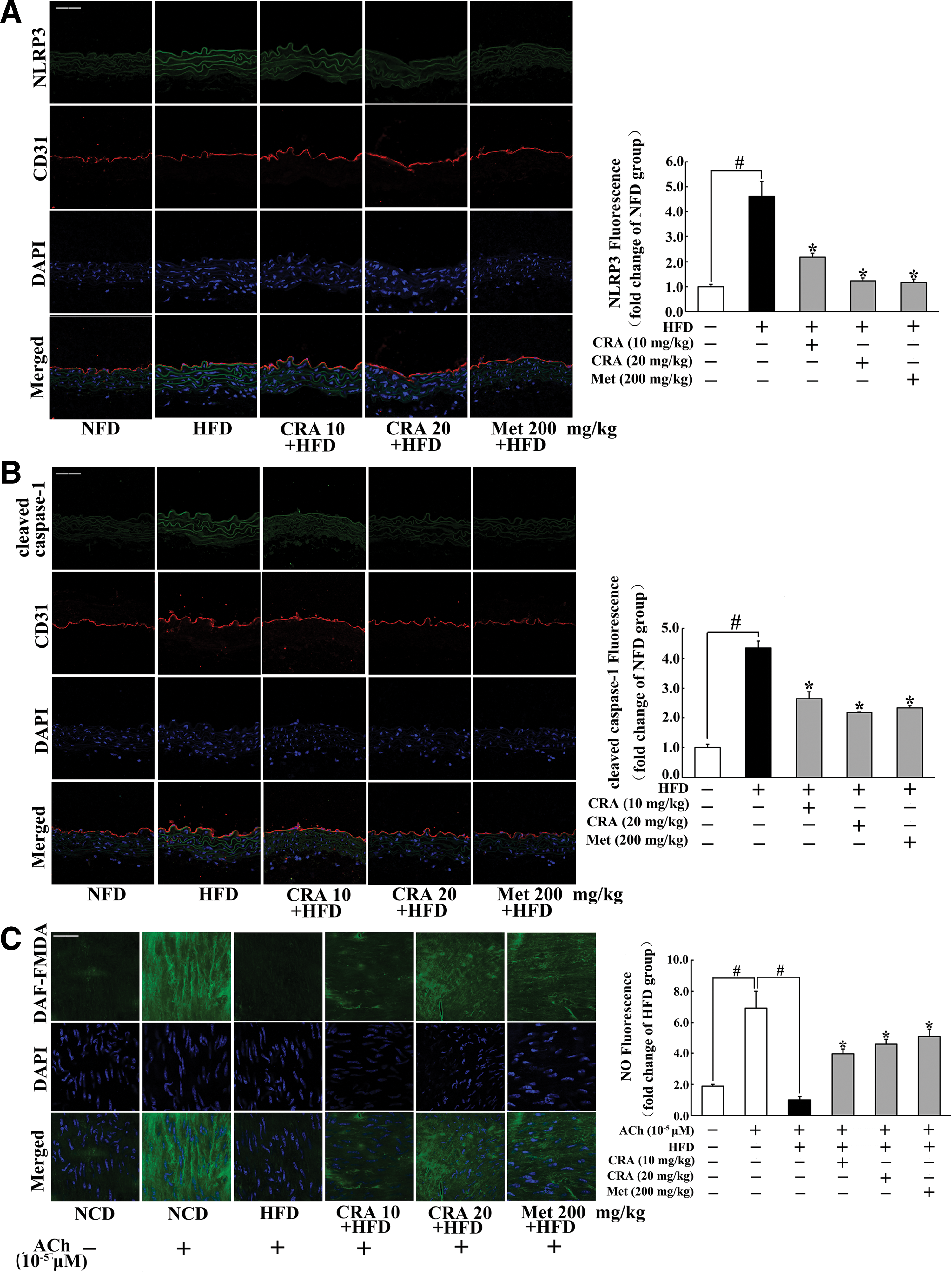
CRA suppressed NLRP3 inflammasome expression in the aorta of HFD-fed mice
Immunofluorescence examination showed that HFD feeding enhanced NLRP3 and cleaved caspase-1 expression in the endothelium indicated by the increased staining, but these alternations were prevented in CRA or metformin-treated mice (Fig. 9A, B). As shown in Figure 9C, CRA and metformin effectively promoted NO production response to acetylcholine (ACh) in the endothelium, demonstrating their action in the protection of endothelial function.
Meanwhile, we observed that levels of blood free fatty acids (FFAs) and total cholesterol (TC) in HFD-fed mice were elevated, while the levels of glucose and triglyceride (TG) in the blood remained unchanged. Oral administration of CRA or metformin reduced the level of FFAs without affecting other metabolic parameters in the blood. No significant difference of body weight gain was observed in CRA or metformin-treated mice compared with the control. These results are shown in the Supplementary Figure S6.
Discussion
As a major source for cellular ROS generation, NADPH oxidases directly catalyze the production of ROS in cellular compartments and this property sets them apart from other known oxidases. In view of the contribution of mitochondrial malfunction to NADPH oxidase activation and ROS production (35), the maintenance of mitochondrial function is a key step to prevent cellular oxidative stress. In the present study, our work in vitro and in vivo demonstrated that Drp1 activation-mediated mitochondrial fission was responsible for NADPH oxidase and NLRP3 inflammasome activation in the endothelium and thereby raised the possibility that pharmacological inhibition of Drp1 activation might be a therapeutic strategy for combating oxidative stress in the endothelium.
Endothelial homeostasis is responsible for the maintenance of vessel function, and it is generally accepted that endothelial dysfunction is considered to be the primary risk for cardiovascular diseases (25). Given the important role of mitochondria in energy metabolism, redox reaction, and cell survival, it is tempting to know whether CRA protects vessel function through regulation of mitochondrial function. Drp1 is a ubiquitous protein responsible for the dynamic regulation of mitochondrial morphology (18). In response to cellular stress, the cytosolic Drp1 is translocated to mitochondria and promotes mitochondrial fission. In contrast, phosphorylation at Drp1 Ser637 can inhibit Drp1 activation and prevent mitochondrial fission (18). CRA enhanced Drp1 phosphorylation at Ser637 under basal and oxidative stress conditions, suggesting its potential action in suppression of Drp1 activation. As an expected result, CRA blocked Drp1 recruitment and prevented PA stimulation-induced mitochondrial fission.
AMPK is a serine/threonine kinase; therefore, its direct regulation should be phosphorylation at the serine/threonine residue of target substrate. In endothelial cells, CRA increased AMPK activity in an LKB1-dependent manner, and silencing of AMPK with siRNA diminished its effects on regulation of Drp1 phosphorylation (Ser637) and mitochondrial fission, demonstrating that Drp1 phosphorylation at Ser637 residue was a direct target for CRA action in prevention of mitochondrial fission by regulation of AMPK activity. These results were consistent with recently published studies, which showed that pharmacological activation of AMPK prevents mitochondrial fission through modification of Drp1 phosphorylation (3, 21).
Compared with the general application of ROS scavenger, prevention of ROS formation by targeting the responsible enzymes could be more effective in combating oxidative stress in endothelial dysfunction (9). The primary catalytic function of NADPH oxidase is the generation of ROS and this property determines its crucial role in oxidative stress. CRA prevented mitochondrial fission by regulation of Drp1 activation and we wondered whether this action contributed to the suppression of NADPH oxidase activation and subsequent ROS production. In comparison with other isoforms of NADPH oxidase, NOX2 has great potential for the treatment of vascular disease because accumulating evidences have shown that pharmacologic inhibition or genetic deletion of NOX2 reduces vascular oxidative stress in several disease models (4, 9, 29). Sustained activation of NOX2 involves translocation of the cytosolic subunits as well as an increased oxidase subunit expression. As PA is a major component of dietary saturated fats and aberrant lipid metabolism can impair mitochondrial homeostasis (14), we stimulated endothelial cells with PA to induce oxidative stress. PA stimulation induced p47phox colocalization in the cytosol, and together with p67phox, the abundance of p47phox protein in the membrane is increased, suggesting the translocation of p47phox and p67phox. Meanwhile, the expression of membrane-located protein gp91phox is increased, further demonstrating the activation of NOX2. CRA inhibited the recruitment of p47phox and p67phox and prevented their association with gp91phox in the membrane, indicating that its antioxidative activity in the endothelium was relative to its inhibition on NOX2.
Silencing of Drp1 with siRNA prevented gp91phox association in the membrane, indicating that Drp1 activation is involved in NADPH oxidase activation. As oxidative stress is associated with mitochondrial function, we silenced gp91phox with siRNA and found preserved Drp1 phosphorylation (Ser637). These results suggested that both Drp1 and NOX2 regulated each other. In this context, the direction regulation of Drp1 by CRA should contribute to blocking the vicious circle between NADPH oxidase and mitochondrial fission. Although our works are focused on NOX2, we should note that in addition to NOX2, other isoforms of NADPH oxidase are also involved in vascular disorders. NOX1 contributes to neointimal formation in the injured artery (20), and NOX4 is responsible for mitochondrial oxidative stress in aging-associated cardiovascular disease (35). Additional works are needed to investigate the role of NADPH oxidase isoforms in vascular diseases.
The NLRP3 inflammasome is a specialized signaling platform critical for the regulation of innate immune and inflammatory responses and its action is to promote IL-1β and IL-18 maturation in a caspase-1-dependent manner. Recent studies have elucidated the role of NLRP3 inflammasome activation in endothelial dysfunction and cardiovascular diseases (21, 34, 40). Its activation is susceptible to ROS (8, 24) and the resultant IL-1β production is responsible for inflammation and several kinds of cell death, including apoptosis in endothelial cells (21), indicating that NLRP3 inflammasome is involved in ROS-associated endothelial dysfunction. In the present study, NLRP3 inflammasome activation occurred in PA-treated endothelial cells, evidenced by NLRP3 and cleaved caspase-1 induction and increased IL-1β and IL-18 secretion. CRA treatment inhibited NLRP3 induction and prevented cleaved caspase-1 activation, and as a downstream result, IL-1β secretion was reduced, demonstrating its action in suppression of NLRP3 inflammasome activation. Meanwhile, mitochondrial fission inhibitor, Mdivi-1, and knockdown of Drp1 reduced NLRP3 expression, suggesting that activated Drp1 and mitochondrial fission were relative to NLRP3 inflammasome activation in response to oxidative stress.
Furthermore, as oxidative stress is tightly associated with inflammation and mitochondrial malfunction, we want to know whether NADPH oxidase was relative to the regulation of Drp1 and NLRP3. Knockdown of gp91phox blocked PA-stimulated Drp1 activation and NLRP3 induction, indicating that NADPH oxidase expression was responsible for mitochondrial fission and NLRP3 inflammasome activation. Taken together, with the observations described above, these results revealed a reciprocal relationship between NADPH oxidase and Drp1 activation under pathological conditions, and both NADPH oxidase and Drp1 regulate and work together to induce NLRP3 inflammasome activation. Although we showed NLRP3 inflammasome activation in a manner dependent of mitochondria, its activation also can be regulated through pathways independent of mitochondria. Autophagy is demonstrated to protect cell survival by suppressing NLRP3 inflammation activation (5, 7), and AMPK activation is able to induce autophagy through the mTOR pathway (31, 36). These events raise the possibility that CRA can inhibit NLRP3 inflammasome activation through the AMPK/mTOR pathway, independent of mitochondria.
In addition to the induction of inflammation, IL-1β is also involved in the initiation of mitochondria-dependent apoptosis through caspase cascades (21, 27). CRA treatment prevented the collapse of mitochondrial membrane potential and inhibited caspase-3 activity and thus reduced apoptotic cells and preserved endothelial ability to produce NO, exhibiting its beneficial regulation of endothelial homeostasis under oxidative stress conditions. Although it is originally proposed that mitochondrial fission can induce apoptosis (1), our finding presented another mechanism for its action in endothelial damage. Inflammation and oxidative stress are involved in the formation of atherosclerotic plaques (17, 23). Endothelium-derived NO regulates endothelial homeostasis through the anti-inflammatory and vasodilator functions, whereas oxidative stress induces vascular disorders via disruption of the NO signaling (11). NADPH oxidases directly catalyze superoxide anions, which can chemically react with inactivate NO, and thus nullify the anti-inflammatory and vasodilator functions of NO. Although previous works found that CRA prevents atherosclerosis and hypertension with anti-inflammatory and antioxidative activities (6, 38), the present study provided evidence that mitochondrial fission and NOX2 oxidase in the endothelium might be the potential therapeutic targets for CRA in the management of vascular diseases.
Evidences mentioned above demonstrated that CRA protected vessel endothelial function by combating oxidative stress in endothelial cells. To confirm this action, we further observed its regulation in HFD-fed mice. Consistent with the action in endothelial cells, oral administration of CRA effectively prevented Drp1 activation by modulation of phosphorylation (Ser637) and inhibited ROS production by downregulation of gp91phox expression in the vessel endothelium of HFD mice. As a result of suppression of ROS-associated NLRP3 inflammasome activation, oral administration of CRA effectively restored NO production in response to ACh. In view of the impact of lipotoxicity on endothelial dysfunction, the inhibitory effect of CRA on the increased blood FFAs should be beneficial for the maintenance of endothelial homeostasis under lipid-challenged conditions. Metformin is an antidiabetic agent with AMPK activity. The beneficial effects of CRA on vessel function could be reproduced by oral administration of metformin, suggesting AMPK activation in CRA action. Thus, our works in vitro and in vivo showed that CRA protected endothelial function by combating oxidative stress.
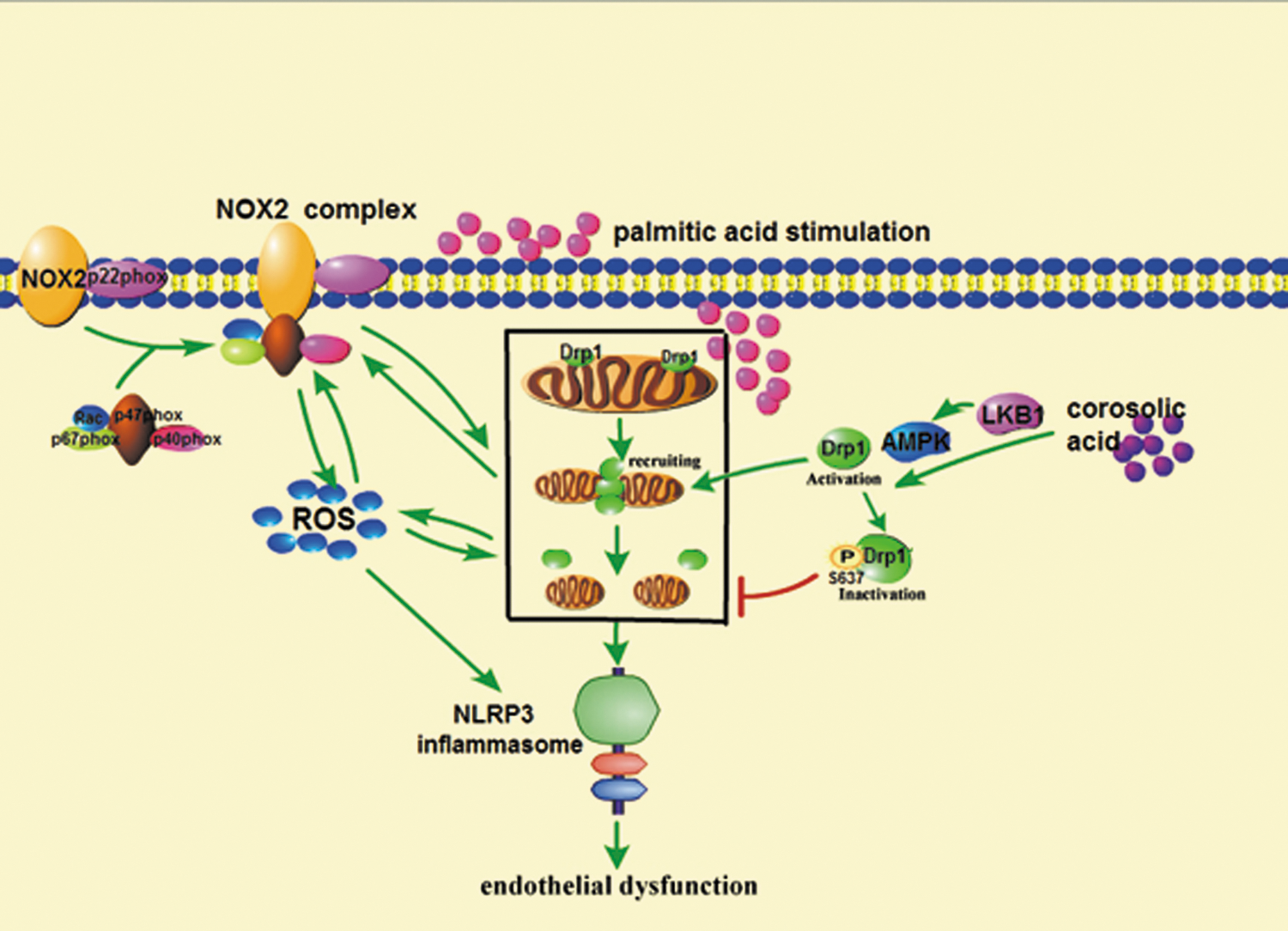
Collectively, our work demonstrated that mitochondrial fission was tightly associated with NADPH oxidase and NLRP3 inflammasome activation in the endothelium, suggesting that modulation of Drp1 phosphorylation should be a therapeutic strategy for suppressing oxidative stress in vessel diseases. CRA prevented mitochondrial fission by regulation of Drp1 phosphorylation (Ser637) in an AMPK-dependent manner and this action contributed to blocking NOX2 oxidase signaling and suppressing NLRP3 inflammasome activation in the endothelium. The proposed working pathway is shown in Figure 10. We should note that this study is focused on the regulation of endothelium homeostasis, whereas NADPH oxidases are also associated with oxidative stress in vessel smooth muscle cells and contribute to macrophage activation and infiltration in the vessel. Although we showed the contribution of Drp1 activation to NOX2 expression, additional experiments will be required to establish their direct connection in the Drp1 KO model in future.
Materials and Methods
Materials
CRA (the purity is ≥98%) was purchased from Chengdu Preferred Biological Technology Co., Ltd. Diphenyleneiodonium chloride (DPI) and Mdivi-1 were from Sigma. Metformin (Met) and Mito-tempol were provided by Beyotime Institute of Biotechnology and Abcam, respectively. These agents were dissolved in dimethyl sulfoxide (DMSO) as a stock solution and the final working concentration of DMSO was <0.1% (v/v). PA (Sinopharm Chemical Rwagent wq Co.) was dissolved in ethanol to prepare 200 mM stock solution and then diluted with medium containing 10% of bovine serum albumin (BSA) at the ratio of 1:19 before use. The following items were purchased from the cited commercial sources: anti-phospho-AMPKα (T172) (2531s), anti-AMPKα (2532s), anti-phospho-ACC (Ser79) (No. 36615), anti-cleaved caspase-3 (Asp175) (5A1E), anti-phospho-LKB1 (S428) (C67A3), and anti-LKB1 (D60C5) (No. 3047), Cell Signaling Technology; anti-cleaved caspase-1/anti-caspase-1 (ab1872), anti-phospho-Drp1 (Ser637) (ab193216), anti-p67phox (ab109366), donkey anti-goat IgG H&L (Alexa Fluor® 647) (ab150131), and donkey anti-goat IgG H&L (Alexa Fluor 488) (ab150129), Abcam; anti-gp91phox (sc-74514) and anti-p47phox, Santa Cruz Biotechnology, Inc.; anti-NLRP3 (NBP2-12446), Novus Biologicals; anti-ACC (BS1377), anti-caspase-3 (BS5644), anti-Drp1 (BS7390), anti-GAPDH (AP0063), goat anti-rabbit IgG (H+L) HRP (BS13278), goat anti-mouse IgG (H+L) HRP (BS12478), and anti-Na+/K+-ATPase α1 (G19) (BS3002), Bioworld Technology; and anti-CD31 (AF3628) and anti-IL-1β antibody (MAB201), R&D.
Cell preparation and culture
Primary RAECs were prepared as previously described (22). In brief, RAECs were isolated from rat thoracic aorta tissue and grown in Dulbecco's minimum essential medium supplemented with 20% fetal bovine serum (FBS), 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate until the cells were outgrowing from the tissue. All of the experiments were performed at passage 3 to 6.
HUVECs (the cell bank of the Chinese Academy of Sciences) were maintained in RPMI 1640 medium supplemented with 10% FBS and antibiotics (100 U/ml penicillin G and 100 μg/ml streptomycin sulfate). All cultured cells were maintained at 37°C in a humidified atmosphere of 5% CO2. After reaching confluence, cells were washed with phosphate-buffered saline (PBS) twice and switched to serum-free medium for 2–4 h before proceeding with further experiments.
To specifically suppress AMPK, Drp1, and gp91phox expression, HUVECs were grown to 80% confluence and then transfected with small interfering RNA (siRNA) duplexes specific for human AMPKα1/2 (sc-45312; Santa Cruz Biotechnology), Drp1 (sc-43732; Santa Cruz Biotechnology), gp91phox (sc-35503; Santa Cruz Biotechnology), or control siRNA (sc-37007) by siRNA transfection reagent (sc-29528), respectively. After transfection, cells were cultured in the medium for 48 h. The cells were treated with indicated agents in the presence or absence of PA (100 μM) for 24 h and then collected and processed for immunofluorescence microscopy of mitochondrial fission, enzyme-linked immunosorbent assay (ELISA) of IL-1β, and Western blot.
Animals
ICR male mice (5 weeks) and Sprague-Dawley rats (200–250 g) were supplied by the Laboratory Animal Center of Nanjing Qinglongshan. The care and treatment of these animals were performed in accordance with the Provisions and General Recommendation of Chinese Experimental Animals Administration Legislation. Animals were housed in a room with a constant temperature (22 C ± 1°C) and a 12-h light–12-h dark cycle and allowed free access to a standard diet and water ad libitum. This study was approved by the Animal Ethics Committee of School of Chinese Materia Medica, China Pharmaceutical University.
HFD feeding
Mice were fed with HFD containing 10% lard, 10% yolk, 1% cholesterol, 0.2% cholate, and 78.8% standard diet simultaneously with administration of CRA (10, 20 mg/kg) or Met (200 mg/kg) daily by oral gavage for 14 days. At the end of the experiment, blood was collected from retinal venous plexus and the thoracic aorta was removed. Blood glucose, nonesterified fatty acid, TGs, and TC were assayed using commercial enzyme kits.
Assay of ROS production
For intracellular ROS detection, RAECs were cultured to 80% confluency and then treated with CRA (0.01, 0.1, 1 μM), DPI (1 μM), mito-TEMPO (20 μM), or Mdivi-1 (10 μM) in the presence or absence of PA (100 μM) for 24 h. After treatment, cells were washed with PBS twice and then kept in the dark with ROS-specific fluorescent probe dye dihydroethidium (Beyotime Institute of Biotechnology) for 0.5 h and DAPI (Sigma) for 5 min at 37°C. After washing, cells were fixed in 4% paraformaldehyde (v/v) for 5 min at 4°C. For the view of mitochondrial ROS, cells were pretreated with 5 μM MitoSox Red (Molecular Probes) and 30 nM MitoTracker Green (Beyotime) for 20 min. To visualize ROS production in the vascular endothelium, the thoracic aortas were incubated with specific primary antibodies (anti-CD31) and secondary antibodies, and then stained with the superoxide oxidized dihydroethidium and DAPI. Images were viewed by confocal scanning microscopy (Zeiss LSM 700).
Mitochondrial fission and mitochondrial membrane potential analysis
For mitochondrial fission assay, confluent RAECs were pretreated with CRA (0.01, 0.1, 1 μM), DPI (1 μM), or mito-TEMPO (20 μM) for 0.5 h and then incubated with PA (100 μM) for 24 h. Meanwhile, HUVECs were transfected and treated with CRA (1 μM) in the presence or absence of PA (100 μM) for 24 h. RAECs or HUVECs were washed with PBS, and then incubated with 200 nM MitoTracker Red CMXRos (Molecular Probes) for 20 min at 37°C. For mitochondrial membrane potential (Δψm), RAECs were loaded with the potentiometric dye 500 nM TMRE for 30 min at 37°C. The structure of mitochondria was viewed by confocal scanning microscopy. Several random fields of cells (≥90 cells per dish) from each sample were evaluated for mitochondrial morphology in three independent experiments.
ELISA assay of IL-1β and IL-18
RAECs were seeded in 48-well plates in the presence of CRA (0.01, 0.1, 1 μM), or Mdivi-1 (10 μM) for 0.5 h and then incubated with PA (100 μM) for 24 h. Meanwhile, HUVECs were transfected and treated with CRA (1 μM) in the presence or absence of PA (100 μM) for 24 h. The supernatant was collected and the concentration of IL-1β and IL-18 was assayed with commercial ELISA kits (Neobioscience Technology Co., Ltd.).
Apoptosis analysis
Confluent RAECs were treated with CRA (0.01, 0.1, 1 μM) or Mdivi-1 (10 μM) and then incubated with PA (100 μM) for 24 h. After incubation, cells were collected and stained with the Annexin V-FITC Apoptosis Detection Kit (KeyGEN Biotech Co., Ltd.). According to the manufacturer's instructions, cellular fluorescence was imaged using flow cytometry analysis with the FACSCalibur Flow Cytometer (BD Biosciences).
Detection of NO production
RAECs were pretreated with CRA or Mdivi-1 at given concentrations for 0.5 h and then incubated with PA (100 μM) for another 0.5 h. After washing with PBS, cells were loaded with a fluorescent probe (NO-specific fluorescent dye: DAF-FMDA) at 37°C for 30 min in the dark. For the observation of NO in the vascular endothelium of HFD-fed mice, the removed thoracic aortas were incubated with 10 μM ACh (Sigma) for 30 min and then stained with DAF-FMDA and DAPI. After washing with PBS, RAECs and thoracic aortas were examined with a Zeiss confocal scanning microscope.
Western blot assay
For protein analysis, cells were lysed in ice-cold RIPA buffer and incubated in an ice bath for 45 min. Protein was obtained by centrifugation at 12,000 g for 20 min at 4°C, and the concentration was measured with the Bicinchoninic Acid Protein Assay Kit (Biosky Biotechnology Corporation). An equal amount of protein was electrophoresed on SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and then blocked at room temperature for 2 h. For immunoblotting, the primary antibodies were used, respectively, at 4°C overnight, followed by incubating with the secondary antibody at room temperature for 2 h. An enhanced ECL kit used to detect signals and Image-Pro Plus 6.0 (IPP 6.0) software to analyze quantitatively by densitometry were used.
Immunofluorescence
For the studies in the aorta from HFD-fed mice, the removed thoracic aortas were fixed with 4% (w/v) paraformaldehyde for 24 h and dehydrated and embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek), and then cut into 8 μm slices. RAECs cultured on coverslips were washed with cold PBS and fixed with 4% paraformaldehyde in PBS for 20 min. The slides and RAECs were permeabilized with 0.2% Triton X-100 and incubated with 5% BSA to block nonspecific staining, and then incubated with specific primary antibodies (anti-CD31, anti-cleaved caspase-1, anti-Drp1, anti-phospho-Drp1, anti-gp91phox, anti-NLRP3, anti-p47phox) overnight at 4°C in a humidified chamber. After several washings, the sections and cells were incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) antibody (Beyotimes, Inc.), Alexa Fluor 488-labeled goat anti-mouse IgG (H+L) antibody (Beyotimes, Inc.), donkey anti-goat IgG H&L (Alexa Fluor 647), or donkey anti-goat IgG H&L (Alexa Fluor 488) for 1 h at 37°C. They were then washed in PBS twice and incubated in DAPI for 5 min at 37°C. The sections and cells were mounted in mounting medium and visualized under a confocal scanning microscope.
Statistical analysis
For quantitative images taken by confocal scanning microscopy, images were quantified densitometrically by Image-Pro Plus 6.0 software. Single-cell fluorescence intensities were assessed from a minimum of 50 cells in three independent experiments. Data were derived by comparing fold change in fluorescence intensity of treated cells versus untreated cells. For the HFD mice, data were derived by comparing fold change of fluorescence intensity in the vessel endothelium.
All of the experimental results are expressed as the mean ± SD (standard deviation), and each experiment was performed a minimum of three times. Statistical significance was determined by ANOVA, and when the results passed the ANOVA test, we performed post hoc multiple comparisons to calculate the relevant p-values. A value of p < 0.05 was considered to be statistically significant.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81373957, 81573567, 81102763, and 81373919) and the Natural Science Foundation of Jiangsu Province (No. BK20131306). The authors greatly appreciate the financial support from the Jiangsu Shuang Chuang team and the priority academic program development of Jiangsu higher education institutions (PAPD). The authors also thank Xiao-Nan Ma, Min-Hui Sun, and Ying-Jian Hou for technical support from Cellular and Molecular Biology Center of China Pharmaceutical University.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.