
Abstract
Introduction
C
CAVD begins with inflammation at the endothelium, leading to ultimate calcium deposition in the valve interstitium (32). The aortic side of the valve (fibrosa) is preferentially calcified, sparing the ventricular side of the tissue. The current hypothesis is that the fibrosa is subjected to oscillatory shear stress, combined with increased mechanical strain, conditions known to increase inflammation and endothelial dysfunction (31, 68, 98). Inflammation at the fibrosa endothelium may result from many systemic factors, including pathological flow, chronic inflammatory disease, or other inflammatory conditions (18, 42). Valve stenosis occurs after initial inflammation, resulting from lipoprotein accumulation, cellular infiltration, and extracellular matrix (ECM) formation (93). Cytokines released by the endothelium promote ECM formation and tissue remodeling in later stages of CAVD, finally leading to calcification within the valve tissue (59, 61).
This review explores the current state of understanding of the aortic valve endothelium, from proteins at the cell surface to transcription factors in the nucleus. While many potential therapeutic targets have been discovered in recent years, nonsurgical treatment options remain elusive. The overall picture of valvular endothelial biology shown in this review points to exciting areas of future research, which may lead to effective treatment and prevention of CAVD.
Aortic Valve Structure and Hemodynamics
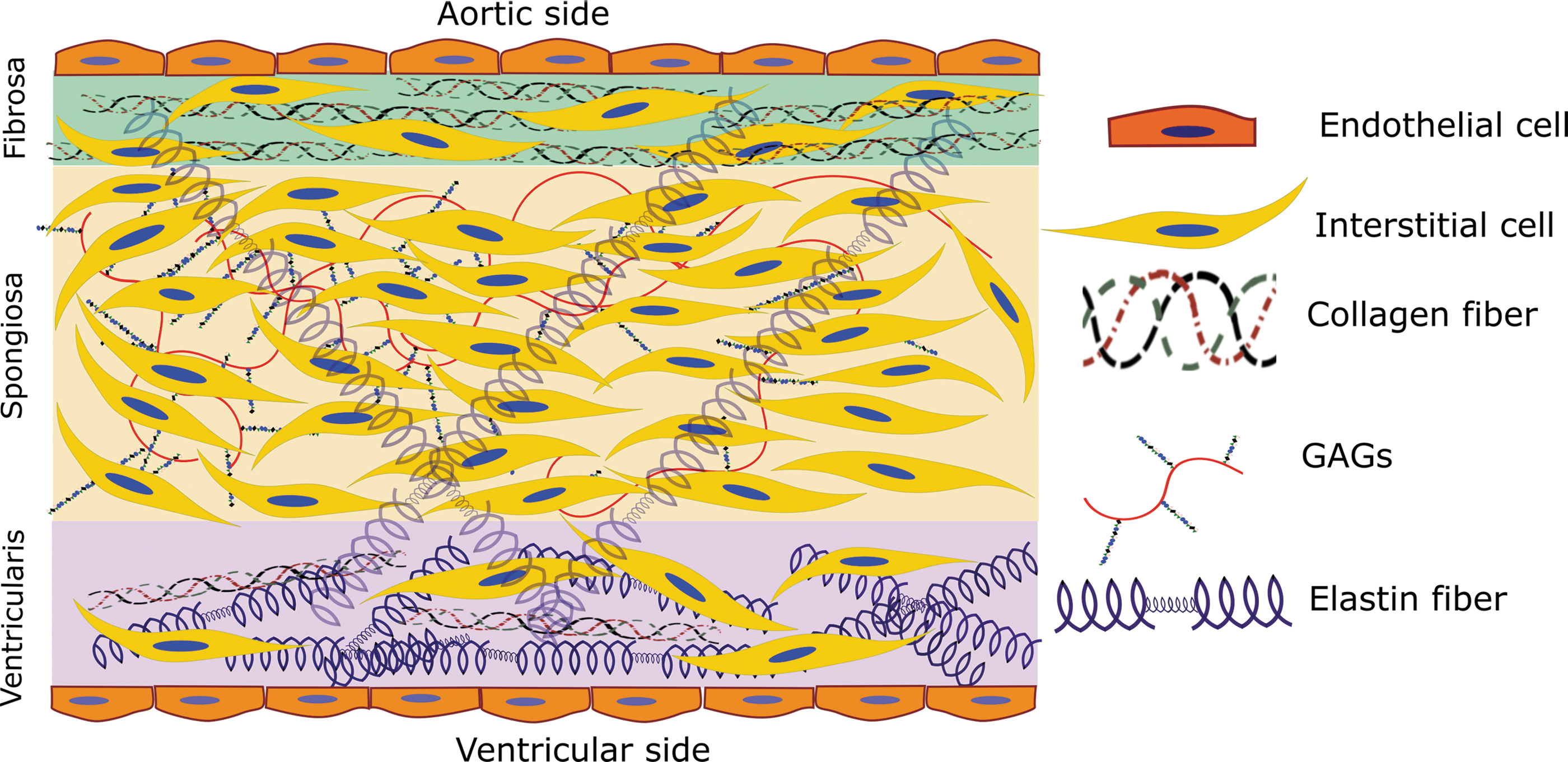
The aortic valve consists of three separate, but highly interactive, layers, which control mechanical and biological function of the tissue. As discussed above, the fibrosa faces the aortic side of the valve, containing endothelial cells at the interface with the blood, and valvular interstitial cells (VICs) below the endothelium, interspersed among a small amount of elastin fibers and circumferential type I and type III fibrillar collagen (Figure 1). The circumferential collagen allows the fibrosa to bear the higher mechanical loads exerted upon it during systole (105). The spongiosa is located between fibrosa and ventricularis and contains VICs, a small amount of elastin fibers, glycosaminoglycans (GAGs), and proteoglycans. The glycosylated components lubricate the fibrosa and ventricularis layers during tissue deformation. Hyaluronan plays a major role in blunting the impact of constant valve movement, binding large amounts of water and forming a foam-like structure in the spongiosa that absorbs energy (109, 132). The ventricularis, at the ventricular side of the valve, is made up of valvular endothelial cells, VICs, collagen, and radially oriented elastin fibers (22, 72, 132). The larger amount of matrix proteins, most especially elastin, within the ventricularis allows for fast and consistent compression during valve opening and closing (109).
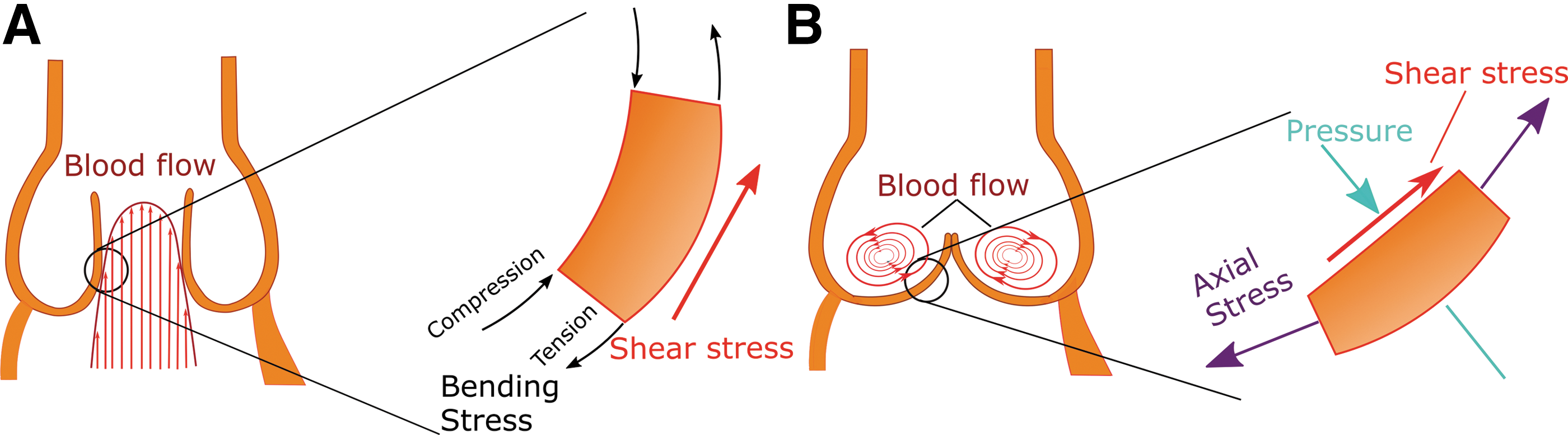
The constant movement of the valve affects biological pathways within the valvular endothelium. The endothelial cells experience a wide array of forces, which are integrated into signaling pathways to translate a biological response to increased stress on the tissue. The following discussion gives an overview of those forces, followed by an overview of the known mechanical signaling in the valve endothelium, and how those pathways may change in CAVD.
There are three types of forces, summarized in Figure 2, exerted upon the aortic valve: (i) Pressure: Differential pressures during the dynamic movement of the aortic valve significantly shape the tissue shape and geometry (8). Low diastolic blood pressure in the ventricle versus stable pressure in the aorta causes the valve to close. The physiological transvalvular pressure remains at 80–120 mmHg. In hypertension, the transvalvular pressure may exceed 180 mmHg (121), contributing to degeneration of the valve tissue. According to findings from the SMART study, systolic blood pressure is correlated with calcification in the valve and vasculature, reflecting the contribution of pressure to general cardiovascular calcifications (118). Increased diastolic blood pressure also contributes significantly to aortic valve calcification in the aging population (58), possibly through synergistic effects on shear stress and cyclic stretch. A wide array of studies (13, 57, 62) have indicated the connection between valvular and systemic pressure on aortic valve calcification, indicating the importance of this parameter in the disease. (ii) Axial and bending stress: Axial stress, perpendicular to the valve face in both the circumferential and radial directions, is critical for prevention of blood regurgitation backward into the ventricle (23). As the aortic valve ages, the highest valvular axial stress is observed near the aortic root, the area at which the first calcium nodules are observed in early valve calcification (111). Thus, a vicious cycle occurs, during which increased axial stress induces valve dysfunction and stenosis, leading to even higher levels of stress on the valve. Valvular bending stress compresses the concave fibrosa layer and increases tension on the convex ventricularis layer, as visualized in Figure 2A (23). (iii) Shear stress: Shear stress, caused by blood flow parallel to the valve surface, occurs in a cycle due to changing blood flow during the cardiac cycle. The ventricularis experiences unidirectional laminar shear stress as blood is ejected from the ventricle to the aorta. The fibrosa experiences disturbed flow and oscillatory shear stress, as shown in Figure 2B, which have been directly linked to onset and progression of CAVD (6, 40, 104). Shear stress regulates endothelial cell function: the cells sense changes in shear stress to determine systemic changes, such as hypertension. For example, laminar shear stress alters cell morphology and cytoskeletal arrangement, resulting in alignment of endothelial cells in the direction of flow (78).
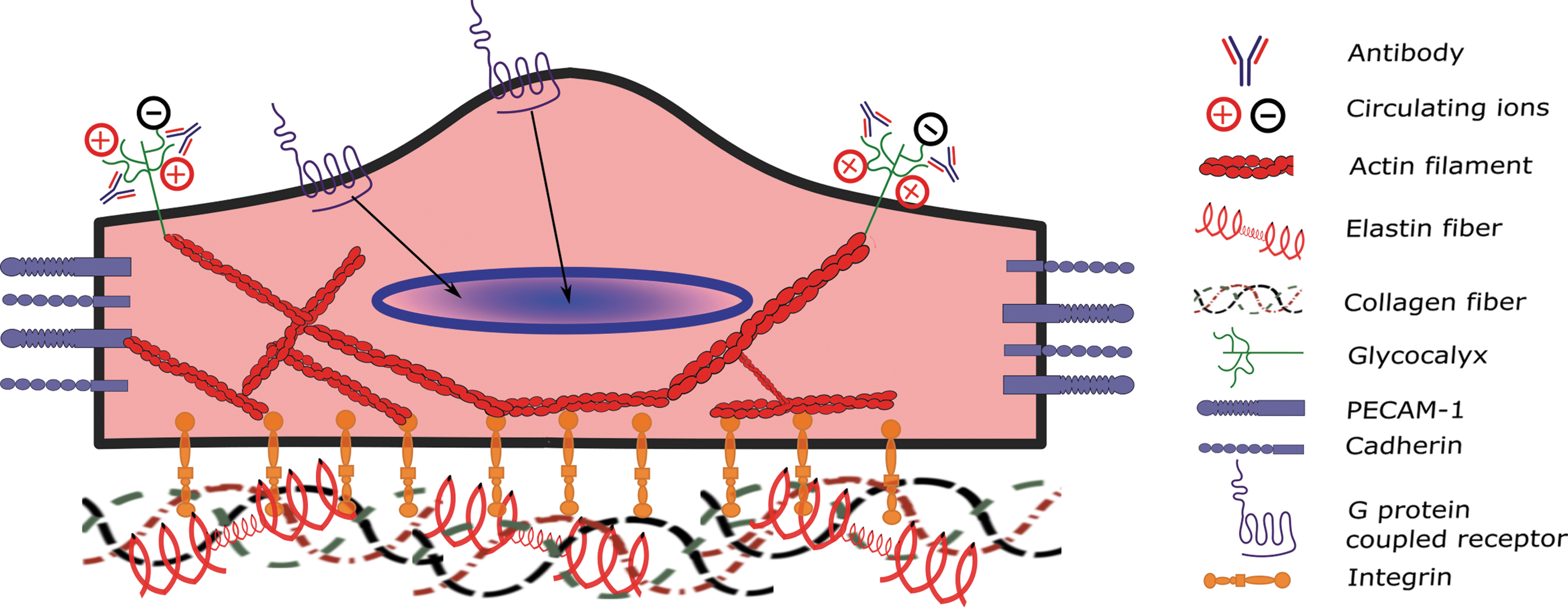
Mechanosensors in the Aortic Valve Endothelium
Endothelial cells utilize a variety of sensing mechanisms to respond to the extracellular mechanical environment, including transmembrane proteins, cytoskeletal proteins, networks of sugars, ion channels, or some combination of these. A vast array of studies have been carried out to further understand mechanosensors in the vascular endothelium, including caveolae, glycocalyx, G protein-coupled receptors (GPCRs), integrins, intercellular junctional proteins, ion channels, and tyrosine kinase receptors (119). However, a relatively small body of work has focused on the mechanical sensing of the valvular endothelial cells. In this study, we describe the current state of research into valvular endothelial mechanosensing, highlighting the major players at the cell surface, including integrins, GPCR, and the glycocalyx. Figure 3 shows a representative image of aortic valve endothelial cells and the major mechanosensors discussed in this article.
Integrins, focal adhesion complexes, and the cytoskeleton
Integrins are a superfamily of transmembrane cell adhesion receptors that have traditionally been known to bind to ligands at the cell surface, within the ECM, and within the cytoplasm. Integrins transduce signals from the extracellular environment to the cell interior; in this way, changes in the concentrations of key signaling molecules in the plasma are communicated to the cell. In addition, integrins have more recently been recognized as sensors of the mechanical environment surrounding the cell, which also results in changes in intracellular signal transduction pathways (139, 147). In addition, integrins receive intracellular signals that regulate their ligand-binding affinity, fine-tuning the communication between the cell membrane and other cellular compartments (117). In coordinating cues from the extracellular environment with intracellular signaling, integrins play an important role in cell adhesion, migration, proliferation, and survival of the cell.
The mechanism by which integrins transmit signals to the cell is dependent on their binding to the cytoskeleton (149). Along with cytoskeletal proteins such as actin, a large number of binding partners colocalize with integrins near the cell surface, forming complexes known as focal adhesions (29, 97). Focal adhesion proteins in these complexes mediate the bidirectional mechanical transduction among the integrins, external stimuli, and the cell (145). Extensive research has found that the integrin–focal adhesion complexes have the ability to sense forces important to the aortic valve, including cyclic stretch (71), shear stress (74), and hypotonic stress (46). The complexes at the focal adhesions are also critical in signaling changes in the cell phenotype in response to mechanical and other cues, including signals for cell and tissue remodeling (51, 112), cell proliferation and apoptosis (112, 135), and cell migration and angiogenesis (74, 127).
Integrins encompass all but one of the families of mechanosensors, but the signals they transmit through focal adhesion complexes are critical for cell phenotype. Other classes transmit signals specific to cell–cell communication and inflammation, and all of the adhesion molecules work in tandem in the valve endothelium to determine the fate of the cell and the entire tissue.
G protein-coupled receptors
Similar to the other transmembrane proteins discussed above, GPCRs bind to a variety of extracellular ligands to activate downstream signaling, namely through G protein-activated signal transduction pathways (65). These receptors have also been shown to be strongly sensitive to changes in flow across the surface of the endothelium (19).
While some work has shown specific methods of subunit recruitment utilized by the GPCR to become activated under mechanical stimuli (30), the role of GPCR in the endothelium has not been fully realized, either in the vasculature or in the valve. Studies by Anger et al. showed that the mechanism by which statins may partially inhibit inflammation in the aortic valve is through GPCRs and their downstream effectors (5). This work demonstrated via array analysis that the extracellular regulated kinase (ERK) activation regulated by GPCRs is highly upregulated in calcified human aortic valve tissue (5). In addition, although the mechanism is unclear, statin therapy inhibits valvular expression of the GPCR regulatory proteins (RGS) (5). While these initial data offer exciting direction for future targeted therapies, new insights have not been delivered for several years. These receptors have known functions in many disease models (28) and thus their potential importance in the valve is clear. More work must be focused here to integrate these mechanical receptors into the lexicon of valvular mechanobiology.
Glycocalyx
The large network of sugars embedded into the cell membrane consists of proteoglycans and glycoproteins, bound to long chains of carbohydrates, called GAGs. The sugars provide a physical barrier between the blood and the endothelial cells (4, 11) and act as a trap where ions and other molecules in the blood may interact with membrane proteins (2). The endothelial glycocalyx significantly changes in shape and height as a result of disturbed flow, possibly leading to changes in its function as a scaffold for signaling molecules (66).
Inflammation also plays a role in the deformation of the glycocalyx, possibly indicating a positive feedback between the oscillatory flow and inflammation and exacerbating proinflammatory signaling (24). Specific studies of the glycocalyx in the valve endothelium show that proinflammatory mediators such as low-density lipoprotein (LDL) and immunoglobulins are more tightly bound to the sugar network in the valves of rabbits fed a high-cholesterol diet (107, 108). The binding of these molecules to the glycocalyx is increased on the fibrosa endothelium, the aortic side of the valve more vulnerable to interstitial calcification. Thorough investigative work by Sarphie (107) indicates the link between shear stress and the composition of the glycocalyx, which may alter the affinity of LDL particles for the cell surface, leading to infiltration and lesion formation. Of note, these studies have not been confirmed or progressed for over a quarter century, leaving a significant gap in our understanding of the glycocalyx and its role in the valve. New technologies may prove useful in defining this role and future therapies, which may change glycocalyx stability and composition. In addition, although the characterization of the glycocalyx has not recently been performed in the valve, its extensive characterization in the vasculature may be easily translated to the valvular environment. With more work focused on this exciting area, we may discover a wide assortment of targets for therapeutic intervention in early stages of valve dysfunction in the future.
Mechanosensitive Genes in Flow-Mediated Valve Biology and Dysfunction
Shear stress is an important regulator of endothelial cell function. The vascular endothelium has been widely studied in this context, and a variety of vascular mechanosensitive genes have been discovered (25, 27, 34, 119). However, there is little known about the role of these mechanosensitive genes in the valvular endothelium. In this section, we review the genes that have been studied in the valvular endothelial cells, including some exciting new opportunities in valve research based on findings in the vasculature. For reference, Figure 4 shows a summary of valvular mechanosensitive genes in both disturbed flow, as seen at the calcifying fibrosa endothelium, and laminar flow as observed at the ventricularis.

Transforming growth factor-β ligand and receptors
The transforming growth factor (TGF)-β family of growth factors regulates a wide variety of cellular responses such as growth, development, immune system regulation, and tissue homeostasis (69). TGF-β signaling is initiated by the binding of TGF-β to TGF-β receptors, type I and type II. These two receptors form a receptor heterocomplex that recruits and phosphorylates R-Smad proteins, causing different signal transduction pathways depending on the Smad protein complexes that have been phosphorylated (53). TGF-β activity increases more than 100-fold in bovine aortic endothelial cells under shear conditions compared with static culture (92), suggesting a role for TGF-β in flow-induced vascular remodeling driven by fluid shear stress in the endothelium.
Villar et al. (125) found that circulating plasma levels of TGF-β1, the most abundant isoform of the TGF-β family, in patients with aortic stenosis were nearly threefold higher than in healthy controls. Yetkin et al. (141) studied patients with tricuspid calcified and stenotic aortic valves and found that the calcified valves presented similarly increased expression of TGF-β1. Furthermore, this work also suggested a link between TGF-β1 and cysteine C, a cysteine protease inhibitor highly expressed in mature osteoblasts, which inhibit bone resorption and appear in osteoblast differentiation (17, 60). These findings suggest the important role of TGF-β in aortic calcification and of TGF-β receptors in aortic valve endothelium as the high levels of this cytokine in blood induce an endothelial cell inflammatory phenotype. TGF-β has also been found to promote endothelial–mesenchymal transition (EMT): ovine aortic valve endothelial cells grown in the presence of TGF-β exhibited higher levels of CD31 (an endothelial cell marker) and α-smooth muscle actin, both EMT markers (96).
The combined evidence from the described studies reflects the importance of TGF-β in the events leading to CAVD. Matrix production and endothelial phenotypic transformation are critical for the events leading to stenosis of the valve, and TGF-β is a critical regulator of these pathologies. In combination with the proteins discussed below, a plethora of targets is available for future therapies.
Bone morphogenetic proteins
Bone morphogenetic proteins (BMPs) are multifunctional growth factors that belong to the TGF-β superfamily (21). These proteins are physiologically observed in bone and they initiate its formation during development and in injury (113). In vascular endothelium, bone morphogenic protein 4 (BMP4) is upregulated by disturbed flow, leading to endothelial inflammation and ultimate dysfunction (20, 113, 114). Additionally, in human atherosclerotic vasculature, both BMP2 and BMP4 expression levels are upregulated (37, 113). Our group has observed endothelial dysfunction and hypertension in mice infused with BMP4 (84), and BMP antagonist treatment protects against atherosclerosis (36, 140). Endothelial cells in the aortic valve may secrete BMPs in response to changes in shear stress (110), and the BMP expression of the interstitial cells may be linked with age-related valvular degeneration (20). Smad 1,5, and 8 are downstream molecules in the BMP signaling pathway and have been associated with onset of aortic stenosis (144). Sucosky et al. (116) studied porcine aortic valve leaflets exposed to physiological and altered shear stress conditions for 48 h ex vivo and found that BMP-4 and TGF-β were increased in pathologically altered conditions, which stimulated endothelial inflammatory response demonstrated by an increase in adhesion molecule expression. These results were validated by Ankeny et al. (6) in human calcified aortic valves and noncalcified aortic valves, which presented higher Smad 1/5/8 phosphorylation and lower BMP antagonists (crossveinless-2/BMPR and noggin) in calcified fibrosa.
These data, in the valve and in other vascular tissues, strongly suggest the importance of the BMP signaling pathway in aortic valve calcification. In designing future therapies for valve stenosis and calcification, the pathological downregulation of BMP antagonists provides an interesting target since this process may contribute to the side-dependent calcification pattern observed in human aortic valves. Any treatment providing some replenishment of physiological levels of these antagonists in the valve might be effective in treatment of valve dysfunction.
Wnt/β-catenin
Wnt proteins are a family of secreted signaling glycoproteins used for short or long-range signaling, which bind to receptors of the Frizzled family to regulate a wide variety of cellular processes such as cell fate determination, motility, stem cell renewal, and cell migration (67, 76, 103). The Wnt/β-catenin pathway (also known as canonical Wnt pathway) is widely known to regulate aortic valve formation and disease (54). β-catenin is a cytosolic protein that is bound to Wnt receptors in the cytosol and is degraded via ubiquitination in the absence of Wnt (1). When Wnt binds to Wnt receptors, β-catenin is stabilized and released to the cytosol, where it accumulates and translocates to the nucleus to activate the transcription of specific genes (76).
Shear stress has been found to regulate the canonical Wnt pathway in endothelial cells, leading to the increased expression of angiopoietin-2, a protein involved in vascular development and repair, in human aortic endothelial cells (HAECs) (73). In patients with symptomatic aortic valve stenosis, modulators of the Wnt signaling pathway, including WIF-1, DKK-1, and sFRP-3, exhibit increased expression in calcified aortic valves, and some modulators positively correlate with valvular calcification, indicating their potential role as biomarkers of aortic valve stenosis (7). Additionally, total levels of β-catenin have been found to be upregulated in human calcified aortic valves (16), calcified valves in a hypercholesterolemic rabbit model (102), and calcified valves in a hypercholesterolemic mouse model (83). This evidence shows the importance of the Wnt canonical pathway in CAVD. Although no evidence has been found directly linking the Wnt/β-catenin pathway and valvular shear stress, the data obtained from HAECs indicate that the abnormal shear stress profile in the fibrosa may regulate this pathway and lead to CAVD.
Notch signaling pathways
Notch is a single transmembrane protein that upon activation, releases a soluble Notch intracellular domain, which translocates to the nucleus and binds to specific sequences in the DNA to regulate gene transcription (9, 39). The Notch signaling pathway plays a critical role in the development and homeostasis of the cardiovascular system (47). While not directly working with the valve endothelium, Theodoris et al. (122) have studied the effect of shear stress in Notch1, a single transmembrane protein of the Notch family, in endothelial cells. Notch1 haploinsufficient (Notch1+/−) and wild-type (Notch1+/+) IPSC-derived ECs were exposed to hemodynamic shear stress. The expression of more than 1000 genes involved in osteogenesis, oxidative stress, and inflammation was dysregulated in the Notch+/− cells, causing an osteogenic and inflammatory phenotype. Interestingly, in the aortic valve, overrepresentation of Notch1 missense variants correlates with increased prevalence of bicuspid aortic valve (BAV) disease in human patients (80). Notch1 in the aortic valve interstitium has been found to repress the activity of Runx2, a transcription regulator of osteoblast cell fate (41), and therefore decreased expression of Notch1 in the aortic valve may lead to accelerated valve calcification. While no direct link is known to relate the disturbed shear stress profile observed at the fibrosa with calcification, the hemodynamic shear stress at the ventricularis may promote Notch1 activity, leading to an antiosteogenic and anti-inflammatory endothelial phenotype and protecting the ventricularis endothelium from dysfunction and calcification.
Nitric oxide signaling and endothelial nitric oxide synthase
Nitric oxide (NO) is a soluble gas with a short half-life (up to 30 s) that is synthetized from the amino acid, L-arginine, by the constitutive calcium–calmodulin-dependent enzyme nitric oxide synthase (NOS) (95), which is considered one of the most importance substances produced in the endothelium as it plays a key role in inflammation, vasodilation, and oxidative stress (142). Decreased NO biosynthesis facilitates vascular inflammation through an increase in lipoprotein oxidation (123).
Endothelial nitric oxide synthase (eNOS) is a shear-sensitive gene, which is upregulated in laminar shear conditions (physiological conditions) and downregulated in low and oscillatory shear conditions (pathophysiological conditions) (94). Bosse et al. (14) studied the role of NO in aortic valve calcification using a coculture system of endothelial cells and aortic valve interstitial cells (AVICs) and found that endothelial cells secrete NO, which is absorbed by AVICs, preventing calcification through regulation of the Notch1 signaling pathway in AVICs. Richards et al. (104) found that in both calcified and noncalcified human aortic valves, the ventricularis side exhibited a threefold higher expression of eNOS compared with the fibrosa. Interestingly, the calcified valves presented lower values of eNOS expression on both sides compared with healthy aortic valves. In the same study, in vitro experiments determined that endothelial secretion of NO decreases myofibroblastic activation, osteoblastic differentiation, and matrix calcification of VICs. El Accaoui et al. (38) have developed a mice model of aortic valve stenosis by knocking down eNOS and found that 30% of these mice had BAVs and that these mice presented fibrosis and calcification at 6 and 18 months of age. To further validate their work, porcine VICs were cocultured with or without valvular endothelial cells and they found that the endothelial cells inhibited profibrotic processes in VICs.
This VIC study shows the importance of eNOS in valvular calcification and provides a clear mechanism linking decreased NO bioavailability to endothelial dysfunction, leading to increased fibrosis and calcification. While NO has been investigated previously as a therapeutic agent, stimulation of endothelial NO production and exogenous NO treatment in the valve still offers an avenue of potential therapy for aortic valve disease.
Reactive oxygen species
Reactive oxygen species (ROS) are partially reduced metabolites of oxygen that possess high oxidizing capabilities and act by damaging DNA and oxidizing lipid and cellular constituents (85). They regulate cell growth, apoptosis, senescence, cell adhesion, and differentiation and are considered key components in inflammatory diseases (43, 120). One of the most important mechanisms of ROS production is the NADPH oxidase complex, which donates an electron to oxygen species in the cell to generate superoxide (12). Both the gene expression and protein levels of NADPH are increased fourfold in oscillatory flow in bovine aortic endothelial cells, which correlates with an increase of oxidized LDL through superoxide (O2 —) modification (55).
Under laminar shear stress, ROS production in human umbilical vein endothelial cells (HUVECs) increases initially and decreases back to baseline levels over time, reflecting the complexity of ROS signaling in the endothelium (26). Both superoxide and hydrogen peroxide expression levels are significantly increased in calcified regions of the valves, whereas noncalcified regions, even in stenosed valves, present similar levels of superoxide and hydrogen peroxide as the tissue of healthy valves (82). ROS signaling, integrated with our understanding of NO, provides an interconnected web, linking the pathological signaling pathways of many mechanical genes expressed in the valve. Finding key components of these pathways may be critical for a full picture of the pathological degeneration of the valve.
ROS play a role further downstream of the endothelium: even in early stages of CAVD, VICs exhibit an accumulation of ROS, suggesting a role in disease development (86, 106, 143). Branchetti et al. (15) collected VICs from patients with and without CAVD and found that cells from stenosed patients possessed ROS-induced DNA damage and impaired DNA repair ability, which could be rescued with antioxidant enzyme treatment. In a thorough study showing the direct role of ROS in the interstitium, Das et al. (33) found that treatments decreasing ROS formation, specifically via decreases in the MAPK-TGF-β pathway, resulted in decreased calcium nodule formation.
Throughout the valve, ROS are important contributors to CAVD, both at the endothelial and interstitial cell layers, and changes in the oxidative state of the valve and the valvular endothelium may contribute significantly to the pathological degeneration of the valve. Further studies into specific species and their regulatory enzymes may be invaluable to determine their validity as therapeutic targets for CAVD.
Interleukins
Interleukins are important inflammatory cytokines released from T lymphocytes, macrophages, monocytes, and endothelial cells, which bind to specific cell receptors and play a role in communication with leukocytes, growth, and cell differentiation (3, 64). Interleukins produced in the endothelium play an important role in several diseases such as atherosclerosis, tumor development, and chronic infections (3, 130). Pathological low shear stress facilitates the increased expression of interleukin-6 (IL-6) and interleukin-1 (IL-1) from the endothelium, causing vascular smooth muscle cells to proliferate in early atherosclerotic plaques (115).
The interleukins also play a role in the valve: IL-6 has been found to be involved in the development and progression of aortic valve disease by affecting the EMT (124). In innovative studies, Mahler et al. (77) utilized a three-dimensional collagen gel to culture porcine aortic valve endothelial cells (PAVECs) and found that PAVECs underwent EMT when treated with IL-6 in a dose-dependent manner, via the Akt/nuclear factor-κβ-dependent pathway. The EMT is a critical step in tissue dysfunction, ultimately leading to valve calcification (133), and thus IL-6 is a potent regulator of the process. Moura et al. (89) conducted a study in patients with asymptomatic moderate to severe aortic stenosis treated with or without rosuvastatin, a statin used to reduce total and LDL cholesterol (75), measuring the expression of IL-6 in the patients before and after the statin treatment. Patients treated with rosuvastatin exhibited a slowed progression of aortic valve stenosis and a fivefold decrease in the serum levels of IL-6, indicating reduced inflammation at the endothelial level. Interleukin-1 is expressed in the endothelium of atherosclerotic plaque and it may be linked to atherogenic inflammation (81). In the frame of aortic valve disease, IL-1 receptor antagonist-deficient mice were studied and it was found that these mice presented an increase of aortic valve thickness compared with wild-type mice. T cells of the IL-1 receptor-deficient mouse were analyzed and they expressed much higher levels of tumor necrosis factor (TNF)-α compared with T cells of wild-type mice; these findings suggest that IL-1 may be inducing inflammation in aortic valve endothelial cells through the TNF-α signaling pathway and this inflammation may play a critical role in development of aortic stenosis (56).
From these studies, in combination with those in vitro, it is clear that abnormal shear stress, as experienced at the fibrosa, increases IL-6, and this interleukin, possibly coupled with others, promotes valve calcification. More studies into how the interleukins may be targeted to change the EMT signature profile may shed light on how to alter endothelial phenotype to alleviate valve calcification therapeutically.
Kruppel-like factors
Kruppel-like factors (KLFs) are members of the zinc finger family of transcription factors, which bind to DNA sequences, including CACC-, GC-, or GT- box elements in promoter and enhancer regions (100). In the vascular endothelium, KLF2 has been widely studied; it is upregulated in high laminar unidirectional shear stress (physiological conditions) and downregulated in low oscillatory shear stress (pathophysiological conditions) (35, 128, 137). In extensive in vitro experiments, human aortic valve endothelial cells (HAVECs) from noncalcified aortic valves exhibited higher KLF2 expression in high unidirectional shear stress (20 dynes/cm2, conditions similar to ventricularis) and lower expression in low and oscillatory shear stress (±5 dynes/cm2, conditions similar to fibrosa), reflecting the fact that the ventricularis side shows higher KLF2 expression and an anti-inflammatory phenotype (50). To study inflammatory markers, including KLF2, in detail, Weinberg et al. (129) created a computational model of the aortic valve hemodynamics to model the exact shear profile experienced by the aortic valve. The simulated shear profiles were then applied to HUVECs, and the expression of KLF2 was found to be significantly higher in the cells that experienced the ventricularis shear profile. In microarray studies by our group, porcine aortic valve fibrosa shows decreased expression of KLF2 and KLF4, indicating their importance in valvular endothelium just as in the vasculature (50). These initial studies may lead to important insights for these anti-inflammatory markers in valvular function and endothelial cell phenotype.
KLF4 is another member of the KLF family that when overexpressed, induces anti-inflammatory and antithrombotic factors such as thrombomodulin and endothelial nitric oxide synthase, whereas knockdown of KLF4 induces inflammation (45, 148). More importantly, KLF4 is known to be upregulated in vascular endothelial cells in laminar shear stress compared with oscillatory shear stress (126, 131). So far, the role of this KLF protein in aortic valve calcification is unknown; however, a study conducted by Maleki et al. (79) analyzed patients with BAV, who are known to develop aortic valvular stenosis more rapidly than patients with tricuspid aortic valve (TAV), finding a significant decrease in KLF4 expression in the aortic region near the valve in BAV patients compared with the TAV patients. The authors postulate that the disturbed flow generated by the BAV is the primary contributor to this decreased KLF4 expression. Therefore, we expect that the same abnormal shear profile will be experienced by the valve and would cause a decrease of KLF4 with the corresponding increase in inflammatory endothelial response.
The KLFs are important protective anti-inflammatory mediators in the valvular endothelium. Therapies developed to increase these factors, possibly along with the many other factors discussed in this review, will have a significant impact on the maintenance of physiological valve function and health.
microRNAs
microRNAs (miRNAs) act as suppressors of protein expression by targeting transcription. miRNAs are small nucleotide sequences up to ∼22 bases in length; their binding to the 3′ untranslated region of mRNA leads to the degradation of that mRNA or inhibition of protein translation (44). miRNAs have been implicated in most diseases, either preserving physiological cell function or promoting pathological signaling and dysfunction. In cardiovascular diseases, major miRNAs have been thoroughly cataloged and analyzed; exciting miRNA targets have been found in atherosclerosis (70, 90), heart failure (63), diabetes (88), and hypertension (91).
Despite their characterization in other cardiovascular systems, miRNAs have not been well characterized in the aortic valve. In the interstitium, miRNA-30b was shown to prevent the signaling necessary for interstitial cells to transform into calcifying cells (146). In addition, miRNA-141 targets TGF-β and BMP-2 signaling, which are critical for interstitial osteoblastic differentiation leading to calcification of the tissue (138). Our group has analyzed miRNAs in the endothelium, and we found a novel family of shear-sensitive miRNAs, which change expression under different shear stress conditions (70). As our recent review indicates, the many studies linking disturbed flow to cardiovascular complications provide important information for other diseases involving laminar and oscillatory flow, including aortic valve stenosis and calcification.
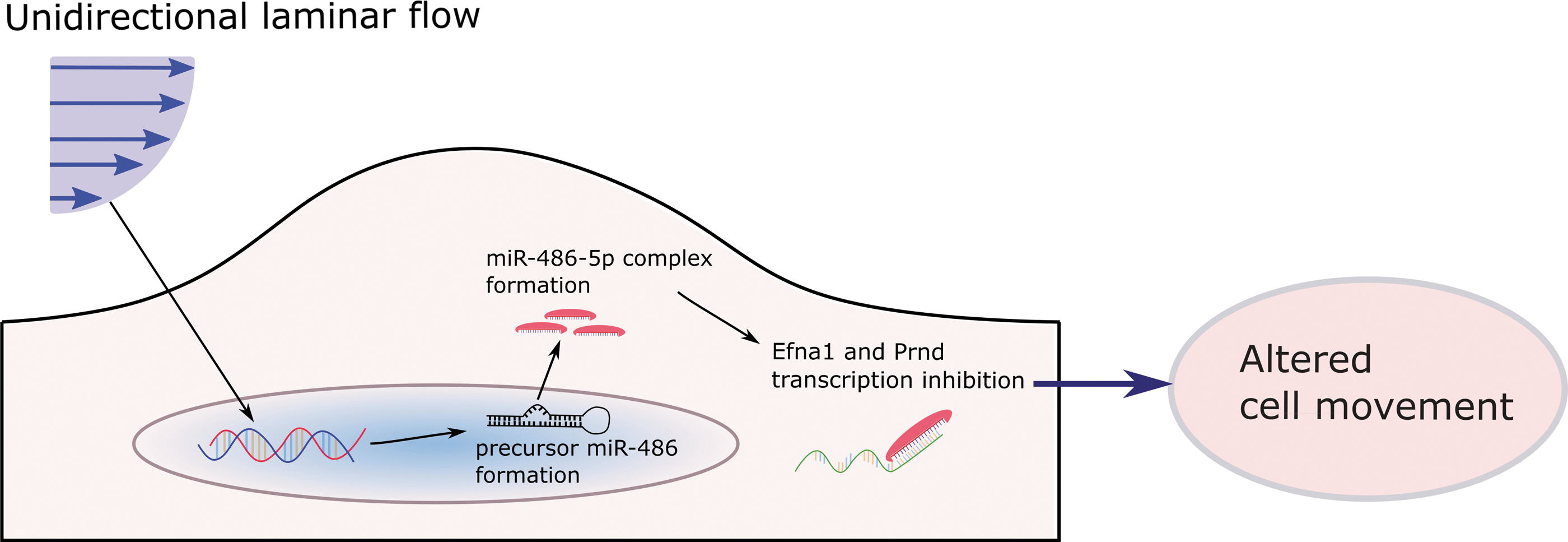
In recent studies, we have performed an HAVEC microarray to find novel miRNAs differentially expressed in the aortic valve endothelium and which are sensitive to disturbed flow (Figure 5). We have discovered many interesting potential targets for therapy, including miRNA-486-5p, which exhibited one of the most dramatic changes in expression between different flow conditions.

In preliminary studies, we have found that miRNA-486-5p in the aortic valve endothelium enhances cell movement in response to shear stress to alter cell phenotype (48, 99). The study conducted in our laboratory (149) showed that Efna1 and Prnd are directly targeted by miR-486-5p. Cell migration was significantly enhanced in HAVECs overexpressing miRNA-486-5p (see Figure 6 for a representation of this potential mechanism). This work reveals the power of microarray to piece together miRNAs, their targets, and ultimate cell fate. However, more work must be performed to confirm these links and their potential in treatment of valve disease. Our group and others have made great strides in the study of miRNAs in the valve. Studies investigating their potential in treatment of valvular dysfunction and stenosis are ongoing and they may reveal insights that provide dramatic benefits for valvular function. These exciting results are only the beginning of our investigation into the role of miRNAs in the aortic valve endothelium; many more potential targets are sure to lead us to a clearer understanding of the disease in the future.
Future Perspectives in CAVD Research and Treatment
Work in aortic valve mechanics and flow-mediated signaling has rapidly advanced in recent years; however, viable options for nonsurgical treatment of CAVD remain elusive. Future research should translate findings in the vascular endothelium to research in the valve, which is a much clearer example of biomechanical force. The aortic valve is a complex machine and an elegant regulator of systemic blood flow. With the latest technological advances in computer and animal models, we are closer than ever to treating and preventing aortic valve stenosis and calcification.